

Abstract
Human tissue kallikrein (KLK) proteases are hormone-like signaling molecules with important functions in cancer pathophysiology. KLK-related peptidase 6 (KLK6), specifically, is highly up-regulated in several types of cancer, where its increased activity promotes cancer invasion and metastasis. This characteristic suggests KLK6 as an attractive target for therapeutic interventions. However, inhibitors that specifically target KLK6 have not yet been reported, possibly because KLK6 shares a high sequence homology and structural similarity with other serine proteases and resists inhibition by many polypeptide inhibitors. Here, we present an innovative combinatorial approach to engineering KLK6 inhibitors via flow cytometry–based screening of a yeast-displayed mutant library of the human amyloid precursor protein Kunitz protease inhibitor domain (APPI), an inhibitor of other serine proteases, such as anionic and cationic trypsins. On the basis of this screening, we generated APPIM17L,I18F,S19F,F34V (APPI-4M), an APPI variant with a KLK6 inhibition constant (Ki) of 160 pm and a turnover time of 10 days. To the best of our knowledge, APPI-4M is the most potent KLK6 inhibitor reported to date, displaying 146-fold improved affinity and 13-fold improved proteolytic stability compared with WT APPI (APPIWT). We further demonstrate that APPI-4M acts as a functional inhibitor in a cell-based model of KLK6-dependent breast cancer invasion. Finally, the crystal structures of the APPIWT/KLK6 and APPI-4M/KLK6 complexes revealed the structural and mechanistic bases for the improved KLK6 binding and proteolytic resistance of APPI-4M. We anticipate that APPI-4M will have substantial translational potential as both imaging agent and therapeutic.
Keywords: protein engineering, directed evolution, proteolysis, protease inhibitor, X-ray crystallography, cell invasion, enzyme inhibition, selective binding, yeast surface display
Introduction
An important family of serine proteases, implicated in various processes in both health and disease, is the family of human tissue kallikreins (KLKs)4 (1). The 15 serine proteases in this family are involved in the tissue-specific proteolysis of numerous endogenous substrates (1–3) and are considered major regulatory proteases with key signaling properties, acting either individually or as part of other proteolytic cascades (4). KLKs can also be employed clinically as markers for the diagnosis and prognosis of cancer (5) and as targets for therapeutic intervention; KLK6, specifically, has been shown to be a key factor in driving the invasion and migration of cancer cells (6–8).
Several independent studies reported that KLK6 expression is associated especially with a shorter overall survival and/or progression/disease-free survival in colon, gastric, lung, and ovarian cancers (9–18). In ovarian cancer, for example, accumulating data suggest that KLK6 is responsible for increased E-cadherin shedding, which in turn increases metastasis and ascites in epithelial ovarian cancer (7, 19, 20). Similarly, in colon cancer, in which KLK6 can be used for both prognosis and diagnosis, it induces the invasiveness and proliferation of cancer cells by down-regulating E-cadherin, thereby interrupting E-cadherin–mediated cell–cell adhesion, a prerequisite for tumor invasiveness and metastasis (14, 16, 21). Moreover, KLK6 appears to be relevant for breast cancer, as high KLK6 expression levels were recently shown to promote the invasiveness of breast cancer cells; KLK6 serum levels were found to be significantly higher in patients with invasive breast cancer, and patients with high KLK6 expression showed poorer survival rates (22–24).
Several studies have investigated how KLK6 is involved in the pathogenesis and metastasis of various types of cancer (21, 24, 25). For instance, KLK6 has been shown to be involved in the progression of melanoma, where it triggers tumor cells and tumor-associated parenchymal cells in the microenvironment of the tumor through the proteolysis and activation of the proteinase-activated receptor PAR1, which leads to the acquisition of a malignant phenotype by facilitating tumor cell invasion and metastasis (8, 26, 27). In addition, in a nonsmall cell lung cancer model system, KLK6 has been shown to promote the proliferation of lung tumor cells, and this effect was facilitated by KLK6 cleavage and activation of PAR2 (28). By degrading various proteins in the extracellular matrix (29), KLK6 has also been shown, in vitro, to increase the metastatic potential of tumor cells (29, 30); concomitantly, both in vitro (6–8) and in vivo (7, 31) studies have established a correlation between the ectopic expression of KLK6 and an increase in tumor proliferation and migration, whereas an RNAi-induced KLK6 suppression reduces the cell growth, proliferation, and invasive potential of tumors (14, 21). These data underlie KLK6 as an attractive target for novel molecules that could inhibit its proteolytic activity as a treatment for various cancers (32–34). Finally, numerous studies indicated that KLK6 may also be involved in the pathogenesis of various neurodegenerative diseases, including Alzheimer's disease, Parkinson's disease, multiple sclerosis, and others (35–41). For example, raising neutralizing KLK6 antibodies in mice exerted overall beneficial effects in models of multiple sclerosis and central nervous system inflammatory disease, suggesting that selectively inhibiting KLK6 is not expected to be considerably toxic.
Despite the numerous studies on human serine proteases, KLK inhibitors are still much sought-after (42–45). As the KLK family comprises 15 different members, many of which contain structurally similar binding pockets, the discovery of either natural or synthetic subtype-selective KLK inhibitors is a challenging task. KLK6 is structurally similar to other members of the KLK family and to other human trypsin-like proteases (46, 47), rendering its specific targeting difficult to achieve. An additional challenge in developing KLK6 inhibitors is its resistance to inhibition by polypeptide serine protease inhibitors, according to several prior reports (48, 49). Indeed, to the best of our knowledge, to date, highly potent, selective, and stable KLK6 inhibitors have not been reported.
Recently, we employed a combinatorial engineering approach that has enabled us to transform the human amyloid precursor protein Kunitz protease inhibitor domain (APPI) into a highly potent inhibitor of mesotrypsin, another member of the human serine protease family. Importantly, the engineered APPI showed a high selectivity toward mesotrypsin (as compared with other serine proteases), whereas the native APPI (APPIWT) bound nonspecifically to various serine proteases, including KLK6, and was susceptible to rapid cleavage and inactivation by mesotrypsin (50). We have previously found that the specificity of APPI toward serine proteases is largely directed by the sequence of its canonical binding loop, whereas its proteolytic stability is strongly influenced by residues buried within its scaffold (51, 52). These findings suggest that both the affinity and the proteolytic stability of APPI can be optimized by using combinatorial screening methods, so as to generate high affinity and stable KLK6 inhibitors based on the protein scaffold of the human APPI.
In this study, we employed the powerful yeast surface display (YSD) technology (53, 54) to screen combinatorial libraries of APPI variants containing mutations in both the active site and the scaffold. Using these libraries, we engineered an APPI that, as compared with its WT counterpart, demonstrates a 14-fold increase in proteolytic stability and a 146-fold higher binding affinity to KLK6, without increasing its binding affinity toward other KLK proteases. These optimized properties were the result of four mutations (two in the binding loop and two in the scaffold), which enabled the engineered APPI variant (termed here APPI-4M) to potently block the invasiveness of KLK6-dependent cancer cells in cell-based assays, in which a high binding affinity and an exceptional protease stability are crucial. Finally, by solving the crystal structure of both the APPIWT/KLK6 and APPI-4M/KLK6 complexes, we elucidated the molecular basis of the improved binding affinity and proteolytic resistance of APPI-4M. This novel KLK6 inhibitor offers promising avenues for cancer therapy and imaging applications, as well as an extremely valuable laboratory reagent for deciphering the specific mechanisms by which KLK6 drives cancer progression.
Results
Constructing and screening the APPI library
To develop APPI variants with a strong affinity to the human KLK6, we used the YSD screening platform (53). We cloned the coding region of APPIWT into a YSD plasmid to present it on the surface of the Saccharomyces cerevisiae yeast fused with the Aga2p/Aga1p system (Fig. 1, A–C). In the YSD plasmid, we placed APPIWT between the Aga2 protein and the C-terminal c-Myc epitope tag and verified the expression of APPIWT by detecting the c-Myc epitope using FACS (Fig. 1D). We first verified the ability of the presented APPIWT to bind a soluble, fluorescently labeled KLK6 (Fig. 1D). Next, we designed two APPI-based libraries, cloned these libraries into the YSD vector, and transformed them into yeast through homologous recombination (see the “Experimental procedures” for details); in one library, termed the APPIWT library (or “random library”), we constructed a randomly mutated version of the APPIWT gene. Random mutagenesis in the entire APPI sequence resulted in an APPI library with 1–3 mutations per clone, throughout the entire APPI sequence, yielding a library size of about 9 × 106 clones. We constructed the second library based on our previously published sequence of APPIM17G,I18F,F34V (50), a clone that demonstrates a superior proteolytic stability to that of APPIWT. We generated this APPIM17G,I18F,F34V-based library using a PCR assembly of NNS-containing oligos combined with an error-prone PCR. The combination of these two mutagenesis strategies in the APPIM17G,I18F,F34V library yielded a library (termed “focused library”) with an average mutagenesis rate of 1–2 amino acid mutations per the overall 56 residues of APPI, with one mutation in the binding loop (positions 11–18, excluding Cys at position 14; Fig. 1C) and the other either within or outside of the binding loop, yielding an experimental library of about 3.5 × 106 different clones. A sequence analysis of the focused library revealed that, as designed, 79% of the sequences contained a single mutation within the canonical binding loop, 7% contained two mutations within this loop, 10% contained a single mutation within the loop and one mutation outside of the loop (a scaffold mutation), and only 1% did not contain any mutation.
Figure 1.

FACS analysis of the different YSD APPI libraries, showing expression and KLK6 binding. A, YSD vector (pCTCON-2) is aligned with the general scheme of the insert: the insert gene consists of the APPI gene, which is flanked by two restriction sites (BamHI and NheI), followed by a linker sequence (LPDKPLAFQDPS) on the 3′ end, along with two pCTCON homologous sequences. B, APPI is displayed on the yeast cell surface as a translational fusion to Aga2p, which is linked to Aga1p by two disulfide bonds. Surface expression is detected by using fluorescence-labeled antibodies binding to the C terminus of the c-Myc epitope tag, whereas target binding is detected using fluorescence-labeled KLK6, via FACS. C, 3D structure of APPI (PDB code 1ZJD). D, expression of APPI and the binding of 10 nm labeled KLK6 were determined in a YSD system for an APPIWT clone, for a library of mutants based on APPIWT, and for a library of mutants based on APPIG17M,I18F,F34V.
As shown in Fig. 1D, the two yeast-displayed APPI libraries were well-expressed and bound KLK6 effectively. The flow-cytometry results indicated that the binding distribution of the APPIWT clone to KLK6 was similar to that of clones in the APPIWT-based library. In contrast to the APPIWT-based random library, the APPIM17G,I18F,F34V-based library (focused library) showed a more diverse binding pattern and displayed clones with various levels of binding to KLK6. Because the focused library showed greater diversity in KLK6 binding, we chose this library for further research.
As a first step toward isolating APPI variants with a high affinity to KLK6, we pre-sorted the focused library to isolate cells with a high-expression signal (Fig. 2A, post S0). Then, we propagated and re-sorted the isolated, high-expressing cells, this time in the presence of a fluorescently-labeled KLK6. We used a diagonal sort gate to select the cell population with a high affinity to KLK6 relative to APPI expression (as assessed by detecting the c-Myc tag), which composed 3.7% of the initial library (Fig. 2A). Such a selection protocol allowed us to decrease the bias for selecting variants with high-expression levels but possibly a low-binding affinity. To isolate clones with the highest intrinsic affinity to KLK6, we repeated this process such that, in each round of sorting, the KLK6 concentration was reduced to increase the selective pressure in each subsequent round. After each sorting round, we analyzed the expression and KLK6-binding levels by using flow cytometry (Fig. 2A, post S1–3). After the final sorting round (S3), this process yielded the sequences of 17 clones, of which eight sequences (47%) contained the mutation G17L within the binding loop; of these, four contained only the G17L mutation and four contained an additional scaffold mutation (S19F). The two most abundant clones in the sequence results, which we chose to use for further individual analyses, were a clone that comprised the single mutation G17L (termed APPIM17L,I18F,F34V) and APPI-4M, a clone that comprised two mutations (G17L and S19F) relative to APPIM17G,I18F,F34V. We used FACS to analyze the ability of these yeast-displayed clones to bind KLK6 and compared their binding with that of the yeast-displayed APPIWT and APPIM17G,I18F,F34V clone (Fig. 2B). This analysis revealed that the binding affinity of both APPIM17L,I18F,F34V and APPI-4M to KLK6 was superior to that of APPIWT and APPIM17G,I18F,F34V (Fig. 2B). Accordingly, we chose these clones for further analysis as purified soluble proteins.
Figure 2.
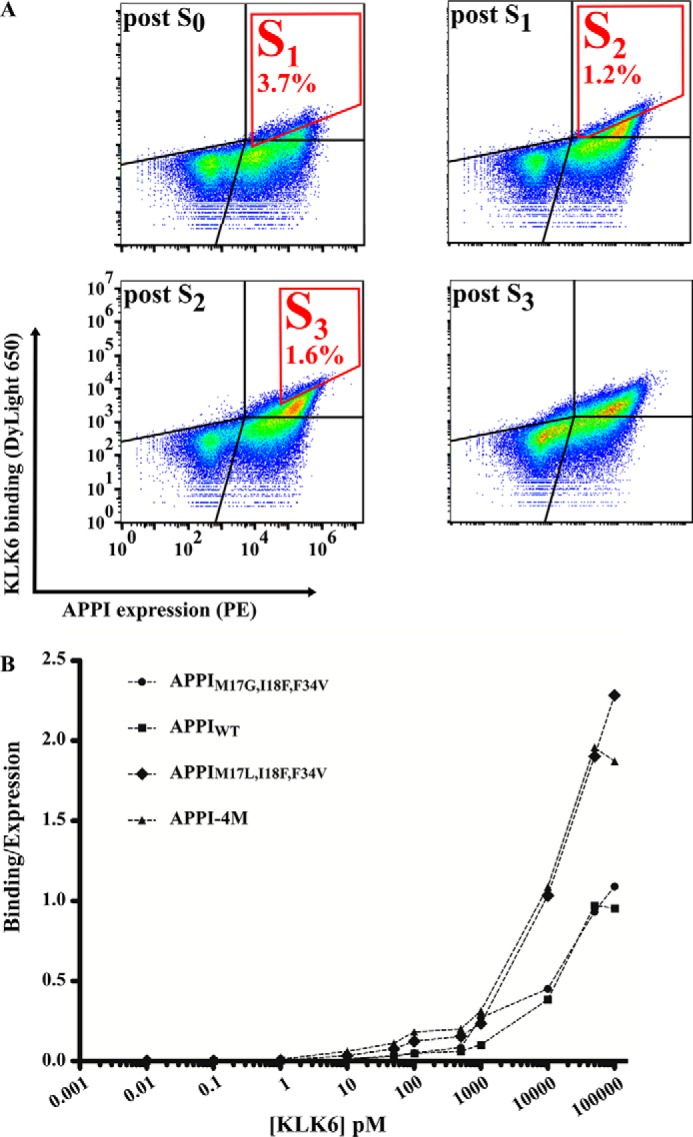
Affinity maturation of the APPI library. A, to enrich the APPI library for high-affinity KLK6 binders, four sequential rounds of FACS sorting were conducted (S1–S3 marked in red). The results of the analysis after each sorting round are presented. S0 represents an initial sorting round intended only for selecting a cell population with high APPI expression, regardless of KLK6 binding. In S1, 500 pm DyLight-650–labeled KLK6 was used to select cells with a high APPI expression and a high KLK6 binding, which comprised 3.7% of the entire cell population. In S2, 50 pm DyLight-650–labeled KLK6 was used to select cells with a high APPI expression and a high KLK6 binding (gated in red), which comprised 1.2% of the entire cell population. In S3, 50 pm DyLight-650–labeled KLK6 was used to select cells with a high APPI expression and a high KLK6 binding, which comprised 1.6% of the entire cell population. B, FACS binding titration curve of KLK6 to yeast cells expressing APPIWT or its three variants. A leftward shift in the curve indicates a higher affinity. The binding signal of KLK6 is normalized to the expression signal of the APPIs.
APPI-4M confers a high affinity to KLK6 and resistance to KLK6-mediated hydrolysis
To further characterize the APPI variants APPIM17L,I18F,F34V and APPI-4M, we expressed and purified the soluble forms of both proteins, as well as the control APPIWT and APPIM17G,I18F,F34V proteins (Fig. 3A). We expressed these proteins in Pichia pastoris as described previously (50), measured the dissociation constant (KD) of the most potent variant, APPI-4M, and compared it with that of APPIWT by using surface plasmon resonance (SPR). The KD value for APPIM17L,I18F,F34V could not be measured because there was not a good fit for the SPR data. The KD value for APPI-4M was 36 pm, which was 145.7-fold lower (i.e. better) than that of APPIWT (5290 pm) (Fig. 3B and Table 1). This improvement was due to both an increase in the association rate (kon = (3.17 ± 0.01) × 105 m−1 s−1 for APPI-4M versus (1.96 ± 0.01) × 105 m−1 s−1 for APPIWT) and, more significantly, a 90-fold decrease in the dissociation rate (koff = (1.15 ± 0.06) × 10−5 s−1 for APPI-4M versus (104 ± 0.20) × 10−5 for APPIWT).
Figure 3.

Binding and inhibition kinetics of KLK6 by APPIWT and APPI-4M. A, purification of soluble APPI variants. Left, size-exclusion chromatography for APPI-4M. Arrows indicate correlations between the elution volume and size, according to known standards. Right, SDS-PAGE analysis of the purified APPI variants on a 15% polyacrylamide gel under reducing conditions. B, surface plasmon resonance binding experiment, in which 2 μg of either APPIWT (top panel) or APPI-4M (lower panel) was mounted as the ligands on a GLC chip. Six KLK6 concentrations (0–10 nm; represented by different colors) were used as the analytes. C, slow tight–binding inhibition of KLK6 catalytic activity by APPIWT (left, 1 nm KLK6) and APPI-4M (right, 100 nm KLK6). The Ki value of the reaction was calculated by using Equation 1 (see under “Experimental procedures”). V0 represents the uninhibited rate, and Vi represents the rate in the presence of APPI. In the APPI-4M plot, the dashed regression line indicates the slope of the inhibition by APPIWT. D, formation of complexes between KLK6 and APPI-4M. The SDS-PAGE was performed under reducing conditions and with KLK6/APPI molar ratios of 1:0, 1:1, 1:5, and 1:10. Note the formation of stable, higher molecular weight complexes in lanes loaded with KLK6/APPI-4M but not with KLK6/APPIWT complexes.
Table 1.
Inhibition, binding, and hydrolysis constants of APPIWT and APPI-4M
| APPI variant | Ki a | KDb | konb | koffb | Turnover timec |
|---|---|---|---|---|---|
| nm | nm | m−1 s−1 | s−1 | h | |
| APPIWT | 2.24 ± 0.11d | 5.29 | (1.96 ± 0.01) × 105 | (10.40 ± 0.02) × 10−4 | 16.62 ± 0.05 |
| APPI-4M | 0.16 ± 0.06 | 0.04 | (3.17 ± 0.01) × 105 | (1.15 ± 0.06) × 10−5 | 230.07 ± 40.90 |
a Data were determined by slow tight binding inhibition assays.
b Data were determined by SPR.
c Data were determined by HPLC.
d Data were previously reported in Ref. 50.
We next determined the inhibition constants, Ki, using a method appropriate for quantifying tight, slow-binding inhibition in KLK6 assays versus the peptide substrate BOC-FSR-AMC (49), according to Equation 1 (see under “Experimental procedures”). Both the APPIM17L,I18F,F34V and the APPI-4M variants demonstrated a considerably improved inhibition activity compared with APPIWT and APPIM17G,I18F,F34V (Fig. 3C), with Ki values of 326.1 ± 5.6 pm for APPIM17L,I18F,F34V and 160.1 ± 59.2 pm for APPI-4M, as compared with 2240 ± 110 pm for APPIWT and 1190 ± 120 pm for APPIM17G,I18F,F34V. Thus, APPIM17L,I18F,F34V and APPI-4M showed a 7- and a 14-fold improvement in inhibiting KLK6, respectively, as compared with the APPIWT (Table 1).
To further evaluate the stability of the APPI-4M/KLK6 interaction, we employed an SDS-PAGE analysis. Surprisingly, we found that, unlike the APPIWT/KLK6 complex, the APPI-4M/KLK6 complex is resistant to dissociation under reducing, denaturing conditions (Fig. 3D). The higher molecular weight band in APPI-4M/KLK6 incubations versus the APPIWT/KLK6 incubations describes the formation of a stable covalent complex, which may indicate the formation of a more thermodynamically stable acyl–enzyme intermediate (see “Discussion”).
To test whether the APPI-4M variant is indeed more resistant to KLK6 proteolysis than APPIWT, we incubated each protein with KLK6 (90 and 300 h for APPIWT and APPI-4M, respectively), and we used an HPLC-based time course hydrolysis assay to measure the concentration of intact APPI over time, and from that, we calculated the turnover time (1/kcat) of each protein (Fig. 4 and Table 1). This analysis revealed that APPI-4M is 13-fold more proteolytically stable than APPIWT (with turnover times of 230.07 ± 40.90 and 16.62 ± 0.05 h, respectively, Table 1), such that, over the 90-h incubation period, APPIWT shifted from mostly intact to mostly cleaved, whereas APPI-4M showed only a negligible change.
Figure 4.
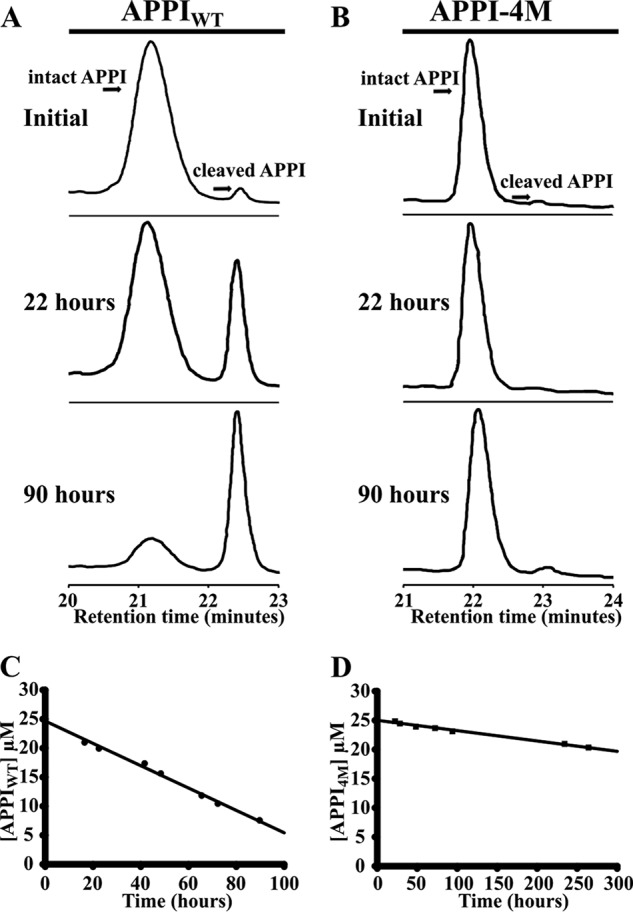
Hydrolysis of APPIWT (A and C) and APPI-4M (B and D) by KLK6, as determined by HPLC. The depletion of intact APPI over time was quantified by integrating the HPLC peak, which represents the intact APPI variant, at eight different time points (examples shown in A and B), and then determining the rate of depletion by linear regression (C and D). APPI concentrations were 25 μm for each variant, and the molar ratio of KLK6 to APPI was 1:8.
APPI-4M is more potent than APPIWT toward KLK6, but not toward other serine proteases
To evaluate the potency of APPI-4M toward KLK6, we quantified the inhibition constants of both APPIWT and APPI-4M toward KLK6 and toward three other structurally and functionally similar serine proteases, including anionic trypsin, cationic trypsin, and coagulation factor XIa (FXIa) (Table 2). As compared with APPIWT, APPI-4M demonstrated both a 14-fold increase in inhibition potency toward KLK6 and a weaker inhibition potency toward the other serine proteases (namely, a 40-, a 27-, and a 1.7-fold weaker inhibition potency toward anionic trypsin, cationic trypsin, and FXIa, respectively). Moreover, using SPR, we measured the binding affinities of the two APPI variants to KLK6, as compared with their binding to KLK1, KLK2, KLK3, KLK4, and KLK5. This analysis revealed that the affinity of APPI-4M toward KLK6 was ∼146-fold higher (i.e. a lower KD) than that of APPIWT, whereas its affinity toward the other KLKs was considerably lower and either did not significantly differ or was only marginally better than that of APPIWT (Table 3).
Table 2.
Inhibition (Ki) of APPIWT and APPI-4M toward various human serine proteases
| KLK6 | Anionic trypsin | Cationic trypsin | FXIa | |
|---|---|---|---|---|
| pm | pm | pm | pm | |
| APPIWT | (2240 ± 110) | (1.74 ± 0.05) | (6.27 ± 1.01) | (410 ± 14) |
| APPI-4M | (160 ± 60) | (69.8 ± 8.0) | (168 ± 17.5) | (724.8 ± 33.7) |
| Fold difference | 14 | 0.025 | 0.037 | 0.58 |
Table 3.
Binding affinity (measured by SPR) of APPIWT and APPI-4M toward kallikreins
ND means binding was not detected; NA means not available.
| APPI-KLK | KD | kon | koff |
|---|---|---|---|
| nm | m−1 s−1 | s−1 | |
| APPIWT–KLK6 | 5.29 | (1.96 ± 0.01) × 105 | (104 ± 0.20) ± 10−5 |
| APPI-4M–KLK6 | 0.04 | (3.17 ± 0.01) × 105 | (1.15 ± 0.06) × 10−5 |
| APPIWT–KLK1 | ND | NA | NA |
| APPI-4M–KLK1 | ND | NA | NA |
| APPIWT–KLK2 | 0.55 | (2.59 ± 0.05) × 104 | (1.41 ± 0.02) × 10−5 |
| APPI-4M–KLK2 | 0.44 | (2.53 ± 0.04) × 104 | (1.10 ± 0.02) × 10−5 |
| APPIWT–KLK3 | ND | NA | NA |
| APPI-4M–KLK3 | ND | NA | NA |
| APPIWT–KLK4 | 10.08 | (3.81 ± 0.05) × 104 | (4.13 ± 0.02) × 10−4 |
| APPI-4M–KLK4 | 2.34 | (7.39 ± 0.04) × 104 | (1.73 ± 0.02) × 10−4 |
| APPIWT–KLK5 | 3.24 | (1.31 ± 0.02) × 104 | (4.26 ± 0.01) × 10−5 |
| APPI-4M–KLK5 | 1.97 | (3.46 ± 0.05) × 104 | (6.80 ± 0.12) × 10−5 |
APPI-4M inhibits KLK6-mediated cell invasion and migration
To determine the ability of APPI-4M to inhibit the KLK6-dependent invasive behavior of cancer cells, we used human BT-20 cells, a metastatic breast cancer cell line that secretes high levels of KLK6 and low levels of other KLKs (29). Notably, BT-20 cells previously demonstrated a reduction in cell invasiveness in the presence of an anti-KLK6 antibody in a Boyden chamber assay (29), in which the cells migrate through a Matrigel-coated membrane and are visualized by light microscopy. Using this assay, we found that BT-20 cells treated with 10, 50, or 100 nm APPI-4M showed a significant decrease in invasiveness (namely a 20, 24, and 37% decrease, respectively) in the number of cells that migrated through the Matrigel. In contrast, cells treated with APPIWT showed no significant decrease in invasiveness, as compared with untreated control. Cells transfected with a KLK6-specific siRNA showed a 50% decrease in the number of cells that migrated through the Matrigel (Fig. 5, A–C) compared with a scrambled siRNA. Importantly, in an XTT viability assay (Fig. 5D), APPI-4M did not affect the viability of BT-20 cells, indicating that the observed decrease in the number of invading cells in the Boyden chamber assay was not due to cell death but rather was due to the ability of APPI-4M to inhibit the invasiveness of the BT-20 cells.
Figure 5.

APPI-4M decreases the invasiveness of BT-20 breast cancer cells, but not their viability. The invasive ability of BT-20 cells in the presence of APPI-4M was evaluated by using a Matrigel-coated Boyden chamber. The cells that successfully invaded the Matrigel 36 h after plating were stained and quantified. A, representative images of the invading cells treated with either buffer (vehicle), APPIWT, or APPI-4M (100 nm). siRNA-transfected cells were used as a control. B, reverse-transcriptase PCR (RT-PCR) analysis was conducted in BT-20 breast cancer cells that were transfected with KLK6 siRNA. After the cells were transfected with siKLK6 or with a small-interfering control (scrambled siRNA) for 48 h, whole RNA was extracted, and the RT-PCR was performed. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. C, quantification of invading cells. C is control (scrambled siRNA); K is KLK6–siRNA. Bars represent means ± S.D. of three biological repetitions. D, quantification of cell viability using the XTT assay, 36 h after incubation with APPIWT or APPI-4M. Bars represent means ± S.D. of three biological repetitions. E, quantification of migrating cells (see F), normalized to untreated cells (control). Bars represents the mean (± S.D.) of triplicate experiments. *, p < 0.05; **, p < 0.01 (Student's t test, as compared with untreated control). F, BT-20 cells were scratched by removing a strip across the well. Then, the cells were treated with APPI-4M for 24 h. The area free from cells was counted.
To further examine the ability of APPI-4M to inhibit the migration of BT-20 cells, we used a wound-healing assay, in which we estimated the percentage of recolonization of the scratched surface 24 h after cell wounding (Fig. 5, E and F). Treating the cells with APPI-4M (100 nm) significantly inhibited their migration, as compared with untreated cells.
Crystal structure of the APPI-4M/KLK6 complex reveals optimized interactions
To elucidate the mechanism of binding and inhibition of APPI-4M to KLK6, and to compare it with that of APPIWT, we crystallized each inhibitor in complex with KLK6 and solved the crystal structures. Data collection and refinement statistics are reported in Table 4. The crystal structure of KLK6/APPIWT was solved from a crystal that diffracted with an effective resolution of 1.95 Å (see under “Experimental procedures” for more information). The crystal structure of KLK6/APPI-4M was solved from a crystal that diffracted to a maximum resolution of 2.3 Å. The co-crystallization of KLK6 with APPIWT or with the APPI-4M variant produced a crystal form containing one molecule of KLK6 bound to a molecule of APPI, which occupied the active site in the classic inhibitory binding mode of canonical or Laskowski-mechanism inhibitors (55, 56), in which the binding loop of the inhibitor occupies (roughly) the S3–S3′ substrate-binding subsites in a substrate-like manner (Fig. 6A). The crystal lattice asymmetric unit also contained an additional molecule of APPI that had undergone proteolysis of the reactive site Arg-15–Ala-16 bond (Fig. 6A); this cleaved form, designated APPI*, is composed of two protein chains connected by two disulfide bonds. In the cleaved APPI* molecule, a nearly 180° rotation in the Cys-14 ψ angle (relative to the intact APPI molecule) allows the formation of a salt bridge between the Arg-15 side chain and the Asp-203 residue of KLK6, thus stabilizing the crystal lattice, whereas the Cys-14 carbonyl forms a hydrogen bond with the N terminus of Ala-16 (Fig. 6B). Notably, crystals of the APPIWT/KLK6 complex grew over the course of 7 weeks, whereas crystals of the APPI-4M/KLK6 complex did not appear until 6 months after the initial setup of crystallization drops. Given the apparent requirement for a stoichiometric equivalent of cleaved APPI for formation of the crystal lattice, it is likely that the extended duration required to form the APPI-4M/KLK6 complex crystal reflects its much greater resistance to proteolysis.
Table 4.
Crystallographic and refinement statistics
Values in parentheses correspond to the highest resolution shell of the data. r.m.s.d. is root mean square deviation. Imposing a resolution cutoff of 1.95Å for the KLK6/APPIWT data, results in 1.44 I/σI, multiplicity of 12.8 completeness of 99.07% and CC1/2 of 0.747 in the highest resolution shell.
| KLK6/APPIWT | KLK6/APPI-4M | |
|---|---|---|
| Data collection | ||
| X-ray source | ESRF ID30B | ESRF ID23-1 |
| Wavelength (Å) | 0.969 | 0.976 |
| Dmin (Å) | 46.10–1.85 (1.92–1.85) | 46.5–2.3 (2.4–2.3) |
| Space group | P212121 | P212121 |
| Cell dimensions | ||
| a, b, c (Å) | 59.543, 77.702, 92.207 | 58.058, 77.752, 91.130 |
| α, β, γ (°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Total reflections | 73,717 | 37,715 |
| Unique reflections | 36,867 | 18,865 |
| Rpim (%) | 3.12 (64.19) | 5.26 (35.21) |
| Completeness (%) | 99.52 (96.37) | 99.14 (95.6) |
| I/σI | 13.48 (1.08) | 8.51 (2.13) |
| Multiplicity | 2.0 (2.0) | 2.0 (2.0) |
| CC½ | 0.999 (0.574) | 0.996 (0.760) |
| Refinement | ||
| Refinement resolution (Å) | 46.10–1.85 | 46.520–2.296 |
| Total reflections used | 36,851 | 18,856 |
| r.m.s.d. bond lengths (Å) | 0.006 | 0.007 |
| r.m.s.d. bond angles (°) | 0.87 | 0.91 |
| Ramachandran plot | ||
| Favored (%) | 97.83 | 98.14 |
| Outliers (%) | 0.00 | 0.00 |
| Rwork/Rfree (%) | 18.48/22.60 | 17.47/22.56 |
| Protein atoms | 2565 | 2555 |
| Solvent molecules | 195 | 142 |
| Average B-factor (Å2) | 42.03 | 34.16 |
| Protein | 41.81 | 34.1 |
| Solvent | 44.82 | 35.25 |
| Wilson B factor (Å2) | 28.97 | 28.31 |
| PDB codes | 5NX1 | 5NX3 |
Figure 6.

Crystal structure reveals an inhibited KLK6/APPI-4M complex and a cleaved product APPI-4M*. A, co-crystal structure of KLK6 with the APPI-4M variant shows one molecule of KLK6 (gray) bound to a molecule of APPI-4M occupying the active site in the inhibitory mode (cyan). The crystal structure also reveals an additional APPI-4M molecule that has undergone proteolysis at Arg-15–Ala-16 to yield the product APPI-4M*, which is comprised of two protein chains (yellow and orange) connected by two disulfide bonds. B, cleaved APPI-4M* molecule reveals a large rotation of the Cys-14 ψ angle (relative to the intact APPI-4M), allowing the formation of a salt bridge between the Arg-15 side chain and the Asp-203 residue of KLK6, thus stabilizing the crystal lattice, whereas the Cys-14 carbonyl forms a hydrogen bond with the Ala-16 N terminus. C, Arg-15 of the intact APPI-4M was observed in both the expected “up” conformation, in which it forms a salt bridge with KLK6 Asp-189 (dotted yellow lines), and a “down” conformation, which is similar to that previously reported in the superposed structure of APPI (green) bound to chymotrypsin (white) (PDB code 1CA0).
Surprisingly, in the APPI-4M/KLK6 complex, the Arg-15 P1 residue of the inhibitory APPI molecule was present in two conformations: the expected “up” conformation, featuring a salt bridge with the KLK6 specificity pocket residue Asp-189, and a “down” conformation, which is not normally observed in complexes of enzymes with trypsin-like specificity (Fig. 6C). This “down” conformation was also the dominant species observed in the APPIWT/KLK6 crystal. The “down” conformation of Arg-15 observed in these structures is similar to the conformation reported previously for the noncognate binding of APPI to chymotrypsin (PDB code 1CA0) (Fig. 6C). Unlike KLK6, chymotrypsin possesses a Ser-189 instead of Asp-189 and cannot form a salt bridge in the specificity pocket. It is likely that the “down” conformation of Arg-15 observed in the APPI/KLK6 complexes is an artifact of the acidic crystallization conditions, which may have resulted in a partial protonation of Asp-189. This perturbation of the P1/S1 subsite interaction is a localized phenomenon, which is not expected to impact the extended interactions of the other contact residues at the interface.
The crystal structures reveal an optimization of the contacts between the residues of the APPI-4M binding loop and the primed side subsites of KLK6. In the APPIWT/KLK6 complex, the APPIWT residues Met-17, Ile-18, and Ser-19 interact with the S2′, S3′, and S4′ subsites of KLK6, respectively (Fig. 7A). These subsites are largely shaped by His-39, Leu-40, and Leu-41, residues that are unique to KLK6 and are poorly conserved between KLKs and among other trypsin-like proteases. In the APPI-4M/KLK6 complex, the hydrophobic interactions between APPI-4M and the primed side subsites of KLK6 are optimized (Fig. 7B). Phe-18 makes more extensive contacts with the S3′ subsite, whereas Phe-19 forms a ring-stacking interaction with His-39 of KLK6, thus locking this residue into a single side chain conformation. The crystal structures also reveal differences in the intramolecular packing within APPIWT and APPI-4M (Fig. 8, A and B), which may further stabilize the conformation of the binding loop and maximize the interactions between the two proteins. Residues Leu-17, Phe-19, and Val-34 of APPI-4M form a hydrophobic cluster that fills the primed side of the KLK6 substrate-binding crevice (Fig. 8B), featuring more extensive hydrophobic interactions with KLK6 Phe-151 and Leu-40 than those observed in the APPIWT/KLK6 complex. In the APPI-4M complex, Phe-19 also forms a ring-stacking interaction with His-39 of KLK6 (Fig. 8, B and D). The mutated residues Phe-18 and Phe-19 of APPI-4M can wrap around the ridge formed by KLK6 residues 39–41 (Fig. 8D), thus forming a more extensive contact interface than that in the corresponding region of the APPIWT/KLK6 complex (Fig. 8C).
Figure 7.

Binding loop mutations of APPI-4M optimize interactions with the primed side subsites of KLK6. A, in the complex of KLK6 (gray surface) with APPIWT (cyan sticks), APPI residues Met-17, Ile-18, and Ser-19 interact with the S2′, S3′, and S4′ subsites of KLK6, respectively, which are shaped by His-39, Leu-40, and Leu-41 (salmon surface patch). B, in the APPI-4M/KLK6 complex, Phe-18 of APPI-4M makes more extensive contacts with the S3′ subsite of KLK6, whereas Phe-19 forms a ring-stacking interaction with His-39 of KLK6, thus locking this residue into a single side-chain conformation. Leu-17 and Phe-19 residues of APPI-4M also form intramolecular hydrophobic contacts, which may help to stabilize the conformation of the binding loop.
Figure 8.

Four APPI mutations optimize intra- and intermolecular packing interactions with KLK6. A and B, side view of the primed side of the substrate-binding cleft between KLK6 residues 39–41 (salmon surfaces) and Phe-151 (lavender) portrays van der Waals packing interactions in complexes with APPIWT (cyan) (A) and APPI-4M (mutated residues shown in yellow) (B). The mutated residues Leu-17, Phe-19, and Val-34 of APPI-4M form a hydrophobic cluster that fills the crevice and form hydrophobic interactions with KLK6 residues Phe-151 and Leu-40, whereas Phe-19 also forms a ring-stacking interaction with KLK6 His-39. C and D, rotated view, revealing how the mutated residues Phe-18 and Phe-19 of APPI-4M wrap around the ridge formed by KLK6 residues 39–41 (D), thus forming a more extensive contact interface than in the APPIWT/KLK6 complex (C).
Discussion
We developed a potent, four-mutant KLK6 inhibitor by employing the powerful YSD technology to engineer an APPI scaffold that demonstrates an increased proteolytic stability and a higher binding affinity toward KLK6. To develop this unique inhibitor, we generated two libraries: one with multiple random mutations in the entire protein scaffold (termed “random library”), and another more focused library, with multiple random mutations in the entire protein scaffold, having at least a single mutation in the APPI/KLK6 interface (termed “focused library”). The focused library was constructed based on our previously published APPIM17G,I18F,F34V sequence. As expected, the focused library, when displayed on yeast, showed a superior affinity to KLK6 than the random library. This result is in agreement with previous data showing that the sequence of the canonical binding loop largely determines the affinity and specificity of APPI to its target serine proteases (50) and implies that generating diversity within the APPI-binding loop, as opposed to randomizing the entire protein sequence, has more potential to improve the binding affinity. Sequencing results from three KLK6 affinity maturation screens of the focused library showed no consensus sequence for the selected clones; nevertheless, a repeating mutation at position 17, G17L, located in the inhibition loop, was identified in ∼50% of the clones. The two most abundant YSD variants, namely APPIG17L,I18F,F34V and APPIM17L,I18F,S19F,F34V (APPI-4M), displayed the highest affinity toward KLK6, as measured by flow cytometry (data not shown).
To better characterize our most potent variant, APPI-4M, we purified this protein in its soluble form and tested its binding and inhibition potencies, as well as its stability toward cleavage by KLK6, and we compared these parameters to those of APPIWT. Our mutant outperformed APPIWT in all these parameters. When tested in binding assays using SPR, our mutant demonstrated a sub-nanomolar affinity with not only a higher kon value, reflecting initial molecular recognition by KLK6, but also a lower koff rate compared with APPIWT, reflecting long-term complex stability. This finding has an important clinical implication, as a slow koff rate may facilitate a durable pharmacological potency of a drug in vivo (57, 58). Another important issue that we addressed was proteolytic stability. We show that KLK6 can cleave APPIWT within several hours, whereas APPI-4M proteolysis is observed only on a time scale of days. The turnover of APPI as a substrate of KLK6 is further emphasized by the presence of the product molecule APPI* in our crystal structures, cleaved at the Arg-15–Ala-16 reactive-site bond. We have previously demonstrated that APPI* is severely compromised as a protease inhibitor, and thus this cleavage serves as a mechanism for inhibitor inactivation (59). As APPI-4M showed both a slow koff rate and a high stability toward KLK6 cleavage, it may have prolonged on-target residence time and extended potency, leading to superior efficacy in vivo (60, 61).
In SDS-PAGE analyses of complexes formed between KLK6 and APPI-4M, but not between KLK6 and APPIWT, we observed a shift of KLK6 to a higher molecular weight in the presence of the inhibitor, suggesting the formation of a covalent complex that cannot be dissociated by reducing, denaturing conditions. This finding was surprising, because, although such covalent complexes are a hallmark of the unrelated serpin family of serine protease inhibitors, which form stable acyl–enzyme complexes with their target proteases (49, 62, 63), the canonical inhibitors, including APPI, follow a different mechanism, in which the stable complex is a tightly but reversibly bound Michaelis complex (55, 64). Previous studies of the chymotrypsin inhibitor 2, which belongs to a different family of canonical inhibitors, identified the rapid formation of a covalent acyl–enzyme by SDS-PAGE, but this species accumulated to represent only a small fraction of the inhibited enzyme due to the reversibility of acylation and the comparatively greater thermodynamic stability of the Michaelis complex (65, 66). Thus, to the best of our knowledge, our observation represents the first detection of a covalent species formed by an inhibitor of the Kunitz family, as well as the only example of any canonical inhibitor forming a covalent complex with an inhibited enzyme, in which the covalent complex represented the major species. However, the crystal structure of the KLK6/APPI-4M complex did not reveal a stable covalent complex, most likely because the Michaelis complex/acyl–enzyme equilibrium was shifted by local electrostatic alterations in the active site due to the low pH of the crystallization process. Nevertheless, we speculate that the improved inhibitory potency and slower dissociation rate of APPI-4M relative to APPIWT may have been achieved, at least in part, through enhanced enzyme/inhibitor interactions that stabilize the conformation of the acyl–enzyme or the acylation transition state in the KLK6/APPI-4M complex, resulting in the thermodynamic stabilization of the acyl–enzyme relative to the Michaelis complex. The slow rate of formation of fully cleaved APPI-4M* indicates that the hydrolysis of the acyl–enzyme remains rate-limiting in APPI proteolysis, as is characteristic of canonical inhibitors (65), rendering APPI-4M a strong inhibitor rather than a good substrate.
In addition to the clinical importance of the stability and affinity of an inhibitor, its lower potency toward other serine proteases is another highly significant factor for in vivo applications. Notably, APPI can inhibit several human proteases, including trypsin, chymotrypsin, plasmin, and FXIa (67, 68); these proteases may act as modulators of in vivo APPI concentrations (i.e. KLK6 competitors) (69). We found that, as compared with APPIWT, the APPI-4M mutant showed a substantially greater binding affinity toward KLK6, whereas its affinities toward cationic trypsin, anionic trypsin, and FXIa were decreased. In addition, our SPR results show that the binding affinity of the APPI-4M mutant toward KLK6 is stronger than toward the other KLKs, despite the high sequence and structure similarities among the KLKs (34). This enhanced affinity is compelling, especially considering that our YSD sorting strategy explicitly selected only for KLK6 binding and did not incorporate counterselection strategies. Our crystal structures reveal altered interactions of APPI-4M at the enzyme interface, primarily involving the optimization of primed side interactions with the S2′, S3′, and S4′ subsites of KLK6. Notably, these subsites are largely shaped by KLK6 residues 39–41, representing a stretch of sequence that is highly unique to KLK6 and is poorly conserved between KLKs and other trypsin-like proteases. Indeed, the improved affinity of APPI-4M toward KLK6 versus other KLKs suggests that an optimization of the S2′–S4′ subsite contacts may represent a general strategy for developing selective KLK6 inhibitors (70–73).
Over the past decade, KLK6 has emerged as a significant player in various stages of cancer development, and it has been associated with cell malignancy in multiple cancers, including melanoma, ovarian, colon, gastric, and breast cancers (8, 19, 21, 23). Previous studies from the Mori laboratory demonstrated that siRNA silencing of KLK6 expression in gastric cancer cells can inhibit a variety of malignant phenotypes, implicating KLK6 as a therapeutic target in cancer (14). In breast cancer, Gosh et al. (29) have identified BT-20 cells as a highly metastatic breast cancer cell line, which expresses high levels of KLK6. It was found that efficient inhibition by a KLK6 antibody has led to striking suppression of BT-20 cellular invasion in a Matrigel transwell assays (29). Similarly, we found that treating BT-20 cells with APPI-4M, but not with APPIWT, inhibits their invasion capabilities. We show, using siRNA transfection, that BT-20 cell invasiveness indeed depends on KLK6 expression. Importantly, APPI-4M did not inhibit the proliferation/viability of these cells, suggesting that, in these cells, KLK6 is responsible for invasion but not proliferation. Our study thus suggests that targeting KLK6 with the high-affinity, high-stability APPI-4M can offer therapeutic benefits in breast cancer, and it further identifies useful models for evaluating the efficacy of KLK6 inhibitors (e.g. in combination with cytotoxic or cytostatic chemotherapeutic agents).
Another important aspect of this study is that it provides the first proof-of-principle for an innovative approach, developed here, to engineer protein-based, therapeutic-oriented inhibitors with improved target affinity and greater proteolytic stability. Despite the well-known advantages of protein-based drugs (e.g. their lower immunogenicity and toxicity relative to small-molecule drugs) (74), they often suffer from two main caveats. First, the naturally occurring protein on which the drug is based may be specific to several structurally similar targets (indeed, in our case, the WT APPI similarly inhibits several target proteins) (75), which may cause severe side effects in a therapeutic context. Second, they are typically susceptible to proteolytic degradation in the body, which limits their effectiveness. Our engineered APPI-4M protein showed a dramatic improvement in both these aspects relative to the WT APPI. Hence, the strategy developed here to engineer this potent stable inhibitor may have far-reaching implications, as it can potentially enable the production of other improved protein-based therapeutics. We also emphasize that, beyond the translational potential of this specific inhibitor, a significant advance of this work lies in the structural elucidation of key interactions responsible for modifying inhibitor affinity, insights that will be beneficial for future efforts toward KLK inhibitor development.
Experimental procedures
Reagents
Synthetic oligonucleotides were obtained from Integrated DNA Technologies (San Jose, CA). Restriction enzymes and polymerases were purchased from New England Biolabs (Ipswich, MA), and dNTPs were purchased from Jena Bioscience (Jena, Germany). The EBY100 yeast strain and yeast surface display (YSD) plasmid (pCTCON) were gifts from the laboratory of Dane Wittrup (Massachusetts Institute of Technology). Bacterial plasmid extraction and purification kits were purchased from RBC Bioscience (New Taipei City, Taiwan), and yeast plasmid extraction kits were purchased from Zymo Research (Irvine, CA). The methylotrophic yeast P. pastoris strain GS115 and its expression vector (pPIC9K) were purchased from Invitrogen. The phycoerythrin-conjugated anti-mouse antibody was purchased from Sigma. The mouse anti-c-Myc antibody (Ab-9E10) was obtained from Abcam (UK). Factor-XIa and its substrate S-2366 (Chromogenix) were obtained from Hematologic Technologies Inc. (Essex Junction, VT) and from Diapharma (West Chester Township, OH), respectively. The kallikrein-6 substrate BOC-Phe-Ser-Arg-AMC was obtained from Bachem (Bubendorf, Switzerland). Kallikrein-1 and Kallikrein-5 were obtained from ProSpec (Rehovot, Israel).
Generating the combinatorial APPI libraries
The synthesis and cloning of the DNA encoding the APPIWT gene and APPI random library are described elsewhere (50). Briefly, for the APPI random library, we constructed a randomly mutated version of the APPI gene by an error-prone PCR, using nucleotide analogues and a low-fidelity Taq polymerase, with the APPIWT gene as a template. The APPI focused library was constructed based on our previously published APPIM17G,I18F,F34V sequence (50). This library was generated by PCR assembly combined with error-prone PCR protocols, using a total of 10 overlapping oligonucleotides. In addition to generating random mutations in the entire APPI sequence (error-prone randomization) and further incorporating random mutations in specific residues, seven of the 10 oligonucleotides were individually randomized by using NNS-degenerated codons at specific positions within the APPI inhibitory binding loop region (positions 11–18, excluding the Cys residue at position 14; Fig. 1D). The APPI random and focused DNA libraries were amplified and transformed into yeast through homologous recombination and sequenced at the DNA Microarray and Sequencing Unit ((DMSU), NIBN, BGU), and their yield was evaluated by dilution plating, as described previously (53).
Synthesis and cloning of the APPI library
The APPI focused library was constructed based on our previously published APPIM17G,I18F,F34V sequence (50) with a peptide linker (NH3+–APPI-LPDKPLAFQDPS-COO−), using codons optimized for S. cerevisiae and P. pastoris usage. The library was generated by PCR assembly using the following 10 overlapping oligonucleotides (5′–3′): 1) GGTGGTTCTGGTGGTGGTGGTTCTGGTGGTGGTGGTTCTGCTAGCGAAGTTTGTTCTGAACAAGCTG; 2) GAAGTTTGTTCTGAACAAGCTGAANNSGGTCCATGTAGAGCTGGTTTTTCTAGATGGTATTTCGATGTTACTG; 3) GAAGTTTGTTCTGAACAAGCTGAAACTNNSCCATGTAGAGCTGGTTTTTCTAGATGGTATTTCGATGTTACTG; 4) GAAGTTTGTTCTGAACAAGCTGAAACTGGTNNSTGTAGAGCTGGTTTTTCTAGATGGTATTTCGATGTTACTG; 5) GAAGTTTGTTCTGAACAAGCTGAAACTGGTCCATGTNNSGCTGGTTTTTCTAGATGGTATTTCGATGTTACTG; 6) GAAGTTTGTTCTGAACAAGCTGAAACTGGTCCATGTAGANNSGGTTTTTCTAGATGGTATTTCGATGTTACTG; 7) GAAGTTTGTTCTGAACAAGCTGAAACTGGTCCATGTAGAGCTNNSTTTTCTAGATGGTATTTCGATGTTACTG; 8) GAAGTTTGTTCTGAACAAGCTGAAACTGGTCCATGTAGAGCTGGTNNSTCTAGATGGTATTTCGATGTTACTG; 9) GATGGTATTTCGATGTTACTGAAGGTAAATGTGCTCCATTCGTCTATGGTGGTTGTGGTGGTAATAGAAATAATTTCGATACTGAAGAATATTGTATGGCTGTTTGTGGTTCTGCTATTGGATCCTTGCCAGATAAACCATTGGCTTTCC; 10) GAGCTATTACAAGTCCTCTTCAGAAATAAGCTTTTGTTCAGATGGATCTTGGAAAGCCAATGGTTTATC.
The synthetic library was assembled by a set of three PCRs, using low-fidelity Taq polymerase, as follows: in the first reaction, oligos 2–9 were incorporated using error-prone PCR to generate both a site-specific saturation randomization of the binding loop residues (using NNS codons, where N = A/C/G/T and S = C/G) and an arbitrary randomization of their neighboring residues, ranging between Glu-10 and Gly-38 on the sequence, chosen for their involvement in the KLK6 hypothesized interaction interface. The error-prone PCR was optimized to 15× PCR doublings using low-fidelity Taq polymerase, 1% nucleotide analogues, and 2 mm MgCl2. Next, the assembled fragment was amplified by PCR using 5′-GAAGTTTGTTCTGAACAAGCTG-3′ and 5′-GGAAAGCCAATGGTTTATC-3′ primers, followed by a final PCR using oligos 1 and 10. The final PCR-assembled fragment was gel-purified and cloned into a pCTCON vector via transformation by electroporation of EBY100 yeast cells and homologous recombination with the linearized vector (digested with NheI and BamHI), as described previously (53). Random and site-specific mutagenesis using error-prone PCR and NNS degenerate codons in the APPI sequence yielded a library of ∼3.5 × 106 variants, as estimated by dilution plating on selective SDCAA plates (15% agar, 2% dextrose, 1.47% sodium citrate, 0.429% citric acid monohydrate, 0.67% yeast nitrogen base, and 0.5% casamino acids). Sequencing results (obtained from the DNA Microarray and Sequencing Unit (DMSU) of the National Institute for Biotechnology in the Negev (NIBN), Ben-Gurion University of the Negev (BGU), Israel) revealed an average amino acid mutation rate of 1–2 mutations per 56 amino acids of the APPI sequence (data not shown).
Production of recombinant trypsins
The recombinant human anionic trypsinogen and the human cationic trypsinogen were expressed in Escherichia coli, extracted from inclusion bodies, refolded, purified, and activated with bovine enteropeptidase, as described previously (52, 76).
Production of the human kallikrein 6
Recombinant pro-KLK6 was expressed and purified from a virus/insect cell line system Sf21 at the Protein Expression and Purification Core Facility, EMBL Heidelberg, Germany, as described previously (46). The pro-enzyme was activated with enterokinase and purified, and its concentration was estimated by UV-visible absorbance at 280 nm with an extinction coefficient (ϵ280) of 34.67 × 103 m−1 cm−1 in a DeNovix DS-11 spectrophotometer (DeNovix, Wilmington, DE). KLK6 was labeled with the DyLight-650 amine-reactive dye according to the manufacturer's protocol (ThermoFisher Scientific, Waltham, MA), at a ratio of 1:5 (KLK6/dye).
Construction and cloning of the expression vector pPIC9K-APPI
The human cDNA of APPIWT, based on a published sequence (PDB code 1ZJD), was amplified by PCR using a Phusion DNA polymerase with an upstream primer: 5′-AGCGTATACGTAGACTATAAGGATGACGACGACAAAGAATTCGAAGTTTGTTCTGAACAAGCTG-3′ and a downstream primer; 5′-ATAGTTTAGCGGCCGCATGATGGTGGTGATGGTGCCTAGGAATAGCAGAACCACAAACAGC-3′. The resulting construct included four restriction sites and two epitope tags (FLAG and HIS6), as follows: SnaBI-FLAG–EcoRI-APPIWT–AvrII-HIS6–NotI. The obtained DNA fragment was digested with SnaBI and NotI and subcloned by using the same restriction sites as for the Pichia expression vector pPIC9K. Next, the recombinant expression plasmid was used as a template for the construction of the APPI variants, as follows: the cDNA of each variant was amplified by PCR using an upstream primer 5′-CGGAGCGAATTCGAAGTTTGTTCTGAACAAGCTG-3′ and a downstream primer 5′-CGCTACCCTAGGAATAGCAGAACCACAAACAGC-3′. The resulting construct included the restriction sites EcoRI and AvrII. The obtained DNA fragment was digested with EcoRI and AvrII and subcloned using the same restriction sites of the template vector. Finally, all sequences of the recombinant expression plasmids were confirmed by a DNA sequencing analysis (DMSU, NIBN, BGU). Expression vectors were linearized by SacI digestion and used to transform P. pastoris strain GS115 by electroporation. This process resulted in the insertion of the construct at the AOX1 (alcohol oxidase) locus of P. pastoris, thereby generating a His+ Mut+ phenotype. Transformants were selected for the His+ phenotype on a 2% agar containing regeneration dextrose biotin (RDB: 18.6% sorbitol, 2% dextrose, 1.34% yeast nitrogen base, 4 × 10−5 % biotin, and 0.005% each of l-glutamic acid, l-methionine, l-lysine, l-leucine, and l-isoleucine) and allowed to grow for 2 days at 30 °C. The cells were harvested from the plates and subjected to further selection for a high copy number by their ability to grow on 2% agar containing 1% yeast extract, 2% peptone, 2% dextrose medium, and the antibiotics G418 (Geneticin, 4 mg/ml, Invitrogen). To verify the direct insertion of the construct at the AOX1 locus of P. pastoris, the genomic DNA of the highest APPI-expressing colony from each APPI variant was extracted and amplified by PCR with an AOX1 upstream primer 5′-GACTGGTTCCAATTGACAAGC-3′ and an AOX1 downstream primer 5′-GCAAATGGCATTCTGACATCC-3′. The resulting linear DNA was gel-purified, and its correct sequence was confirmed by DNA sequencing analysis (DMSU, NIBN, BGU).
Flow cytometry analysis and cell sorting
The yeast-displayed APPI library was grown in an SDCAA selective medium (2% dextrose, 0.67% yeast nitrogen base, 0.5% BactoTM casamino acids, 1.47% sodium citrate, 0.43% citric acid monohydrate, pH 4.5) and induced for expression with an SGCAA selective medium (similar to SDCAA, but with 2% galactose instead of dextrose) according to an established protocol (53). The expression of the yeast-displayed proteins was detected by labeling the cells with the 9E10 mouse anti-c-Myc antibody (Abcam, Cambridge, UK) in a 1:50 ratio. Next, the cells were washed and resuspended in a goat anti-mouse secondary antibody, which was conjugated to phycoerythrin (Sigma) in a 1:50 ratio in a solution containing 50 m Tris-HCl, 100 mm NaCl, and 0.2% BSA, pH 7.3. The KLK6 binding signal and the APPI expression signal were analyzed by dual-color flow cytometry (Accuri C6; BD Biosciences).
Cell sorting of the APPI library was conducted with a FACS Aria III BD Biosciences (Ilse Katz Institute for Nanoscale Science and Technology, BGU), as described in Fig. 1. Briefly, 30 × 106 yeast cells were sorted first, and cells demonstrating a high APPI expression signal were isolated to select for high-expressing clones (sort S0). Following each successive sort, the number of yeast cells used was at least 10-fold in excess relative to the number of sorted cells from the previous sort. Several clones from each round of sorting were sequenced.
The same labeling procedure was used for the YSD-based titration curves of the individual clones, and the binding of different APPI clones to KLK6 was evaluated at KLK6 concentrations ranging from 100 fm to 10 nm. Each binding measurement was normalized to the APPI expression signal of the same clone.
Large-scale expression and purification of APPI proteins
P. pastoris GS115-APPI clones were first inoculated into 50 ml of BMGY (1% yeast extract, 2% peptone, 0.23% potassium phosphate monobasic, 1.18% potassium phosphate dibasic, 1.34% yeast nitrogen base, 4 × 10−5 % biotin, and 1% glycerol) to an A600 = 10.0 (108 cells/ml), followed by scaling-up to 500 ml of BMGY, until an A600 = 10.0 was reached (overnight growth at 30 °C with shaking at 300 rpm). The cells were harvested by centrifugation and resuspended in 1 liter of BMMY (similar to BMGY, but with 0.5% methanol instead of glycerol) to an A600 = 5.0 to induce expression, and were grown at 30 °C with shaking at 300 rpm. Methanol was added to a final concentration of 2% every 24 h to maintain induction. Following 5 days of induction, the culture was centrifuged again, and the supernatant, containing the secreted recombinant inhibitors, was prepared for purification by nickel-immobilized metal-affinity chromatography. The supernatant containing the recombinant APPI was filtered through a 0.22-μm Steritop bottle-top filter (Millipore, Billerica, MA). The filtered supernatant was adjusted to 10 mm imidazole and 0.5 m NaCl at pH 8.0 and left to stand overnight at 4 °C. Then, a second filtration step was performed to remove any additional precipitation. The resulting supernatant was loaded onto a HisTrap 5-ml column (GE Healthcare, UK) at a flow rate of 0.7 ml/min for 24 h, washed with a washing buffer (20 mm sodium phosphate, 0.5 m NaCl, and 10 mm imidazole; pH 8.0), and eluted with an elution buffer (similar to the washing buffer, but with 0.5 m imidazole) in an ÄKTA pure instrument (GE Healthcare). The eluted inhibitors were concentrated, and the buffer was replaced with 50 mm Tris-HCl and 100 mm NaCl, pH 7.3, in a 3-kDa MWCO Vivaspin concentrator (GE Healthcare). Gel-filtration chromatography was performed with a Superdex 75 16/600 column (GE Healthcare) equilibrated with 50 mm Tris-HCl and 100 mm NaCl, pH 7.3, at a flow rate of 1 ml/min on an ÄKTA start instrument (GE Healthcare). An SDS-PAGE analysis was used to confirm the purity of the proteins. The correct mass of the pure proteins was validated by using a MALDI-TOF REFLEX-IV (Bruker, Billerica, MA) mass spectrometer (The Ilse Katz Institute for Nanoscale Science and Technology, BGU). Purification yields for all APPI clones were 0.8–50 mg per 1-liter culture flask.
SPR studies
The affinity of binding between the APPI variants and KLKs (KLK1, KLK2, KLK3, KLK4, KLK5, and KLK6) was detected by using SPR spectroscopy on a ProteOn XPR36 system (Cytometry, Proteomic and Microscopy Unit, NIBN, BGU). APPIWT and APPI-4M were immobilized on the surface of a GLC chip by using the amine-coupling reagents sulfo-NHS (0.1 m N-hydroxysuccinimide) and EDC (0.4 m 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide, Bio-Rad). APPIWT and APPI-4M (2 μg) were each covalently immobilized on the chip in 10 mm sodium acetate buffer, pH 4.0, to give 237.4 and 439.9 response units, respectively. A single flow channel immobilized with a BSA (3 μg) served as a background control. Each KLK was diluted in PBS with 0.05% Tween 20 to 0.625, 1.25, 2.5, 5, or 10 nm in 400 μl, and it was circulated for 600 s at a flow rate of 25 μl/min, followed by 2400 s of dissociation at a flow rate of 25 μl/min. The response was monitored as a function of time at 25 °C. The kinetic constants, kon and koff, and the equilibrium affinity constant KD between the APPI variants and KLKs were determined by using the 1:1 Langmuir model (77).
Inhibition studies
The inhibition constant Ki of APPIWT and its variants, APPIM17G,I18F,F34V, APPIM17L,I18F,F34V, and APPI-4M, in complex with KLK6 was determined according to a previously described methodology (50, 76) with minor modifications. Briefly, tight binding experiments were conducted at a fixed concentration of the substrate BOC-Phe-Ser-Arg-AMC (1 mm). These assays were performed at 37 °C in the presence of various concentrations of inhibitor, using a Synergy2 microplate spectrophotometer (BioTek, Winooski, VT). KLK6 (50 μl) and APPI (50 μl) were mixed and equilibrated in a 96-well microplate (Greiner, Kremsmünster, Austria) prior to the addition of the substrate (50 μl). For the APPIWT and APPIM17G,I18F,F34V reactions, the KLK6 concentration was 1 nm, and the concentration range of APPI proteins was 0–50 nm. For the APPIM17L,I18F,F34V and APPI-4M reactions, no detectable activity of KLK6 was observed in the presence of the inhibitors, and therefore, the concentration of KLK6 was changed to 100 nm, and the concentration range of the APPIs was 419–1600 nm. The fluorescent signal of the reactions was monitored in an Infinite 200 PRO NanoQuant microplate reader (Tecan, Männedorf, Switzerland), set at 355 nm for excitation and 460 nm for emission. Reactions including APPIWT and APPIM17G,I18F,F34V were followed for 4 h, and those including APPIM17L,I18F,F34V and APPI-4M were followed for 20 min to determine the initial steady-state rates of substrate hydrolysis. The inhibition constants were calculated by using Equation 1, as described previously (76),
| (Eq. 1) |
where Vi and V0 are the steady-state rates in the presence and absence of the inhibitor, respectively; Km is the Michaelis constant for substrate cleavage; and [S]0 and [I]0 are the initial concentrations of the substrate and inhibitor, respectively. The affinity of the inhibitors was evaluated by conducting similar inhibition studies using cationic trypsin, anionic trypsin, and FXIa; briefly, tight binding experiments were conducted at a fixed concentration of 145 μm Z-GPR-pNA as a substrate for cationic trypsin and anionic trypsin or with 600 μm S-2366 as a substrate for FXIa. For the experiments with cationic and anionic trypsins, the concentrations of the inhibitors ranged between 0 and 80 nm, and the concentration of the enzyme was 0.1 nm. For the experiment with FXIa, the concentrations of the inhibitor ranged between 2 and 10 nm, and the concentration of the enzyme was 0.125 nm. The enzyme (8 μl), inhibitor (8 μl), and buffer (144 μl) were preincubated at room temperature for 60 min in a solution of 100 mm Tris-HCl and 1 mm CaCl2, pH 8.0, for cationic and anionic trypsins and 50 mm Tris-HCl, pH 7.6, 150 mm NaCl, 5 mm CaCl2, and 0.1% BSA for FXIa. The reactions were then initiated by diluting the mixture of enzyme and inhibitor into a pre-equilibrated microplate (nonbinding 96-well; Greiner, Kremsmünster, Austria) containing a buffer (152 μl) and a substrate (8 μl). The reactions were conducted at 25 °C and monitored spectroscopically for 2 h. The concentrations of cationic trypsin and anionic trypsin were quantified by active-site titration using pNPGB, which serves as both an irreversible trypsin inhibitor and a substrate. The concentrations of FXIa and KLK6 were determined by using UV-visible absorbance at 280 nm, with extinction coefficients (ϵ280) of 214.4 × 103 m−1 cm−1 for FXIa and 34.67 × 103 m−1 cm−1 for KLK6. The concentrations of the chromogenic substrates Z-GPR-pNA and S-2366 were determined in an end-point assay from the change in the absorbance (plateau after complete hydrolysis) that is obtained by the release of p-nitroaniline. The concentrations of the APPI variants were determined by titration with pre-titrated bovine trypsin and the substrate l-BAPA, as described previously (76). The reaction buffers used in this study were as follows: for KLK6 experiments, 50 mm Tris-HCl, 100 mm NaCl, and 0.2% BSA, pH 7.3; for trypsin experiments, 100 mm Tris-HCl and 1 mm CaCl2, pH 8.0; and for FXIa experiments, 50 mm Tris-HCl, 150 mm NaCl, 5 mm CaCl2, and 0.1% BSA, pH 7.6. Calculations were performed by using Km values (mean ± S.D.) of 329.33 ± 1.44 μm for KLK6, 22.8 ± 1.9 μm for cationic trypsin, 10.7 ± 0.7 μm for anionic trypsin, and 361.3 ± 12.1 μm for FXIa, as determined from at least three Michaelis-Menten kinetic experiments with triplicates (50). The results of the inhibition studies are reported as mean ± S.D.
APPI/KLK6 complex formation in SDS-PAGE
This assay was performed as described previously, with minor modifications (49). KLK6 (5 μm) was incubated for 1 h with APPIWT or APPI-4M, at 1:1, 1:5, or 1:10 molar ratios, in a solution of 50 mm Tris-HCl and 100 mm NaCl, pH 7.3, at 37 °C. Following this incubation, samples were prepared in a reducing sample buffer containing 1% SDS, 10% glycerol, 10 mm Tris-HCl, 0.2 mm EDTA, 80 mm DTT, 0.01% bromphenol blue, pH 6.8, run on an SDS-PAGE, and stained with Coomassie Blue.
APPI hydrolysis studies
The cleavage of intact APPI variants (between residues Arg-15 and Ala-16) in time-course incubations with KLK6 was monitored by high-performance LC (HPLC; Waters, Milford, MA), as described previously (50, 59), with minor modifications. Briefly, KLK6 was incubated with APPIWT or APPI-4M in 30-μl aliquots in a solution of 50 mm Tris-HCl and 100 mm NaCl, pH 7.3, at 37 °C; the concentration of the inhibitor was 25 μm, and the concentration of KLK6 was 3.28 μm. For analysis by HPLC, the aliquots were withdrawn at periodic intervals (over 90 h for APPIWT and 300 h for APPI-4M), and the samples were immediately quenched by acidification with 70 μl of 0.3 m HCl. The samples were resolved on a 50 × 2.0-mm Jupiter 4 μm 90 Å C12 column (Phenomenex, Torrance, CA) with a gradient of 0–100% acetonitrile in 0.1% TFA at a flow rate of 0.6 ml/min over 50 min. Intact inhibitors were quantified by peak integration of absorbance traces, monitored at 210 nm. The initial rates of hydrolysis over time were obtained by linear regression, using a minimum of seven data points within the initial linear phase of the reaction. The reported hydrolysis rates for each inhibitor represent the average (± S.D.) of three independent experiments.
Cells
BT-20 cells were purchased from the American Type Culture Collection (ATCC). The cells were maintained in Eagle's minimum essential medium supplemented with 10% FBS, 1 mm sodium pyruvate, 2 mm l-glutamine, and 1% penicillin/streptomycin.
siRNA transfection
BT-20 breast cancer cells (also known as HTB-19 cells) were plated at 80% confluency 1 day before transfection and then transfected with KLK6-specific siRNA or scrambled nonspecific siRNA using the TriFECTa RNAi kit (IDT, San-Jose, CA) with the Lipofectamine 3000 reagent (Invitrogen) according to the manufacturer's instructions, dosing the cells twice with siRNA (45 pmol) with a 24-h interval.
RNA extraction and RT-PCR analysis
Cells were lysed using TRIzol (Invitrogen), and total RNA was isolated according to the manufacturer's instructions. After the RNA was quantified, 750 ng of RNA were used for cDNA synthesis by the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA), according to the manufacturer's instructions. Each cDNA sample was amplified using the Phusion Hot Start Flex polymerase (New England Biolabs, Beverly, MA). The following primers were used: KLK6 (sense, 5′-GCA AGA CAG CAG ATG GTG AT-3′; antisense, 5′-CAC TTG GCC TGA ATG GTT TT-3′) and GAPDH (sense, 5′-GAG TCA ACG GAT TTG GTC GT-3′; antisense, 5′-TTG ATT TTG GAG GGA TCT CG-3′). GAPDH was used as a reaction control. The PCR-amplified cDNA was subjected to agarose gel electrophoresis. The DNA bands were imaged with the Vilber Fusion automatic gel imaging analysis system (Vilber, Marne La Vallée, France).
Cell invasion assay
The Boyden chamber invasion assay was performed as described previously (29) with the following modifications. The tumorigenic breast cancer cell line BT-20 (HTB19), which secretes KLK6, was used. Cells were cultured in complete culture medium, grown to 70% confluency, harvested, and suspended in a serum-free medium. Three hours prior to conducting the assay, inserts (ThinCert, 8 μm pore size; Greiner BioOne, Kremsmünster, Austria) were coated with 30 μg of Matrigel (Corning, Corning, NY). Cells (1.5 × 104) were seeded in each insert. A solution containing either APPI-4M (10, 50, or 100 nm), APPIWT (10, 50, or 100 nm), or as a control 50 mm Tris-HCl and 100 mm NaCl, pH 7.3, was added to each insert to reach a total volume of 200 μl. Each condition was performed in triplicate. Additional controls included KLK6-specific siRNA-transfected cells and scrambled nonspecific siRNA-transfected cells, seeded in the same conditions. Culture media (600 μl) with 10% FBS were used as a chemoattractant in the lower chambers. The chambers were incubated in a CO2 incubator at 37 °C for 36 h, and then the invasive cells were fixed and stained with the Dipp Kwik Differential Stain kit (American Mastertech Scientific, Lodi, CA). The migrated cells were counted (five fields per insert) with an EVOS FL cell imaging system (ThermoFisher Scientific) at a ×10 magnification. The reported results are the mean (±S.E.) of three biological repetitions. Statistical analysis was conducted with the Student's t test.
Cell viability assay
Cell viability was evaluated by using the XTT cell proliferation kit (Biological Industries, Israel), according to the manufacturer's instructions, in a 96-well plate. The cells were cultured in a culture medium, grown to 70% confluency, harvested, and replaced in a serum-free medium, and 1.5 × 104 cells were seeded in each well. APPI-4M or APPIWT (10, 50, or 100 nm), 50 mm Tris-HCl, and 100 mm NaCl, pH 7.3, as a control, was added to each well to reach a total volume of 200 μl, in triplicate. The plate was incubated in a CO2 incubator at 37 °C for 36 h, and cell viability was measured according to the manufacturer's instructions. The reported results are the mean (± S.E.) of three biological repetitions. Statistical analysis was done using the Student's t test.
Wound-healing assay
BT-20 cells (1 × 105) were cultured as confluent monolayers in a 24-well plate. After reaching full confluency, a scratch was formed by removing a strip of cells across the well using a p200 pipette tip. The scratched monolayers were then washed twice to remove nonadherent cells, and then 500 μl of a 2% FBS medium, containing either APPI-4M (100 nm) or a 2% FBS medium (control), was added. The wells were photographed with an EVOS FL cell imaging system at a ×4 magnification, both immediately after cell wounding and again after 24 h. The experiment was performed in triplicate, and the images were analyzed using ImageJ. Error bars in the figures represent the standard deviation (S.D.). Statistical significance was determined by Student's t test, with p < 0.05 considered significant.
Protein crystallization, data collection, structure determination, and refinement
KLK6 was mixed with either APPIWT or APPI-4M in a 1:5 molar ratio and subjected to crystallization trials using the sitting drop vapor diffusion method. Initial crystallization conditions were screened by using the Index screening kit (Hampton Research, Aliso Viejo, CA) at 293 K. Each drop contained a mixture of a 0.3-μl crystallization solution and 0.3 μl of the APPIWT/KLK6 or APPI-4M/KLK6 complex. For the APPIWT/KLK6 complex, one crystal grew after 40 days in a drop containing 0.2 m ammonium sulfate, 0.1 m BisTris, pH 5.5, and 25% PEG 3350. The crystal was harvested and flash-cooled in liquid nitrogen prior to data collection. X-ray diffraction data were collected at 100 K in beamline ID30B at the European Synchrotron Radiation Facility (ESRF, Grenoble, France). The crystal diffracted to a maximum resolution of 1.85 Å with a low mean I/σI and multiplicity (1.08 and 2.0, respectively). However, the effective resolution of this structure was considered to be 1.95 Å. The last shell (1.95–2.02) had a mean I/σI of 1.44 and a multiplicity of 12.8. The crystal belongs to the P212121 space group, with unit cell dimensions of a 59.543 Å, b 77.702 Å, and c 92.207 Å, and it contains one molecule of KLK6 and two molecules of APPIWT in the asymmetric unit. For the APPI-4M/KLK6 complex, one crystal grew after 6 min in a drop containing 0.1 m tri-sodium citrate (pH 5.6), 20% 2-propanol, and 20% PEG 4000. The crystal was harvested and flash-cooled in liquid nitrogen prior to data collection. X-ray diffraction data were collected at 100 K in beamline ID23-1 at the ESRF to a maximum resolution of 2.3 Å. The crystal belongs to the P212121 space group, with unit cell dimensions of a 58.058 Å, b 77.752 Å, and c 91.130 Å, and it contains one molecule of KLK6 and two molecules of APPI-4M in the asymmetric unit. The X-ray data of both the APPIWT/KLK6 and the APPI-4M-/KLK6 crystals were processed, merged, and scaled by XDS (78). Data collection statistics are provided in Table 4. Phase acquisitions and structural determination were performed by molecular replacement using Phaser (79) from the CCP4 Program Suite (80). Protein Data Bank (PDB) codes 1AAP and 1LO6 were used as the search models. Refinement was performed using Phenix.refine (81), and alternating rounds of model building and manual corrections were performed by COOT (82). The coordinates and structure factors were submitted to the PDB under the accession codes 5NX1 for APPIWT/KLK6 and 5NX3 for APPI-4M/KLK6.
Author contributions
A. Sananes and N. P. conceptualization; A. Sananes and A. Shahar data curation; A. Sananes, I. C., A. Shahar, E. S. R., and N. P. formal analysis; A. Sananes, E. S. R., and N. P. validation; A. Sananes, A. Shahar, and N. P. investigation; A. Sananes, I. C., A. Shahar, A. H., and E. D. V. methodology; A. Sananes, E. S. R., and N. P. writing-original draft; A. Sananes, I. C., A. Shahar, A. K. M., E. S. R., and N. P. writing-review and editing; E. S. R., A. K. M., and N. P. resources; E. S. R., A. K. M., and N. P. funding acquisition; N. P. supervision; N. P. visualization.
Acknowledgments
We thank Dr. Alon Zilka and Dr. Uzi Hadad for their technical assistance. FACS experiments were performed at the Ilse Katz Institute for Nanoscale Science and Technology, BGU. The structural studies were performed on beamlines ID23-1 and ID30-B at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. We are grateful to Christoph Mueller-Dieckmann for providing assistance in using those beamlines. We thank Eitan Rabinovich for providing KLK4.
This work was supported in part by the European Research Council “Ideas Program” ERC-2013-StG Contract Grant 336041, the Prostate Cancer Foundation (to N. P.), and the DKFZ-MOST Contract Grant GR2495 (to N. P. and A. K. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (codes 5NX1 and 5NX3) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- KLK
- human tissue kallikrein
- APPI
- amyloid precursor protein Kunitz protease inhibitor domain
- BGU
- Ben-Gurion University of the Negev
- DMSU
- DNA Microarray and Sequencing Unit
- FBS
- fetal bovine serum
- FXIa
- factor XIa
- SPR
- surface plasmon resonance
- NIBN
- National Institute of Biotechnology in the Negev
- YSD
- yeast surface display
- PDB
- Protein Data Bank
- Z
- benzyloxycarbonyl
- pNA
- p-nitroaniline
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- oligo
- oligonucleotide
- BOC
- t-butyloxycarbonyl
- AMC
- 4-methylcoumaryl-7-amide
- YSD
- yeast surface display.
References
- 1. Prassas I., Eissa A., Poda G., and Diamandis E. P. (2015) Unleashing the therapeutic potential of human kallikrein-related serine proteases. Nat. Rev. Drug Discov. 14, 183–202 10.1038/nrd4534 [DOI] [PubMed] [Google Scholar]
- 2. Emami N., and Diamandis E. P. (2007) New insights into the functional mechanisms and clinical applications of the kallikrein-related peptidase family. Mol. Oncol. 1, 269–287 10.1016/j.molonc.2007.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marceau F., and Regoli D. (2004) Bradykinin receptor ligands: therapeutic perspectives. Nat. Rev. Drug Discov. 3, 845–852 10.1038/nrd1522 [DOI] [PubMed] [Google Scholar]
- 4. Oikonomopoulou K., Diamandis E. P., and Hollenberg M. D. (2010) Kallikrein-related peptidases: proteolysis and signaling in cancer, the new frontier. Biol. Chem. 391, 299–310 [DOI] [PubMed] [Google Scholar]
- 5. Kontos C. K., and Scorilas A. (2012) Kallikrein-related peptidases (KLKs): a gene family of novel cancer biomarkers. Clin. Chem. Lab. Med. 50, 1877–1891 [DOI] [PubMed] [Google Scholar]
- 6. Henkhaus R. S., Gerner E. W., and Ignatenko N. A. (2008) Kallikrein 6 is a mediator of K-RAS-dependent migration of colon carcinoma cells. Biol. Chem. 389, 757–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Klucky B., Mueller R., Vogt I., Teurich S., Hartenstein B., Breuhahn K., Flechtenmacher C., Angel P., and Hess J. (2007) Kallikrein 6 induces E-cadherin shedding and promotes cell proliferation, migration, and invasion. Cancer Res. 67, 8198–8206 10.1158/0008-5472.CAN-07-0607 [DOI] [PubMed] [Google Scholar]
- 8. Krenzer S., Peterziel H., Mauch C., Blaber S. I., Blaber M., Angel P., and Hess J. (2011) Expression and function of the kallikrein-related peptidase 6 in the human melanoma microenvironment. J. Invest. Dermatol. 131, 2281–2288 10.1038/jid.2011.190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Christodoulou S., Alexopoulou D. K., Kontos C. K., Scorilas A., and Papadopoulos I. N. (2014) Kallikrein-related peptidase-6 (KLK6) mRNA expression is an independent prognostic tissue biomarker of poor disease-free and overall survival in colorectal adenocarcinoma. Tumour Biol. 35, 4673–4685 10.1007/s13277-014-1612-y [DOI] [PubMed] [Google Scholar]
- 10. Vakrakou A., Devetzi M., Papachristopoulou G., Malachias A., Scorilas A., Xynopoulos D., and Talieri M. (2014) Kallikrein-related peptidase 6 (KLK6) expression in the progression of colon adenoma to carcinoma. Biol. Chem. 395, 1105–1117 [DOI] [PubMed] [Google Scholar]
- 11. Kolin D. L., Sy K., Rotondo F., Bassily M. N., Kovacs K., Brezden-Masley C., Streutker C. J., and Yousef G. M. (2014) Prognostic significance of human tissue kallikrein-related peptidases 6 and 10 in gastric cancer. Biol. Chem. 395, 1087–1093 [DOI] [PubMed] [Google Scholar]
- 12. Hoffman B. R., Katsaros D., Scorilas A., Diamandis P., Fracchioli S., Rigault de la Longrais I. A., Colgan T., Puopolo M., Giardina G., Massobrio M., and Diamandis E. P. (2002) Immunofluorometric quantitation and histochemical localisation of kallikrein 6 protein in ovarian cancer tissue: a new independent unfavourable prognostic biomarker. Br. J. Cancer 87, 763–771 10.1038/sj.bjc.6600533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kountourakis P., Psyrri A., Scorilas A., Camp R., Markakis S., Kowalski D., Diamandis E. P., and Dimopoulos M. A. (2008) Prognostic value of kallikrein-related peptidase 6 protein expression levels in advanced ovarian cancer evaluated by automated quantitative analysis (AQUA). Cancer Sci. 99, 2224–2229 10.1111/j.1349-7006.2008.00942.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nagahara H., Mimori K., Utsunomiya T., Barnard G. F., Ohira M., Hirakawa K., and Mori M. (2005) Clinicopathologic and biological significance of kallikrein 6 overexpression in human gastric cancer. Clin. Cancer Res. 11, 6800–6806 10.1158/1078-0432.CCR-05-0943 [DOI] [PubMed] [Google Scholar]
- 15. Nathalie H. V., Chris P., Serge G., Catherine C., Benjamin B., Claire B., Christelle P., Briollais L., Pascale R., Marie-Lise J., and Yves C. (2009) High kallikrein-related peptidase 6 in non-small cell lung cancer cells: an indicator of tumour proliferation and poor prognosis. J. Cell Mol. Med. 13, 4014–4022 10.1111/j.1582-4934.2009.00763.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ogawa K., Utsunomiya T., Mimori K., Tanaka F., Inoue H., Nagahara H., Murayama S., and Mori M. (2005) Clinical significance of human kallikrein gene 6 messenger RNA expression in colorectal cancer. Clin. Cancer Res. 11, 2889–2893 10.1158/1078-0432.CCR-04-2281 [DOI] [PubMed] [Google Scholar]
- 17. Petraki C. D., Gregorakis A. K., Vaslamatzis M. M., Papanastasiou P. A., Yousef G. M., Levesque M. A., and Diamandis E. P. (2006) Prognostic implications of the immunohistochemical expression of human kallikreins 5, 6, 10 and 11 in renal cell carcinoma. Tumour Biol. 27, 1–7 10.1159/000090150 [DOI] [PubMed] [Google Scholar]
- 18. Shan S. J., Scorilas A., Katsaros D., and Diamandis E. P. (2007) Transcriptional upregulation of human tissue kallikrein 6 in ovarian cancer: clinical and mechanistic aspects. Br. J. Cancer 96, 362–372 10.1038/sj.bjc.6603556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Diamandis E. P., Scorilas A., Fracchioli S., Van Gramberen M., De Bruijn H., Henrik A., Soosaipillai A., Grass L., Yousef G. M., Stenman U. H., Massobrio M., Van Der Zee A. G., Vergote I., and Katsaros D. (2003) Human kallikrein 6 (hK6): a new potential serum biomarker for diagnosis and prognosis of ovarian carcinoma. J. Clin. Oncol. 21, 1035–1043 10.1200/JCO.2003.02.022 [DOI] [PubMed] [Google Scholar]
- 20. Sawada K., Mitra A. K., Radjabi A. R., Bhaskar V., Kistner E. O., Tretiakova M., Jagadeeswaran S., Montag A., Becker A., Kenny H. A., Peter M. E., Ramakrishnan V., Yamada S. D., and Lengyel E. (2008) Loss of E-cadherin promotes ovarian cancer metastasis via α5-integrin, which is a therapeutic target. Cancer Res. 68, 2329–2339 10.1158/0008-5472.CAN-07-5167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim J. T., Song E. Y., Chung K. S., Kang M. A., Kim J. W., Kim S. J., Yeom Y. I., Kim J. H., Kim K. H., and Lee H. G. (2011) Up-regulation and clinical significance of serine protease kallikrein 6 in colon cancer. Cancer 117, 2608–2619 10.1002/cncr.25841 [DOI] [PubMed] [Google Scholar]
- 22. Ehrenfeld P., Manso L., Pavicic M. F., Matus C. E., Borquez C., Lizama A., Sarmiento J., Poblete M. T., Bhoola K. D., Naran A., and Figueroa C. D. (2014) Bioregulation of kallikrein-related peptidases 6, 10 and 11 by the kinin B1 receptor in breast cancer cells. Anticancer Res. 34, 6925–6938 [PubMed] [Google Scholar]
- 23. Mangé A., Dimitrakopoulos L., Soosaipillai A., Coopman P., Diamandis E. P., and Solassol J. (2016) An integrated cell line-based discovery strategy identified follistatin and kallikrein 6 as serum biomarker candidates of breast carcinoma. J. Proteomics 142, 114–121 10.1016/j.jprot.2016.04.050 [DOI] [PubMed] [Google Scholar]
- 24. Sidiropoulos K. G., Ding Q., Pampalakis G., White N. M., Boulos P., Sotiropoulou G., and Yousef G. M. (2016) KLK6-regulated miRNA networks activate oncogenic pathways in breast cancer subtypes. Mol. Oncol. 10, 993–1007 10.1016/j.molonc.2016.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. White N. M., Mathews M., Yousef G. M., Prizada A., Fontaine D., Ghatage P., Popadiuk C., Dawson L., and Doré J. J. (2009) Human kallikrein related peptidases 6 and 13 in combination with CA125 is a more sensitive test for ovarian cancer than CA125 alone. Cancer Biomark 5, 279–287 10.3233/CBM-2009-0113 [DOI] [PubMed] [Google Scholar]
- 26. Borgoño C. A., and Diamandis E. P. (2004) The emerging roles of human tissue kallikreins in cancer. Nat. Rev. Cancer 4, 876–890 10.1038/nrc1474 [DOI] [PubMed] [Google Scholar]
- 27. Melnikova V. O., Villares G. J., and Bar-Eli M. (2008) Emerging roles of PAR-1 and PAFR in melanoma metastasis. Cancer Microenviron. 1, 103–111 10.1007/s12307-008-0002-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Michel N., Heuzé-Vourc'h N., Lavergne E., Parent C., Jourdan M. L., Vallet A., Iochmann S., Musso O., Reverdiau P., and Courty Y. (2014) Growth and survival of lung cancer cells: regulation by kallikrein-related peptidase 6 via activation of proteinase-activated receptor 2 and the epidermal growth factor receptor. Biol. Chem. 395, 1015–1025 [DOI] [PubMed] [Google Scholar]
- 29. Ghosh M. C., Grass L., Soosaipillai A., Sotiropoulou G., and Diamandis E. P. (2004) Human kallikrein 6 degrades extracellular matrix proteins and may enhance the metastatic potential of tumour cells. Tumour Biol. 25, 193–199 10.1159/000081102 [DOI] [PubMed] [Google Scholar]
- 30. Avgeris M., Mavridis K., and Scorilas A. (2010) Kallikrein-related peptidase genes as promising biomarkers for prognosis and monitoring of human malignancies. Biol. Chem. 391, 505–511 [DOI] [PubMed] [Google Scholar]
- 31. Prezas P., Arlt M. J., Viktorov P., Soosaipillai A., Holzscheiter L., Schmitt M., Talieri M., Diamandis E. P., Krüger A., and Magdolen V. (2006) Overexpression of the human tissue kallikrein genes KLK4, 5, 6, and 7 increases the malignant phenotype of ovarian cancer cells. Biol. Chem. 387, 807–811 [DOI] [PubMed] [Google Scholar]
- 32. Caliendo G., Santagada V., Perissutti E., Severino B., Fiorino F., Frecentese F., and Juliano L. (2012) Kallikrein protease activated receptor (PAR) axis: an attractive target for drug development. J. Med. Chem. 55, 6669–6686 10.1021/jm300407t [DOI] [PubMed] [Google Scholar]
- 33. Sotiropoulou G., and Pampalakis G. (2012) Targeting the kallikrein-related peptidases for drug development. Trends Pharmacol. Sci. 33, 623–634 10.1016/j.tips.2012.09.005 [DOI] [PubMed] [Google Scholar]
- 34. Swedberg J. E., de Veer S. J., and Harris J. M. (2010) Natural and engineered kallikrein inhibitors: an emerging pharmacopoeia. Biol. Chem. 391, 357–374 [DOI] [PubMed] [Google Scholar]
- 35. Bando Y., Hagiwara Y., Suzuki Y., Yoshida K., Aburakawa Y., Kimura T., Murakami C., Ono M., Tanaka T., Jiang Y. P., Mitrovi B., Bochimoto H., Yahara O., and Yoshida S. (2018) Kallikrein 6 secreted by oligodendrocytes regulates the progression of experimental autoimmune encephalomyelitis. Glia 66, 359–378 10.1002/glia.23249 [DOI] [PubMed] [Google Scholar]
- 36. Patra K., Soosaipillai A., Sando S. B., Lauridsen C., Berge G., Møller I., Grøntvedt G. R., Bråthen G., Begcevic I., Moussaud S., Minthon L., Hansson O., Diamandis E. P., White L. R., and Nielsen H. M. (2018) Assessment of kallikrein 6 as a cross-sectional and longitudinal biomarker for Alzheimer's disease. Alzheimers Res. Ther. 10, 9 10.1186/s13195-018-0336-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Scarisbrick I. A., Radulovic M., Burda J. E., Larson N., Blaber S. I., Giannini C., Blaber M., and Vandell A. G. (2012) Kallikrein 6 is a novel molecular trigger of reactive astrogliosis. Biol. Chem. 393, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Scarisbrick I. A., Yoon H., Panos M., Larson N., Blaber S. I., Blaber M., and Rodriguez M. (2012) Kallikrein 6 regulates early CNS demyelination in a viral model of multiple sclerosis. Brain Pathol. 22, 709–722 10.1111/j.1750-3639.2012.00577.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ashby E. L., Kehoe P. G., and Love S. (2010) Kallikrein-related peptidase 6 in Alzheimer's disease and vascular dementia. Brain Res. 1363, 1–10 10.1016/j.brainres.2010.09.017 [DOI] [PubMed] [Google Scholar]
- 40. Hebb A. L., Bhan V., Wishart A. D., Moore C. S., and Robertson G. S. (2010) Human kallikrein 6 cerebrospinal levels are elevated in multiple sclerosis. Curr. Drug Discov. Technol. 7, 137–140 10.2174/157016310793180611 [DOI] [PubMed] [Google Scholar]
- 41. Tatebe H., Watanabe Y., Kasai T., Mizuno T., Nakagawa M., Tanaka M., and Tokuda T. (2010) Extracellular neurosin degrades α-synuclein in cultured cells. Neurosci. Res. 67, 341–346 10.1016/j.neures.2010.04.008 [DOI] [PubMed] [Google Scholar]
- 42. Krishnan R., Zhang E., Hakansson K., Arni R. K., Tulinsky A., Lim-Wilby M. S., Levy O. E., Semple J. E., and Brunck T. K. (1998) Highly selective mechanism-based thrombin inhibitors: structures of thrombin and trypsin inhibited with rigid peptidyl aldehydes. Biochemistry 37, 12094–12103 10.1021/bi980840e [DOI] [PubMed] [Google Scholar]
- 43. Katz B. A., Sprengeler P. A., Luong C., Verner E., Elrod K., Kirtley M., Janc J., Spencer J. R., Breitenbucher J. G., Hui H., McGee D., Allen D., Martelli A., and Mackman R. L. (2001) Engineering inhibitors highly selective for the S1 sites of Ser190 trypsin-like serine protease drug targets. Chem. Biol. 8, 1107–1121 10.1016/S1074-5521(01)00084-9 [DOI] [PubMed] [Google Scholar]
- 44. Katz B. A., Clark J. M., Finer-Moore J. S., Jenkins T. E., Johnson C. R., Ross M. J., Luong C., Moore W. R., and Stroud R. M. (1998) Design of potent selective zinc-mediated serine protease inhibitors. Nature 391, 608–612 10.1038/35422 [DOI] [PubMed] [Google Scholar]
- 45. Liebeschuetz J. W., Jones S. D., Morgan P. J., Murray C. W., Rimmer A. D., Roscoe J. M., Waszkowycz B., Welsh P. M., Wylie W. A., Young S. C., Martin H., Mahler J., Brady L., and Wilkinson K. (2002) PRO_SELECT: combining structure-based drug design and array-based chemistry for rapid lead discovery. 2. The development of a series of highly potent and selective factor Xa inhibitors. J. Med. Chem. 45, 1221–1232 10.1021/jm010944e [DOI] [PubMed] [Google Scholar]
- 46. Bernett M. J., Blaber S. I., Scarisbrick I. A., Dhanarajan P., Thompson S. M., and Blaber M. (2002) Crystal structure and biochemical characterization of human kallikrein 6 reveals that a trypsin-like kallikrein is expressed in the central nervous system. J. Biol. Chem. 277, 24562–24570 10.1074/jbc.M202392200 [DOI] [PubMed] [Google Scholar]
- 47. Gomis-Rüth F. X., Bayés A., Sotiropoulou G., Pampalakis G., Tsetsenis T., Villegas V., Avilés F. X., and Coll M. (2002) The structure of human prokallikrein 6 reveals a novel activation mechanism for the kallikrein family. J. Biol. Chem. 277, 27273–27281 10.1074/jbc.M201534200 [DOI] [PubMed] [Google Scholar]
- 48. Bayés A., Tsetsenis T., Ventura S., Vendrell J., Aviles F. X., and Sotiropoulou G. (2004) Human kallikrein 6 activity is regulated via an autoproteolytic mechanism of activation/inactivation. Biol. Chem. 385, 517–524 [DOI] [PubMed] [Google Scholar]
- 49. Magklara A., Mellati A. A., Wasney G. A., Little S. P., Sotiropoulou G., Becker G. W., and Diamandis E. P. (2003) Characterization of the enzymatic activity of human kallikrein 6: Autoactivation, substrate specificity, and regulation by inhibitors. Biochem. Biophys. Res. Commun. 307, 948–955 10.1016/S0006-291X(03)01271-3 [DOI] [PubMed] [Google Scholar]
- 50. Cohen I., Kayode O., Hockla A., Sankaran B., Radisky D. C., Radisky E. S., and Papo N. (2016) Combinatorial protein engineering of proteolytically resistant mesotrypsin inhibitors as candidates for cancer therapy. Biochem. J. 473, 1329–1341 10.1042/BJ20151410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Salameh M. A., Soares A. S., Navaneetham D., Sinha D., Walsh P. N., and Radisky E. S. (2010) Determinants of affinity and proteolytic stability in interactions of Kunitz family protease inhibitors with mesotrypsin. J. Biol. Chem. 285, 36884–36896 10.1074/jbc.M110.171348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Salameh M. A., Soares A. S., Hockla A., Radisky D. C., and Radisky E. S. (2011) The P(2)′ residue is a key determinant of mesotrypsin specificity: engineering a high-affinity inhibitor with anticancer activity. Biochem. J. 440, 95–105 10.1042/BJ20110788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chao G., Lau W. L., Hackel B. J., Sazinsky S. L., Lippow S. M., and Wittrup K. D. (2006) Isolating and engineering human antibodies using yeast surface display. Nat. Protoc. 1, 755–768 10.1038/nprot.2006.94 [DOI] [PubMed] [Google Scholar]
- 54. Angelini A., Chen T. F., de Picciotto S., Yang N. J., Tzeng A., Santos M. S., Van Deventer J. A., Traxlmayr M. W., and Wittrup K. D. (2015) Protein engineering and selection using yeast surface display. Methods Mol. Biol. 1319, 3–36 10.1007/978-1-4939-2748-7_1 [DOI] [PubMed] [Google Scholar]
- 55. Laskowski M. Jr., and Kato I. (1980) Protein inhibitors of proteinases. Annu. Rev. Biochem. 49, 593–626 10.1146/annurev.bi.49.070180.003113 [DOI] [PubMed] [Google Scholar]
- 56. Bode W., and Huber R. (1992) Natural protein proteinase inhibitors and their interaction with proteinases. Eur. J. Biochem. 204, 433–451 10.1111/j.1432-1033.1992.tb16654.x [DOI] [PubMed] [Google Scholar]
- 57. Copeland R. A. (2016) The drug-target residence time model: a 10-year retrospective. Nat. Rev. Drug Discov. 15, 87–95 10.1038/nrd.2015.18 [DOI] [PubMed] [Google Scholar]
- 58. Gooljarsingh L. T., Fernandes C., Yan K., Zhang H., Grooms M., Johanson K., Sinnamon R. H., Kirkpatrick R. B., Kerrigan J., Lewis T., Arnone M., King A. J., Lai Z., Copeland R. A., and Tummino P. J. (2006) A biochemical rationale for the anticancer effects of Hsp90 inhibitors: slow, tight binding inhibition by geldanamycin and its analogues. Proc. Natl. Acad. Sci. U.S.A. 103, 7625–7630 10.1073/pnas.0602650103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Salameh M. A., Robinson J. L., Navaneetham D., Sinha D., Madden B. J., Walsh P. N., and Radisky E. S. (2010) The amyloid precursor protein/protease nexin 2 Kunitz inhibitor domain is a highly specific substrate of mesotrypsin. J. Biol. Chem. 285, 1939–1949 10.1074/jbc.M109.057216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Copeland R. A. (2010) The dynamics of drug-target interactions: drug-target residence time and its impact on efficacy and safety. Expert Opin. Drug Discov. 5, 305–310 10.1517/17460441003677725 [DOI] [PubMed] [Google Scholar]
- 61. Bradshaw J. M., McFarland J. M., Paavilainen V. O., Bisconte A., Tam D., Phan V. T., Romanov S., Finkle D., Shu J., Patel V., Ton T., Li X., Loughhead D. G., Nunn P. A., Karr D. E., et al. (2015) Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat. Chem. Biol. 11, 525–531 10.1038/nchembio.1817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Longas M. O., and Finlay T. H. (1980) The covalent nature of the human antithrombin III–thrombin bond. Biochem. J. 189, 481–489 10.1042/bj1890481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wilczynska M., Fa M., Karolin J., Ohlsson P. I., Johansson L. B., and Ny T. (1997) Structural insights into serpin-protease complexes reveal the inhibitory mechanism of serpins. Nat. Struct. Biol. 4, 354–357 10.1038/nsb0597-354 [DOI] [PubMed] [Google Scholar]
- 64. Otlewski J., Jelen F., Zakrzewska M., and Oleksy A. (2005) The many faces of protease-protein inhibitor interaction. EMBO J. 24, 1303–1310 10.1038/sj.emboj.7600611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Radisky E. S., and Koshland D. E. (2002) A clogged gutter mechanism for protease inhibitors. Proc. Natl. Acad. Sci. U.S.A. 99, 10316–10321 10.1073/pnas.112332899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Radisky E. S., Kwan G., Karen Lu C. J., and Koshland D. E. (2004) Binding, proteolytic, and crystallographic analyses of mutations at the protease-inhibitor interface of the subtilisin BPN′/chymotrypsin inhibitor 2 complex. Biochemistry 43, 13648–13656 10.1021/bi048797k [DOI] [PubMed] [Google Scholar]
- 67. Kitaguchi N., Takahashi Y., Oishi K., Shiojiri S., Tokushima Y., Utsunomiya T., and Ito H. (1990) Enzyme specificity of proteinase inhibitor region in amyloid precursor protein of Alzheimer's disease: different properties compared with protease nexin I. Biochim. Biophys. Acta 1038, 105–113 10.1016/0167-4838(90)90017-A [DOI] [PubMed] [Google Scholar]
- 68. Smith R. P., Higuchi D. A., and Broze G. J. (1990) Platelet coagulation factor XIa-inhibitor, a form of Alzheimer amyloid precursor protein. Science 248, 1126–1128 10.1126/science.2111585 [DOI] [PubMed] [Google Scholar]
- 69. López-Otín C., and Bond J. S. (2008) Proteases: multifunctional enzymes in life and disease. J. Biol. Chem. 283, 30433–30437 10.1074/jbc.R800035200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhang K., Nelson K. M., Bhuripanyo K., Grimes K. D., Zhao B., Aldrich C. C., and Yin J. (2013) Engineering the substrate specificity of the DhbE adenylation domain by yeast cell surface display. Chem. Biol. 20, 92–101 10.1016/j.chembiol.2012.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. de Veer S. J., Furio L., Swedberg J. E., Munro C. A., Brattsand M., Clements J. A., Hovnanian A., and Harris J. M. (2017) Selective substrates and inhibitors for Kallikrein-related peptidase 7 (KLK7) shed light on KLK proteolytic activity in the stratum corneum. J. Invest. Dermatol. 137, 430–439 10.1016/j.jid.2016.09.017 [DOI] [PubMed] [Google Scholar]
- 72. de Veer S. J., Swedberg J. E., Brattsand M., Clements J. A., and Harris J. M. (2016) Exploring the active site binding specificity of kallikrein-related peptidase 5 (KLK5) guides the design of new peptide substrates and inhibitors. Biol. Chem. 397, 1237–1249 [DOI] [PubMed] [Google Scholar]
- 73. de Veer S. J., Wang C. K., Harris J. M., Craik D. J., and Swedberg J. E. (2015) Improving the selectivity of engineered protease inhibitors: optimizing the P2 prime residue using a versatile cyclic peptide library. J. Med. Chem. 58, 8257–8268 10.1021/acs.jmedchem.5b01148 [DOI] [PubMed] [Google Scholar]
- 74. Craik D. J., Fairlie D. P., Liras S., and Price D. (2013) The future of peptide-based drugs. Chem. Biol. Drug Des. 81, 136–147 10.1111/cbdd.12055 [DOI] [PubMed] [Google Scholar]
- 75. Liebler D. C., and Guengerich F. P. (2005) Elucidating mechanisms of drug-induced toxicity. Nat. Rev. Drug Discov. 4, 410–420 10.1038/nrd1720 [DOI] [PubMed] [Google Scholar]
- 76. Salameh M. A., Soares A. S., Hockla A., and Radisky E. S. (2008) Structural basis for accelerated cleavage of bovine pancreatic trypsin inhibitor (BPTI) by human mesotrypsin. J. Biol. Chem. 283, 4115–4123 10.1074/jbc.M708268200 [DOI] [PubMed] [Google Scholar]
- 77. Edwards P. R., Gill A., Pollard-Knight D. V., Hoare M., Buckle P. E., Lowe P. A., and Leatherbarrow R. J. (1995) Kinetics of protein-protein interactions at the surface of an optical biosensor. Anal. Biochem. 231, 210–217 10.1006/abio.1995.1522 [DOI] [PubMed] [Google Scholar]
- 78. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]