

Abstract
Leishmania species are intracellular protozoan pathogens that have evolved to successfully infect and deactivate host macrophages. How this deactivation is brought about is not completely understood. Recently, microRNAs (miRNAs) have emerged as ubiquitous regulators of macrophage gene expression that contribute to shaping the immune responses to intracellular pathogens. Conversely, several pathogens have evolved the ability to exploit host miRNA expression to manipulate host-cell phenotype. However, very little is known about the mechanisms used by intracellular pathogens to drive changes in host-cell miRNA abundance. Using miRNA expression profiling of Leishmania donovani–infected human macrophages, we show here that Leishmania infection induced a genome-wide down-regulation of host miRNAs. This repression occurred at the level of miRNA gene transcription, because the synthesis rates of primary miRNAs were significantly decreased in infected cells. miRNA repression depended on the host macrophage transcription factor c-Myc. Indeed, the expression of host c-Myc was markedly up-regulated by Leishmania infection, and c-Myc silencing reversed the miRNA suppression. Furthermore, c-Myc silencing significantly reduced intracellular survival of Leishmania, demonstrating that c-Myc is essential for Leishmania pathogenesis. Taken together, these findings identify c-Myc not only as being responsible for miRNA repression in Leishmania-infected macrophages but also as a novel and essential virulence factor by proxy that promotes Leishmania survival.
Keywords: microRNA (miRNA), microRNA biogenesis, Leishmania, protozoan, infection, macrophage, Myc (c-Myc), transcription regulation, virulence factor
Introduction
The leishmaniases are vector borne protozoan infections, widely distributed in tropical and subtropical regions of the world. These chronic and frequently fatal infections affect an estimated 12 million people worldwide with ∼1.3 million new cases each year (1). Leishmania donovani, the focus of this report, causes visceral leishmaniasis with severe morbidity and mortality. There are no effective vaccines, and current treatments have limited efficacy and significant toxicities (2). Truly novel approaches and paradigms are needed to advance drug and vaccine development.
Leishmania infect mononuclear phagocytes in their mammalian hosts where they replicate within phagolysosomes. A central aspect of Leishmania pathogenesis is the modification of macrophage phenotype, where Leishmania interfere with their host-cell gene expression and signal transduction to inhibit macrophage activation, which could lead to their destruction (3–7). How Leishmania modify macrophage phenotype is a focus of intense interest, and one possibility to consider is by targeting host miRNAs.2 miRNAs are small noncoding RNAs that play ubiquitous and important roles in the regulation of gene expression at the post-transcriptional level. Thus, targeting of the host miRNA machinery by Leishmania could constitute a powerful mechanism for modifying host-cell phenotype. By disrupting miRNA expression, Leishmania could act on many target genes simultaneously. In the present study, we used miRNA expression profiling to define the effects of L. donovani on the expression of human macrophage miRNAs and to identify the underlying mechanisms involved.
Here, we show that infection of human monocyte-derived macrophages (HMDMs) with L. donovani brought about the down-regulation of a group of 19 host miRNAs whose genes are widely distributed throughout the genome. Repression of miRNA expression took place at the level of gene transcription as the synthesis rates of primary miRNAs were down-regulated in infected cells. This repression was host transcription factor c-Myc–dependent because the expression of c-Myc itself was markedly up-regulated by infection, and miRNA repression was reversed by c-Myc silencing. To our knowledge, this is the first report to identify a mechanism of how the miRNA machinery is targeted during Leishmania infection. Moreover, and of special interest, c-Myc silencing also brought about a dramatic reduction in the intracellular survival of Leishmania. Taken together, these findings identify c-Myc as not only acting to bring about genome-wide repression of host miRNAs, but also playing an essential role in promoting Leishmania survival. These findings identify c-Myc as a novel virulence factor by proxy contributing to Leishmania pathogenesis.
Results
Leishmania infection represses host miRNAs expression in human macrophages
To profile the expression of miRNAs in the context of Leishmania infection, we used the Nanostring nCounter human v2 miRNA expression assay. HMDMs were infected or not with L. donovani for 24, 48, and 72 h. 800 miRNAs are available for detection in this assay. After background subtraction and normalization of the data, 46 miRNAs (hereafter called the “group of 46”) were reproducibly detected (average > 50 counts) in all samples from all three donors (Fig. 1A). As shown in Fig. 1B, a large majority of these miRNAs (35 of 46) appeared to be down-regulated in infected cells (log2 ratio < 0). The remaining 11 miRNAs did not display a consistent expression in the three donors. Statistical analysis revealed that the expression of 19 miRNAs (hereafter called the “group of 19”) from the group of 46 were significantly down-regulated at one or more of the infection time points (Fig. 1C). Remarkably, no miRNAs were observed to be up-regulated in infected cells.
Figure 1.

miRNAs are largely down-regulated in L. donovani–infected human macrophages. A–C, human monocytes were purified from buffy coats of three healthy donors, differentiated into macrophages, and infected with L. donovani stationary phase promastigotes (Ld) at a multiplicity of infection of 20:1 or left uninfected (Control). RNA was extracted at 24, 48, and 72 h postinfection and sent to Nanostring for miRNA expression analysis. A, heat map (agglomerative cluster) showing z scores of all 46 expressed miRNAs in each of the three donors (D1, D2, and D3). B, log2 ratios of all 46 expressed miRNAs in infected over uninfected cells (mean, n = 3 donors). C, log2 ratios of significantly changed miRNAs in infected over uninfected cells (the group of 19, all down-regulated) (mean, n = 3 donors; one-way ANOVA). D and E, RNA was extracted from infected or uninfected macrophages and analyzed by RT–qPCR using primers for six representative miRNAs, with the RNA U6 as a reference gene. D, data are expressed as log2 of fold change, compared with control cells (means ± S.D., n = 3 donors; one-way ANOVA). E, Pearson correlation between mean expression of miRNAs obtained by RT–qPCR and Nanostring (n = 3 donors each). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
The group of 19 miRNAs is shown in Table 1. Further inspection revealed that the repressed miRNAs came from genes on 11 different chromosomes and 17 different loci (miR-23 and miR-93 are from the same cluster, cluster 9; let-7a-3 and let-7b are from cluster 3). Moreover, all types of miRNA were represented: there were 36% intergenic miRNAs, 52% intronic and 10 exonic, which resembles the general distribution of all miRNAs (∼40% intergenic, 40% introns, 10% exons, and 10% “mixed” miRNAs (1)).
Table 1.
The group of 19 miRNAs found to be down-regulated in Leishmania-infected HMDMs
In bold type are the miRNAs selected for confirmation by qPCR and further study.
| ID | Accession | Genomic location | Type | Host gene | Cluster |
|---|---|---|---|---|---|
| hsa-mir-34a-5p | MI0000268 | Chr1: 9211836 [–] | Intergenic | ||
| hsa-mir-15b-5p | MI0000438 | Chr3: 160122376 [+] | Intron | SMC4 | Cluster 28 |
| hsa-mir-191-5p | MI0000465 | Chr3: 49058142 [–] | Intron | DALRD3 | Cluster 35 |
| hsa-let-7g-5p | MI0000433 | Chr3: 52302377 [–] | Intron | WDR82 | |
| hsa-mir-378a-3p | MI0000786 | Chr5: 149112388 [+] | Intron | PPARGC1B | |
| hsa-mir-25-3p | MI0000082 | Chr7: 99691266 [–] | Intron | MCM7 | Cluster 9 |
| hsa-mir-93-5p | MI0000095 | Chr7: 99691470 [–] | Intron | MCM7 | Cluster 9 |
| hsa-mir-181a-2-5p | MI0000269 | Chr9: 127454721 [+] | Intron | MIR181A2HG | Cluster 31 |
| hsa-let-7d-5p | MI0000065 | Chr9: 96941116 [+] | Intergenic | Cluster 1 | |
| hsa-mir-148b-3p | MI0000811 | Chr12: 54731000 [+] | Intron | COPZ1 | |
| hsa-let-7i-5p | MI0000434 | Chr12: 62997466 [+] | Intergenic | ||
| hsa-mir-15a-5p | MI0000069 | Chr13: 50623337 [–] | Intron | DLEU2 | Cluster 27 |
| hsa-mir-132-3p | MI0000449 | Chr17: 1953302 [–] | Intergenic | Cluster 21 | |
| hsa-mir-142-3p | MI0000458 | Chr17: 56408679 [–] | Intergenic | ||
| hsa-mir-23a-3p | MI0000079 | Chr19: 13947473 [–] | Intergenic | Cluster 45 | |
| hsa-let-7a-3-5p | MI0000062 | Chr22: 46508629 [+] | Exon | MIRLET7BHG | Cluster 3 |
| hsa-let-7b-5p | MI0000063 | Chr22: 46509566 [+] | Exon | MIRLET7BHG | Cluster 3 |
| hsa-mir-98-5p | MI0000100 | ChrX: 53583302 [–] | Intron | HUWE1 | Cluster 6 |
| hsa-mir-223-3p | MI0000300 | ChrX: 65238712 [+] | Intergenic |
To validate the Nanostring results, 6 representative miRNAs from the group of 19 were selected from distinct regions of the genome and their expression was tested by RT–qPCR. The results from qPCR analysis (Fig. 1D) confirmed the down-regulation of miRNAs observed previously, and the Nanostring results and RT–qPCR data were clearly tightly correlated (Fig. 1E).
Moreover, the extracted RNA was also analyzed using an Agilent Bioanalyzer. This showed that despite some differences in the distributions of RNA sizes after infection, there was no detectable down-regulation of all small RNAs (Fig. S1). This eliminated the concern that what we observed in infected cells might be nonspecific.
To explore how the observed down-regulation in miRNA expression could affect host macrophage phenotype in the context of infection, we searched for predicted target mRNAs of the group of 19 miRNAs, using TargetScan (2). The 2950 predicted targets were then analyzed for Gene Ontology (GO) and KEGG pathways (Fig. S2). In the GO term enrichment analysis, the most enriched biological processes were “biological regulation,” “metabolic process,” and “response to stimulus,” closely followed by “cell communication,” which make intuitive sense when considered in the context of a response to an infection (Fig. S2A). With regard to cellular compartments, interestingly “membrane” was the most enriched, consistent with the role of membrane proteins in uptake of Leishmania and also in the initiation of cell signaling responses (Fig. S2B). Among the significantly enriched KEGG pathways (Fig. S2D), a number of cellular pathways previously implicated in Leishmania pathogenesis were identified, such as the “MAPK signaling pathway,” “regulation of actin cytoskeleton,” and “endocytosis.” Based upon this analysis, it became clear that miRNA repression by Leishmania has the potential to profoundly affect macrophage cell regulation and host defense and that understanding the mechanisms involved is an important objective.
Leishmania infection reduces the transcription rate of macrophage primary miRNAs
Based on these striking results, we aimed at understanding the regulatory mechanism taking place in infected cells and leading to this genome-wide repression of miRNAs. To the best of our knowledge, such genome-wide repression of host miRNAs as we observed here has not previously been reported in the context of infection by any other nonviral intracellular pathogens, be they protozoan or bacterial. It is noteworthy that in a model of human cytomegalovirus infection, the stability of miR-17-92 cluster miRNAs was reduced by a viral noncoding RNA, accounting for their reduced abundance (3). Otherwise, global decreases in miRNA expression have been associated with several types of cancers. In these studies, the down-regulation was attributed variably to mutations or deletions in the miRNA processing proteins Dicer and Drosha (4–7), to hypermethylation of miRNA genes (8), to mutations or suppression of the protein Exportin-5 (9, 10), or to transcriptional repression of miRNA genes by oncogenic transcription factors (11).
The findings that the down-regulated miRNAs were very diverse in terms of the chromosome localization of their genes and loci and their type, suggested to us that it was worthwhile to look for a regulatory mechanism that might be common to all of these miRNAs. One potential explanation for the observed genome-wide down-regulation of miRNAs could be that one or more of the proteins involved in miRNA processing (Fig. S3A) was affected by infection: down-regulated, inhibited, or sequestered in a compartment other than where it acts. First, we investigated the abundance of the major regulatory proteins involved in the biosynthesis of miRNAs. The expression of the proteins DGCR8, Dicer, TRBP, and Ago2 in fact were all unchanged by infection, both at 24 and 48 h (Fig. S3, B and C). Surprisingly, Drosha was unchanged at 24 h but was strongly up-regulated at 48 h postinfection.
Drosha and DGCR8 need to be present in the nucleus to process primary miRNAs (pri-miRNAs); therefore, we examined whether Leishmania infection might block the nuclear localization of these proteins. Using confocal microscopy, the nuclear versus cytosolic localization of the proteins was measured in infected and noninfected cells, and no differences were observed (Fig. S3, D and E). Taken together, these findings indicate that broad-based down-regulation of miRNAs by Leishmania infection cannot be explained either by decreased expression of miRNA-processing proteins or by restricted cytosolic localization of Drosha and DGCR8, such that they are not available for nuclear processing of pri-miRNAs.
An alternative mechanism to explain down-regulation of mature miRNAs in infected cells could be faster rates of miRNA decay. To examine whether the stabilities of miRNAs were reduced in the context of infection, actinomycin D was used to inhibit transcription. RNA from both control and infected cells (24 h of infection) was then collected at 0, 6, and 24 h postaddition of actinomycin D, and RT–qPCR was done to measure the decay rates of the six representative miRNAs. Fig. 2A shows the amount of each miRNA at each time point in control and infected cells ± actinomycin. The slope for each curve was then calculated, and these slope values are shown in Fig. 2B. Inspection of these results led to several conclusions. First of all, as expected, the slopes of actinomycin D–treated control cells were lower (more negative) than those in DMSO-treated control cells, showing the decay of miRNAs when their synthesis is blocked and validating this model. Second, when comparing actinomycin D–treated samples, thereby looking only at decay, the slopes were similar between control and infected cells, indicating that miRNAs did not have faster decay rates in infected cells. Finally, when control and infected cells were treated only with DMSO, therefore, taking into account both synthesis and decay, the slopes were lower in infected cells as compared with control cells. By default, this pointed toward a de facto defect in miRNA synthesis in infected cells, because as discussed above, decay rates per se were not changed by infection. Taken together, these results showed no differences in the stabilities of mature miRNAs in the context of infection but rather identified a defect in synthesis of miRNAs in infected cells.
Figure 2.

The stability of mature miRNAs is not decreased in infected macrophages. A, human monocyte-derived macrophages were either infected (Ld) or not for 24 h. Subsequently, the cells were incubated with actinomycin D (Act D) to inhibit transcription, allowing us to look at decay rates of mature miRNAs, whereas DMSO was used as a control. RNA was collected after 6 and 24 h of treatment, and RT–qPCR was performed on the six representative miRNAs. Represented here is the fold change of miRNA expression at each time point, normalized to DMSO treatment (means ± S.D., n = 4 donors). B, the slope of each individual curve was measured and is presented here (means ± S.E.). Statistical analysis was done using a two-way ANOVA comparing each treatment. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
To examine further whether the synthesis of miRNAs was down-regulated in infected cells, the levels of five pri-miRNAs and five precursor miRNAs (pre-miRNAs) from the group of 19 were assessed by RT–qPCR. The results indeed showed that pri-miRNAs and pre-miRNA levels were down-regulated in infected cells (Fig. 3, A and B, respectively), as was earlier shown be the case for the levels of their cognate mature miRNAs (Fig. 1D). This observation was found to be the case for all pri- and pre-miRNAs examined at 24 h and a majority of them at 48 h postinfection. These findings indicated a defect in transcription rates of pri-miRNAs as contributing to the genome-wide down-regulation of mature miRNAs in infected cells.
Figure 3.

Infected macrophages display down-regulated pri-miRNAs and pre-miRNAs, and lower transcription rates of pri-miRNAs. Monocytes were purified, differentiated into macrophages, and infected with L. donovani (Ld) for 24 and 48 h. A and B, total RNA was extracted and analyzed by RT–qPCR using primers for five pri-miRNAs and five pre-miRNAs. The data are represented as log2 fold change in infected cells compared with uninfected cells (means ± S.D., n = 4 donors; two-tailed t test). A, pri-miRNAs. B, pre-miRNAs. C–F, Leishmania-infected [Ld] (24 h) and control (C) cells were treated with ethynyl uridine for 15, 30, and 60 min. Total RNA was collected, and Click-iT technology was used to pull down nascent RNA. RT–qPCR was performed on total and nascent RNA for four pri-miRNAs (pri-miR-98 was not detected consistently in nascent RNA) and three control genes (mean, n = 4 donors). C and D, newly synthesized RNAs (fold change, normalized to total RNA and t = 0) in control and infected cells (mean, n = 4 donors). E and F, the slope of the synthesis rate was assessed for each gene in control and infected cells (means ± S.D., n = 4 donors; two-tailed t test). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To examine directly the transcription rates of pri-miRNAs, RNA in infected and control cells was metabolically labeled using ethynyl uridine (EU) and then collected after 15, 30, and 60 min of EU treatment. The newly synthesized RNA (labeled with EU) was pulled down using Click-iT technology and analyzed by RT–qPCR. This allowed us to look in real time at the synthesis rates of four pri-miRNAs (pri-miR-98 was not detected in nascent RNA) and three control RNAs (Fig. 3, C and D). The slopes of these curves were measured for each gene and each donor, in infected and control cells, and are presented normalized to control cells (Fig. 3, E and F). The miRNAs selected for this experiment were of different transcriptional types: miR-15a and miR-15b are intronic miRNAs (their transcription depends on their cognate host gene), let-7a is an exonic miRNA, and miR-34a is intergenic, in which case miRNAs are transcribed on their own. Despite the variability between donors, the transcription rates were reduced by half in infected cells for all four pri-miRNAs without regard to their transcriptional type (Fig. 3E). This experiment shows that the transcription rates of pri-miRNAs were significantly lower in infected cells as compared with control cells.
The main factors that could explain decreased transcription rates are either epigenetic control or transcription factors being differentially expressed. In some cancer cells, miRNAs have been shown to be repressed by CpG island hypermethylation (12, 13). In analyzing the data from an article that looked at DNA methylation changes in Leishmania-infected macrophages (14), we found that the 125 sites linked to the group of 19 miRNAs identified in our study did not change in their methylation status in response to infection (confirmed by A. Marr et al.)3 (Fig. S4). This suggests that the transcription of pri-miRNA in infected cells was not inhibited by any changes in DNA methylation.
The host transcription factor c-Myc is up-regulated during Leishmania infection and is linked to miRNA repression
Reduced transcription rates of pri-miRNAs in Leishmania-infected cells could have been due to aberrant activity of a transcription factor, and c-Myc was of particular interest. C-Myc has pleiotropic effects on cell growth, proliferation, differentiation, and apoptosis, mediated by its ability to regulate—either positively or negatively—the expression of thousands of genes including many miRNAs (15). Moreover, c-Myc was shown to participate in the global down-regulation of miRNAs observed in lymphoma cells (11). Fig. 4A shows the miRNAs that are known to be either induced or repressed by c-Myc. In regard to the miRNAs that have been shown to be repressed by c-Myc, 11 of them (highlighted in bold in Fig. 4A) belong to the group of 19 miRNAs that were down-regulated by Leishmania infection: let-7g, let-7d, let-7i, let-7a, let-7b, miR-223, miR-23a, miR-15a, miR-181a, miR-34a, and miR-98 (11, 16) (Fig. 4A). Moreover, c-Myc is known to induce Drosha (17), the expression of which was markedly up-regulated at 48 h postinfection (Fig. S3C). Finally, c-Myc plays a role in alternative activation of macrophages (18), which is recognized to promote Leishmania survival (19). Based upon these considerations, we investigated whether c-Myc might be responsible for the down-regulation of miRNAs observed in infected cells.
Figure 4.

c-Myc is up-regulated in infected macrophages. A, schematic representation of either c-Myc repression or induction of miRNAs as reported in the literature (11, 16). In bold are shown the miRNA members of the group of 19 down-regulated by Leishmania infection. B, HMDMs were infected or not for 24 and 48 h, and Leishmania-infected (Ld) and control (C) cells were analyzed by Western blotting with probes for c-Myc. Densitometry analysis was used to measure c-Myc expression, normalized to actin (means ± S.D., n = 4 donors; two-tailed t test. **, p < 0.01. C, HMDMs were infected for 24 h, and both control and infected cells were treated with cycloheximide for 20, 40, and 60 min. Western blotting was performed to assess the decay rates of c-Myc, followed by densitometry analysis, and normalization to actin (means ± S.D., n = 3 donors; nonlinear regression fit).
Using Western blotting, we found that c-Myc was strongly up-regulated in infected cells, both at 24 and 48 h postinfection (Fig. 4B). c-Myc is a highly unstable protein and is regulated mostly through alterations of its stability (20). Using cycloheximide to block translation, we established that the stability of c-Myc is increased in infected cells, accounting at least in part for its increased abundance (Fig. 4C).
To examine the possibility that c-Myc induction in infected cells could result in the repression of transcription, the levels of classical targets of c-Myc repression were assessed by RT–qPCR. c-Myc is known to repress transcription of the cyclin-dependent kinase inhibitors CDKN1A (p21CIP1/WAF1), CDKN1B (p27KIP1), and CDKN2B (p15INK4B) (21–24). Indeed, all three cyclin-dependent kinase inhibitors were down-regulated in infected cells, consistent with c-Myc induction bringing about the down-regulation of c-Myc–repressed targets (Fig. S5, C–E).
Transcriptional repression by c-Myc has been described to take place indirectly through interactions with two zinc finger transcription factors, Miz-1 and Sp1 (25–27) (Fig. S5A). These factors bind to core promoters and stimulate transcription in the absence of c-Myc. Binding of c-Myc interferes with transcriptional activation by Miz-1 and Sp1. It is believed that c-Myc may also repress transcription through interactions with other zinc finger transcription factors, such as nuclear factor Y (NF-Y) (28), yin yang 1(YY1) (29), and transcription factor II-I (TFII-I) (30). Moreover, the c-Myc homolog N-Myc was shown to form a repression complex with both Sp1 and Miz-1 and to recruit the histone deacetylase HDAC1 to induce a repressed chromatin state at the TRKA and p75NTR promoters (31) (Fig. S5B). According to the BIOGRID database of protein–protein interactions, c-Myc also interacts with Sp1, Miz-1, and HDAC1 and could therefore function similarly (MYC result summary in BioGRID database; https://thebiogrid.org/110694/summary/homo-sapiens/myc.html).4
To examine this possibility, promoters of the group of 19 miRNAs (for intergenic miRNAs) or their host genes (for intronic miRNAs) were analyzed computationally to determine whether c-Myc could regulate their transcription through one of the mechanisms discussed above. Of note, two pairs of the 19 miRNAs are from the same loci, expressed under the same promoter (miR-25 and miR-93, let-7a-3 and let-7b); therefore, a total of 17 distinct promoters were analyzed (Table S1). First, we predicted binding sites in all 17 of the promoters for Miz-1, in 16 of 17 for Sp1, and in only 7 for c-Myc (Fig. 5A). Next, we explored whether the computational predictions were supported by in vivo binding data from ChIP-seq. ChIP-seq data indicated that individually, c-Myc, Sp1, and Miz-1 bound respectively to 16, 15, and 14 of the 17 promoters (Fig. 5B). Therefore, a large majority of our promoters have binding sites for and are bound in vivo by Sp1 and Miz-1, which supports our model of direct binding of these two transcription factors to the promoters. Moreover, it is worth noting that although only 7 promoters of 17 have a predicted binding site for c-Myc (Fig. 5A), 16 of them were shown to bind c-Myc in vivo (Fig. 5B), supporting the hypothesis of indirect binding of c-Myc to the various promoters. We then looked at whether ChIP-seq data for c-Myc and the other two transcription factors overlapped in the promoters, which would support the model in which they work as complexes. Indeed, c-Myc and Sp1 ChIP-seq data overlapped at 15 of the promoters, whereas overlaps occurred at 14 of the promoters for c-Myc and Miz-1 and at 12 of the promoters for all three transcription factors (Fig. 5C). Altogether, our analysis supports a model in which c-Myc binds indirectly to the repressed promoters through complexes with either Sp1 or Miz-1 (or both) and acts as a transcriptional repressor of the group of 19.
Figure 5.
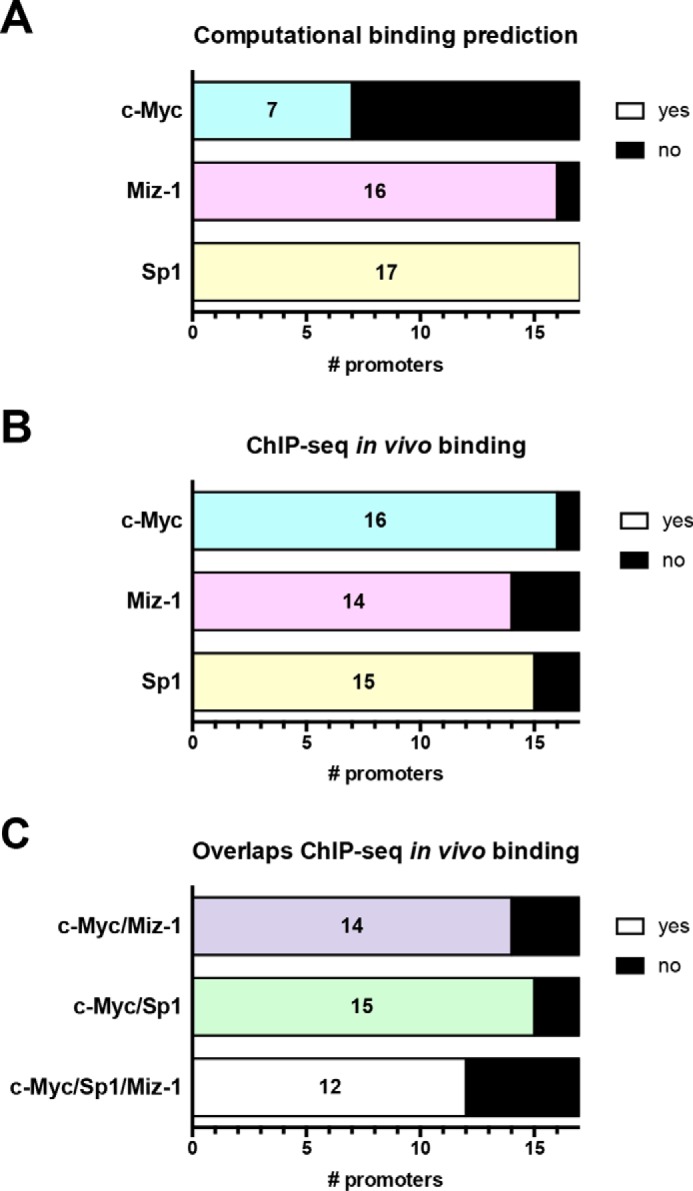
Computational binding predictions and in vivo binding data from ChIP-seq support an indirect model of repression by c-Myc through Sp1 and Miz-1. A, the promoters of miRNAs (for intergenic miRNAs) or their host genes (intronic miRNAs) (see Table S1) from the group of 19 were analyzed for computational binding prediction of c-Myc, Miz-1, and Sp1. B, using ChIP-seq data, in vivo binding for the three transcription factors was analyzed in all 17 promoters. C, the promoters were analyzed for overlaps between c-Myc and Miz-1, c-Myc, and Sp1 and c-Myc/Miz-1/Sp1 ChIP-seq data. A–C, the numbers indicate the number of promoters with a predicted binding site or overlapping ChIP-seq data.
To seek evidence to link c-Myc to miRNA repression, macrophages were incubated with either control (scrambled) or c-Myc siRNAs and then infected with Leishmania for 24 h. c-Myc siRNAs did not affect cell viability or infection rates (Fig. S6, A and B). The cells were then lysed, and Western blotting was used to confirm knockdown of c-Myc. Three c-Myc siRNAs were used: siRNA-A had a negligible effect, whereas both siRNA-B and siRNA-C down-regulated c-Myc to the same extent in control cells, but in infected cells siRNA-C was more efficacious that siRNA-B (Fig. 6A). In parallel, RNA was collected, and RT–qPCR was performed to assess the levels of the six representative miRNAs from the group of 19. c-Myc siRNA-B led to a partial recovery of miRNA levels in infected cells, whereas c-Myc siRNA-C induced a strong up-regulation of the miRNAs of interest (Fig. 6B). These results show that c-Myc levels and miRNA levels were inversely correlated: siRNA-B caused a modest down-regulation of c-Myc and modest increases in miRNAs, whereas siRNA-C caused a strong down-regulation of c-Myc along with robust recoveries of miRNA levels. Thus, knockdown of c-Myc protein abrogated the repression of miRNA levels caused by infection.
Figure 6.

c-Myc knockdown and inhibition reverse miRNA down-regulation and Drosha up-regulation. A, HMDMs were treated for 48 h with control (Scrambled) and c-Myc siRNAs (siRNA A, B, and C), followed by infection for 24 h, lysis, and analysis by Western blotting for c-Myc and actin. Densitometry analysis results are shown below (means ± S.D., n = 3 donors; two-tailed t test). B, RNA was collected from siRNA-treated cells (siRNA B and C), both infected (Ld) and control, and RT–qPCR was performed to assess the expression of U6 and six miRNAs. Levels of miRNA expression are depicted as log2 fold change (FC) compared with scrambled and normalized to U6 (means ± S.D., n = 3 donors; two-way ANOVA comparing each condition). C, HMDMs were treated with c-Myc inhibitor 10058-F4 (F4, 25 or 50 μm) or DMSO for 24 h and then infected for 24 h. Drosha expression was assessed by Western blotting (means ± S.D., n = 3 donors; two-tailed t test). D, RNA was extracted from 10058-F4–treated cells, both infected (Ld) and control cells, and analyzed by RT–qPCR. The expression levels of six miRNAs are depicted as log2 fold change compared with DMSO and normalized to U6 (means ± S.D., n = 4 donors; two-way ANOVA). *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
To confirm this link between c-Myc and miRNA repression, we used an orthogonal approach consisting of a c-Myc specific inhibitor, compound 10058-F4. The latter blocks c-Myc/MAX interactions and also down-regulates the expression of c-Myc after prolonged exposure (33), which was confirmed with our cells (Fig. S6C). The cells were treated with either 25 or 50 μm 10058-F4 and then infected for 24 h, after which RT–qPCR was performed to assess the levels of the six representative miRNAs from the group of 19. Treatment with 10058-F4 did not affect macrophage viability, infection rates, or Leishmania viability (Fig. S6, D–F). As expected, inhibiting c-Myc led to a strong down-regulation of Drosha in both infected and uninfected cells (Fig. 6C). Concerning miRNA abundance, uninfected cells treated with 50 μm of 10058-F4 displayed higher levels of miRNAs than untreated cells (Fig. 6D). Most importantly, infected cells treated with either 25 or 50 μm of 10058-F4 showed levels of miRNAs similar to uninfected cells (Fig. 6D). These findings show that the inhibition of c-Myc with 10058-F4 reversed the down-regulation of miRNAs caused by infection.
Down-regulation of c-Myc attenuates Leishmania survival
The results above linked c-Myc to Leishmania–mediated down-regulation of miRNAs (Fig. 6, B and D). Given this broad-based, c-Myc–dependent repression of host-cell miRNAs, it seemed likely that this would have pleiotropic and important effects on macrophage phenotype. In particular, this raised the question of what impact this might have on the intracellular survival of Leishmania. Furthermore, it seemed likely that the up-regulation of c-Myc itself—independent of any effects on miRNA repression—would have consequences for macrophage biology, especially considering that c-Myc plays a role in macrophage alternative activation (33), which is recognized to promote Leishmania survival (18).
We therefore assessed Leishmania intracellular survival in macrophages treated with either c-Myc siRNAs or c-Myc inhibitor 10058-F4. Silencing of c-Myc in HMDMs by siRNAs decreased Leishmania counts by half (Fig. 7A). Similarly, inhibition of c-Myc by 10058-F4 led to a 10-fold reduction in parasite counts (Fig. 7B). Together, these results show that c-Myc is essential to the intracellular survival of Leishmania.
Figure 7.
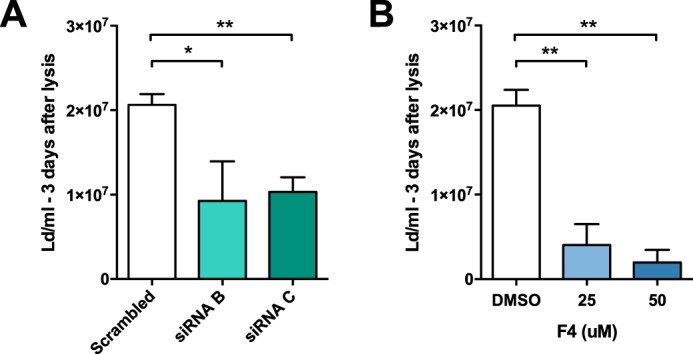
c-Myc knockdown and inhibition attenuate Leishmania survival. A, HMDMs were treated for 48 h with control (Scrambled) and c-Myc siRNAs (siRNA B and C), followed by infection for 24 h. A parasite rescue assay as described under “Experimental procedures” was performed (means ± S.D., n = 3 donors; two-tailed t test). B, HMDMs were treated with c-Myc inhibitor 10058-F4 (F4, 25 or 50 μm) or DMSO for 24 h and then infected for 24 h. A parasite rescue assay was performed (means ± S.D., n = 4 donors; two-tailed t test). *, p < 0.05; **, p < 0.01.
Discussion
The results presented above identify c-Myc as a novel Leishmania virulence factor by proxy. c-Myc is hijacked by Leishmania to drive genome-wide repression of host miRNAs (Table 1 and Fig. 1) and is essential for parasite survival (Fig. 7). Moreover, this is the first elucidation of a mechanism leading to miRNA modulation during Leishmania infection, whereby the parasites hijack a host transcription factor, resulting in transcriptional repression and ultimately down-regulation of many miRNAs in the infected cell.
A limited number of other studies have investigated the impact of Leishmania infection on macrophage miRNA expression, looking at different species of Leishmania and different host-cell types (34–38). Each of these studies identified both up- and down-regulated miRNAs, but there was no commonality among them. Also, no mechanism was identified to explain these changes. In this report, we show that host miRNA expression is broadly down-regulated by infection with L. donovani, with a group of 19 miRNAs being significantly down-regulated in infected cells (Fig. 1). Our findings that the down-regulated miRNAs in the group of 19 were diverse in terms of chromosomal localization of their genes and loci and their types (Table 1) suggested that this likely involved a novel and versatile mechanism of gene repression that is activated in Leishmania-infected cells. In pursuit of such a mechanism, we found that the down-regulation of miRNAs in Leishmania-infected cells was not due to any change in either the abundance or subcellular localization of critical proteins involved in miRNA biogenesis (Fig. S3). Neither could this be accounted for by decreased stability of mature miRNAs (Fig. 2). On the other hand, levels of both pri- and pre-miRNAs were decreased in infected cells in concert with their cognate mature miRNAs (Fig. 3, A and B). Consistent with these findings, the transcription rates of pri-miRNAs were significantly repressed by Leishmania infection (Fig. 3E).
These findings raised the specter of a potential role for a transcriptional repressor activated by Leishmania, and for a number of reasons our search for such a candidate led us to c-Myc. First, it is becoming evident that a predominant consequence of activation of c-Myc is widespread repression of miRNA biosynthesis (11). Consistent with this, we found that 11 members of the group of 19 miRNAs had previously been shown to be repressed by c-Myc in other models (Fig. 4A) (11, 16). Second, c-Myc is known to induce Drosha (39), the expression of which was markedly up-regulated at 48 h postinfection (Fig. S3C). Third, c-Myc is known to promote alternative activation of macrophages. In fact, c-Myc controls the induction of a subset of genes associated with alternative activation of macrophages (18), and the latter is recognized to promote Leishmania survival (19). Taken together, these findings made c-Myc an excellent candidate for repression of miRNA expression in our system. Indeed, we found that c-Myc expression itself was dramatically increased in Leishmania-infected cells (Fig. 4B), seemingly by increasing the stability of c-Myc (Fig. 4C). Furthermore, using either c-Myc inhibitor 10058-F4 or siRNAs to silence c-Myc, we were able to rescue miRNA expression in infected cells and restore them to control levels (Fig. 6, B and D).
These findings establish a direct link between Leishmania-mediated up-regulation of host c-Myc and repression of select miRNAs (Figs. 4 and 5). c-Myc is thought to regulate miRNAs through both transcriptional and post-transcriptional mechanisms (11, 40). Here, the results suggest that the genome-wide down-regulation of miRNAs seen in infected cells is due to transcriptional regulation by c-Myc (Fig. 3). Although the mechanisms by which c-Myc activates gene transcription are well known, less is known about how c-Myc represses transcription of target genes, including miRNAs. It is thought that c-Myc indirectly represses transcription by interacting with the zinc finger transcription factors Sp1 and Miz-1, which bind to core promoters (21, 26, 31). The potential roles of Miz-1 and Sp1 were strongly inferred by bioinformatic analysis of the promoters of the group of 19 miRNAs (Table S1). Indeed, direct binding of Sp1 and Miz-1 to promoters was supported both by computational prediction and in vivo ChIP-seq data (Fig. 5, A and B). On the other hand, indirect binding of c-Myc was suggested by ChIP-seq results, in the absence of binding sites for c-Myc in a majority of the various promoters. Finally, overlaps between c-Myc and the two zinc finger transcription factors were highly prevalent among the group of 19 promoters, supporting the hypothesis that they work as complexes (Fig. 5C). Altogether, these results support a model in which c-Myc binds indirectly to the promoters through Miz-1 and/or Sp1 to repress the transcription of pri-miRNAs.
A number of miRNAs are known to be induced by c-Myc (41) (Fig. 4A), and interestingly, none of these were increased upon Leishmania infection. This was the case despite the finding that c-Myc itself was increased (Fig. 4B) and suggests that an additional mechanism(s) regulates their expression. Furthermore, although c-Myc acts as a regulator of miRNAs, its expression itself can be repressed in turn by a number of miRNAs (42). Notable among these is miR-34a, which was down-regulated in Leishmania-infected cells (Fig. 1), perhaps explaining the reciprocal increase in c-Myc we observed (Fig. 4B).
It is informative to consider how this c-Myc–driven strategy may otherwise be used by Leishmania to affect cell regulation leading to altered macrophage phenotype. The specific effects of miRNA repression in Leishmania-infected cells are not known at this time. However, in cancer cells, it has been hypothesized that the global down-regulation of miRNAs contributes to neoplastic transformation by allowing for increased expression of critical proteins with oncogenic potential (43). A Gene Ontology analysis on the multiple putative target mRNAs of the group of 19 provided further insight into the impact on host cells of miRNA repression by Leishmania (Fig. S2). This exercise identified thousands of putative target genes for the group of 19, playing roles in biological regulation, metabolic processes, and several signaling pathways, suggesting that c-Myc–dependent miRNA repression in Leishmania-infected cells is highly likely to lead to pleiotropic effects on macrophage cell regulation and cell phenotype that promote persistent infection.
In addition to the discovery that c-Myc functions as a transcriptional miRNA repressor in Leishmania-infected cells is the parallel discovery reported here that c-Myc is a novel Leishmania virulence factor by proxy that is essential for parasite survival. This conclusion is based on the findings that either c-Myc silencing or its inhibition brought about dramatic reductions in the intracellular survival of Leishmania (Fig. 7, A and B). Taken together, these findings identify c-Myc as playing an essential role in promoting Leishmania survival.
The novelty of this finding is highlighted by a review of the literature searching for c-Myc and infection of human macrophages by intracellular pathogens. This revealed only one report in which c-Myc was found to be up-regulated in primary macrophages infected with several species of mycobacteria (Bacillus Calmette-Guérin, Mycobacterium avium, Mycobacterium chelonae, or Mycobacterium kansasii) (44). However, this was associated with suppression of M. avium survival, the converse of what we report here where c-Myc promoted Leishmania survival.
As mentioned earlier, in the course of these studies, our attention was directed to c-Myc partly because it is a marker of M2 macrophages and is required for alternative activation of macrophages (18). In contrast, c-Myc is inhibited by the pro-inflammatory agonists lipopolysaccharide and interferon-γ, which induce classically activated (M1) macrophages (45). Although macrophages exist in a continuum between M1 and M2 activation profiles, a clear correlation exists in Leishmania infection, in which classical activation is linked to enhanced microbicidal functions (production of TNFα, nitric oxide, and reactive oxygen species) and parasite killing, whereas alternative activation leads to parasite survival and disease progression (19). This suggests the hypothesis that the induction of c-Myc is part of Leishmania-induced alternative activation pathway of macrophages, favoring parasite survival. Conversely, when c-Myc is inhibited by 10058-F4 or siRNAs, this may block alternative activation of macrophages and in turn suppress Leishmania survival (Fig. 7, A and B).
In summary, the results of the present study identify for the first time c-Myc as a novel Leishmania virulence factor by proxy. This host transcription factor c-Myc is induced by infection and appears to bring about genome-wide repression of host miRNAs. c-Myc is also shown to be essential to support persistent infection. Given the central role of c-Myc as an oncogene, it has been and continues to be the focus of many attempts to develop inhibitors for therapeutic purposes in cancer (46). We anticipate that it should be possible to capitalize on this work in the cancer clinic to identify novel therapeutics for leishmaniasis.
Experimental procedures
Ethics statement
Buffy coats from healthy donors were obtained from Canadian Blood Services Network Centre for Applied Development with review and approval from Canadian Blood Services Research Ethics (reference 2015.035). The source of human cells did not disclose the identity of the human donors, thereby anonymizing all data to the investigators. This study abides by the Declaration of Helsinki principles.
All work with animal in this study was reviewed and approved by the University of British Columbia Animal Care Committee (protocol license number A14-0218). The animal care and use protocol adhered to the standards and regulations provided by the Canadian Council on Animal Care in Science.
Purification and culture of human cells
The monocytes were enriched from buffy coats by a centrifugation in Ficoll gradient (GE Healthcare) followed by thorough washings in PBS. The cells were then incubated for 1 h at 37 °C in 5% CO2 in culture flasks (Corning), and nonadherent cells were washed away with HBSS. Adherent cells were collected using a cell scraper, and monocytes were counted using Tuerk's solution. The cells were plated at a density of 0.75 × 106 cells/ml in multiwell plates in RPMI medium supplemented with 10% fetal bovine serum (Gibco), 2 mm l-glutamine (StemCell), and 100 units/ml penicillin/streptomycin (StemCell). Differentiation into macrophages was induced by addition of 10 ng/ml GM–CSF (StemCell). After 3 days, the cells were washed with HBSS and given fresh supplemented RPMI medium containing GM–CSF. The cells were washed again on day 6, rested in supplemented RPMI medium without GM–CSF, and used for experiments on day 7.
Parasite culture and infections
L. donovani Sudan strain S2 was obtained from Dr. Kwang Poo Chang (Rockefeller University, New York, NY). Parasite virulence was maintained by regular passages through Syrian golden hamsters. Promastigotes were routinely cultured in the lab and passaged every 3 days for a maximum of 20–25 passages. Promastigotes were cultured at 26 °C in M199 (Sigma–Aldrich) supplemented with 10% heat-inactivated fetal bovine serum (Gibco), 10 mm HEPES (Stemcell), 6 μg/ml hemin (Sigma–Aldrich), 10 μg/ml folic acid (Sigma–Aldrich), 2 mm l-Glutamine (Stemcell), 100 units/ml penicillin/streptomycin (Stemcell), and 100 mm adenosine (Sigma–Aldrich).
Infection was performed with day 5 stationary phase promastigotes. Briefly, the cells were counted, washed in HBSS, resuspended in HBSS, and used to infect human cells. Infection rate and parasite rescue were assessed as described previously (47).
RNA extraction
For mature miRNAs studies, RNA was collected from cell pellets using the miRCURY RNA isolation kit Cell and Plant (Exiqon), known to enrich for small RNAs, following the manufacturer's instructions. Total RNAs (for primary and precursor miRNAs, control mRNAs, and c-Myc targets) were extracted using GeneJET RNA purification kit (Thermo), following the manufacturer's instructions.
The RNA was eluted in nuclease-free water, and the concentration of RNA was determined by nanodrop. A DNase treatment was performed with DNase I in solution (Thermo Scientific). 1 μg of eluted RNA was treated with 1 unit of DNase I in a total volume of 15 μl and incubated for 30 min at 37 °C before addition of 1 μl of 50 mm EDTA and incubation at 65 °C for 10 min.
Nanostring assay, data analysis, and mRNA target prediction
Purified RNA, extracted with the Exiqon kit was diluted to 50 ng/μl with nuclease-free water and sent to Nanostring (Seattle, WA) for analysis of the expression of 800 human miRNAs with the Nanostring nCounter human v2 miRNA expression assay. Data analysis was performed using the nSolver2.0 Analysis software (Nanostring). Background subtraction was done using the mean of the negative controls +2 standard deviations. The data were normalized by computing a normalization factor using the geometric mean of all genes except housekeeping genes. 46 miRNAs were detectably expressed in all samples (average > 50 counts). Agglomerative clustering (heat map) of the 46 expressed miRNAs was generated using nSolver's built-in analysis feature, applying linkage type “average” and metric type “Euclidean distance.”
Target prediction was performed for the miRNAs that showed a significantly (p < 0.05) different expression in infected cells. The validated target module of the TargetScan (48) (minimum of one 8-mer binding site, only targets with conserved binding sites) were used to predict targets for the 19 significantly down-regulated miRNAs. The list of target genes was further analyzed with the WEB-based gene set analysis toolkit (WebGestalt) (49) to look for enrichment of KEGG pathways and GO terms. The KEGG pathway enrichment analysis was performed using the homosapiens genome as a reference gene set, the hypergeometric statistical method with the Benjamin and Hochberg multiple test adjustment, retrieving the top 10 enriched pathways with a minimum of 5 genes per category. This analysis was expanded by setting the p value at <0.01 as a threshold. The GO slim analysis was performed using WebGestalt's default settings.
Quantitative real-time PCR
For mature miRNAs, 10 ng of DNase-treated RNA (extracted with the Exiqon kit) were used for cDNA synthesis using the universal cDNA synthesis kit II (Exiqon) according to the manufacturer's instructions. The resulting cDNA was diluted 80× in nuclease-free water, and ROX reference dye (Life Technologies) was added, at a dilution of 50×. Diluted cDNA was used as input for qPCR, using the EXILENT SYBR Green master mix kit (Exiqon) and miRNA-specific primers (Tables 2 and 3). Real-time PCR amplification was performed using the StepOne Plus System (ABI).
Table 2.
Commercial primers used in RT–qPCR
| Commercial primers | Company | Product | Reference |
|---|---|---|---|
| U6 | Exiqon | microRNA LNATM PCR primer sets | 203907 |
| miR-98-5p | Exiqon | microRNA LNATM PCR primer sets | 204640 |
| miR-15a-5p | Exiqon | microRNA LNATM PCR primer sets | 204066 |
| let-7a-5p | Exiqon | microRNA LNATM PCR primer sets | 205727 |
| miR-148b-3p | Exiqon | microRNA LNATM PCR primer sets | 204047 |
| miR-378a-3p | Exiqon | microRNA LNATM PCR primer sets | 205946 |
| miR-34a-5p | Exiqon | microRNA LNATM PCR primer sets | 204486 |
| pre-miR-15a | Qiagen | MiScript precursor assay | MP00001043 |
| pri-miR-98 | Exiqon | Custom LNATM oligonucleotides | 531396 |
Table 3.
Custom primers used in RT–qPCR (Invitrogen custom DNA oligonucleotides)
| Custom primers | Sequences: forward and reverse | Reference |
|---|---|---|
| GAPDH | Forward: ACCACAGTCCATGCCATCAC | Ref. 58 |
| Reverse: TCCACCACCCTGTTGCTGTA | ||
| 18S | Forward: CAAGACGGACCAGAGCGAAA | Ref. 59 |
| Reverse: GGCGGGTCATGGGAATAAC | ||
| β-Actin | Forward: CCAACCGCGAGAAGATGA | Ref. 60 |
| Reverse: TCCATCACGATGCCAGTG | ||
| c-Myc | Forward: AATGAAAAGGCCCCCAAGGTAGTTATCC | Ref. 61 |
| Reverse: GTCGTTTCCGCAACAAGTCCTCTTC | ||
| pri-let-7a | Forward: CCTGGATGTTCTCTTCACTG | Ref. 62 |
| Reverse: GCCTGGATGCAGACTTTTCT | ||
| pre-let-7a | Forward: AGGTAGTAGGTTGTATAGTTTTAGG | Ref. 62 |
| Reverse: TAGGAAAGACAGTAGATTGTATAGT | ||
| pri-miR-15a | Forward: CTAAGGCACTGCTGACATTGCT | Ref. 63 |
| Reverse: GTAGCAGCACATAATGGTTTGTGG | ||
| pri-miR-15b | Forward: CATGCTACAGTCAAGATGCGAATC | Ref. 64 |
| Reverse: CGTGCTGCTAGAGTGGAACAAGT | ||
| pre-miR-15b | Forward: GGCCTTAAAGTACTGTAGC | Ref. 65 |
| Reverse: CCTTAAATTTCTAGAGCAGC | ||
| pri-miR-34a | Forward: CCTCCAAGCCAGCTCAGTTG | Ref. 66 |
| Reverse: TGACTTTGGTCCAATTCCTGTTG | ||
| pre-miR-34a | Forward: TGGCAGTGTCTTAGCTGGTTG | Ref. 62 |
| Reverse: GGCAGTATACTTGCTGATTGCTT | ||
| pre-miR-98 | Forward: GGTAGTAAGTTGTATTGTTGTGGG | Ref. 62 |
| Reverse: TATAGTTATCTTCTAATTGGGGCC | ||
| CDKN2B (p15INK4b) | Forward: ATCCCAACGGAGTCAACCG | Ref. 67 |
| Reverse: CTGCCCATCATCATGACCT | ||
| CDKN1A (p21Waf1) | Forward: TGTCCGTCAGAACCCATGC | Ref. 68 |
| Reverse: AAAGTCGAAGTTCCATCGCTC | ||
| CDKN1B (p27Kip1) | Forward: AACGTGCGAGTGTCTAACGG | Ref. 32 |
| Reverse: CCCTCTAGGGGTTTGTGATTCT |
For analysis of longer RNA (primary and precursor miRNAs, control RNAs, and c-Myc targets), 10 ng of RNA (extracted with the Thermo kit) were used for cDNA synthesis using the OneScript cDNA synthesis kit (ABM) according to the manufacturer's instructions. The cDNA was diluted 80× in nuclease-free water and used as input for qPCR, using the EvaGreen 2× qPCR MasterMix-ROX and primers (Tables 2 and 3).
Click-iT labeling of nascent RNA
Differentiated macrophages were infected for 24 h, and uninfected cells were used as control. Uninternalized parasites were removed by washing the cells thoroughly. Fresh complete RPMI medium was added, as well as 0.5 mm ethynyl uridine (Abcam). Nontreated cells were collected as baseline (t = 0). After 15, 30, and 60 min, the cells were collected and lysed for total RNA extraction using a GeneJET RNA purification kit (Thermo).
The Click-iT nascent RNA capture kit was used according to the manufacturer's instructions. Briefly, 1 μg of RNA was biotinylated using 0.5 mm biotin azide. The RNA was then precipitated overnight. 400 ng of biotinylated RNA was bound to streptavidin beads, which were washed thoroughly. cDNA synthesis was done using the RNA captured on the beads as template and the SuperScript VILO cDNA synthesis kit (Thermo). The cDNA was then released from the beads and used for qPCR (called “nascent”). In parallel, total RNA from the same samples was reverse-transcribed and used for qPCR as normalization. Calculations for newly synthesized RNA were based on the ΔΔCt method (50) as follows.
| (Eq. 1) |
| (Eq. 2) |
| (Eq. 3) |
Western blotting
Frozen pellets of cells were lysed on ice using protein lysis buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% Triton-X-100, 1 mm EDTA, 2.5 mm sodium orthovanadate, 1 mm β-glycerophosphate, 1 mm phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, 5 μg/ml leupeptin). Lysates were incubated on ice for 5 min and then passed 10 times through a 27-gauge needle for mechanic disruption. Lysates were centrifuged at 13,000 × gfor 5 min, before addition of 4× Laemmli loading buffer, and boiled for 7 min. Samples were run on SDS-PAGE gels (7.5–15%), followed by semidry transfer to nitrocellulose membranes. Immunoblotting was performed with primary antibodies against Drosha (Abcam ab183732), DGCR8 (Abcam ab191875), Dicer (Santa Cruz sc-30226), TRBP (Abcam ab42018), Ago2 (Abcam ab186733), c-Myc (Cell Signaling 5605), and Actin (Sigma–Aldrich A2066). The blots were incubated with secondary antibody anti-rabbit HRPO (ABM), followed by developing with Supersignal West Femto Chemoluminescence Substrate (Thermo). Densitometric analysis was performed in ImageJ, and the results are expressed normalized to actin.
Confocal microscopy
Human macrophages grown on coverslips were infected with L. donovani for 24 and 48 h (in which case they were washed at 24 h postinfection to remove unbound parasites) or kept uninfected as control. Intracellular immunofluorescence staining was performed following Abcam's guidelines: fixation was done with 2% polyformaldehyde in PBS and permeabilization with 0.3% Triton X-100 in PBS. Anti-Drosha (Abcam ab183732) was used at 1:100 and anti-DGCR8 (Abcam ab191875) was used at 1:1000, whereas the secondary antibody, anti-rabbit Alexa Fluor 594 (Life Technologies), was used at 1:250. The coverslips were mounted on slides using ProLong Diamond antifade mountant with DAPI (Thermo Fisher). The slides were then analyzed using a Zeiss LSM 780 confocal microscope (Carl Zeiss, Thornwood, NY). Controls for staining were performed using only the secondary antibody, as well as full staining with both primary and secondary antibodies on uninfected macrophages, both of which led to no detectable fluorescence. ImageJ was used to determine the fluorescence intensity in the nucleus versus the cytoplasm: DAPI-positive regions were selected as “nucleus,” and the intensity of the DGCR8 or Drosha staining was measured in the DAPI regions, whereas the staining in the rest of the image was measured for cytoplasmic localization.
siRNA transfection and 10058-F4 treatment
The siRNAs—three c-Myc siRNAs duplexes and one scrambled negative control—were obtained from ORIGENE (SR321047). After 6 days of treatment with GM–CSF, differentiated macrophages were given a transfection mix containing siRNAs at a final concentration of 200 nm and the transfection reagent HiPerfect (Qiagen). The cells were incubated with siRNAs for 6 h, and then fresh medium was added to the wells. After 48 h of siRNA treatment, the cells were washed with HBSS and infected or rested in culture medium for 24 h.
The c-Myc inhibitor 10058-F4 was purchased from Sigma (reference F3680). GM–CSF differentiated macrophages were treated with 25 μm or 50 μm of 10058-F4 for 24 h, at which point the medium was removed and fresh medium containing 10058-F4 or not was added, as well as Leishmania for the infected samples. DMSO was used as vehicle control, at a final concentration of 0.1%.
Bioinformatics analysis of promoters
The ChIP-seq data were retrieved from the ReMap database (51), for c-Myc and Sp1, and the ENCODE Consortium (52), for Miz-1. The computational DNA-binding models were downloaded from JASPAR (53), for c-Myc and Sp1, and HOCOMOCO (54), for Miz-1. Gene annotations, including the chromosome, strand, and the transcription start and end positions, were obtained using the UCSC Table Browser data retrieval tool (55). Note that all data refers to the build 37 of the Genome Reference Consortium human genome (GRCh37). Promoters were defined as the ±500-bp regions around gene transcription start positions. Overlaps between the promoter regions and ChIP-seq data were calculated using BEDTools (56). Transcription factor–binding sites were predicted using FIMO with default parameters (57).
Statistical analysis
Statistical analysis was performed using GraphPad Prism v.5 and v.6; p values equal to or less than 0.05 were considered statistically significant. One-way and two-way ANOVA were used, as well as two-tailed t tests, as described in each figure legend.
Author contributions
L. C., U. L., O. F., and N. E. R. conceptualization; L. C., U. L., and O. F. data curation; L. C., U. L., and O. F. formal analysis; L. C., U. L., and W. W. W. validation; L. C. and U. L. investigation; L. C. and U. L. visualization; L. C., U. L., and O. F. methodology; L. C. writing-original draft; L. C. and U. L. project administration; L. C., U. L., O. F., W. W. W., and N. E. R. writing-review and editing; W. W. W. and N. E. R. resources; W. W. W. and N. E. R. supervision; W. W. W. and N. E. R. funding acquisition.
Supplementary Material
Acknowledgments
We thank A. Marr and R. McMaster for providing the DNA methylation results.
Note added in proof
In the version of this article that was published as a Paper in Press on June 22, 2018, the values of the overlapping promoters for c-Myc with Miz-1 and Sp1 were inadvertently switched in Fig. 5C. This error has now been corrected and does not affect the results or conclusions of this work.
This work was funded by Grant MOP-125879 from the Canadian Institutes for Health Research. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Table S1 and Figs. S1–S6.
A. K. Marr and W. R. McMaster, personal communication.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- miRNA
- microRNA
- HMDM
- human monocyte-derived macrophage
- qPCR
- quantitative PCR
- GO
- Gene Ontology
- KEGG
- Kyoto Encyclopedia of Genes and Genomes
- EU
- ethynyl uridine
- HBSS
- Hanks' balanced salt solution
- GM–CSF
- granulocyte/macrophage colony-stimulating factor
- DAPI
- 4′,6′-diamino-2-phenylindole
- ANOVA
- analysis of variance.
References
- 1. Kim V. N., Han J., and Siomi M. C. (2009) Biogenesis of small RNAs in animals. Nat. Rev. Mol. Cell Biol. 10, 126–139 10.1038/nrn2572,10.1038/nrm2632 [DOI] [PubMed] [Google Scholar]
- 2. Agarwal V., Bell G. W., Nam J.-W., and Bartel D. P. (2015) Predicting effective microRNA target sites in mammalian mRNAs. eLife 4, e05005 10.7554/eLife.05005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lee S., Song J., Kim S., Kim J., Hong Y., Kim Y., Kim D., Baek D., and Ahn K. (2013) Selective degradation of host microRNAs by an intergenic HCMV noncoding RNA accelerates virus production. Cell Host Microbe 13, 678–690 10.1016/j.chom.2013.05.007 [DOI] [PubMed] [Google Scholar]
- 4. Wu D., Hu Y., Tong S., Williams B. R., Smyth G. K., and Gantier M. P. (2013) The use of miRNA microarrays for the analysis of cancer samples with global miRNA decrease. RNA 19, 876–888 10.1261/rna.035055.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Muralidhar B., Goldstein L. D., Ng G., Winder D. M., Palmer R. D., Gooding E. L., Barbosa-Morais N. L., Mukherjee G., Thorne N. P., Roberts I., Pett M. R., and Coleman N. (2007) Global microRNA profiles in cervical squamous cell carcinoma depend on Drosha expression levels. J. Pathol. 212, 368–377 10.1002/path.2179 [DOI] [PubMed] [Google Scholar]
- 6. Martello G., Rosato A., Ferrari F., Manfrin A., Cordenonsi M., Dupont S., Enzo E., Guzzardo V., Rondina M., Spruce T., Parenti A. R., Daidone M. G., Bicciato S., and Piccolo S. (2010) A microRNA targeting dicer for metastasis control. Cell 141, 1195–1207 10.1016/j.cell.2010.05.017 [DOI] [PubMed] [Google Scholar]
- 7. Rupaimoole R., Wu S. Y., Pradeep S., Ivan C., Pecot C. V., Gharpure K. M., Nagaraja A. S., Armaiz-Pena G. N., McGuire M., Zand B., Dalton H. J., Filant J., Miller J. B., Lu C., Sadaoui N. C., et al. (2014) Hypoxia-mediated downregulation of miRNA biogenesis promotes tumour progression. Nat. Commun. 5, 5202 10.1038/ncomms6202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang W., Wang Y. E., Zhang Y., Leleu X., Reagan M., Zhang Y., Mishima Y., Glavey S., Manier S., Sacco A., Jiang B., Roccaro A. M., and Ghobrial I. M. (2014) Global epigenetic regulation of microRNAs in multiple myeloma. PLoS One 9, e110973 10.1371/journal.pone.0110973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Melo S. A., Moutinho C., Ropero S., Calin G. A., Rossi S., Spizzo R., Fernandez A. F., Davalos V., Villanueva A., Montoya G., Yamamoto H., Schwartz S. Jr., and Esteller M. (2010) A genetic defect in exportin-5 traps precursor microRNAs in the nucleus of cancer cells. Cancer Cell 18, 303–315 10.1016/j.ccr.2010.09.007 [DOI] [PubMed] [Google Scholar]
- 10. Sun H.-L., Cui R., Zhou J., Teng K.-Y., Hsiao Y.-H., Nakanishi K., Fassan M., Luo Z., Shi G., Tili E., Kutay H., Lovat F., Vicentini C., Huang H.-L., Wang S.-W., et al. (2016) ERK activation globally downregulates miRNAs through phosphorylating exportin-5. Cancer Cell 30, 723–736 10.1016/j.ccell.2016.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chang T.-C., Yu D., Lee Y.-S., Wentzel E. A., Arking D. E., West K. M., Dang C. V., Thomas-Tikhonenko A., and Mendell J. T. (2008) Widespread microRNA repression by Myc contributes to tumorigenesis. Nat. Genet. 40, 43–50 10.1038/ng.2007.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weber B., Stresemann C., Brueckner B., and Lyko F. (2007) Methylation of human microRNA genes in normal and neoplastic cells. Cell Cycle 6, 1001–1005 10.4161/cc.6.9.4209 [DOI] [PubMed] [Google Scholar]
- 13. Suzuki H., Maruyama R., Yamamoto E., and Kai M. (2012) DNA methylation and microRNA dysregulation in cancer. Mol. Oncol. 6, 567–578 10.1016/j.molonc.2012.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Marr A. K., MacIsaac J. L., Jiang R., Airo A. M., Kobor M. S., and McMaster W. R. (2014) Leishmania donovani infection causes distinct epigenetic DNA methylation changes in host macrophages. PLoS Pathog. 10, e1004419 10.1371/journal.ppat.1004419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tao J., Zhao X., and Tao J. (2014) c-MYC–miRNA circuitry. Cell Cycle 13, 191–198 10.4161/cc.27646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gao P., Tchernyshyov I., Chang T.-C., Lee Y.-S., Kita K., Ochi T., Zeller K. I., De Marzo A. M., Van Eyk J. E., Mendell J. T., and Dang C. V. (2009) c-Myc suppression of miR-23 enhances mitochondrial glutaminase and glutamine metabolism. Nature 458, 762–765 10.1038/nature07823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang X., Zhao X., Gao P., and Wu M. (2013) c-Myc modulates microRNA processing via the transcriptional regulation of Drosha. Sci. Rep. 10.1038/srep01942 10.1038/srep01942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pello O. M., De Pizzol M., Mirolo M., Soucek L., Zammataro L., Amabile A., Doni A., Nebuloni M., Swigart L. B., Evan G. I., Mantovani A., and Locati M. (2012) Role of c-MYC in alternative activation of human macrophages and tumor-associated macrophage biology. Blood 119, 411–421 10.1182/blood-2011-02-339911 [DOI] [PubMed] [Google Scholar]
- 19. Liu D., and Uzonna J. E. (2012) The early interaction of Leishmania with macrophages and dendritic cells and its influence on the host immune response. Front. Cell. Infect. Microbiol. 2, 83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sears R. C. (2004) The life cycle of c-myc: from synthesis to degradation. Cell Cycle 3, 1133–1137 [PubMed] [Google Scholar]
- 21. Wu S., Cetinkaya C., Munoz-Alonso M. J., von der Lehr N., Bahram F., Beuger V., Eilers M., Leon J., and Larsson L.-G. (2003) Myc represses differentiation-induced p21CIP1 expression via Miz-1-dependent interaction with the p21 core promoter. Oncogene 22, 351–360 10.1038/sj.onc.1206145 [DOI] [PubMed] [Google Scholar]
- 22. Yang W., Shen J., Wu M., Arsura M., FitzGerald M., Suldan Z., Kim D. W., Hofmann C. S., Pianetti S., Romieu-Mourez R., Freedman L. P., and Sonenshein G. E. (2001) Repression of transcription of the p27 Kip1 cyclin-dependent kinase inhibitor gene by c-Myc. Oncogene 20, 1688–1702 10.1038/sj.onc.1204245 [DOI] [PubMed] [Google Scholar]
- 23. Seoane J., Pouponnot C., Staller P., Schader M., Eilers M., and Massagué J. (2001) TGFβ influences Myc, Miz-1 and Smad to control the CDK inhibitor p15INK4b. Nat. Cell Biol. 3, 400–408 10.1038/35070086 [DOI] [PubMed] [Google Scholar]
- 24. Qi Y., Tu Y., Yang D., Chen Q., Xiao J., Chen Y., Fu J., Xiao X., and Zhou Z. (2007) Cyclin a but not cyclin D1 is essential for c-myc-modulated cell-cycle progression. J. Cell. Physiol. 210, 63–71 10.1002/jcp.20816 [DOI] [PubMed] [Google Scholar]
- 25. Peukert K., Staller P., Schneider A., Carmichael G., Hänel F., and Eilers M. (1997) An alternative pathway for gene regulation by Myc. EMBO J. 16, 5672–5686 10.1093/emboj/16.18.5672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gartel A. L., Ye X., Goufman E., Shianov P., Hay N., Najmabadi F., and Tyner A. L. (2001) Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc. Natl. Acad. Sci. U.S.A. 98, 4510–4515 10.1073/pnas.081074898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Walz S., Lorenzin F., Morton J., Wiese K. E., von Eyss B., Herold S., Rycak L., Dumay-Odelot H., Karim S., Bartkuhn M., Roels F., Wüstefeld T., Fischer M., Teichmann M., Zender L., et al. (2014) Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 511, 483–487 10.1038/nature13473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Izumi H., Molander C., Penn L. Z., Ishisaki A., Kohno K., and Funa K. (2001) Mechanism for the transcriptional repression by c-Myc on PDGF β-receptor. J. Cell Sci. 114, 1533–1544 [DOI] [PubMed] [Google Scholar]
- 29. Shrivastava A., Saleque S., Kalpana G. V., Artandi S., Goff S. P., and Calame K. (1993) Inhibition of transcriptional regulator Yin-Yang-1 by association with c-Myc. Science 262, 1889–1892 10.1126/science.8266081 [DOI] [PubMed] [Google Scholar]
- 30. Roy A. L., Carruthers C., Gutjahr T., and Roeder R. G. (1993) Direct role for Myc in transcription initiation mediated by interactions with TFII-I. Nature 365, 359–361 10.1038/365359a0 [DOI] [PubMed] [Google Scholar]
- 31. Iraci N., Diolaiti D., Papa A., Porro A., Valli E., Gherardi S., Herold S., Eilers M., Bernardoni R., Della Valle G., and Perini G. (2011) A SP1/MIZ1/MYCN repression complex recruits HDAC1 at the TRKA and p75NTR promoters and affects neuroblastoma malignancy by inhibiting the cell response to NGF. Cancer Res. 71, 404–412 10.1158/0008-5472.CAN-10-2627,10.1158/1538-7445.AM2011-404 [DOI] [PubMed] [Google Scholar]
- 32. Xu Y., Zhong C., Ding S., Huang H., and Shen Z. (2015) MicroRNA-221 promotes human non-small cell lung cancer cell H460 growth. Int. J. Clin. Exp. Med. 8, 2024–2030 [PMC free article] [PubMed] [Google Scholar]
- 33. Huang M.-J., Cheng Y.-C., Liu C.-R., Lin S., and Liu H. E. (2006) A small-molecule c-Myc inhibitor, 10058-F4, induces cell-cycle arrest, apoptosis, and myeloid differentiation of human acute myeloid leukemia. Exp. Hematol. 34, 1480–1489 10.1016/j.exphem.2006.06.019 [DOI] [PubMed] [Google Scholar]
- 34. Lemaire J., Mkannez G., Guerfali F. Z., Gustin C., Attia H., Sghaier R. M., Sysco-Consortium, Dellagi K., Laouini D., and Renard P. (2013) MicroRNA expression profile in human macrophages in response to Leishmania major infection. PLoS Negl. Trop. Dis. 7, e2478 10.1371/journal.pntd.0002478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Frank B., Marcu A., de Oliveira Almeida Petersen A. L., Weber H., Stigloher C., Mottram J. C., Scholz C. J., and Schurigt U. (2015) Autophagic digestion of Leishmania major by host macrophages is associated with differential expression of BNIP3, CTSE, and the miRNAs miR-101c, miR-129, and miR-210. Parasit. Vectors 8, 404 10.1186/s13071-015-0974-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tiwari N., Kumar V., Gedda M. R., Singh A. K., Singh V. K., Gannavaram S., Singh S. P., and Singh R. K. (2017) Identification and characterization of miRNAs in response to Leishmania donovani infection: delineation of their roles in macrophage dysfunction. Front. Microbiol. 8, 314 10.3389/fmicb.2017.00314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Singh A. K., Pandey R. K., Shaha C., and Madhubala R. (2016) MicroRNA expression profiling of Leishmania donovani–infected host cells uncovers the regulatory role of MIR30A-3p in host autophagy. Autophagy 12, 1817–1831 10.1080/15548627.2016.1203500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Muxel S. M., Laranjeira-Silva M. F., Zampieri R. A., and Floeter-Winter L. M. (2017) Leishmania (Leishmania) amazonensis induces macrophage miR-294 and miR-721 expression and modulates infection by targeting NOS2 and l-arginine metabolism. Sci. Rep. 7, 44141 10.1038/srep44141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang X., Zhao X., Gao P., and Wu M. (2013) c-Myc modulates microRNA processing via the transcriptional regulation of Drosha. Sci. Rep. 3, 1942 10.1038/srep01942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Piskounova E., Polytarchou C., Thornton J. E., LaPierre R. J., Pothoulakis C., Hagan J. P., Iliopoulos D., and Gregory R. I. (2011) Oncogenic Lin28A and Lin28B inhibit let-7 microRNA biogenesis by distinct mechanisms. Cell. 147, 1066–1079 10.1016/j.cell.2011.10.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Psathas J. N., and Thomas-Tikhonenko A. (2014) MYC and the art of microRNA maintenance. Cold Spring Harb. Perspect. Med. 4, a014175 10.1101/cshperspect.a014175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jackstadt R., and Hermeking H. (2015) MicroRNAs as regulators and mediators of c-MYC function. Biochim. Biophys. Acta 1849, 544–553 10.1016/j.bbagrm.2014.04.003 [DOI] [PubMed] [Google Scholar]
- 43. Kumar M. S., Lu J., Mercer K. L., Golub T. R., and Jacks T. (2007) Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat. Genet. 39, 673–677 10.1038/ng2003 [DOI] [PubMed] [Google Scholar]
- 44. Yim H. C., Li J. C., Pong J. C., and Lau A. S. (2011) A role for c-Myc in regulating anti-mycobacterial responses. Proc. Natl. Acad. Sci. U.S.A. 108, 17749–17754 10.1073/pnas.1104892108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu L., Lu Y., Martinez J., Bi Y., Lian G., Wang T., Milasta S., Wang J., Yang M., Liu G., Green D. R., and Wang R. (2016) Proinflammatory signal suppresses proliferation and shifts macrophage metabolism from Myc-dependent to HIF1α-dependent. Proc. Natl. Acad. Sci. U.S.A. 113, 1564–1569 10.1073/pnas.1518000113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Whitfield J. R., Beaulieu M.-E., and Soucek L. (2017) Strategies to inhibit Myc and their clinical applicability. Front. Cell Dev. Biol. 5, 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Colineau L., Clos J., Moon K.-M., Foster L. J., and Reiner N. E. (2017) Leishmania donovani chaperonin 10 regulates parasite internalization and intracellular survival in human macrophages. Med. Microbiol. Immunol. 206, 235–257 10.1007/s00430-017-0500-7 [DOI] [PubMed] [Google Scholar]
- 48. Lewis B. P., Burge C. B., and Bartel D. P. (2005) Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20 10.1016/j.cell.2004.12.035 [DOI] [PubMed] [Google Scholar]
- 49. Zhang B., Kirov S., and Snoddy J. (2005) WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 33, W741–W748 10.1093/nar/gki475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔC(T) method. Methods 25, 402–408 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 51. Chèneby J., Gheorghe M., Artufel M., Mathelier A., and Ballester B. (2018) ReMap 2018: an updated atlas of regulatory regions from an integrative analysis of DNA-binding ChIP-seq experiments. Nucleic Acids Res. 46, D267–D275 10.1093/nar/gkx1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. ENCODE Project Consortium. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Khan A., Fornes O., Stigliani A., Gheorghe M., Castro-Mondragon J. A., van der Lee R., Bessy A., Chèneby J., Kulkarni S. R., Tan G., Baranasic D., Arenillas D. J., Sandelin A., Vandepoele K., Lenhard B., et al. (2018) JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 46, D260–D266 10.1093/nar/gkx1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kulakovskiy I. V., Vorontsov I. E., Yevshin I. S., Sharipov R. N., Fedorova A. D., Rumynskiy E. I., Medvedeva Y. A., Magana-Mora A., Bajic V. B., Papatsenko D. A., Kolpakov F. A., and Makeev V. J. (2018) HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 46, D252–D259 10.1093/nar/gkx1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Karolchik D., Hinrichs A. S., Furey T. S., Roskin K. M., Sugnet C. W., Haussler D., and Kent W. J. (2004) The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 32, D493–D496 10.1093/nar/gkh103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Quinlan A. R., and Hall I. M. (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Grant C. E., Bailey T. L., and Noble W. S. (2011) FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018 10.1093/bioinformatics/btr064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nothelfer K., Obermayr F., Belz N., Reinartz E., Bareiss P. M., Bühring H.-J., Beschorner R., and Just L. (2016) Expression of the Wnt receptor Frizzled-4 in the human enteric nervous system of infants. Stem Cells Int. 2016, 1–12 10.1155/2016/9076823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wisskirchen C., Ludersdorfer T. H., Müller D. A., Moritz E., and Pavlovic J. (2011) The cellular RNA helicase UAP56 is required for prevention of double-stranded RNA formation during influenza A virus infection. J. Virol. 85, 8646–8655 10.1128/JVI.02559-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rzymski T., Paantjens A., Bod J., and Harris A. L. (2008) Multiple pathways are involved in the anoxia response of SKIP3 including HuR-regulated RNA stability, NF-κB and ATF4. Oncogene 27, 4532–4543 10.1038/onc.2008.100 [DOI] [PubMed] [Google Scholar]
- 61. Guo X., Zhang R., Liu J., Li M., Song C., Dovat S., Li J., and Ge Z. (2015) Characterization of LEF1 high expression and novel mutations in adult acute lymphoblastic leukemia. PLoS One 10, e0125429 10.1371/journal.pone.0125429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jiang J., Lee E. J., Gusev Y., and Schmittgen T. D. (2005) Real-time expression profiling of microRNA precursors in human cancer cell lines. Nucleic Acids Res. 33, 5394–5403 10.1093/nar/gki863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. von Brandenstein M., Pandarakalam J. J., Kroon L., Loeser H., Herden J., Braun G., Wendland K., Dienes H. P., Engelmann U., Engelmann U., and Fries J. W. (2012) MicroRNA 15a, inversely correlated to PKCα, is a potential marker to differentiate between benign and malignant renal tumors in biopsy and urine samples. Am. J. Pathol. 180, 1787–1797 10.1016/j.ajpath.2012.01.014 [DOI] [PubMed] [Google Scholar]
- 64. Zhou R., Hu G., Liu J., Gong A.-Y., Drescher K. M., and Chen X.-M. (2009) NF-cκB p65-dependent transactivation of miRNA genes following Cryptosporidium parvum infection stimulates epithelial cell immune responses. PLoS Pathog. 5, e1000681 10.1371/journal.ppat.1000681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhu B., Ye J., Ashraf U., Li Y., Chen H., Song Y., and Cao S. (2016) Transcriptional regulation of miR-15b by c-Rel and CREB in Japanese encephalitis virus infection. Sci. Rep. 6, 22581 10.1038/srep22581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Suzuki H. I., Yamagata K., Sugimoto K., Iwamoto T., Kato S., and Miyazono K. (2009) Modulation of microRNA processing by p53. Nature 460, 529–533 10.1038/nature08199 [DOI] [PubMed] [Google Scholar]
- 67. Amatori S., Persico G., and Fanelli M. (2017) Real-time quantitative PCR array to study drug-induced changes of gene expression in tumor cell lines. J. Cancer Metastasis Treat. 3, 90 10.20517/2394-4722.2017.22 [DOI] [Google Scholar]
- 68. Huang Q., Li L., Lin Z., Xu W., Han S., Zhao C., Li L., Cao W., Yang X., Wei H., and Xiao J. (2016) Identification of preferentially expressed antigen of melanoma as a potential tumor suppressor in lung adenocarcinoma. Med. Sci. Monit. 22, 1837–1842 10.12659/MSM.895642 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.