

Abstract
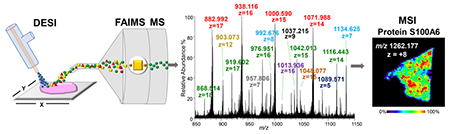
Analysis of large biomolecules including proteins has proven challenging using ambient ionization mass spectrometry imaging techniques. Here, we have successfully optimized desorption electrospray ionization mass spectrometry (DESI-MS) to detect intact proteins directly from tissue sections and further integrated DESI-MS to a high field asymmetric waveform ion mobility (FAIMS) device for protein imaging. Optimized DESI-FAIMS-MS parameters were used to image mouse kidney, mouse brain, and human ovarian and breast tissue samples, allowing detection of 11, 16, 14, and 16 proteoforms, respectively. Identification of protein species detected by DESI-MS was performed on-tissue by top-down ultraviolet photodissociation (UVPD) and collision induced dissociation (CID) as well as using tissue extracts by bottom-up CID and top-down UVPD. Our results demonstrate that DESI-MS imaging is suitable for the analysis of the distribution of proteins within biological tissue sections.
Mass spectrometry (MS) imaging is a powerful tool to investigate the spatial distribution of molecular species directly from tissue samples.1,2 Matrix assisted laser desorption/ionization (MALDI) is the most widely used MS imaging technique, which has been extensively explored to image and characterize metabolites, lipids, and proteins from biological tissue sections.1,3 Ambient ionization MS techniques have become increasingly used for biological tissue imaging as they allow analysis to be performed in the open environment with minimal sample preparation requirements, which is appealing for clinical applications.2,4 Desorption electrospray ionization (DESI) MS imaging, for example, is the most widely used ambient ionization MS technique, which has been broadly used for tissue imaging.4
DESI-MS imaging has been successfully used to analyze biological tissue sections allowing efficient desorption and ionization of lipids and metabolites that are diagnostic of cancer including breast,5 ovarian,6 brain,7 and others.4 Although typically used for small molecule analysis, a few studies have described optimization of DESI-MS for protein analysis from nonbiological substrates.8–10 For example, DESI-MS has been recently used to desorb membrane proteins in their native conformations from planar surfaces.10 However, inefficient desorption of large biomolecules and chemical noise arising from the complex tissue matrix have impeded detection of proteins directly from biological tissue sections by DESI-MS. Recently, ambient ionization MS using liquid extraction techniques, such as nanospray desorption electrospray ionization (nano-DESI),11 liquid extraction surface analysis (LESA),12 and the liquid microjunction surface sampling probe (LMJ-SSP),13 were applied to image proteins from biological tissue sections. In the latter two studies, protein analysis was enhanced by integrating ion mobility separation into the workflow, which allowed selective transmission of protein ions and reduced chemical noise in the mass spectra.12,13 Here, we describe the optimization of DESI-MS imaging for protein analysis and further coupling of DESI to a high field asymmetric waveform ion mobility (FAIMS) device for imaging proteins from biological tissue sections, indicating that this approach could be used for top-down proteomics studies in various biomedical applications.
We first evaluated the effectiveness of a washing step with organic solvents on enhancing protein detection, which is commonly performed in MALDI-MS imaging experiments to remove endogenous lipids and biological salts that may affect efficiency of protein desorption and ionization.14 Detailed methods are provided in the Supporting Information. DESI-MS imaging was performed on an unwashed mouse kidney tissue section in the positive ion mode using pure ACN as the solvent and typical DESI-MS lipid imaging parameters (Table S1).15 Ions identified as triacylglycerols and glycerophosphocholines were detected at high relative abundances (Figure 1a),15 while multiply charged protein ions were not seen. Next, a solvent system of ACN–H2O (80:20) (v/v) with 0.2% formic acid previously reported to enhance protein desorption by nano-DESI was used for analysis of unwashed tissue sections, at a flow rate of 5 μL/min.11 Similar lipid species were detected at high relative abundances in addition to multiply charged ions at low abundances that were tentatively identified as protein species (Figure 1b). The lipid washing step was then performed on an adjacent mouse kidney tissue section followed by DESI-MS imaging analysis at the same parameters (Figure 1c). While the washing step was effective at removing lipids, the mass spectra obtained presented low total ion abundance of the multiple charged ions. Previous studies have reported that the desorption of protein standards from glass slides by DESI-MS is dependent on the spray angle and spray-to-surface distance.10,16 Thus, we performed optimization of DESI spray parameters for protein detection by tuning the angle, spray-to-sample distance, and sample-to-inlet distance to 55°, 3.5 mm, and 2.5 mm, respectively. Performance was evaluated by the improvement in the total ion abundance of m/z 938.117, later identified as an alpha-globin proteoform with an asparagine to a lysine substitution. While protein ions were detected in mouse kidney tissue at various spray-to-sample and sample-to-inlet distances, proteins were not detected above a S/N = 3 using spray angles other than 55°, indicating that protein desorption and detection is more strongly dependent on the spray angle than other source parameters. At these optimized parameters, the alpha-globin proteoform was detected with a S/N = 27.9 (average of n = 3 tissue sections, n = 3 lines/tissue section, n = 20 mass spectra/line), as well as 10 other distinct protein species (Figure 1d).
Figure 1.

Representative positive ion mode DESI mass spectra obtained from mouse kidney tissue sections that were (a) unwashed, using lipid DESI-MS parameters and pure ACN as the solvent system, (b) unwashed, using lipid DESI-MS parameters with ACN–H2O 80:20 (v/v) and 0.2% formic acid as the solvent, (c) washed, using lipid DESI-MS parameters and ACN–H2O 80:20 (v/v) with 0.2% formic acid as the solvent, (d) washed, using protein DESI-MS parameters and ACN–H2O 80:20 (v/v) with 0.2% formic acid as the solvent, and (e) washed, using protein DESI-FAIMS-MS parameters and ACN–H2O 80:20 (v/v) with 0.2% formic acid as the solvent. Mass spectra are an average of 25 scans. (f) On-tissue UVPD mass spectrum of m/z 938.117 from a mouse kidney tissue section (average of n = 80 mass spectra). PCS, P-score, and sequence coverage are reported for the protein ion identified as an alpha-globin proteoform.
In an effort to further increase the S/N of proteins, we integrated FAIMS to the DESI-MS imaging source and mass spectrometer interface as we have previously described.13 Two-dimensional FAIMS sweep experiments were performed to determine the optimal dispersion field (DF = 180 Td) and compensation field (CF = +1.0 Td) for protein detection. Under the optimized FAIMS parameters, a S/N = 32.1 was achieved for the alpha-globin proteoform (average of n = 3 tissue sections, n = 3 lines/tissue section, n = 20 mass spectra/line), as well as detection of 10 other distinct protein species (Table S2 and Figure 1e). The addition of FAIMS increased the S/N for all the proteins detected, thus improving image contrast and quality (Figure S1). The increase in the S/N of protein ions was due to the substantial filtering of interfering background species (68% decrease), including abundant solvent peaks and reduction of chemical noise (43% decrease), despite an overall drop (30%) in the absolute intensities of protein ions (Figure S2). Therefore, all further experiments were performed using the optimized FAIMS parameters. Spray voltage and transfer capillary temperature were also tuned to optimal values of 1 kV and 300 °C for protein detection, respectively, using the optimized DESI-FAIMS parameters (Figures S3 and S4). For more details on the effect of FAIMS in the data and the optimization approach, please see the Supporting Information.
Top-down and bottom-up protein-sequencing methods were explored to identify the proteins detected. On-tissue CID was performed by isolating and fragmenting protein ions while directly profiling the tissue sections using DESI-MS alone (no FAIMS). Fragmentation of the 13+ charge state isotope envelope of the ion at m/z 1,153.298 (MM = 15 085 Da, Figure S5) by CID allowed identification of this ion as the alpha globin protein (16% sequence coverage) in mouse kidney tissue sections. Other proteoforms of alpha-globin were detected at high relative abundances, which are likely associated with the highly vascularized nature of the kidney tissue. In an effort to obtain higher sequence coverage, UVPD was integrated with DESI-MS for on-tissue protein fragmentation (no FAIMS).17 The proteoform of alpha-globin presenting an asparagine to a lysine substitution (MM = 14 985 Da) used for optimization was identified by on-tissue UVPD of m/z 938.114 (16+ charge state, 32% sequence coverage) (Figure 1f and Figure S6). Hemoglobin α (m/z 1009.335, 15+ charge state) was identified from normal human ovarian tissue using on-tissue UVPD (sequence coverage of 20%). These results demonstrate feasibility of UVPD for the identification of abundant proteins detected using DESI-MS from biological tissue sections, and to the best of our knowledge represent the first application of UVPD for on-tissue protein identification. Nevertheless, further optimization of this integrated approach is needed for fragmentation and identification of lower abundant protein species. Top-down UVPD and bottom-up proteomics were also performed on protein extracts obtained from the tissues analyzed to assist in the identification of low abundance protein ions. Sequence coverage for the proteins identified and the respective method used are provided in Table S3.
Next, we applied the optimized DESI-FAIMS approach to image proteins from biological tissue sections. As shown in Figure 2a, DESI-FAIMS allowed imaging of a variety of proteins from mouse brain tissue sections at distinct spatial distributions within the histologic structures of the brain. Figure 2b shows representative 2D DESI-FAIMS ion images of selected protein ions. A distinct cluster of ions centered at m/z 707.068 (20+ charge state, MM = 14,211 Da), identified as myelin basic protein (MBP) isoform 8 was observed at higher relative abundances within the white matter of the brain, while unidentified protein ions at m/z 992.680 (8+ charge state) and m/z 985.265 (11+ charge state) were distributed throughout the tissue section. The relative abundance of alpha globin was higher in the outer portion of the mouse brain tissue sections, likely correlating to regions containing blood vessels and arteries.11 The spatial distribution of the protein ions detected was reproducible across multiple mouse brain tissue sections (Figure S7), and in agreement with what was previously reported by nanoDESI and LMJ-SSP.11,13 Further, multimodal DESI imaging was successfully performed to obtain both lipid and protein information from the same mouse brain tissue section by first imaging the tissue in negative ion mode for lipid analysis, followed by a lipid-washing step, and then positive ion mode protein analysis using the DESI-FAIMS system (for more information, please see Supporting Information and Figures S8–S10).
Figure 2.
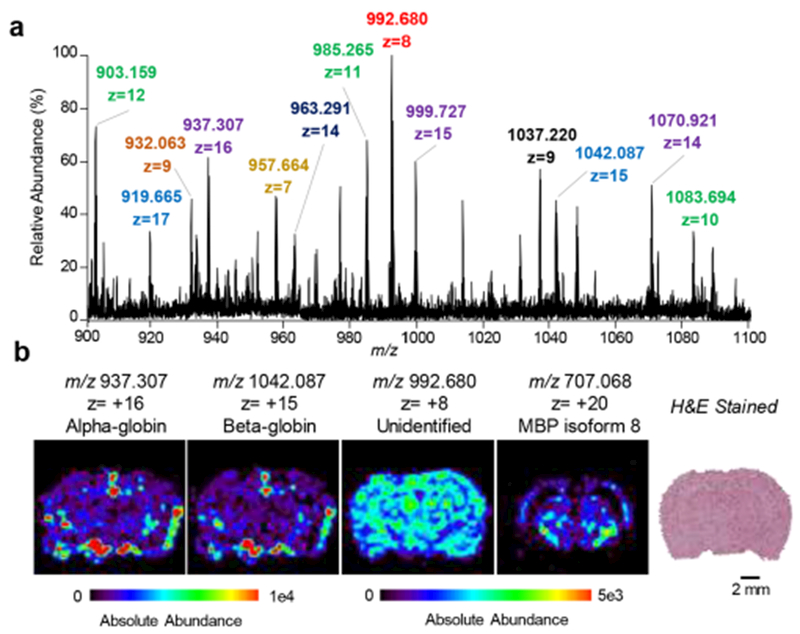
(a) Representative protein DESI mass spectrum of a washed mouse brain tissue section with optimized FAIMS parameters. Mass spectra are an average of 25 scans. Different charge states of the same protein species are denoted by same colored labels. (b) DESI-MS ion images of selected protein species as well as an optical image of a serial H&E stained mouse brain tissue section are shown.
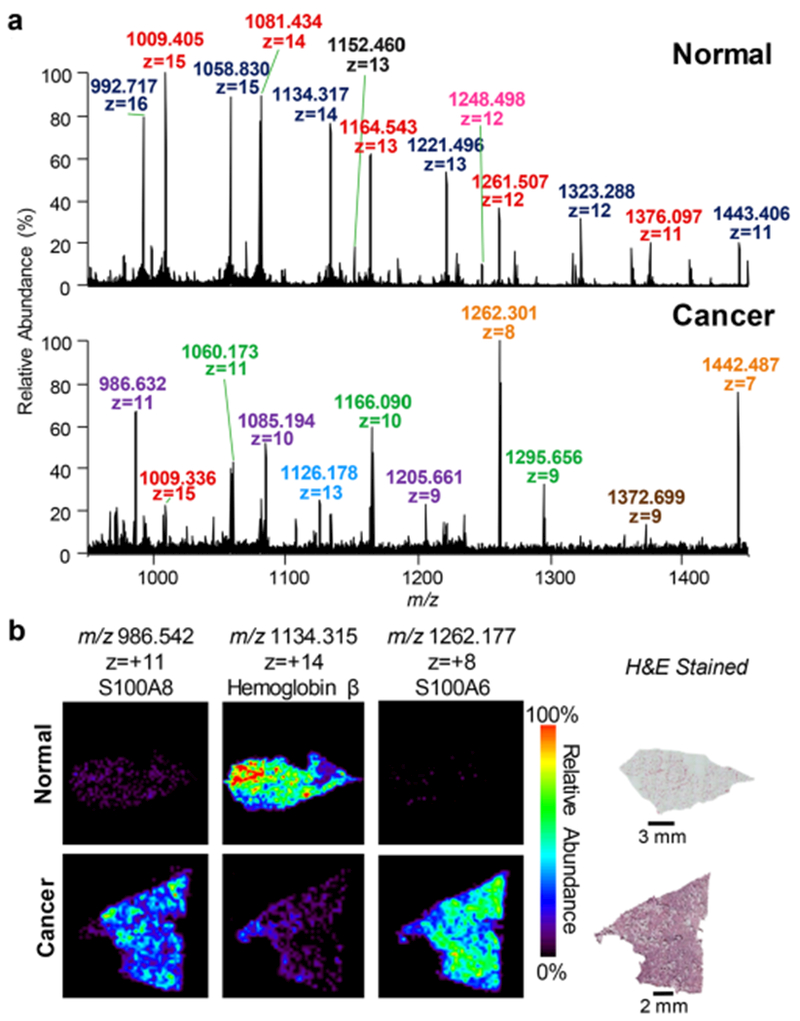
We next employed DESI-FAIMS to image proteins from human normal and cancer tissue sections. As shown in Figure 3a, the mass spectra obtained from normal ovarian and high-grade serous ovarian cancer (HGSC) tissue sections showed distinct relative abundances of protein ions. Hemoglobin β (m/z 1443.318; 11+ charge state; MM = 15 998 Da), for example, was observed at high relative abundances in healthy ovarian tissue samples, while the S100A6 protein (m/z 1442.339; 7+ charge state; MM = 10 180 Da), was observed at high relative abundances in HGSC tissue. Note that although differing by ~1 m/z value, hemoglobin β and S100A6 were clearly resolved in the mass spectra (Figure 3b) and identified using a top-down approach. Increased abundance of S100A6 has been previously reported in a variety of human cancers.18 DESI-FAIMS ion images enabled clear visualization of protein ions within the heterogeneous regions of a single tissue sample (Figure 3c), which corroborates with previous findings.13,19
Figure 3.

(a) Representative protein DESI-MS spectra of normal and HGSC ovarian tumor with optimized FAIMS parameters. Mass spectra are averages of 10 scans. Different charge states of the same protein species are denoted by same colored labels. (b) Zoomed in mass spectra of normal and HGSC samples, highlighting the observance of m/z 1442.339, identified as S100A6, within the cancer tissue. (c) DESI-MS ion images obtained from normal, pure cancer, and mixed normal/cancer ovarian tissue sections. Ion images are in the same scale. H&E stained images are of a serial ovarian tissue sections as protein conditions are not histologically compatible.
DESI-FAIMS-MS imaging also allowed detection of several proteins from human normal and Her2-ductal carcinoma breast tissue samples (Figure 4). For example, profilin-1 at m/z 1 152.460 (13+ charge state, MM = 15 054 Da) and hemoglobin α at m/z 1 009.536 (15+ charge state) were observed at higher relative abundance in a normal breast tissue. On the other hand, S100 proteins including S100A4 at m/z 1058.893 (11+ charge state, MM = 11 279 Da), S100A8 at m/z 986.633 (11+ charge state, MM = 10 835 Da), and S100A11 at m/z 1 166.091 (10+ charge state, MM = 11 740 Da), were seen at higher relative abundances in breast cancer tissue (Figure 4b). Upregulation of members of the S100 family of proteins is known to occur in breast cancer and has been reported by MALDI-MS imaging and other techniques.20–22 Galectin-1 (m/z 1 126.177, 13+ charge state, MM = 14 716 Da), previously associated with Her2-cancer stromal tissue,23 was also detected by DESI-FAIMS-MS at higher relative abundances in the Her2-cancer tissue analyzed.
Figure 4.
(a) Representative protein DESI-MS spectra of normal and breast cancer samples with optimized FAIMS parameters. Mass spectra are averages of 20 scans. Different charge states of the same protein species are denoted by same colored labels. (b) DESI-MS ion images obtained from normal and breast cancer tissue sections. Ion images are in the same scale. H&E stained images are of a serial breast tissue sections as protein conditions are not histologically compatible.
In conclusion, we describe the successful optimization of DESI for protein detection and further coupling to a FAIMS device for protein imaging directly from biological tissue sections. Addition of FAIMS at parameters optimized for protein transmission reduced the mass spectra noise and transmission of background ions, resulting in higher S/N of protein ions and thus improved imaging contrast and quality. We further demonstrate on-tissue top-down protein identification using UVPD and CID for identification of abundant protein ions detected by DESI-MS. While this study shows a noteworthy advancement for DESI-MS imaging, it represents an initial step toward in-depth tissue proteomics applications. Most protein species detected are highly abundant in biological tissues, such as hemoglobin and S100 proteins. Thus, additional optimization is needed to improve the desorption efficiency of lower abundant proteins. Although protein coverage by DESI-MS imaging remains vastly poorer to the coverage achieved through traditional LC–MS/MS of tissue extracts and MALDI-MS imaging workflows,24,25 the ability to rapidly image intact proteins from tissue sections with minimal sample preparation and under ambient conditions suggests DESI-MS as a promising tool for top-down proteomics, with potential applications in cancer imaging and diagnosis.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by The Welch Foundation (Grants F-1895 LSE and F-1155 J.S.B.) and the National Institutes of Health (Grant R00CA190783 to L.S.E. and Grant R21CA191664 to J.S.B.). Tissue samples were provided by the Cooperative Human Tissue Network (supported by the NCI) and the MD Anderson Cancer Center Tissue Bank. We thank Dr. Chandandeep Nagi and Dr. Wendong Yu for pathological evaluation and the University of Texas Proteomics Facility (funded by the UT System) for protein identification experiments. We also thank Owlstone Medical (Cambridge, UK) for providing a custom-made FAIMS capillary, and Marta Sans for assistance with experiments.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge via the Internet at The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.8b00967.
Detailed materials, methods, and supporting results; tables presenting DESI-MS parameters, DESI-FAIMS S/N comparison, summary of detected and identified proteins, and DESI-FAIMS optimization data; figures showing DESI mass spectra at various optimization parameters, protein images of mouse brain with and without FAIMS, DESI mass spectra of kidney tissue with and without FAIMS, voltage and temperature optimization mass spectra, replicates of DESI-MS imaging, and MS/MS data of proteins (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Schwamborn K; Caprioli RM Mol. Oncol 2010, 4, 529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Wu C; Dill AL; Eberlin LS; Cooks RG; Ifa DR Mass Spectrom. Rev 2013, 32, 218–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kriegsmann M; Casadonte R; Kriegsmann J; Dienemann H; Schirmacher P; Kobarg JH; Schwamborn K; Stenzinger A; Warth A; Weichert W Mol. Cell. Proteomics 2016, 15, 3081–3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Ifa DR; Eberlin LS Clin. Chem 2016, 62, 111–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Calligaris D; Caragacianu D; Liu X; Norton I; Thompson CJ; Richardson AL; Golshan M; Easterling ML; Santagata S; Dillon DA; Jolesz FA; Agar NYR Proc. Natl. Acad. Sci. U. S. A 2014, 111, 15184–15189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Sans M; Gharpure K; Tibshirani R; Zhang J; Liang L; Liu J; Young JH; Dood RL; Sood AK; Eberlin LS Cancer Res. 2017, 77, 2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Jarmusch AK; Pirro V; Baird Z; Hattab EM; Cohen-Gadol AA; Cooks RG Proc. Natl. Acad. Sci. U. S. A 2016, 113, 1486–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Honarvar E; Venter AR J. Am. Soc. Mass Spectrom 2017, 28, 1109–1117. [DOI] [PubMed] [Google Scholar]
- (9).Shin YS; Drolet B; Mayer R; Dolence K; Basile F Anal. Chem 2007, 79, 3514–3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ambrose S; Housden NG; Gupta K; Fan J; White P; Yen HY; Marcoux J; Kleanthous C; Hopper JTS; Robinson CV Angew. Chem., Int. Ed 2017, 56, 14463–14468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Hsu CC; Chou PT; Zare RN Anal. Chem 2015, 87, 11171–11175. [DOI] [PubMed] [Google Scholar]
- (12).Griffiths RL; Creese AJ; Race AM; Bunch J; Cooper HJ Anal. Chem 2016, 88, 6758–6766. [DOI] [PubMed] [Google Scholar]
- (13).Feider CL; Elizondo N; Eberlin LS Anal. Chem 2016, 88, 11533–11541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Seeley EH; Oppenheimer SR; Mi D; Chaurand P; Caprioli RM J. Am. Soc. Mass Spectrom 2008, 19, 1069–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Eberlin LS; Ferreira CR; Dill AL; Ifa DR; Cooks RG Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2011, 1811, 946–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Takats Z; Wiseman JM; Cooks RG J. Mass Spectrom 2005, 40, 1261–1275. [DOI] [PubMed] [Google Scholar]
- (17).Klein DR; Holden DD; Brodbelt JS Anal. Chem 2016, 88, 1044–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Bresnick AR; Weber DJ; Zimmer DB Nat. Rev. Cancer 2015, 15, 96–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Delcourt V; Franck J; Leblanc E; Narducci F; Robin YM; Gimeno JP; Quanico J; Wisztorski M; Kobeissy F; Jacques JF; Roucou X; Salzet M; Fournier I EBioMedicine 2017, 21, 55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Cross SS; Hamdy FC; Deloulme JC; Rehman I Histopathology 2005, 46, 256–269. [DOI] [PubMed] [Google Scholar]
- (21).McKiernan E; McDermott EW; Evoy D; Crown J; Duffy MJ Tumor Biol. 2011, 32, 441–450. [DOI] [PubMed] [Google Scholar]
- (22).Sanders ME; Dias EC; Xu BJ; Mobley JA; Billheimer D; Roder H; Grigorieva J; Dowsett M; Arteaga CL; Caprioli RM J. Proteome Res 2008, 7, 1500–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Grosset AA; Labrie M; Vladoiu MC; Yousef EM; Gaboury L; St-Pierre Y Oncotarget 2016, 7, 18183–18203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Kompauer M; Heiles S; Spengler B Nat. Methods 2017, 14, 90–96. [DOI] [PubMed] [Google Scholar]
- (25).Palma CD; Grassi ML; Thome CH; Ferreira GA; Albuquerque D; Pinto MT; Melo FUF; Kashima S; Covas DT; Pitteri SJ; Faca VM Mol. Cell. Proteomics 2016, 15, 906–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.