

Abstract
The initiation and progression of liver cancer, including hepatocellular carcinoma and intrahepatic cholangiocarcinoma, are dependent on its tumor microenvironment. Immune cells are key players in the liver cancer microenvironment and show complicated crosstalk with cancer cells. Emerging evidence has shown that the functions of immune cells are closely related to cell metabolism. However, the effects of metabolic changes of immune cells on liver cancer progression are largely undefined. In this review, we summarize the recent findings of immunometabolism and relate these findings to liver cancer progression. We also explore the translation of the understanding of immunometabolism for clinical use.
Keywords: Cholangiocarcinoma, Hepatocellular carcinoma, Tumor microenvironment, Local immune status, Metabolite
Core tip: The liver microenvironment provides a special place for initiation and progression of liver cancer, in which immune cells play a vital role. On the one hand, immunosuppression leads to tumor survival and progression; on the other hand, the instigation of tumor metabolites and signal molecules to immune cells makes the state of immunosuppression further strengthened. Intensive studies of the metabolic state of immune cells in the tumor microenvironment is beneficial to our understanding of the regulation of pro-tumor patterns, and to provide theoretical basis and guidance for immunometabolic therapies for liver cancer.
INTRODUCTION
Liver cancer is one of the most common malignancies and has the third leading mortality rate and seventh leading incidence rate worldwide[1]. To date, with over 42000 cases diagnosed and 30000 deaths in the United States per year, a continuous increase in both morbidity and mortality is observed in liver cancer[2]. Unfortunately, the prognosis of advanced liver cancer is still poor despite the many treatments that have been developed in the past few decades. Thus, novel approaches to treat liver cancer are urgently needed.
The tumor microenvironment (TME) plays critical roles in tumor development and is characterized by complicated components, including various types of non-tumoral cells and non-cellular materials[3]. TMEs can be largely distinct among different types of cancers and among different patients with the same type of cancer. Within a patient, the TME can consistently change as the tumor progresses and is influenced by the physiopathological conditions of the patient. Thus, it is theoretically impossible to identify the precise state of the TME. However, under certain conditions, the TME can be specialized to show typical traits, and in particular, this specialized TME may affect tumor progression. Understanding these particular TMEs, if not all of them, can outline the crosstalk between the TME and tumor cells and facilitate the development of novel strategies for tumor treatment[4].
Pathologically, liver cancer mainly includes hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma (ICC). The two types of liver cancers share a similar hepatic microenvironment, but may have different TMEs due to the various biological characteristics of the tumor cells. For instance, in a considerable proportion of cases, ICC harbors isocitrate dehydrogenase (IDH) 1 and IDH2 mutations[5], which are rare in HCC[6]. IDH1 and IDH2 are important enzymes involved in cellular metabolism. Therefore, IDH1/2 mutations can substantially influence the metabolite profiles in cells and TMEs. In this case, the roles of the TME in liver cancer progression may be greatly altered. However, knowledge in this field is poor and scattered.
Immune cells are key players in liver cancer progression, and their recruitment and functions are profoundly affected by the TME[7]. Other molecules in the TME, such as fatty acids and glucose, are able to regulate the metabolism, phenotype and function of immune cells[8,9]. The term “immunometabolism” has recently been proposed and indicates the functional intracellular metabolic alterations that occur within immune cells[10]. These affected immune cells, in turn, could have significant effects on adjacent tumor cells. In this minireview, we collected evidence of TME heterogeneity resulting from the different biological features of cancer cells and functional changes of immune cells resulting from an altered TME and how the TME affects tumor progression via metabolically regulated immune cells.
CHARACTERISTICS OF IMMUNE CELLS IN LIVER CANCER MICROENVIRONMENT
With genetic and epigenetic changes, hepatoma cells express specific tumor-associated antigens, such as α-fetoprotein (AFP), glypican-3 (GPC3) and melanoma-associated gene-A1 (MAGE-A1), which can be taken up by antigen-presenting cells and presented to T cells, resulting in a cytotoxic reaction to eliminate cancerous cells[11,12]. However, immunosuppressive factors and immune-inhibitory checkpoint molecules inhibit anti-tumor reactions and create a special microenvironment to facilitate tumor progression[12]. Almost all types of immune cells are deeply involved in the TME of liver cancer (Figure 1), including macrophages, Kupffer cells, neutrophils, T cells, B cells, innate lymphoid cells (ILCs), dendritic cells (DCs), natural killer (NK) cells, natural killer T (NKT) cells, and myeloid-derived suppressor cells (MDSCs)[13-18].
Figure 1.
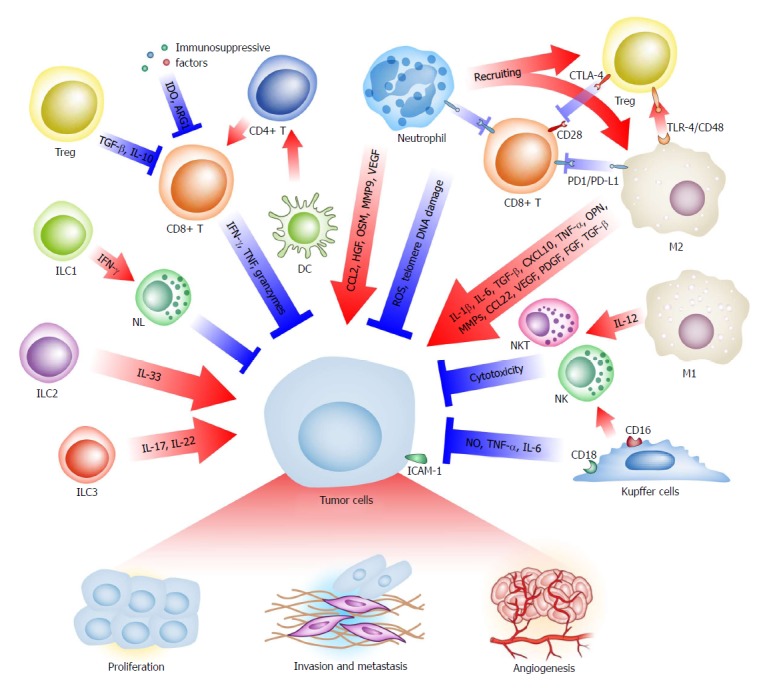
The immune cells in the tumor microenvironment regulate liver cancer progression. Many types of immune cells in the TME show pro- or anti-tumoral effects on the liver cancer cells by cell-specific mechanisms. Complicated crosstalk between immune cells is also common. TME: Tumor microenvironment; ILC: Innate lymphoid cell; NKT: Natural killer T.
Macrophages and neutrophils
Macrophages display remarkable heterogeneity in liver cancer for various reasons, such as the cell origin (resident Kupffer cells and recruited monocyte-derived macrophages), stimulating signals (i.e., microbes, cell debris) and functional phenotype (i.e., inflammatory, anti-inflammatory). Notably, macrophages can play two or more contradictory roles in a particular TME. As guardians, macrophages are normally activated by inflammatory signals and together with CD4+ T cells eliminate precancerous senescent cells[19]. Kupffer cells directly show cytotoxicity against tumor cells[20-22]. In addition, macrophage-derived interleukin (IL)-12 hampers tumor progression by activating NK and NKT cells[23,24]. As an accomplice, however, tumor-associated macrophages (TAMs, most of which are M2-polarized macrophages), induced by IL-4 and tumor growth factor (TGF)-β, accumulate in liver cancer and are correlated with the poor prognosis of patients[25,26]. Importantly, these macrophages express the immune checkpoint protein programmed cell death-ligand 1 (PD-L1, also known as B7-H1), which inactivates CD8+ T cells[13,27,28]. Via other immunosuppressive signals, such as Toll-like receptor (TLR) 4 and CD48/2B4, M2-polarized macrophages promote the recruitment of regulatory T cells (Tregs) and suppress the activity of NK cells[29-31]. Moreover, these macrophages can secrete various tumor proliferation-promoting cytokines, such as IL-1β, IL-6, TGF-β, C-X-C motif chemokine (CXCL) 10, invasion and metastasis-promoting factors like tumor necrosis factor (TNF)-α, osteopontin (OPN), matrix metalloproteinases (MMPs), C-C Motif chemokine ligand (CCL) 22, and proangiogenic growth factors, like vascular endothelial growth factor (VEGF), platelet derived growth factor (PDGF), fibroblast growth factor (FGF), and TGF-β, to build a tumor-prone inflammatory microenvironment[3,26,32-35].
Similar to macrophages, neutrophils also have diverse functions at different stages of liver cancer progression. In the case of a hepatic infection or injury, neutrophils gather at the wound site together with macrophages to eliminate pathogens and necrotic materials. Additionally, neutrophils stimulate reactive oxygen species (ROS) and telomere DNA damage in hepatocytes, mediating neoplasia and progression[36]. Mirroring macrophage plasticity, a pro-tumoral phenotype of tumor-associated neutrophils (TANs) is proposed[37,38]. Despite biomarkers of this subtype, immunosuppression is the most central function of TANs. The immunosuppressive molecule PD-L1 is regularly displayed in TANs[39] and recruits macrophages and Treg cells to the liver cancer TME and induces impaired anti-tumoral immunity[14]. The infiltrating TAN density and neutrophil-lymphocyte ratio is reported to be a predictor of outcome, chemotherapy resistance, and recurrence risk[40-42]. Furthermore, neutrophils promote tumor progression by secreting cytokines and other functional molecules, such as CCL2 for tumor growth, hepatocyte growth factor (HGF) and oncostatin M (OSM) for metastasis, and MMP9 and VEGF for angiogenesis[38,43-47].
T cells
CD8+ T cells are the most important executors of adaptive immunity against neoplasms, including liver cancer. Unfortunately, the TME transforms these ‘warriors’ into ‘servants’. Compared with the normal liver, tumor tissue has a lower density of CD8+ T cells and a higher density of Tregs. The ratio of CD8+ T cells to Tregs typically indicates a poor prognosis[48-50]. Recent studies suggest that interferon (IFN)-γ, TNF and granzyme secretion by CD8+ cytotoxic T lymphocytes (CTLs) represent a common cytotoxic reaction against tumors[51,52]. Tregs, characterized by CD4, CD25, cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and forkhead box P3 (FoxP3) expression, can eliminate IL-2 via its receptor subunit CD25, downregulate CD80 and CD86 and conjugate to the co-stimulatory molecule CD28 competitively with CTLA-4 to suppress immune responses. In addition, Tregs secrete TGF-β and IL-10 into the TME to suppress T effector cells[52]. Via a complicated regulatory network, several subtypes of T cells contribute to the immunosuppressive TME.
ILCs
ILCs are recently identified innate immune cells that lack a specific antigen receptor. These cells originate from mucosal-associated lymphoid tissues and act as a sentry of the rapid immune response and regulator of immune homeostasis and inflammation[53]. Mirroring the classification of helper T cells (Th), ILCs are divided into three classes. ILC1s produce Th1-associated cytokines, ILC2s are associated with Th2-associated cytokine release, and ILC3s secrete Th17-associated cytokines[54]. As these three components simultaneously exist in the liver, they are likely to be involved in hepatocarcinogenesis and progression. ILC1s produce IFN-γ to activate NK cells and indirectly participate in cancer immunosurveillance, while ILC3s release IL-17 and IL-22, which may promote tumor growth and angiogenesis[16,55,56]. Moreover, we observed an increasing number of ILC2 in HCC tissue (unpublished data) and an elevated level of IL-33 in both serum and tumor tissue samples from HCC patients[57]. IL-33 can stimulate rapid growth and metastatic progression in breast cancer and cholangiocarcinoma[58,59]. Therefore, the initiation and growth of liver cancer promoted by the ILC2-related IL-33/ST2 axis might be theoretically tenable[60].
ALTERED METABOLISM OF LIVER CANCER AND ITS INFLUENCE ON THE TUMOR MICROENVIRONMENT
With their rapid growth and limited nutrition supply, the survival of tumor cells seems to be in question. However, the tumor is able to reprogram the cellular metabolism for neoplastic proliferation. The abnormal metabolism induced by cancer provides energy and metabolites for cell activity and additionally modifies many related pathways that influence various biological processes[61].
General alterations
The liver is a metabolic organ, and metabolic disruption of the liver can lead to spontaneous hepatocarcinogenesis[62]. The principal metabolic alterations in the liver cancer were profiled by metabolomics analysis, which showed elevated glycolysis, gluconeogenesis and β-oxidation, with reduced tricarboxylic acid cycle (TCA cycle) activity[63,64]. Aerobic glycolysis, also known as the “Warburg effect”, is frequently observed in various tumors. This theory indicates that tumor cells predominantly use glycolysis, even in the presence of sufficient oxygen[65]. For liver cancer, glycolysis also plays a dominating role in glucose metabolism[63]. Accumulating studies have revealed that glycolysis is associated with genetic and epigenetic changes. Activated oncogenes and mutant tumor suppressors are associated with glycolysis. Glucose transporters (GLUTs), glycolysis-related enzyme hexokinase2 (HK2), pyruvate kinase M2 (PKM2), and lactate dehydrogenase (LDH) A are also overexpressed in liver cancer tissue, suggesting an increased glycolysis activity[66]. Moreover, the hypoxic TME and overexpression of β-catenin in liver cancer stabilize hypoxia inducible factor (HIF)-1α to activate glycolytic enzymes[67-70].
Fatty acid β-oxidation is enhanced in liver cancer to overcome the energy shortage and reduce tumor dependence on glucose. Metabolic alteration increases FA biosynthesis and glycerolipid metabolism, leading to fatty acid and lipid accumulation[71,72]. However, the alterations of various types of fatty acids are complicated. Additionally, almost all amino acids are increased in liver cancer due to the reduction of amino acid catabolism[63]. Although these general changes were identified, many other factors may influence the levels of metabolites in TME.
Involvement of specific mutations
Some specific mutations profoundly change the metabolism of liver cancer. The Tumor Cell Genomic Atlas (TCGA) project of HCC demonstrated that IDH and fibroblast growth factor receptor (FGFR) are meaningful mutations in HCC. IDHs are critical enzymes for cell metabolism, particularly the TCA cycle. IDH1/2 mutations convert α-ketogluterate (α-KG) into 2-hydrogluterate (2-HG)[73]. The unusually decreased ratio of α-KG/2-HG can significantly influence tumor cell biology by competing with α-KG and regulating epigenetic expression[74]. However, overproduced 2-HG by tumor cells can also be released into the TME since elevated serum and urinary levels of 2-HG can be detected in patients with IDH1/2-mutated solid tumors[75,76]. Although 2-HG seems to be impermeable, this enzyme acts on stromal cells within the bone marrow microenvironment through an ROS/extracellular signal-regulated kinase (ERK) signaling pathway[77], suggesting that 2-HG may influence immune cells in the TME of liver cancer. This finding is important since IDH-like HCCs (particularly those containing IDH1/2 mutations and those with IDH-like gene expression) are indicative of worse clinical outcomes for undefined reasons[6]. In addition to 2-HG, IDH-like HCCs and IDH1/2 mutated ICCs have other significant metabolic alterations, such as gliomas due to pseudohypoxia, an interrupted Krebs cycle, and epigenetic changes[78]. In addition, IDH gain-of-function mutations also induce the reprogramming of pyruvate and lipid metabolism to maintain cell proliferation and clonogenicity[79,80]. Therefore, the TMEs of these tumors likely have distinct features, and the immune cells within such TMEs may be reprogrammed to have different functions.
FGFR genetic aberrations include mutations, amplifications, and gene fusions, which have been observed in over 10% of liver cancer[81-83]. FGFR2 fusions are active kinases with the highest incidence[84]. These genetic aberrations indirectly affect glucose and lipid metabolism by activating the kinase network[85]. Additionally, the oncogenic FGFR3-transforming acidic coiled-coil containing protein (TACC) 3 fusion activates oxidative phosphorylation and mitochondrial biogenesis, causing a mitochondrial respiration-dependent subtype of tumor cell[86].
Influence of hepatic viruses
Hepatitis viruses, particularly HBV in eastern countries and HCV in western countries, cause specific metabolic alterations during hepatocarcinogenesis and tumor progression. Hepatic virus infections activate many abnormal signaling pathways, which cause aberrant functions and expression of metabolism related enzymes[87]. As a consequence, HBV infection in patients is associated with increased free fatty acids (FFAs) and acyl-carnitines and decreased triglycerides, phospholipids, and sphingomyelins[88]. Thus, in the TME of HBV-related liver cancer, the increased FFAs and decreased glucose may result from increased β-oxidation and glycolysis, respectively. HBV-related inflammation can stimulate the expression of HIF-1α through the PI3K/AKT and mitogen-activated protein kinase (MAPK)-Ras-Raf pathways. The latter up-regulates GLUT1 and glycolytic enzymes, increases the glycolytic flow, and induces ROS accumulation. These alterations eventually cause DNA oxidative damage and malignant transformation[89-92]. HBx activates the adenosine 5’-monophosphate-activated protein kinase (AMPK) and fatty acid oxidation (FAO) pathways as well as the HBx-LXRα-SREBP1/FAS pathway, which allows HCC cells to survive under metabolic stress[93-95]. HCV establishes an insulin-resistant TME in the liver through several mechanisms[96,97]. For instance, HCV protein can induce the overexpression of protein phosphatase 2A (PP2A), which dysregulates hepatic glucose homeostasis by inhibiting AKT and the dephosphorylation of FoxO1[98]. However, the molecular mechanism of viral hepatitis-related metabolic alternations is not well understood. HBV integration into the tumor suppressor gene region causes metabolic alterations. Signaling molecules, such as TGF-β, mammalian target of rapamycin (mTOR), Smad3, and c-Myc, are activated by HBV and HCV, which is closely correlated with tumor progression[87,96].
Participation of other concomitant diseases
Liver cancer has a higher incidence in people with chronic non-infectious liver diseases. Therefore, the metabolic changes of liver cancer are related to cirrhosis and nonalcoholic fatty liver disease (NAFLD), which typically includes lipid anomalies, particularly dysregulated de novo lipogenesis[99]. For instance, peripheral insulin resistance together with enhanced mitochondrial β-oxidation and oxidative stress are the most prominent features of NAFLD and nonalcoholic steatohepatitis (NASH)[100]. Thus, NAFLD-related liver cancer is associated with elevated oxidative metabolism and amplified anaplerosis/cataplerosis[101]. With the increasing content of hepatic FFAs, liver cells may endure a mild respiratory dysfunction. The increasing import of FFAs into the mitochondria is accompanied by an elevated rate of β-oxidation. The overloaded fatty acid β-oxidation triggers the subsequent accumulation of ROS, which further leads to lipid peroxidation and severe mitochondrial oxidative damage[102,103]. These strong oxidizing products promote inflammation, fibrosis, and even carcinogenesis. Lipid metabolism shows an alteration from β-oxidation to ω-oxidation during liver cirrhosis[104]. Intriguingly, a metabolic switch from oxidative phosphorylation to glycolysis in hepatocytes in early-stage cirrhosis may satisfy the extreme energy requirements under such conditions[105]. This evidence demonstrates that concomitant liver diseases or even systemic metabolic diseases, such as diabetes mellitus, can influence the function of immune cells and their functions in promoting tumor progression.
Metabolic reprogramming of local immune cells in liver cancer
The role of metabolism in regulating immune cells has recently aroused general concerns. Evidence collected in several types of solid tumors indicated the importance of tumor immunometabolic reprogramming and suggested a novel and crucial area for future research of liver cancer[106]. The complicated crosstalk between metabolically reprogrammed immune cells and liver cancer cells has been suggested, but the molecular mechanisms need further exploration (Figure 2).
Figure 2.
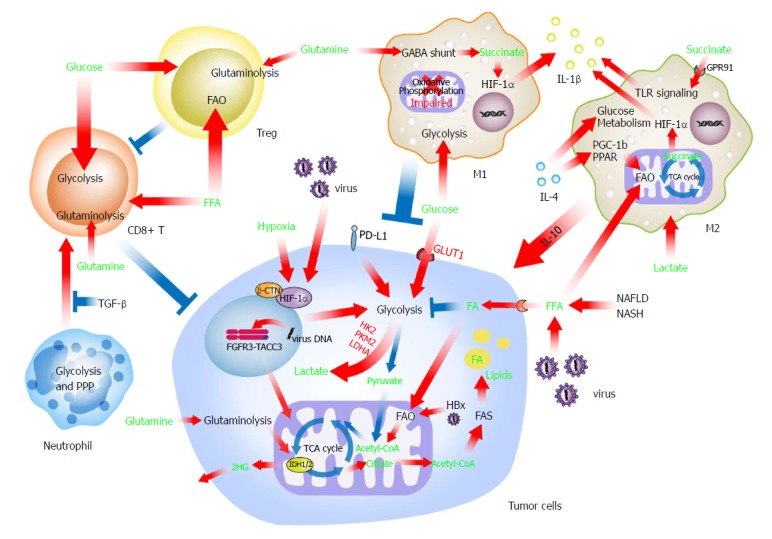
Metabolites in the tumor microenvironment affect the anti-/pro-tumoral functions of immune cells. Energy resources including glucose, glutamine, free fatty acids, and other metabolites such as succinate largely alter the functions of macrophages, neutrophils, and T cells. These metabolically reprogrammed immune cells then have a differed influence on the liver cancer cells compared to the original immune cells. FAO: Fatty acid oxidation.
Because they exist at a considerable quantity, macrophages play a leading role in the crosstalk between liver cancer cells and immune cells. The phenotypes and functions of macrophages are hot topics in the field of immunometabolism. Professor O’Neill’s group and other investigations have conducted several pioneering and important studies to decipher macrophage immunometabolism. Recent evidence has suggested that the energy source (e.g., glucose and fatty acids) participates in the determination of macrophage polarization[107,108]. However, the role of FAO in the alternative activation of macrophages is still under debate. Huang et al[108] initially demonstrated an essential role for FAO in the alternative activation of mouse macrophages, while two groups in Germany and the United States subsequently provided convincing evidence that FAO was indispensable for the M2 polarization of human and mouse macrophages, respectively[109,110]. This discrepancy may partially reflect differences between mouse and human macrophages and the potential off-target effects of etomoxir (a carnitine palmitoyltransferase I A, CPT1A inhibitor used to inhibit FAO by some researchers). Although the role of FAO in macrophage activation has not yet been determined, FAO enhances the functions of M2 polarized macrophages[111].
Generally, M2 polarized macrophages use oxidative phosphorylation, FAO in particular, to obtain the required energy and exert pro-tumor function in various scenarios, including liver cancer[108,112,113]. Mechanistically, IL-4 induces the expression of peroxisome proliferator-activated receptors (PPAR) and PPAR gamma coactivator 1-beta (PGC-1b), which orchestrate alteration of FAO as well as mitochondrial respiration[114]. Similar to liver cancer cells in a background of chronic fatty liver disease, FFA accumulation attenuates mitochondrial respiration and increases FFA uptake via CD36, resulting in a predominant FAO mode in macrophages[115,116]. In addition, IL-4 signaling also increases glucose metabolism by the AKT/mTOR pathway[117-119]. Lactic acid produced by liver cancer cells through glycolysis is another non-negligible issue when considering the metabolic reprogramming of liver cancer-associated macrophages. The polarization of macrophages to an immunosuppressive and pro-tumoral phenotype is mediated by lactic acid via a different, as yet unidentified pathway[119-121] in liver cancer. M2 polarized macrophages also take up insulin growth factor 1 for a self-immunometabolic reprogramming, leading to reduced phagocytosis and lower energy expenditure[122], which may consequently regulate liver cancer progression.
However, M1 polarized macrophages have enhanced glycolytic metabolism and impaired oxidative phosphorylation through the AKT/mTOR/HIF-1α pathway[123]. This association is consistent with the fact that many glycolytic enzymes facilitate inflammatory cytokine production. For example, PKM2, which is critical for glycolysis, activates HIF-1α and induces IL-1β production[124]. Although under some conditions, FAO was also found to support inflammasome activation in M1 polarized macrophages by regulating CPT1A activity[125,126].
Other metabolites in the TME can also influence the phenotype and function of macrophages. Succinate is a known inflammation inducer of macrophages. Succinate enhances IL-1β secretion via succinate receptor 1 (SUCNR1, also known as GPR91)-mediated amplification of TLR signaling[127]. In addition, intracellular succinate, derived from the γ-aminobutyric acid (GABA) shunt and anerplerosis of α-KG upon stimulation of TLR4, enhances IL-1β production through the repurposing of mitochondrial function from ATP production to ROS generation[128,129].
Neutrophils are metabolically similar to M1 polarized macrophages and rely on aerobic glycolysis and the pentose phosphate pathway (PPP) as their principal mode of energy metabolism, by which the formation of neutrophil extracellular traps produces biological effects[130].
During differentiation, a switch from FAO to glycolysis and glutaminolysis triggers the maturation of T effector cells[131]. The rapid supply of ATP helps CTLs to meet their increasing bioenergetic and biosynthetic requirements[132]. Nevertheless, glucose shortage and lactic acid abundance limit the function of CTLs, and the enhancement of FAO preserves the cytotoxicity of CTLs in tumors[133]. By contrast, Treg cells favor FAO rather than glycolysis, by which these cells survive in the persistent low-glucose and hypoxic tumor microenvironment and suppress the tumor-killing function of CTLs[131,134]. Moreover, Treg cells rely on oxidative phosphorylation for energy supply, on FAO for cell differentiation, and on glutaminolysis for cell proliferation[134,135]. Taken together, these observations demonstrate that environment-related metabolism regulates the anti- and pro-tumor functions of tumor-infiltrating lymphocytes.
INFLUENCE OF REPROGRAMMED IMMUNE CELLS ON LIVER CANCER PROGRESSION
Immunometabolic reprogramming has a dual-function in tumor progression. Typically, M1 polarized macrophages facilitate inflammation and the antitumor response via elevated glycolysis, while M2 polarized macrophages play an FAO predominant role, secreting IL-10 to suppress the immune reaction. The switch in the metabolism of TAMs leads to an ample signaling transition by which these cells suppress immune reactions (by presenting PD-1, etc.), accelerate tumor proliferation (by releasing TNF, Wnt signals, etc.), promote angiogenesis (by secreting VEGF, FGF2, etc.), and enhance tumor invasion and metastasis (by MMP9, TGF-β, etc.)[26,136]. We further revealed that IL-1β facilitated epithelial-to-mesenchymal transition and subsequent metastasis in liver cancer, and this effect was mediated by a group of pro-inflammatory M2-like TAMs with an up-regulated level of glycolysis[137]. Interestingly, the function of TANs is opposite that of TAMs under different circumstances. For instance, the absence of TGF-β leads to a TANs-induced anti-tumor response by CTLs, whereas this situation is completely different in the presence of TGF-β[37].
The influence of metabolic reprogramming on lymphocytes differs in the presence of high functional heterogeneity. CTLs enhance glycolysis, glutaminolysis, and even FAO to exert anti-tumoral cytotoxicity, while CD4+ T cells develop into two phenotypes with contrary functions. With a similar switch characterized by up-regulated glycolysis as well as increased glutaminolysis and PPP, Th1 cells induce macrophage- and NK cell-related anti-tumoral responses, whereas Th2 cells induce immunosuppressive reactions[113].
PERSPECTIVES OF LIVER CANCER THERAPY FROM IMMUNOMETABOLISM
Theoretically, it is feasible to target metabolic enzymes or metabolites in immune cells for therapeutic purposes. Indeed, several cancer-related immunometabolic molecules are suitable as potential targets for drug development (Table 1).
Table 1.
Immunometabolic therapies for cancers
| Targets | Agents | Mechanisms | Developments |
| PKM2 | DASA, TEPP46 | Inhibition of HIF1α | Preclinical |
| HIF1α | PX-478, RO7070179, EZN-2968 | Inhibition of HIF1α | Phase 1 |
| PTEN | VO-Ohpic, SF1670 | Inhibition of PI3K | Preclinical |
| PDK | Dichloroacetate | Inhibition of glycolysis | Phase 2 |
| GLUT1 | Ritonavir | Inhibition of glycolysis | FDA-Approved |
| LDH | Gossypol (AT-101), FX11, Galloflavin | Inhibition of glycolysis | Phase 3 |
| CPT1A | Etomoxir | Inhibition of FAO | Preclinical |
| CTLA-4 | Ipilimumab | Checkpoint blockade; Inhibition of FAO | FDA-Approved |
| PD-1/PD-L1 | Nivolumab, Pembrolizumab, Atezolizumab | Checkpoint blockade; Inhibition of FAO | FDA-Approved |
| AMPK | Metformin | Increased FAO; Inhibition of Complex I; Decreased mitochondrial ROS | FDA-Approved |
| mTOR | Temsirolimus, Everolimus | Inhibition of HIF-1α translation | FDA-Approved |
| IDO | Epacadostat | Regulation of tryptophan metabolism; Inhibition of mTORC1 | FDA-Approved |
| IDH1/2 mutations | Ivosidenib (AG-120), IDH305, AG-881, DS-1001b | Inhibition of 2-HG production | Phase 3 |
| FGFR | Regorafenib, Sunitinib, TAS120 | Inhibition multi-targeted kinase | Phase 2 |
| iNOS | L-NMMA, 1400W | Inhibition of NO production | Phase 2 |
| FOXP3 | P60 | suppress NF-κB and NFAT | Preclinical |
PKM2: Pyruvate kinase M2; HIF: Hypoxia inducible factor; LDH: Lactate dehydrogenase; FAO: Fatty acid oxidation; CTLA-4: Cytotoxic T-lymphocyte-associated protein 4; mTOR: Mammalian target of rapamycin; FGFR: Fibroblast growth factor receptor; FoxP3: Forkhead box P3; ROS: Reactive oxygen species.
Considering carbohydrate metabolism, glycolysis is important to activated immune cells. Inhibition of HIF-1α can attenuate TAM/TAN-mediated IL-1β secretion, reduce hypoxic adaptation of tumor cells, and regulate the differentiation and function of lymphocytes[137,138]. The regulation of PI3K signaling by PTEN in immune cells may also show some effects on macrophage activation and Treg cell function with a concurrent influence on glycolysis[139,140].
The mTOR pathway may be a suitable target to regulate immune cells by manipulating cellular lipid metabolism. Inhibition of mTOR by rapamycin can block the development of macrophages and CD4+ T cells in several scenarios, including liver cancer[141,142]. Moreover, FAO plays a predominant role in the metabolism of M2 polarized macrophages. Hence, inhibition of FAO by etomoxir or other methods (e.g., targeted delivery of CPT1A siRNA/shRNA) may limit the immunosuppressive function of M2 macrophages. However, in a resource-deficient microenvironment, CTLs also take up FFA and exert cytotoxicity; thus, the selective increase of FFA import to CTLs enhances the anti-tumoral capacity of these cells[133]. It is also feasible to target proteins that play central roles in metabolic pathways (i.e., iNOS, PKM2, Foxp3, etc.). The difficulty of this strategy is that targeting should be cell specific because the same metabolic pathway can have diverse effects on different immune cells.
Furthermore, drugs targeting tumor specific metabolic changes can also regulate the function of immune cells by altering the metabolites of the tumor. For instance, IDH1/2 inhibitors specifically reduce 2-HG production leading to destabilization of HIF-1α in immune cells, which can cause reduction of IL-1β secretion by macrophages.
CONCLUSION
Immunometabolism has increasingly become a component of immunology in the past decade. The current understanding suggests that a complicated metabolic network regulates the various functions of immune cells. This network can either be functionally oriented or environmentally adapted, but together, these complex interactions lead to immune microenvironment homeostasis, which affects the progression of liver cancer. However, numerous findings indicate that almost all types of immune cells present a self-contradictory function under different conditions, although the mechanisms are still unknown. In the future, more subtypes among immune cells will be defined as single cell detection systems are further developed, which may provide a key for functional heterogeneity. Therefore, liver cancer therapy that targets immunometabolism is a promising approach. Further work is urgently needed to explore the utility of targeting specific immunometabolic events in the liver cancer microenvironment for therapeutic gain.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Conflict-of-interest statement: The authors declare that they have no competing interests.
Peer-review started: April 16, 2018
First decision: May 16, 2018
Article in press: June 27, 2018
P- Reviewer: Ooi LL, Rodríguez-Perálvarez M, Yamasaki T S- Editor: Wang XJ L- Editor: Filipodia E- Editor: Huang Y
Contributor Information
Qi Zhang, Department of Hepatobiliary and Pancreatic Surgery, the Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310009, Zhejiang Province, China; Zhejiang Provincial Key Laboratory of Pancreatic Disease, Hangzhou 310009, Zhejiang Province, China.
Yu Lou, Department of Hepatobiliary and Pancreatic Surgery, the Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310009, Zhejiang Province, China; Zhejiang Provincial Key Laboratory of Pancreatic Disease, Hangzhou 310009, Zhejiang Province, China.
Xue-Li Bai, Department of Hepatobiliary and Pancreatic Surgery, the Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310009, Zhejiang Province, China; Zhejiang Provincial Key Laboratory of Pancreatic Disease, Hangzhou 310009, Zhejiang Province, China.
Ting-Bo Liang, Department of Hepatobiliary and Pancreatic Surgery, the Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310009, Zhejiang Province, China; Zhejiang Provincial Key Laboratory of Pancreatic Disease, Hangzhou 310009, Zhejiang Province, China. liangtingbo@zju.edu.cn.
References
- 1.Fact sheets by Population-Globocan-IARC. IARC 2012. [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 3.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fang H, Declerck YA. Targeting the tumor microenvironment: from understanding pathways to effective clinical trials. Cancer Res. 2013;73:4965–4977. doi: 10.1158/0008-5472.CAN-13-0661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farshidfar F, Zheng S, Gingras MC, Newton Y, Shih J, Robertson AG, Hinoue T, Hoadley KA, Gibb EA, Roszik J, et al. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell Rep. 2017;18:2780–2794. doi: 10.1016/j.celrep.2017.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas Research Network. Cancer Genome Atlas Research Network. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell. 2017;169:1327–1341.e23. doi: 10.1016/j.cell.2017.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014–1022. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yaqoob P. Fatty acids as gatekeepers of immune cell regulation. Trends Immunol. 2003;24:639–645. doi: 10.1016/j.it.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 9.Lawless SJ, Kedia-Mehta N, Walls JF, McGarrigle R, Convery O, Sinclair LV, Navarro MN, Murray J, Finlay DK. Glucose represses dendritic cell-induced T cell responses. Nat Commun. 2017;8:15620. doi: 10.1038/ncomms15620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16:553–565. doi: 10.1038/nri.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flecken T, Schmidt N, Hild S, Gostick E, Drognitz O, Zeiser R, Schemmer P, Bruns H, Eiermann T, Price DA, et al. Immunodominance and functional alterations of tumor-associated antigen-specific CD8+ T-cell responses in hepatocellular carcinoma. Hepatology. 2014;59:1415–1426. doi: 10.1002/hep.26731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prieto J, Melero I, Sangro B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2015;12:681–700. doi: 10.1038/nrgastro.2015.173. [DOI] [PubMed] [Google Scholar]
- 13.Wu K, Kryczek I, Chen L, Zou W, Welling TH. Kupffer cell suppression of CD8+ T cells in human hepatocellular carcinoma is mediated by B7-H1/programmed death-1 interactions. Cancer Res. 2009;69:8067–8075. doi: 10.1158/0008-5472.CAN-09-0901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou SL, Zhou ZJ, Hu ZQ, Huang XW, Wang Z, Chen EB, Fan J, Cao Y, Dai Z, Zhou J. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology. 2016;150:1646–1658.e17. doi: 10.1053/j.gastro.2016.02.040. [DOI] [PubMed] [Google Scholar]
- 15.Shi JY, Gao Q, Wang ZC, Zhou J, Wang XY, Min ZH, Shi YH, Shi GM, Ding ZB, Ke AW, et al. Margin-infiltrating CD20(+) B cells display an atypical memory phenotype and correlate with favorable prognosis in hepatocellular carcinoma. Clin Cancer Res. 2013;19:5994–6005. doi: 10.1158/1078-0432.CCR-12-3497. [DOI] [PubMed] [Google Scholar]
- 16.Dadi S, Chhangawala S, Whitlock BM, Franklin RA, Luo CT, Oh SA, Toure A, Pritykin Y, Huse M, Leslie CS, et al. Cancer Immunosurveillance by Tissue-Resident Innate Lymphoid Cells and Innate-like T Cells. Cell. 2016;164:365–377. doi: 10.1016/j.cell.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gonzalez-Carmona MA, Lukacs-Kornek V, Timmerman A, Shabani S, Kornek M, Vogt A, Yildiz Y, Sievers E, Schmidt-Wolf IG, Caselmann WH, et al. CD40ligand-expressing dendritic cells induce regression of hepatocellular carcinoma by activating innate and acquired immunity in vivo. Hepatology. 2008;48:157–168. doi: 10.1002/hep.22296. [DOI] [PubMed] [Google Scholar]
- 18.Schrader J. The role of MDSCs in hepatocellular carcinoma--in vivo veritas? J Hepatol. 2013;59:921–923. doi: 10.1016/j.jhep.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 19.Eggert T, Wolter K, Ji J, Ma C, Yevsa T, Klotz S, Medina-Echeverz J, Longerich T, Forgues M, Reisinger F, et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell. 2016;30:533–547. doi: 10.1016/j.ccell.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aono K, Isobe K, Nakashima I, Kondo S, Miyachi M, Nimura Y. Kupffer cells cytotoxicity against hepatoma cells is related to nitric oxide. Biochem Biophys Res Commun. 1994;201:1175–1181. doi: 10.1006/bbrc.1994.1829. [DOI] [PubMed] [Google Scholar]
- 21.Saito H, Kurose I, Ebinuma H, Fukumura D, Higuchi H, Atsukawa K, Tada S, Kimura H, Yonei Y, Masuda T, et al. Kupffer cell-mediated cytotoxicity against hepatoma cells occurs through production of nitric oxide and adhesion via ICAM-1/CD18. Int Immunol. 1996;8:1165–1172. doi: 10.1093/intimm/8.7.1165. [DOI] [PubMed] [Google Scholar]
- 22.Li XY, Wu L, Li SW, Zhou WB, Wang MY, Zuo GQ, Liu CA, Ding X. Effect of CD16a, the surface receptor of Kupffer cells, on the growth of hepatocellular carcinoma cells. Int J Mol Med. 2016;37:1465–1474. doi: 10.3892/ijmm.2016.2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rushfeldt C, Sveinbjørnsson B, Seljelid R, Smedsrød B. Early events of hepatic metastasis formation in mice: role of Kupffer and NK-cells in natural and interferon-gamma-stimulated defense. J Surg Res. 1999;82:209–215. doi: 10.1006/jsre.1998.5532. [DOI] [PubMed] [Google Scholar]
- 24.Seki S, Nakashima H, Nakashima M, Kinoshita M. Antitumor immunity produced by the liver Kupffer cells, NK cells, NKT cells, and CD8 CD122 T cells. Clin Dev Immunol. 2011;2011:868345. doi: 10.1155/2011/868345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ding T, Xu J, Wang F, Shi M, Zhang Y, Li SP, Zheng L. High tumor-infiltrating macrophage density predicts poor prognosis in patients with primary hepatocellular carcinoma after resection. Hum Pathol. 2009;40:381–389. doi: 10.1016/j.humpath.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 26.Yeung OW, Lo CM, Ling CC, Qi X, Geng W, Li CX, Ng KT, Forbes SJ, Guan XY, Poon RT, et al. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. J Hepatol. 2015;62:607–616. doi: 10.1016/j.jhep.2014.10.029. [DOI] [PubMed] [Google Scholar]
- 27.Umemoto Y, Okano S, Matsumoto Y, Nakagawara H, Matono R, Yoshiya S, Yamashita Y, Yoshizumi T, Ikegami T, Soejima Y, et al. Prognostic impact of programmed cell death 1 ligand 1 expression in human leukocyte antigen class I-positive hepatocellular carcinoma after curative hepatectomy. J Gastroenterol. 2015;50:65–75. doi: 10.1007/s00535-014-0933-3. [DOI] [PubMed] [Google Scholar]
- 28.Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C, Zheng L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med. 2009;206:1327–1337. doi: 10.1084/jem.20082173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou J, Ding T, Pan W, Zhu LY, Li L, Zheng L. Increased intratumoral regulatory T cells are related to intratumoral macrophages and poor prognosis in hepatocellular carcinoma patients. Int J Cancer. 2009;125:1640–1648. doi: 10.1002/ijc.24556. [DOI] [PubMed] [Google Scholar]
- 30.Wu Y, Kuang DM, Pan WD, Wan YL, Lao XM, Wang D, Li XF, Zheng L. Monocyte/macrophage-elicited natural killer cell dysfunction in hepatocellular carcinoma is mediated by CD48/2B4 interactions. Hepatology. 2013;57:1107–1116. doi: 10.1002/hep.26192. [DOI] [PubMed] [Google Scholar]
- 31.Yang J, Zhang JX, Wang H, Wang GL, Hu QG, Zheng QC. Hepatocellular carcinoma and macrophage interaction induced tumor immunosuppression via Treg requires TLR4 signaling. World J Gastroenterol. 2012;18:2938–2947. doi: 10.3748/wjg.v18.i23.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Capece D, Fischietti M, Verzella D, Gaggiano A, Cicciarelli G, Tessitore A, Zazzeroni F, Alesse E. The inflammatory microenvironment in hepatocellular carcinoma: a pivotal role for tumor-associated macrophages. Biomed Res Int. 2013;2013:187204. doi: 10.1155/2013/187204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wan S, Zhao E, Kryczek I, Vatan L, Sadovskaya A, Ludema G, Simeone DM, Zou W, Welling TH. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology. 2014;147:1393–1404. doi: 10.1053/j.gastro.2014.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wan S, Kuo N, Kryczek I, Zou W, Welling TH. Myeloid cells in hepatocellular carcinoma. Hepatology. 2015;62:1304–1312. doi: 10.1002/hep.27867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol. 2017;17:306–321. doi: 10.1038/nri.2017.11. [DOI] [PubMed] [Google Scholar]
- 36.Wilson CL, Jurk D, Fullard N, Banks P, Page A, Luli S, Elsharkawy AM, Gieling RG, Chakraborty JB, Fox C, et al. NFκB1 is a suppressor of neutrophil-driven hepatocellular carcinoma. Nat Commun. 2015;6:6818. doi: 10.1038/ncomms7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009;16:183–194. doi: 10.1016/j.ccr.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsuda Y, Fukui H, Asai A, Fukunishi S, Miyaji K, Fujiwara S, Teramura K, Fukuda A, Higuchi K. An immunosuppressive subtype of neutrophils identified in patients with hepatocellular carcinoma. J Clin Biochem Nutr. 2012;51:204–212. doi: 10.3164/jcbn.12-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He G, Zhang H, Zhou J, Wang B, Chen Y, Kong Y, Xie X, Wang X, Fei R, Wei L, et al. Peritumoural neutrophils negatively regulate adaptive immunity via the PD-L1/PD-1 signalling pathway in hepatocellular carcinoma. J Exp Clin Cancer Res. 2015;34:141. doi: 10.1186/s13046-015-0256-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xiao WK, Chen D, Li SQ, Fu SJ, Peng BG, Liang LJ. Prognostic significance of neutrophil-lymphocyte ratio in hepatocellular carcinoma: a meta-analysis. BMC Cancer. 2014;14:117. doi: 10.1186/1471-2407-14-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.da Fonseca LG, Barroso-Sousa R, Bento Ada S, Blanco BP, Valente GL, Pfiffer TE, Hoff PM, Sabbaga J. Pre-treatment neutrophil-to-lymphocyte ratio affects survival in patients with advanced hepatocellular carcinoma treated with sorafenib. Med Oncol. 2014;31:264. doi: 10.1007/s12032-014-0264-5. [DOI] [PubMed] [Google Scholar]
- 42.Motomura T, Shirabe K, Mano Y, Muto J, Toshima T, Umemoto Y, Fukuhara T, Uchiyama H, Ikegami T, Yoshizumi T, et al. Neutrophil-lymphocyte ratio reflects hepatocellular carcinoma recurrence after liver transplantation via inflammatory microenvironment. J Hepatol. 2013;58:58–64. doi: 10.1016/j.jhep.2012.08.017. [DOI] [PubMed] [Google Scholar]
- 43.Imai Y, Kubota Y, Yamamoto S, Tsuji K, Shimatani M, Shibatani N, Takamido S, Matsushita M, Okazaki K. Neutrophils enhance invasion activity of human cholangiocellular carcinoma and hepatocellular carcinoma cells: an in vitro study. J Gastroenterol Hepatol. 2005;20:287–293. doi: 10.1111/j.1440-1746.2004.03575.x. [DOI] [PubMed] [Google Scholar]
- 44.He M, Peng A, Huang XZ, Shi DC, Wang JC, Zhao Q, Lin H, Kuang DM, Ke PF, Lao XM. Peritumoral stromal neutrophils are essential for c-Met-elicited metastasis in human hepatocellular carcinoma. Oncoimmunology. 2016;5:e1219828. doi: 10.1080/2162402X.2016.1219828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li XF, Chen DP, Ouyang FZ, Chen MM, Wu Y, Kuang DM, Zheng L. Increased autophagy sustains the survival and pro-tumourigenic effects of neutrophils in human hepatocellular carcinoma. J Hepatol. 2015;62:131–139. doi: 10.1016/j.jhep.2014.08.023. [DOI] [PubMed] [Google Scholar]
- 46.Kuang DM, Zhao Q, Wu Y, Peng C, Wang J, Xu Z, Yin XY, Zheng L. Peritumoral neutrophils link inflammatory response to disease progression by fostering angiogenesis in hepatocellular carcinoma. J Hepatol. 2011;54:948–955. doi: 10.1016/j.jhep.2010.08.041. [DOI] [PubMed] [Google Scholar]
- 47.Xu R, Huang H, Zhang Z, Wang FS. The role of neutrophils in the development of liver diseases. Cell Mol Immunol. 2014;11:224–231. doi: 10.1038/cmi.2014.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guo CL, Yang HC, Yang XH, Cheng W, Dong TX, Zhu WJ, Xu Z, Zhao L. Associations between infiltrating lymphocyte subsets and hepatocellular carcinoma. Asian Pac J Cancer Prev. 2012;13:5909–5913. doi: 10.7314/apjcp.2012.13.11.5909. [DOI] [PubMed] [Google Scholar]
- 49.Fu J, Xu D, Liu Z, Shi M, Zhao P, Fu B, Zhang Z, Yang H, Zhang H, Zhou C, et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology. 2007;132:2328–2339. doi: 10.1053/j.gastro.2007.03.102. [DOI] [PubMed] [Google Scholar]
- 50.Kalathil S, Lugade AA, Miller A, Iyer R, Thanavala Y. Higher frequencies of GARP(+)CTLA-4(+)Foxp3(+) T regulatory cells and myeloid-derived suppressor cells in hepatocellular carcinoma patients are associated with impaired T-cell functionality. Cancer Res. 2013;73:2435–2444. doi: 10.1158/0008-5472.CAN-12-3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garnelo M, Tan A, Her Z, Yeong J, Lim CJ, Chen J, Lim KH, Weber A, Chow P, Chung A, et al. Interaction between tumour-infiltrating B cells and T cells controls the progression of hepatocellular carcinoma. Gut. 2017;66:342–351. doi: 10.1136/gutjnl-2015-310814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ringelhan M, Pfister D, O’Connor T, Pikarsky E, Heikenwalder M. The immunology of hepatocellular carcinoma. Nature Immunology. 2018;19:222–232. doi: 10.1038/s41590-018-0044-z. [DOI] [PubMed] [Google Scholar]
- 53.Yang S, Tian Z, Wu Y, van Velkinburgh JC, Ni B. Pivotal roles of ILCs in hepatic diseases. Int Rev Immunol. 2015;34:509–522. doi: 10.3109/08830185.2015.1008631. [DOI] [PubMed] [Google Scholar]
- 54.Artis D, Spits H. The biology of innate lymphoid cells. Nature. 2015;517:293–301. doi: 10.1038/nature14189. [DOI] [PubMed] [Google Scholar]
- 55.Abe H, Kimura A, Tsuruta S, Fukaya T, Sakaguchi R, Morita R, Sekiya T, Shichita T, Chayama K, Fujii-Kuriyama Y, et al. Aryl hydrocarbon receptor plays protective roles in ConA-induced hepatic injury by both suppressing IFN-γ expression and inducing IL-22. Int Immunol. 2014;26:129–137. doi: 10.1093/intimm/dxt049. [DOI] [PubMed] [Google Scholar]
- 56.Kirchberger S, Royston DJ, Boulard O, Thornton E, Franchini F, Szabady RL, Harrison O, Powrie F. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J Exp Med. 2013;210:917–931. doi: 10.1084/jem.20122308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang P, Liu XK, Chu Z, Ye JC, Li KL, Zhuang WL, Yang DJ, Jiang YF. Detection of interleukin-33 in serum and carcinoma tissue from patients with hepatocellular carcinoma and its clinical implications. J Int Med Res. 2012;40:1654–1661. doi: 10.1177/030006051204000504. [DOI] [PubMed] [Google Scholar]
- 58.Li J, Razumilava N, Gores GJ, Walters S, Mizuochi T, Mourya R, Bessho K, Wang YH, Glaser SS, Shivakumar P, et al. Biliary repair and carcinogenesis are mediated by IL-33-dependent cholangiocyte proliferation. J Clin Invest. 2014;124:3241–3251. doi: 10.1172/JCI73742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jovanovic IP, Pejnovic NN, Radosavljevic GD, Pantic JM, Milovanovic MZ, Arsenijevic NN, Lukic ML. Interleukin-33/ST2 axis promotes breast cancer growth and metastases by facilitating intratumoral accumulation of immunosuppressive and innate lymphoid cells. Int J Cancer. 2014;134:1669–1682. doi: 10.1002/ijc.28481. [DOI] [PubMed] [Google Scholar]
- 60.McHedlidze T, Waldner M, Zopf S, Walker J, Rankin AL, Schuchmann M, Voehringer D, McKenzie AN, Neurath MF, Pflanz S, et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity. 2013;39:357–371. doi: 10.1016/j.immuni.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 62.Kettner NM, Voicu H, Finegold MJ, Coarfa C, Sreekumar A, Putluri N, Katchy CA, Lee C, Moore DD, Fu L. Circadian Homeostasis of Liver Metabolism Suppresses Hepatocarcinogenesis. Cancer Cell. 2016;30:909–924. doi: 10.1016/j.ccell.2016.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang Q, Tan Y, Yin P, Ye G, Gao P, Lu X, Wang H, Xu G. Metabolic characterization of hepatocellular carcinoma using nontargeted tissue metabolomics. Cancer Res. 2013;73:4992–5002. doi: 10.1158/0008-5472.CAN-13-0308. [DOI] [PubMed] [Google Scholar]
- 64.Gingold JA, Zhu D, Lee DF, Kaseb A, Chen J. Genomic Profiling and Metabolic Homeostasis in Primary Liver Cancers. Trends Mol Med. 2018;24:395–411. doi: 10.1016/j.molmed.2018.02.006. [DOI] [PubMed] [Google Scholar]
- 65.Ngo H, Tortorella SM, Ververis K, Karagiannis TC. The Warburg effect: molecular aspects and therapeutic possibilities. Mol Biol Rep. 2015;42:825–834. doi: 10.1007/s11033-014-3764-7. [DOI] [PubMed] [Google Scholar]
- 66.Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009;23:537–548. doi: 10.1101/gad.1756509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shang RZ, Qu SB, Wang DS. Reprogramming of glucose metabolism in hepatocellular carcinoma: Progress and prospects. World J Gastroenterol. 2016;22:9933–9943. doi: 10.3748/wjg.v22.i45.9933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang Q, Bai X, Chen W, Ma T, Hu Q, Liang C, Xie S, Chen C, Hu L, Xu S, et al. Wnt/β-catenin signaling enhances hypoxia-induced epithelial-mesenchymal transition in hepatocellular carcinoma via crosstalk with hif-1α signaling. Carcinogenesis. 2013;34:962–973. doi: 10.1093/carcin/bgt027. [DOI] [PubMed] [Google Scholar]
- 69.Kitamura K, Hatano E, Higashi T, Narita M, Seo S, Nakamoto Y, Yamanaka K, Nagata H, Taura K, Yasuchika K, et al. Proliferative activity in hepatocellular carcinoma is closely correlated with glucose metabolism but not angiogenesis. J Hepatol. 2011;55:846–857. doi: 10.1016/j.jhep.2011.01.038. [DOI] [PubMed] [Google Scholar]
- 70.Jia YY, Zhao JY, Li BL, Gao K, Song Y, Liu MY, Yang XJ, Xue Y, Wen AD, Shi L. miR-592/WSB1/HIF-1α axis inhibits glycolytic metabolism to decrease hepatocellular carcinoma growth. Oncotarget. 2016;7:35257–35269. doi: 10.18632/oncotarget.9135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jiang J, Nilsson-Ehle P, Xu N. Influence of liver cancer on lipid and lipoprotein metabolism. Lipids Health Dis. 2006;5:4. doi: 10.1186/1476-511X-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Budhu A, Roessler S, Zhao X, Yu Z, Forgues M, Ji J, Karoly E, Qin LX, Ye QH, Jia HL, et al. Integrated metabolite and gene expression profiles identify lipid biomarkers associated with progression of hepatocellular carcinoma and patient outcomes. Gastroenterology. 2013;144:1066–1075.e1. doi: 10.1053/j.gastro.2013.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang H, Ye D, Guan KL, Xiong Y. IDH1 and IDH2 mutations in tumorigenesis: mechanistic insights and clinical perspectives. Clin Cancer Res. 2012;18:5562–5571. doi: 10.1158/1078-0432.CCR-12-1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Turkalp Z, Karamchandani J, Das S. IDH mutation in glioma: new insights and promises for the future. JAMA Neurol. 2014;71:1319–1325. doi: 10.1001/jamaneurol.2014.1205. [DOI] [PubMed] [Google Scholar]
- 75.Borger DR, Goyal L, Yau T, Poon RT, Ancukiewicz M, Deshpande V, Christiani DC, Liebman HM, Yang H, Kim H, et al. Circulating oncometabolite 2-hydroxyglutarate is a potential surrogate biomarker in patients with isocitrate dehydrogenase-mutant intrahepatic cholangiocarcinoma. Clin Cancer Res. 2014;20:1884–1890. doi: 10.1158/1078-0432.CCR-13-2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fathi AT, Nahed BV, Wander SA, Iafrate AJ, Borger DR, Hu R, Thabet A, Cahill DP, Perry AM, Joseph CP, et al. Elevation of Urinary 2-Hydroxyglutarate in IDH-Mutant Glioma. Oncologist. 2016;21:214–219. doi: 10.1634/theoncologist.2015-0342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen JY, Lai YS, Tsai HJ, Kuo CC, Yen BL, Yeh SP, Sun HS, Hung WC. The oncometabolite R-2-hydroxyglutarate activates NF-κB-dependent tumor-promoting stromal niche for acute myeloid leukemia cells. Sci Rep. 2016;6:32428. doi: 10.1038/srep32428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang C, Moore LM, Li X, Yung WK, Zhang W. IDH1/2 mutations target a key hallmark of cancer by deregulating cellular metabolism in glioma. Neuro Oncol. 2013;15:1114–1126. doi: 10.1093/neuonc/not087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Izquierdo-Garcia JL, Viswanath P, Eriksson P, Cai L, Radoul M, Chaumeil MM, Blough M, Luchman HA, Weiss S, Cairncross JG, et al. IDH1 Mutation Induces Reprogramming of Pyruvate Metabolism. Cancer Res. 2015;75:2999–3009. doi: 10.1158/0008-5472.CAN-15-0840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bogdanovic E. IDH1, lipid metabolism and cancer: Shedding new light on old ideas. Biochim Biophys Acta. 2015;1850:1781–1785. doi: 10.1016/j.bbagen.2015.04.014. [DOI] [PubMed] [Google Scholar]
- 81.Arai Y, Totoki Y, Hosoda F, Shirota T, Hama N, Nakamura H, Ojima H, Furuta K, Shimada K, Okusaka T, et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology. 2014;59:1427–1434. doi: 10.1002/hep.26890. [DOI] [PubMed] [Google Scholar]
- 82.Cheng AL, Shen YC, Zhu AX. Targeting fibroblast growth factor receptor signaling in hepatocellular carcinoma. Oncology. 2011;81:372–380. doi: 10.1159/000335472. [DOI] [PubMed] [Google Scholar]
- 83.Churi CR, Shroff R, Wang Y, Rashid A, Kang HC, Weatherly J, Zuo M, Zinner R, Hong D, Meric-Bernstam F, et al. Mutation profiling in cholangiocarcinoma: prognostic and therapeutic implications. PLoS One. 2014;9:e115383. doi: 10.1371/journal.pone.0115383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sia D, Tovar V, Moeini A, Llovet JM. Intrahepatic cholangiocarcinoma: pathogenesis and rationale for molecular therapies. Oncogene. 2013;32:4861–4870. doi: 10.1038/onc.2012.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wu YM, Su F, Kalyana-Sundaram S, Khazanov N, Ateeq B, Cao X, Lonigro RJ, Vats P, Wang R, Lin SF, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013;3:636–647. doi: 10.1158/2159-8290.CD-13-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Frattini V, Pagnotta SM, Tala, Fan JJ, Russo MV, Lee SB, Garofano L, Zhang J, Shi P, Lewis G, et al. A metabolic function of FGFR3-TACC3 gene fusions in cancer. Nature. 2018;553:222–227. doi: 10.1038/nature25171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chiang CH, Huang KC. Association between metabolic factors and chronic hepatitis B virus infection. World J Gastroenterol. 2014;20:7213–7216. doi: 10.3748/wjg.v20.i23.7213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schoeman JC, Hou J, Harms AC, Vreeken RJ, Berger R, Hankemeier T, Boonstra A. Metabolic characterization of the natural progression of chronic hepatitis B. Genome Med. 2016;8:64. doi: 10.1186/s13073-016-0318-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Agani F, Jiang BH. Oxygen-independent regulation of HIF-1: novel involvement of PI3K/AKT/mTOR pathway in cancer. Curr Cancer Drug Targets. 2013;13:245–251. doi: 10.2174/1568009611313030003. [DOI] [PubMed] [Google Scholar]
- 90.Kasuno K, Takabuchi S, Fukuda K, Kizaka-Kondoh S, Yodoi J, Adachi T, Semenza GL, Hirota K. Nitric oxide induces hypoxia-inducible factor 1 activation that is dependent on MAPK and phosphatidylinositol 3-kinase signaling. J Biol Chem. 2004;279:2550–2558. doi: 10.1074/jbc.M308197200. [DOI] [PubMed] [Google Scholar]
- 91.Tanaka H, Yamamoto M, Hashimoto N, Miyakoshi M, Tamakawa S, Yoshie M, Tokusashi Y, Yokoyama K, Yaginuma Y, Ogawa K. Hypoxia-independent overexpression of hypoxia-inducible factor 1alpha as an early change in mouse hepatocarcinogenesis. Cancer Res. 2006;66:11263–11270. doi: 10.1158/0008-5472.CAN-06-1699. [DOI] [PubMed] [Google Scholar]
- 92.Xia L, Mo P, Huang W, Zhang L, Wang Y, Zhu H, Tian D, Liu J, Chen Z, Zhang Y, et al. The TNF-α/ROS/HIF-1-induced upregulation of FoxMI expression promotes HCC proliferation and resistance to apoptosis. Carcinogenesis. 2012;33:2250–2259. doi: 10.1093/carcin/bgs249. [DOI] [PubMed] [Google Scholar]
- 93.Wang MD, Wu H, Huang S, Zhang HL, Qin CJ, Zhao LH, Fu GB, Zhou X, Wang XM, Tang L, et al. HBx regulates fatty acid oxidation to promote hepatocellular carcinoma survival during metabolic stress. Oncotarget. 2016;7:6711–6726. doi: 10.18632/oncotarget.6817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kim K, Kim KH, Kim HH, Cheong J. Hepatitis B virus X protein induces lipogenic transcription factor SREBP1 and fatty acid synthase through the activation of nuclear receptor LXRalpha. Biochem J. 2008;416:219–230. doi: 10.1042/BJ20081336. [DOI] [PubMed] [Google Scholar]
- 95.Kim SY, Kim JK, Kim HJ, Ahn JK. Hepatitis B virus X protein sensitizes UV-induced apoptosis by transcriptional transactivation of Fas ligand gene expression. IUBMB Life. 2005;57:651–658. doi: 10.1080/15216540500239697. [DOI] [PubMed] [Google Scholar]
- 96.Dai CY, Yeh ML, Huang CF, Hou CH, Hsieh MY, Huang JF, Lin IL, Lin ZY, Chen SC, Wang LY, et al. Chronic hepatitis C infection is associated with insulin resistance and lipid profiles. J Gastroenterol Hepatol. 2015;30:879–884. doi: 10.1111/jgh.12313. [DOI] [PubMed] [Google Scholar]
- 97.Kawaguchi Y, Mizuta T. Interaction between hepatitis C virus and metabolic factors. World J Gastroenterol. 2014;20:2888–2901. doi: 10.3748/wjg.v20.i11.2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bernsmeier C, Calabrese D, Heim MH, Duong HT. Hepatitis C virus dysregulates glucose homeostasis by a dual mechanism involving induction of PGC1α and dephosphorylation of FoxO1. J Viral Hepat. 2014;21:9–18. doi: 10.1111/jvh.12208. [DOI] [PubMed] [Google Scholar]
- 99.Lee S, Mardinoglu A, Zhang C, Lee D, Nielsen J. Dysregulated signaling hubs of liver lipid metabolism reveal hepatocellular carcinoma pathogenesis. Nucleic Acids Res. 2016;44:5529–5539. doi: 10.1093/nar/gkw462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML, Clore JN. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120:1183–1192. doi: 10.1053/gast.2001.23256. [DOI] [PubMed] [Google Scholar]
- 101.Satapati S, Kucejova B, Duarte JA, Fletcher JA, Reynolds L, Sunny NE, He T, Nair LA, Livingston KA, Fu X, et al. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J Clin Invest. 2015;125:4447–4462. doi: 10.1172/JCI82204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nassir F, Ibdah JA. Role of mitochondria in nonalcoholic fatty liver disease. Int J Mol Sci. 2014;15:8713–8742. doi: 10.3390/ijms15058713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fromenty B, Robin MA, Igoudjil A, Mansouri A, Pessayre D. The ins and outs of mitochondrial dysfunction in NASH. Diabetes Metab. 2004;30:121–138. doi: 10.1016/s1262-3636(07)70098-8. [DOI] [PubMed] [Google Scholar]
- 104.Fujisawa K, Takami T, Matsumoto T, Yamamoto N, Sakaida I. Profiling of the circadian metabolome in thioacetamide-induced liver cirrhosis in mice. Hepatol Commun. 2017;1:704–718. doi: 10.1002/hep4.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nishikawa T, Bellance N, Damm A, Bing H, Zhu Z, Handa K, Yovchev MI, Sehgal V, Moss TJ, Oertel M, et al. A switch in the source of ATP production and a loss in capacity to perform glycolysis are hallmarks of hepatocyte failure in advance liver disease. J Hepatol. 2014;60:1203–1211. doi: 10.1016/j.jhep.2014.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Andrejeva G, Rathmell JC. Similarities and Distinctions of Cancer and Immune Metabolism in Inflammation and Tumors. Cell Metab. 2017;26:49–70. doi: 10.1016/j.cmet.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, Knapp B, Haas R, Schmid JA, Jandl C, et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. 2012;15:813–826. doi: 10.1016/j.cmet.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Huang SC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, Beatty W, Love-Gregory L, Lam WY, O’Neill CM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol. 2014;15:846–855. doi: 10.1038/ni.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Namgaladze D, Brüne B. Fatty acid oxidation is dispensable for human macrophage IL-4-induced polarization. Biochim Biophys Acta. 2014;1841:1329–1335. doi: 10.1016/j.bbalip.2014.06.007. [DOI] [PubMed] [Google Scholar]
- 110.Nomura M, Liu J, Rovira II, Gonzalez-Hurtado E, Lee J, Wolfgang MJ, Finkel T. Fatty acid oxidation in macrophage polarization. Nat Immunol. 2016;17:216–217. doi: 10.1038/ni.3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang Q, Wang HR, Mao CY, Sun M, Dominah G, Chen L, Zhuang Z. Fatty acid oxidation contributes to IL-1β secretion in M2 macrophages and promotes macrophage-mediated tumor cell migration. Mol Metab. 2018;94:27–35. doi: 10.1016/j.molimm.2017.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Wagner RA, Greaves DR, Murray PJ, Chawla A. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006;4:13–24. doi: 10.1016/j.cmet.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Biswas SK. Metabolic Reprogramming of Immune Cells in Cancer Progression. Immunity. 2015;43:435–449. doi: 10.1016/j.immuni.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 114.Covarrubias AJ, Aksoylar HI, Yu J, Snyder NW, Worth AJ, Iyer SS, Wang J, Ben-Sahra I, Byles V, Polynne-Stapornkul T, et al. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. Elife. 2016:5. doi: 10.7554/eLife.11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lim JE, Chung E, Son Y. A neuropeptide, Substance-P, directly induces tissue-repairing M2 like macrophages by activating the PI3K/Akt/mTOR pathway even in the presence of IFNγ. Sci Rep. 2017;7:9417. doi: 10.1038/s41598-017-09639-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhou L, Wang Q, Yin P, Xing W, Wu Z, Chen S, Lu X, Zhang Y, Lin X, Xu G. Serum metabolomics reveals the deregulation of fatty acids metabolism in hepatocellular carcinoma and chronic liver diseases. Anal Bioanal Chem. 2012;403:203–213. doi: 10.1007/s00216-012-5782-4. [DOI] [PubMed] [Google Scholar]
- 117.Nath A, Li I, Roberts LR, Chan C. Elevated free fatty acid uptake via CD36 promotes epithelial-mesenchymal transition in hepatocellular carcinoma. Sci Rep. 2015;5:14752. doi: 10.1038/srep14752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Feingold KR, Shigenaga JK, Kazemi MR, McDonald CM, Patzek SM, Cross AS, Moser A, Grunfeld C. Mechanisms of triglyceride accumulation in activated macrophages. J Leukoc Biol. 2012;92:829–839. doi: 10.1189/jlb.1111537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559–563. doi: 10.1038/nature13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ruan GX, Kazlauskas A. Lactate engages receptor tyrosine kinases Axl, Tie2, and vascular endothelial growth factor receptor 2 to activate phosphoinositide 3-kinase/Akt and promote angiogenesis. J Biol Chem. 2013;288:21161–21172. doi: 10.1074/jbc.M113.474619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ohashi T, Aoki M, Tomita H, Akazawa T, Sato K, Kuze B, Mizuta K, Hara A, Nagaoka H, Inoue N, et al. M2-like macrophage polarization in high lactic acid-producing head and neck cancer. Cancer Sci. 2017;108:1128–1134. doi: 10.1111/cas.13244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Spadaro O, Camell CD, Bosurgi L, Nguyen KY, Youm YH, Rothlin CV, Dixit VD. IGF1 Shapes Macrophage Activation in Response to Immunometabolic Challenge. Cell Rep. 2017;19:225–234. doi: 10.1016/j.celrep.2017.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38:633–643. doi: 10.1016/j.immuni.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, van den Bosch MW, Quinn SR, Domingo-Fernandez R, Johnston DG, et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015;21:65–80. doi: 10.1016/j.cmet.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Moon JS, Nakahira K, Chung KP, DeNicola GM, Koo MJ, Pabón MA, Rooney KT, Yoon JH, Ryter SW, Stout-Delgado H, et al. NOX4-dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages. Nat Med. 2016;22:1002–1012. doi: 10.1038/nm.4153. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 126.Moon JS, Hisata S, Park MA, DeNicola GM, Ryter SW, Nakahira K, Choi AMK. mTORC1-Induced HK1-Dependent Glycolysis Regulates NLRP3 Inflammasome Activation. Cell Rep. 2015;12:102–115. doi: 10.1016/j.celrep.2015.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 127.Littlewood-Evans A, Sarret S, Apfel V, Loesle P, Dawson J, Zhang J, Muller A, Tigani B, Kneuer R, Patel S, et al. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J Exp Med. 2016;213:1655–1662. doi: 10.1084/jem.20160061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tourlomousis P, Däbritz JHM, Gottlieb E, Latorre I, et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell. 2016;167:457–470.e13. doi: 10.1016/j.cell.2016.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496:238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Azevedo EP, Rochael NC, Guimarães-Costa AB, de Souza-Vieira TS, Ganilho J, Saraiva EM, Palhano FL, Foguel D. A Metabolic Shift toward Pentose Phosphate Pathway Is Necessary for Amyloid Fibril- and Phorbol 12-Myristate 13-Acetate-induced Neutrophil Extracellular Trap (NET) Formation. J Biol Chem. 2015;290:22174–22183. doi: 10.1074/jbc.M115.640094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Siska PJ, Rathmell JC. T cell metabolic fitness in antitumor immunity. Trends Immunol. 2015;36:257–264. doi: 10.1016/j.it.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chang CH, Curtis JD, Maggi LB Jr, Faubert B, Villarino AV, O’Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, Weber JD, Pearce EJ, Jones RG, Pearce EL. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239–1251. doi: 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, Filisio F, Giles-Davis W, Xu X, Karakousis GC, et al. Enhancing CD8+ T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell. 2017;32:377–391.e9. doi: 10.1016/j.ccell.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wawman RE, Bartlett H, Oo YH. Regulatory T Cell Metabolism in the Hepatic Microenvironment. Front Immunol. 2018;8:1889. doi: 10.3389/fimmu.2017.01889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Beier UH, Angelin A, Akimova T, Wang L, Liu Y, Xiao H, Koike MA, Hancock SA, Bhatti TR, Han R, et al. Essential role of mitochondrial energy metabolism in Foxp3+ T-regulatory cell function and allograft survival. FASEB J. 2015;29:2315–2326. doi: 10.1096/fj.14-268409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Shirabe K, Mano Y, Muto J, Matono R, Motomura T, Toshima T, Takeishi K, Uchiyama H, Yoshizumi T, Taketomi A, et al. Role of tumor-associated macrophages in the progression of hepatocellular carcinoma. Surg Today. 2012;42:1–7. doi: 10.1007/s00595-011-0058-8. [DOI] [PubMed] [Google Scholar]
- 137.Zhang J, Zhang Q, Lou Y, Fu Q, Chen Q, Wei T, Yang J, Tang J, Wang J, Chen Y, et al. Hypoxia-inducible factor-1α/interleukin-1β signaling enhances hepatoma epithelial-mesenchymal transition through macrophages in a hypoxic-inflammatory microenvironment. Hepatology. 2018;67:1872–1889. doi: 10.1002/hep.29681. [DOI] [PubMed] [Google Scholar]
- 138.Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, de Zoeten EF, Cambier JC, Stenmark KR, Colgan SP, et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci. 2012;109:E2784–E2793. doi: 10.1073/pnas.1202366109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chang M, Hamilton JA, Scholz GM, Elsegood CL. Glycolytic control of adjuvant-induced macrophage survival: role of PI3K, MEK1/2, and Bcl-2. J Leukoc Biol. 2009;85:947–956. doi: 10.1189/jlb.0908522. [DOI] [PubMed] [Google Scholar]
- 140.Sharma MD, Shinde R, McGaha TL, Huang L, Holmgaard RB, Wolchok JD, Mautino MR, Celis E, Sharpe AH, Francisco LM, et al. The PTEN pathway in Tregs is a critical driver of the suppressive tumor microenvironment. Sci Adv. 2015;1:e1500845. doi: 10.1126/sciadv.1500845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chen W, Ma T, Shen XN, Xia XF, Xu GD, Bai XL, Liang TB. Macrophage-induced tumor angiogenesis is regulated by the TSC2-mTOR pathway. Cancer Res. 2012;72:1363–1372. doi: 10.1158/0008-5472.CAN-11-2684. [DOI] [PubMed] [Google Scholar]
- 142.Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature. 2013;499:485–490. doi: 10.1038/nature12297. [DOI] [PMC free article] [PubMed] [Google Scholar]