

Abstract
Benzodiazepines are clinically relevant drugs that bind to GABAA neurotransmitter receptors at the α+/γ2– interfaces and thereby enhance GABA-induced chloride ion flux leading to neuronal hyperpolarization. However, the structural basis of benzodiazepine interactions with their high-affinity site at GABAA receptors is controversially debated in the literature, and in silico studies led to discrepant binding mode hypotheses. In this study, computational docking of diazepam into α+/γ2– homology models suggested that a chiral methyl group, which is known to promote preferred binding to α5-containing GABAA receptors (position 3 of the seven-membered diazepine ring), could possibly provide experimental evidence that supports or contradicts the proposed binding modes. Thus, we investigated three pairs of R and S isomers of structurally different chemotypes, namely, diazepam, imidazobenzodiazepine, and triazolam derivatives. We used radioligand displacement studies as well as two-electrode voltage clamp electrophysiology in α1β3γ2-, α2β3γ2-, α3β3γ2-, and α5β3γ2-containing GABAA receptors to determine the ligand binding and functional activity of the three chemotypes. Interestingly, both imidazobenzodiazepine isomers displayed comparable binding affinities, while for the other two chemotypes, a discrepancy in binding affinities of the different isomers was observed. Specifically, the R isomers displayed a loss of binding, whereas the S isomers remained active. These findings are in accordance with the results of our in silico studies suggesting the usage of a different binding mode of imidazobenzodiazepines compared to those of the other two tested chemotypes. Hence, we conclude that different chemically related benzodiazepine ligands interact via distinct binding modes rather than by using a common binding mode.
Since the discovery of the first benzodiazepine chlordiazepoxide in the late 1950s, this class of drugs has emerged rapidly. In 2008, >5% of the U.S. adult population used benzodiazepines1 in the treatment of conditions such as insomnia, anxiety, and acute epileptic seizures or as presurgery medication. Benzodiazepines act by positive allosteric modulatory (PAM) enhancement of GABA-induced chloride ion flux through GABAA receptors.2 GABAA receptors are pentameric ligand-gated ion channels, and a majority of these receptors are composed of two α subunits, two β subunits, and one γ subunit,3 typically γ2, as approximately 75–80% of all GABAA receptors contain γ2.4 Nearly all of the clinically relevant benzodiazepine drugs target all GABAA receptor subtypes equally. However, in the past few years, research has focused on the role of α5-containing GABAA receptors. Although those receptors are rare (<5% of total) with the distribution restricted to hippocampus,5 they are considered promising drug targets for both positive and negative allosteric modulators6 and could be useful for the treatment of cognitive and/or mood disorders.
For structure-guided development of subtype-selective ligands, knowledge of their binding modes in the pocket is required. Classical benzodiazepines, such as diazepam, midazolam, and similar compounds, bind at the α+/γ– interface of α1-, α2-, α3-, or α5- and γ2-containing receptors. However, the exact molecular interactions of benzodiazepines at the GABAA receptor subunits have not been well characterized and remain under debate in the literature. It is generally assumed that all benzodiazepines interact with the receptor in a “common” binding mode. In silico studies, however, led to controversial hypotheses about the exact nature of this common binding mode.7−10 Current studies suggest three possible common binding mode poses (CBM I, CBM II, and CBM III) for binding of diazepam to the pocket.11 Richter et al. favor CBM I because it is supported by experimental evidence and could be used to identify further high-affinity ligands.11 In contrast, Middendorp et al.12 found evidence of CBM II and were also able to identify a number of active compounds by a virtual screening approach using this molecular orientation. This seemingly paradoxical situation can be reconciled by the pseudosymmetry that relates these two binding modes’ pharmacophores for diazepam.13
In the study presented here, we aimed to find experimental evidence that favored one binding mode or the other. A closer examination of binding modes BM I and BM II found in our previous study11 suggested that substitutions on the seven-membered diazepine ring could interfere with the BM II poses more severely than with the BM I poses. We therefore selected three scaffolds for docking and three corresponding pairs of substituted enantiomers for experimental testing. For the in silico docking, an improved homology model could be used, because the crystal structure of the human β3-homopentameric GABAA receptor was resolved by Miller and Ariescu in 201414 and was selected as a template.
This in silico docking confirmed the crucial position at the methyl group of the seven-membered diazepine ring. This methyl group, when oriented in the R configuration, is known to promote α5 subtype selectivity of imidazobenzodiazepines such as SH-053-2′F-R-CH3.15 Here, we predict that the very same methyl group in this position could additionally be used as a chemical reporter to provide insights into the molecular interaction profile of benzodiazepine ligands and provide a means to rank candidate binding modes for given specific ligands and chemotypes.
We investigated a set of three stereoisomeric pairs in radioligand displacement assays and with the two-electrode voltage clamp method. Interestingly, our data gave diverging results for the different chemical scaffolds, suggesting that different benzodiazepine ligands use distinct binding modes rather than a common binding mode. In the accompanying manuscript,13 we also present evidence of different binding mode usage based on an orthogonal mutational approach.
Results and Discussion
Bound State Models Reveal a Crucial Methyl Group
A closer inspection of our previous examination of docking of diazepam to the α+/γ– interface of GABAA receptors11 indicated that substitutions on the seven-membered diazepine ring of the benzodiazepine scaffold could more severely interfere with the published benzodiazepine binding mode BM II binding pose than with the BM I binding pose (see Figure 1). We reasoned that this fact could possibly be used to find experimental evidence supporting one binding mode or the other.
Figure 1.

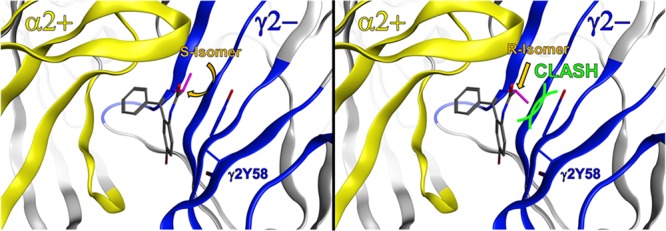
General BM I and BM II orientations of the benzodiazepine core scaffold at the extracellular α+/γ2– site. Color codes for the protein ribbon: yellow for the α subunit and blue for the γ2 subunit. Binding site forming segments (strands and loops A–F) are labeled with Arabic letters. (a and b) BM I orientation of the benzodiazepine core scaffold at the α+/γ2– interface. The fused phenyl ring is colored orange, while the pendant phenyl ring is colored red. The crucial methyl group is colored purple, indicating sufficient space for the R isomer and the S isomer. (c and d) BM II orientation of the benzodiazepine core scaffold at the α+/γ2– interface. The color coding of the ligand is as in panels a and b. The two phenyl rings changed positions if we compared the BM I and BM II orientations. The crucial methyl group is colored purple, indicating the R isomer has a steric clash with segment G of the γ2 subunit while the S isomer does not. Green curved lines indicate van der Waals radii, which illustrate the steric clash.
In this study, we therefore selected three pairs of stereoisomers for our binding and electrophysiological studies, each with a chiral methyl group at position 3 of the seven-membered diazepine ring in the R or S configuration. Our test compounds belong to three different structural classes (see Figure 2): a pair of imidazobenzodiazepines, SH-I-048B and SH-I-047 (termed 1-S and 1-R, respectively), a pair of triazolam derivatives, SH-TS-CH3 and SH-TR-CH316 (termed 2-S and 2-R, respectively), and a pair of diazepam derivatives, SH-I-030 and SH-I-053B (termed 3-S and 3-R, respectively).
Figure 2.

Chemical structures of the three stereoisomer pairs. The S isomers are depicted in the left column, and the corresponding R isomers in the right column. IUPAC nomenclature as well as the numbering of the C atoms in the structures is shown in Figure S1.
To correlate the computational results with the binding studies, we updated and revisited the in silico approach for the prediction and validation of binding modes. Because there is no crystal structure of hetero-oligomeric GABAA receptors, we have to rely on homology modeling based on the published β3 homo-oligomer.14 We chose for the computational study to investigate the α2+/γ2– interface as being representative for the “diazepam-sensitive” class of benzodiazepine binding sites due to the rising interest in α2-selective PAM ligands, in the treatment of not only anxiety disorders but also schizophrenia, depression, and pain disorders.5,17
In terms of expected model accuracy, the α1+, α2+, and α3+ interfaces are more closely related to the experimentally determined β3+/β3– interface than the α5+/γ2– interface, and reliabilities of all three would be expected to be comparable. For the docking procedure, we employed the three achiral scaffolds from the six molecules, which we studied experimentally, and added the respective (R)- and (S)-methyl groups to the candidate binding modes. This procedure avoids excessive false positive in silico docking solutions for the experimentally tested compounds. In addition, docking the unsubstituted parent compounds of the chiral ligands facilitates direct comparison with the previous docking studies in which mainly achiral compounds have been studied. Subsequent in silico derivatization of the docked ligands into the six test compounds led to predictions that were tested in the experimental part of this study.
Computational docking of the three achiral parent ligands into our α2+/γ2– homology model using GOLD18 resulted in the two previously reported binding mode orientations, BM I and BM II,11 for all ligands tested. Careful analysis of the two binding modes supported our initial hypothesis that the introduction of a methyl group into the seven-membered diazepine ring could serve as tool for examining binding mode orientations of different benzodiazepine ligands. In detail, the methyl group at the diazepine ring leads to two distinct isomers, namely, an R isomer and an S isomer. According to our model, the methyl groups of both isomers are pointing in the direction of the more flexible loop C if the ligands bind in a BM I orientation, and thus, both isomers should be active (see panels a and b of Figure 3). On the contrary, if the ligands use a BM II orientation, methyl groups of the two isomers are pointing in substantially different directions. While for the S isomer the methyl group is also directed toward the flexible loop C (see Figure 3c), the methyl group of the R isomer is directed toward the more rigid segment G of the γ2 subunit, which leads to a steric clash with a bulky tyrosine residue (γ2Tyr58) (see Figure 3d). Hence, in silico docking leads to the prediction that for ligands using BM I, R and S isomer analogues should both be binding with high or at least intermediate affinity and differ only mildly in affinity, while for BM II binding scaffolds, the R isomer should be inactive or display a marked drop in affinity compared to that of the S isomer.
Figure 3.

Distinct binding modes (BM I or BM II) of tested compounds 1-S, 1-R, 3-S, and 3-R at the α2+/γ2– interface. (a and b) Compounds 1-S and 1-R are both compatible with BM I in the binding pocket (α2 subunit, yellow; γ2 subunit, blue; amino acids that were set flexible in the docking are rendered as sticks and labeled). The orientation of the methyl is colored purple. (c) For compound 3-S in BM II, the methyl group (purple) is sterically compatible (α2 subunit, yellow; γ2 subunit, blue). (d) For compound 3-R in BM II, the methyl group (purple) is not tolerated and clashes with the γ2Tyr58 on segment G in the binding pocket (α2 subunit, yellow; γ2 subunit, blue); thus, this ligand is predicted to show reduced binding affinity. Green curved lines indicate van der Waals radii, which illustrate the steric clash.
Distinct Binding Activity of R Stereoisomers
To test our predictions, we performed radioligand displacement studies and compared the potencies of our six chiral benzodiazepine ligands for the inhibition of binding of [3H]flunitrazepam to α2β3γ2 recombinant rat GABAA receptor subtypes (see Figure 4).
Figure 4.
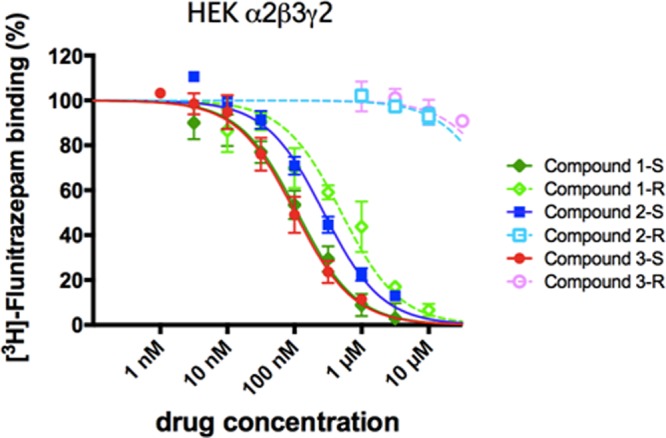
Inhibition of binding of [3H]flunitrazepam to recombinant α2β3γ2 GABAA receptor subtypes. Membranes from HEK-293 cells transfected with the GABAA receptor subunit combination α2β3γ2 were incubated with 2 nM [3H]flunitrazepam in the presence of various concentrations of the six BZ site ligands. Data shown are the means ± the standard error of the mean of three independent experiments each performed in duplicate.
While for the S isomers (compounds 1-S, 2-S, and 3-S) intermediate to high binding affinities for the recombinant receptors were observed, which is in accordance with the results of our docking studies, the R isomers showed diverse binding behavior. Interestingly, only imidazobenzodiazepine compound 1-R retained its potential to displace the radioligand [3H]flunitrazepam, whereas compounds 2-R and 3-R were inactive, suggesting nonbinding. Generally, the S isomers display affinities higher than those of their R analogues.
To exclude a binding behavior unique to α2-containing receptors, we also performed similar radioligand displacement studies on other receptor combinations such as recombinantly expressed rat GABAA receptor subtypes α1β3γ2, α3β3γ2, and α5β3γ2 (see Figure S2). The obtained Ki values are listed in Table 1.
Table 1. Affinities of Benzodiazepine Binding Site Ligands for Recombinant GABAA Receptorsa.
|
Ki |
||||
|---|---|---|---|---|
| compound | α1β3γ2 | α2β3γ2 | α3β3γ2 | α5β3γ2 |
| 1-S (SH-I-048B) | 190 ± 55 nM | 67 ± 9 nM | 136 ± 24 nM | 17 ± 5 nMb |
| 1-R (SH-I-047) | 273 ± 41 nM | 253 ± 31 nM | 501 ± 79 nM | 56 ± 8 nMc |
| 2-S (SH-TS-CH3) | 663 ± 21 nM | 164 ± 15 nM | 656 ± 110 nM | 80 ± 4 nMd |
| 2-R (SH-TR-CH3) | >10 μM | >10 μM | >10 μM | >10 μM |
| 3-S (SH-I-030) | 64 ± 2 nM | 61 ± 10 nM | 102 ± 7 nM | 31 ± 5 nM |
| 3-R (SH-I-053B) | >10 μM | >10 μM | >10 μM | >10 μM |
The concentrations of drugs resulting in half-maximal inhibition of specific [3H]flunitrazepam binding (IC50) shown in Figure S2 were converted to Ki values by using the Cheng–Prusoff relationship19 and the respective published KD values for [3H]flunitrazepam binding (nanomolar).20 Data presented are means ± the standard error of the mean of three independent experiments eachy performed in duplicate. Statistical evaluation was performed with one-way analysis of variance (ANOVA) followed by post hoc Tukey’s multiple-comparison test.
p < 0.01 compared to α1.
p < 0.05 compared to α1.
p < 0.001 compared to α1.
Compounds 1-S, 2-S, 3-S, and 1-R potently replaced [3H]flunitrazepam in all receptor subtypes, while compounds 2-R and 3-R failed to displace the radioligand in all receptors tested (see Table 1). However, we did observe differences in the potency rank order. Although compound 1-S showed a significant preference for α5-containing receptors (>4-fold), it also exhibited substantial affinity at the other receptor subtypes that decreased in the following order: α5 > α2 > α3 = α1. Its R isomeric analogue compound 1-R possessed slightly higher α5 selectivity (∼5-fold); however, it displayed slightly lower affinities in the tested receptor subtypes. Triazolam-like compound 2-S showed significant higher binding affinities in α2 and α5 GABAAR subtypes than in α1 and α3 subtypes. Diazepam-like compound 3-S showed low subtype selectivity. Compounds 2-R and 3-R failed to displace [3H]flunitrazepam binding at all GABAA receptor subtypes investigated even at high micromolar concentrations.
Modulatory Effects Support Heterogeneous Activity Profiles of R Isomers
Radioligand displacement assays provide information about the binding of a compound to only a specific binding site. To exclude the possibility that compounds 2-R and 3-R bind at a site different from the [3H]flunitrazepam binding site (and are therefore unable to compete with the radioligand), we decided to determine the efficacies of all our test compounds using two-electrode voltage clamp recordings on α2β3γ2-mRNA-treated Xenopus laevis oocytes (see Figure 5).
Figure 5.
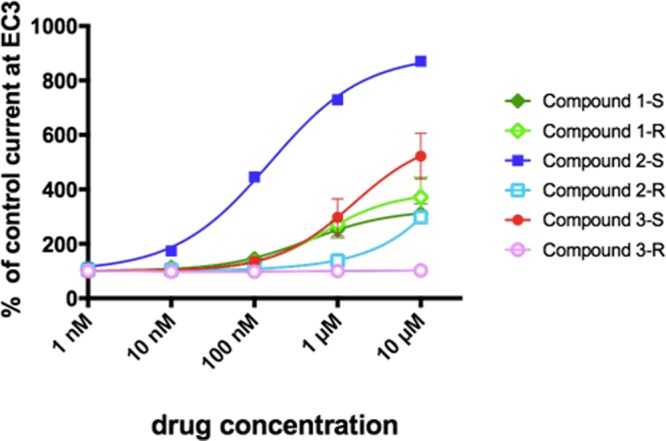
Concentration–response curves of drug-induced enhancement of GABA currents. X. laevis oocytes were treated with mRNA for α2β3γ2-GABAA receptors and studied using two-electrode voltage clamp recordings. GABA-induced currents (3% of maximum) were modulated by addition of drug. Data are means ± the standard error of the mean of three independent experiments.
Electrophysiological experiments supported our radioligand binding assay findings. Both imidazobenzodiazepines 1-S and 1-R as well as compounds 2-S and 3-S interacted with GABA receptors. Two-voltage clamp recordings provide us with the additional knowledge that those drugs behaved as positive allosteric modulators as they potentiated the GABA-induced chloride ion flux. On the other hand, compounds 2-R and 3-R displayed complete loss of activity in the oocyte assay at drug concentrations of ≤1 μM, indicating that they indeed do not interact with GABA receptors. Nevertheless, compound 2-R showed a slight positive modulation of the GABA-induced currents at concentrations of 10 μM. Because at those concentrations the drug does not yet displace [3H]flunitrazepam (see Figure 4 and Figure S2), this modulatory interaction is most likely mediated through a binding site different from the classical benzodiazepine binding site at the α+/γ– interface. One putative binding site could be the α+/β– interface, as described by Ramerstorfer et al., because compounds that interact with both sites, the α+/β– site and the BZ site, are known (e.g., pyrazoloquinolinones).21 This hypothesis is supported by the fact that the previously published compound SH-I-048A,22 which is structurally very similar to compound 3-S, displayed a biphasic concentration–response curve in two-voltage clamp recordings, also indicating an additional interaction site for benzodiazepine ligands.
In addition to the analysis of α2β3γ2-containing GABAA receptors, we also performed similar electrophysiological studies of other receptor combinations such as α1β3γ2, α3β3γ2, and α5β3γ2 (see Figure S3). In accordance with the previous data, compounds 1-S, 2-S, 3-S, and 1-R displayed a positive modulation in all receptor subtypes, while compounds 2-R and 3-R remained mainly modulatory silent.
In this study, we used a combined approach comprising computational ligand docking, radioligand binding assays, and functional two-electrode voltage clamp assays to revisit the common binding mode hypothesis for diazepam-derived ligands versus imidazobenzodiazepines with different substituents on the imidazole ring.
Overall, we found that our chiral benzodiazepine ligand 1-R, representing an ester-substituted imidazobenzodiazepine, binds to the GABAA receptor benzodiazepine binding site. This suggests a preference for the CBM I binding mode as described by Richter et al.11 In contrast, the two R isomer compounds derived from diazepam or from triazolam are no longer able to bind to the benzodiazepine binding site. For these compounds, the CBM II binding mode seems to apply, as suggested by Middendorp et al.12 In the accompanying manuscript,13 an orthogonal approach to the one used here was employed, where an artificial benzodiazepine binding site was engineered into binary α1β3 receptors. In this study, imidazobenzodiazepines interacted with the artificial site in a manner completely different from that of diazepam, which was also investigated with computational models and in turn interpreted as a sign of different binding site usage.
Our results are in line with previous experiments, in which it was demonstrated that the binding affinity, efficacy, and in vivo activity of a series of ligands with a chiral S-CH3 group at C-4 are different from those of the R-CH3 group.23,24 For instance, the R isomer of SH-I-048A22 [which is structurally very similar to the compound 3-S/3-R pair (SH-I-030/SH-I-053B) tested in this study] was almost completely inactive in oocyte assays.22 On the other hand, the previously published compounds SH-053-2′F-S-CH3 and SH-053-2′F-R-CH325 (of which compounds 1-S and 1-R, respectively, are derivatives) both bind to GABAA receptors with high affinity, but the R isomer SH-053-2′F-R-CH3 displays 3–5-fold lower affinity in α1-, α3-, and α5-containing receptors and even 30-fold lower affinity in α2-containing receptors compared to those of the S isomer.23
The compounds tested have a rather low affinity for the benzodiazepine binding site of GABAA receptors. In addition, if we compare the substitutions on the basic benzodiazepine core (scaffold), the pairs of compounds differ at several positions. In Figure 2, the top row (scaffold 1) and the middle row (scaffold 2) differ in three substituents and are thus not a systematic variation on the scaffold (see also Figure S4). The middle row (scaffold 2) differs from the bottom row (scaffold 3) in two positions, which again is not a systematic variation. However, the Br versus ethinyl has little impact on affinity;7 it can therefore be assumed that the presence or absence of the five-membered ring on the “north” end of the molecule is the dominant difference. It still would not be appropriate to generalize to other similar compounds and look for structure–activity relationships (SARs) based on these compounds; our results do indicate in fact that chemically similar compounds can display strikingly dissimilar binding properties and ligand similarity-based SAR extrapolations might fail.
The existence of a “common” binding mode for all benzodiazepine ligands was until now well accepted in spite of the fact that the structure of those ligands can vary dramatically. The flunitrazepam-like compounds possess a 1,4-benzodiazepine nucleus with a 5-phenyl substituent, the midazolam-like compounds an imidazo ring with a 5-phenyl substituent, and the flumazenil-like compounds such as Ro15-4513 and Ro15-1788 an imidazo ring without a 5-phenyl substituent. It is therefore not surprising that some experimental evidence points to the fact that some drugs interact with GABAA receptors differently than others. In particular, the family of imdiazobenzodiazepines seems to possess structural binding requirements (such as the interaction with γ2Ala79) distinct from those of the classical benzodiazepines,26,27 which is supported by our experiments.
In summary, we can conclude that our data suggest that there is not one single “common” binding mode for benzodiazepines in general; there are at least two different ones.
Methods
Competition Binding Assays and Electrophysiological Experiments
Radioligand displacement assays were performed in HEK-293 cells transfected with recombinant rat αxβ3γ2 receptors (x = 1, 2, 3, or 5) using 2 nM [3H]flunitrazepam and 1 nM to 10 μM investigated compound as described previously.20 Two-electrode voltage clamp recordings were performed on X. laevis oocytes treated with mRNA for the desired subunit combination at a GABA concentration eliciting 3% of the maximum current.21
Molecular Docking
The homology model of the extracellular α2+/γ2– interface (BZ site) was based on 4COF14 using Modeller28 as described previously.29 Molecular docking of the three ligands without their chiral methyl groups at position 3 (compounds 1–3 as depicted in Figure S1) was performed using GOLD.18 The putative binding site was defined by a cutoff distance of 11.5 Å around residue α2Ser204 of the C-loop of the α2 subunit. In addition, we selected 10 amino acids with flexible side chains (γ2Tyr58, γ2Phe77, γ2Thr142, α2His101, α2Tyr159, α2Ile202, α2Ser204, α2Ser205, α2Thr206, and α2Tyr209) and set a soft potential to increase the backbone flexibility of the C-loop (α2Ser204, α2Ser205, α2Thr206, and α2Gly207). To ensure convergence of the sampling, 100 genetic algorithm runs were performed. The ligands were built in MOE using the M conformation of the seven-membered ring that is supported by experimental studies. Before the docking run, the ligand was energetically minimized using the MMFF94 force field. The docking poses were rescored with the ChemScore fitness function implemented in GOLD. MOE Builder was used to derivatize the docked parent compounds with an (S)- or (R)-methyl at position 3.
Investigated Compounds
The synthesis of compounds 1-S (SH-I-048B), 1-R (SH-I-047), 2-S (SH-TS-CH3), 2-R (SH-TR-CHR), 3-S (SH-I-030), and 3-R (SH-I-053B) can be found in ref (16) and the Ph.D. Thesis of S. Huang.30
Acknowledgments
The authors thank J. Ramerstorfer and Z. Varagic for their support. J.M.C. acknowledges the National Institutes of Health for generous financial support (MH-096463, HL-118561, and NS-076517). This project is funded by FWF W1232 MolTag (DCBS), P27746 (DCBS), and the Medical University of Vienna.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.8b00144.
IUPAC nomenclature of the structures used in this study (Figure S1), [3H]flunitrazepam displacement assays with recombinant α1β3γ2, α2β3γ2, α3β3γ2, and α5β3γ2 GABAA receptors (Figure S2), two-electrode voltage clamp recordings on oocytes expressing different GABAA receptors (α1β3γ2, α2β3γ2, α3β3γ2, and α5β3γ2) (Figure S3), and comparison of the substitutions on the basic benzodiazepine core scaffold of the compound pairs investigated in this study (Figure S4) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Olfson M.; King M.; Schoenbaum M. (2015) Benzodiazepine use in the United States. JAMA Psychiatry 72, 136–142. 10.1001/jamapsychiatry.2014.1763. [DOI] [PubMed] [Google Scholar]
- Sieghart W. (1995) Structure and pharmacology of GABAA receptor subtypes. Pharmacol. Rev. 47, 181–234. [PubMed] [Google Scholar]
- Sieghart W. (2015) Allosteric Modulation of GABAA Receptors via Multiple Drug-Binding Sites. Adv. Pharmacol. 72, 53–96. 10.1016/bs.apha.2014.10.002. [DOI] [PubMed] [Google Scholar]
- Sieghart W.; Ernst M. (2005) Heterogeneity of GABAA Receptors: Revived Interest in the Development of Subtype-selective Drugs. Curr. Med. Chem.: Cent. Nerv. Syst. Agents 5, 217–242. 10.2174/1568015054863837. [DOI] [Google Scholar]
- Rudolph U.; Mohler H. (2014) GABAA receptor subtypes: Therapeutic potential in Down syndrome, affective disorders, schizophrenia, and autism. Annu. Rev. Pharmacol. Toxicol. 54, 483–507. 10.1146/annurev-pharmtox-011613-135947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrini G.; Ciciani G. (2013) Benzodiazepine receptor ligands: a patent review (2006–2012). Expert Opin. Ther. Pat. 23, 843–866. 10.1517/13543776.2013.782005. [DOI] [PubMed] [Google Scholar]
- Clayton T.; Chen J. L.; Ernst M.; Richter L.; Cromer B. A.; Morton C. J.; Ng H.; Kaczorowski C. C.; Helmstetter F. J.; Furtmuller R.; Ecker G.; Parker M. W.; Sieghart W.; Cook J. M. (2007) An updated unified pharmacophore model of the benzodiazepine binding site on GABAA receptors: correlation with comparative models. Curr. Med. Chem. 14, 2755–2775. 10.2174/092986707782360097. [DOI] [PubMed] [Google Scholar]
- Mokrab Y.; Bavro V. N.; Mizuguchi K.; Todorov N. P.; Martin I. L.; Dunn S. M.; Chan S. L.; Chau P. L. (2007) Exploring ligand recognition and ion flow in comparative models of the human GABA type A receptor. J. Mol. Graphics Modell. 26, 760–774. 10.1016/j.jmgm.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Ci S.; Ren T.; Su Z. (2008) Investigating the putative binding-mode of GABA and diazepam within GABAA receptor using molecular modeling. Protein J. 27, 71–78. 10.1007/s10930-007-9109-9. [DOI] [PubMed] [Google Scholar]
- Berezhnoy D.; Gibbs T. T.; Farb D. H. (2009) Docking of 1,4-benzodiazepines in the α1/γ2 GABAA receptor modulator site. Mol. Pharmacol. 76, 440–450. 10.1124/mol.109.054650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter L.; de Graaf C.; Sieghart W.; Varagic Z.; Morzinger M.; de Esch I. J.; Ecker G. F.; Ernst M. (2012) Diazepam-bound GABAA receptor models identify new benzodiazepine binding-site ligands. Nat. Chem. Biol. 8, 455–464. 10.1038/nchembio.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middendorp S. J.; Puthenkalam R.; Baur R.; Ernst M.; Sigel E. (2014) Accelerated discovery of novel benzodiazepine ligands by experiment-guided virtual screening. ACS Chem. Biol. 9, 1854–1859. 10.1021/cb5001873. [DOI] [PubMed] [Google Scholar]
- Siebert D. C. B., Bampali K., Puthenkalam R., Varagic Z., Sarto-Jackson I., Scholze P., Sieghart W., Mihovilovic M., Schnürch M., and Ernst M. (2018) Engineered benzodiazepine binding site reveals functional conservation of allosteric GABAA receptor binding sites. Submitted to ACS Chem. Biol. [DOI] [PubMed] [Google Scholar]
- Miller P. S.; Aricescu A. R. (2014) Crystal structure of a human GABAA receptor. Nature 512, 270–275. 10.1038/nature13293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton T.; Poe M. M.; Rallapalli S.; Biawat P.; Savic M. M.; Rowlett J. K.; Gallos G.; Emala C. W.; Kaczorowski C. C.; Stafford D. C.; Arnold L. A.; Cook J. M. (2015) A Review of the Updated Pharmacophore for the α5 GABAA Benzodiazepine Receptor Model. Int. J. Med. Chem. 2015, 430248. 10.1155/2015/430248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook J. M., Edwankar R. V., Edwankar C. R., Huang S., Jain H. D., Yang J., Rivas F., and Zhou H. (2010) Selective anticonvulsant agents and their uses. U.S. Patent 2010/0261711 A1.
- Rudolph U.; Knoflach F. (2011) Beyond classical benzodiazepines: novel therapeutic potential of GABAA receptor subtypes. Nat. Rev. Drug Discovery 10, 685–697. 10.1038/nrd3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S.; Thornton J. M. (1997) Prediction of protein-protein interaction sites using patch analysis. J. Mol. Biol. 272, 133–143. 10.1006/jmbi.1997.1233. [DOI] [PubMed] [Google Scholar]
- Cheng Y.; Prusoff W. H. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108. 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Stamenic T. T.; Poe M. M.; Rehman S.; Santrac A.; Divovic B.; Scholze P.; Ernst M.; Cook J. M.; Savic M. M. (2016) Ester to amide substitution improves selectivity, efficacy and kinetic behavior of a benzodiazepine positive modulator of GABAA receptors containing the α5 subunit. Eur. J. Pharmacol. 791, 433–443. 10.1016/j.ejphar.2016.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramerstorfer J.; Furtmuller R.; Sarto-Jackson I.; Varagic Z.; Sieghart W.; Ernst M. (2011) The GABAA receptor α+β- interface: a novel target for subtype selective drugs. J. Neurosci. 31, 870–877. 10.1523/JNEUROSCI.5012-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obradovic A.; Joksimovic S.; Poe M. M.; Ramerstorfer J.; Varagic Z.; Namjoshi O.; Batinic B.; Radulovic T.; Markovic B.; Roth B. L.; Sieghart W.; Cook J. M.; Savic M. M. (2014) SH-I-048A, an in vitro non-selective super-agonist at the benzodiazepine site of GABAA receptors: the approximated activation of receptor subtypes may explain behavioral effects. Brain Res. 1554, 36–48. 10.1016/j.brainres.2014.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer B. D.; Licata S. C.; Edwankar R. V.; Wang Z. J.; Huang S.; He X.; Yu J.; Zhou H.; Johnson E. M. Jr.; Cook J. M.; Furtmuller R.; Ramerstorfer J.; Sieghart W.; Roth B. L.; Majumder S.; Rowlett J. K. (2010) Anxiolytic-like effects of 8-acetylene imidazobenzodiazepines in a rhesus monkey conflict procedure. Neuropharmacology 59, 612–618. 10.1016/j.neuropharm.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savic M. M.; Majumder S.; Huang S.; Edwankar R. V.; Furtmuller R.; Joksimovic S.; Clayton T. Sr.; Ramerstorfer J.; Milinkovic M. M.; Roth B. L.; Sieghart W.; Cook J. M. (2010) Novel positive allosteric modulators of GABAA receptors: do subtle differences in activity at α1 plus α5 versus α2 plus α3 subunits account for dissimilarities in behavioral effects in rats?. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 34, 376–386. 10.1016/j.pnpbp.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savic M. M.; Huang S.; Furtmuller R.; Clayton T.; Huck S.; Obradovic D. I.; Ugresic N. D.; Sieghart W.; Bokonjic D. R.; Cook J. M. (2008) Are GABAA receptors containing α5 subunits contributing to the sedative properties of benzodiazepine site agonists?. Neuropsychopharmacology 33, 332–339. 10.1038/sj.npp.1301403. [DOI] [PubMed] [Google Scholar]
- Kucken A. M.; Wagner D. A.; Ward P. R.; Teissere J. A.; Boileau A. J.; Czajkowski C. (2000) Identification of benzodiazepine binding site residues in the γ2 subunit of the GABAA receptor. Mol. Pharmacol. 57, 932–939. [PubMed] [Google Scholar]
- Kucken A. M.; Teissere J. A.; Seffinga-Clark J.; Wagner D. A.; Czajkowski C. (2003) Structural requirements for imidazobenzodiazepine binding to GABAA receptors. Mol. Pharmacol. 63, 289–296. 10.1124/mol.63.2.289. [DOI] [PubMed] [Google Scholar]
- Sali A.; Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815. 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Puthenkalam R.; Hieckel M.; Simeone X.; Suwattanasophon C.; Feldbauer R. V.; Ecker G. F.; Ernst M. (2016) Structural Studies of GABAA Receptor Binding Sites: Which Experimental Structure Tells us What?. Front. Mol. Neurosci. 9, 44. 10.3389/fnmol.2016.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S. (2007) Synthesis of Optically Active Subtype Selective Benzodiazepine Receptor Ligands. Ph.D. Thesis, University of Wisconsin—Milwaukee, Milwaukee, WI. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.