

Abstract
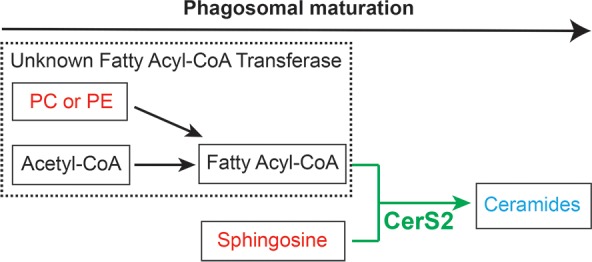
Phagocytosis is an evolutionarily conserved biological process where pathogens or cellular debris are cleared by engulfing them in a membrane-enclosed cellular compartment called the phagosome. The formation, maturation, and subsequent degradation of a phagosome is an important immune response essential for protection against many pathogens. Yet, the global lipid profile of phagosomes remains unknown, especially as a function of their maturation in immune cells. Here, we show using mass spectrometry based quantitative lipidomics that the ceramide class of lipids, especially very long chain ceramides, are enriched on maturing phagosomes with a concomitant decrease in the biosynthetic precursors of ceramides. We thus posit a new function for the enzyme ceramide synthase during phagocytosis in mammalian macrophages. Biochemical assays, cellular lipid feeding experiments, and pharmacological blockade of ceramide synthase together show that this enzyme indeed controls the flux of ceramides on maturing phagosomes. We also find similar results in the primitive eukaryote Dictyostelium discoideum, suggesting that ceramide enrichment may be evolutionarily conserved and likely an indispensible step in phagosome maturation.
In 1884, the phagocyte, a type of immune cell, was discovered and found to engulf, digest, and clear bacterial particles.1 Phagocytosis is the process of internalization of solid particles (pathogens or cellular debris) ≥ 500 nm by immune cells, predominantly macrophages. Over the past century, this universally conserved process has been extensively studied and shown to be a critical component of both the innate and adaptive mammalian immune system.2 Once internalized, these particles acquire membranes from the cellular plasma membrane and undergo a choreographed sequence of events broadly referred to as “phagosomal maturation,” culminating in fusion of the phagosomes with lysosomes. During maturation, the phagosomal lumen decreases in pH from 7 to 4.5 from an early phagosome (EP) to finally fuse with lysosome (phago-lysosome) via a late phagosome (LP) intermediate on the maturation pathway.3−5 During this transformation, the maturing phagosome changes profoundly in its protein and lipid composition via interactions and functional crosstalk with the endocytic pathway and inputs from the trans-golgi-network.
Over the past two decades, several elegant proteomics studies have shown the role of specific proteins in the maturation process, and these in turn have served as unique markers for the different stages in the phagosomal maturation process.2 For example, the GTPase Rab5 and early endosome antigen 1 (EEA1) localize to the EP and drive the EP to mature into a LP. The GTPase Rab7 and lysosomal associated membrane protein 1 (LAMP1) are found enriched on LPs and are critical for the formation of the phago-lysosome. Further studies in characterizing these proteins have shown that the process of phagosomal maturation is spatiotemporally regulated and highly interdependent.2 For instance, the acquisition of Rab5 onto the EP is a prerequisite for recruiting Rab7 in order to proceed with the maturation process. The localization of EPs is mostly peripheral, while that of LPs is toward the perinuclear region of the cell, where the lysosomes reside.6 Though the initial engulfment of a foreign particle mostly involves remodeling of the actin cytoskeleton, phagosome maturation requires transport of the phagosome on the microtubule cytoskeleton. While there exist several global proteomics studies that describe the role of different proteins during phagosomal maturation, there are only a handful of studies reported that describe the lipid profiles of maturing phagosomes, and these, too, only describe a specific lipid class, and not the global lipid profile.7 One such study describes the role and enrichment of different phosphoinositides at different stages of phagosomal maturation.8
We find that cholesterol is enriched on LPs, where it forms membrane lipid raft domains, and deregulated cholesterol metabolism impairs phagosomal maturation.9−11 Recently, we have shown that the motor protein dynein is enriched and clustered on these cholesterol rich lipid microdomains on LPs, and this in turn efficiently facilitates the unidirectional motion of LPs toward the lysosome.9,12 As a corollary to the aforementioned work, here, we set out to quantitatively determine the lipid composition of EPs and LPs. We show by biochemical assays, fatty acid feeding, and cellular pharmacological studies that the enzyme ceramide synthase controls the flux of ceramides during phagosomal maturation and leads to a very pronounced increase of very long-chain ceramides on LPs. This increase, we believe, is universally conserved because it is replicated in both mammalian cells (RAW264.7 macrophages) and in an early eukaryote (Dictyostelium discoideum). Finally, pharmacological blockade of ceramide synthase hampers the process of phagosomal maturation, suggesting an important role for this enzyme in the orchestration of phagocytosis.
Results and Discussion
Lipidomic Characterization of Maturing Phagosomes
To comparatively analyze the lipid profiles, herein referred to as the lipidome of EP and LP, we first extracted lipids from EPs and LPs and established a LC-MS/MS based method to exhaustively analyze >400 unique lipid species from 22 lipid classes in tandem (Figure 1A, Supporting Information Table 1).13−17 These lipid classes broadly encompassed neutral lipids, phospholipids and their lyso-versions, sterols and their esters, and sphingolipids.13−19 EPs and LPs were prepared and purified using established protocols from the mammalian RAW264.7 macrophages, and their purity was assessed to ensure these EP and LP preparations were devoid of any cellular membrane contamination (e.g., ER membrane, Golgi membrane), by previously established Western blot analysis.6,9 As reported earlier, quantitative lipidomics recapitulated a >3-fold increase in cholesterol on LPs compared to EPs, giving further confidence in our phagosomal preparations (Figure 1B, Supporting Information Table 1).9,10 Interestingly, we found that ceramides were highly enriched on LPs, and this enrichment was the most profound for very long chain (VLC) fatty acid chain (≥C22) containing ceramide species (Figure 1C, Supporting Information Table 1). Additionally, there was a concomitant decrease in the level of sphingosine (Figure 1D, Supporting Information Table 1) and VLC fatty acid bearing phosphatidylcholine (PC) and phosphatidylethanolamine (PE) phospholipids (Figure 1E, Supporting Information Table 1). Surprisingly, we also find that two free polyunsaturated fatty acids (PUFAs), namely, arachidonic acid (C20:4) and docasahexenoic acid (C22:6), are enriched on LPs, and there is a concomitant decrease in the concentrations of PUFA containing PC and PE lipids on LPs (Supporting Information Table 1). Besides these lipids, no other major lipid class was found to change significantly between the EP and LP groups (Supporting Information Table 1). As a control, to assess whether the changes in ceramides were EP and LP specific, and not a cellular event during phagosomal maturation, we measured ceramide levels in RAW264.7 macrophages at 0.5 and 4 h post feeding of beads and compared these concentrations to “no bead” treated RAW264.7 macrophages. We found no change in the concentration of cellular ceramides in this experiment, suggesting that ceramide changes from our last experiment were indeed EP and LP specific (Supporting Information Figure 1).
Figure 1.

Lipidomic characterization of EPs and LPs derived from RAW264.7 mouse macrophages. (A) Heat map plot showing the different lipid classes assessed by comparative LC-MS/MS analysis for EPs and LPs from RAW264.7 mouse macrophages. The heat map plot represents an average of fold changes (LP/EP) on a log 2 scale for different lipids from a particular lipid class. Blue and red color changes show enrichment of a particular lipid class on EP and LP respectively. Data represent six biological replicates per group. See Supporting Information Table 1 for complete data sets. Concentration of (B) cholesterol, (C) ceramides, (D) sphingosine, and (E) very long chain (VLC) containing phosphatidylcholine (PC) and phosphatidylethanolamine (PE) phospholipids from EPs and LPs. **P < 0.01, ***P < 0.001 for LP group versus EP group by Student's t test.
Since phagocytosis is an evolutionarily conserved immune process, we decided to perform a similar quantitative lipidomics experiment using a primitive eukaryote as a model system. Dictyostelium discoideum was chosen for this, because we earlier reconstituted the microtubule motor dependent motion of EPs and LPs purified from this soil-living amoeba and found that the clustering of dynein into lipid raft domains transports LPs toward fusion with lysosomes.9 The phytosterols (stigmasterol and sitosterol, as Dictyostelium discoideum does not have any cholesterol) and VLC fatty acid containing ceramides were found most enriched on LPs, while the sphingolipid precursors (sphingosine and sphinganine) and VLC fatty acid containing PCs were found enriched on EPs (Supporting Information Figure 2, Supporting Information Table 1). Given the striking increase in VLC fatty acid containing ceramides and a concomitant decrease in sphingosine and VLC containing phospholipids (PC, PE) on LPs in both RAW264.7 macrophages and Dictyostelium discoideum cells, we hypothesized that an enzyme capable of biosynthesizing ceramides from sphingosine and phospholipid-derived fatty acid precursors was likely causing ceramide accumulation on the phagosome membrane during their maturation. On the basis of the lipidomics data, we also postulated that this enzyme has a preference for biosynthesizing VLC fatty acid containing ceramides.
Role of Ceramide Synthase in Phagocytosis
Mammals have an integral membrane enzyme, ceramide synthase (CerS), that biosynthesizes ceramides from sphingosine and fatty acyl-coenzyme A (CoA) (Figure 2A). There are six isoforms of ceramide synthase (CerS1–6) in mammals, with different tissue distributions and fatty acyl-CoA substrate preferences.20 Both large-scale gene expression studies (http://biogps.org/;21Supporting Information Figure 3) and literature precedence support CerS2 being the major ceramide synthase in RAW264.7 macrophages,22 but its detection at a protein level and enzymatic activity in RAW264.7 macrophages remains lacking. Interestingly, biochemical characterization of recombinant CerS2 has shown that this enzyme prefers VLC fatty acyl-CoA as substrates, to form VLC fatty acid containing ceramides.23 This CerS2 substrate profile matches our lipidomic profile, where we see the highest enrichment for VLC containing ceramide species on LPs (e.g., C22:0, C24:0 ceramides; Figure 1C, Supporting Information Table 1). To test whether CerS2 was indeed the dominant ceramide synthase in RAW264.7 cells, we performed ceramide synthase activity assays using a VLC-CoA (C22:0-CoA) substrate on membrane lysates of RAW264.7 cells using fumonisin B1 (referred as fumonisin), a fungal natural product, which is a potent broad spectrum ceramide synthase inhibitor.24−26 In these assays, we found that the membrane lysates of RAW264.7 cells had robust ceramide synthase activity forming C22:0 ceramide (a characteristic feature of CerS2), and this activity was potently inhibited by fumonisin treatment (5 μM, 30 min; Figure 2B). We also confirmed by Western blot analysis that CerS2 is indeed expressed abundantly in the membrane lysates of RAW264.7 cells (Figure 2C). Having established CerS2 as the major ceramide synthase in RAW264.7 cells, we wanted to assess whether this enzyme is present on EPs and LPs, as maturing phagosomes acquire their membranes predominantly from cellular components that they interact with during their journey toward lysosomes (e.g., plasma membrane, ER membrane).4
Figure 2.

Identification of CerS2 as major ceramide synthase in RAW264.7 mouse macrophages. (A) The reaction catalyzed by ceramide synthase. (B) Ceramide synthase in vitro activity assays on membrane lysates from RAW264.7 mouse macrophages treated with fumonisin (5 μM, 30 min) or DMSO, showing robust inhibition of ceramide synthase activity following fumonisin treatment. As a control, denatured membrane proteomes were used. Data represent mean ± SEM for three biological replicates per group. (C) Western blot analysis confirming abundant expression of CerS2 in RAW264.7 membrane lysates. As a positive control, membrane lysates from HEK293T were used. A total of 40 μg of lysate was loaded for all samples in this analysis, and actin was used as a loading control. The Western blot analysis was performed on five biological replicates with reproducible results.
Toward this, EPs and LPs were assessed for CerS2 content by Western blot analysis. Very interestingly and contrary to our initial expectations, we found that EPs had significantly more CerS2 than LPs (∼3-fold; Figure 3A). Consistent with the enriched CerS2 on EPs, we found that the ceramide synthase activity was ∼3 fold higher on EPs, compared to LPs (Figure 3B). As a control, we measured the cellular levels of CerS2 in RAW264.7 cells, during this treatment, and found no changes in CerS2 cellular levels, suggesting that the change in CerS2 level was EP and LP specific (Supporting Information Figure 1). Next, we fed RAW264.7 cells with the unnatural long chain fatty acid, heptadecenoic acid (C17:1 FFA, 1 mM, 4 h), and found that the C17:1 FFA was mostly incorporated into cellular PC and PE pools in accordance with previous studies (Supporting Information Figure 4).27 We then prepared EPs and LPs by feeding silica beads to cells containing C17:1-PC and PE from C17:1 FFA feeding. The lipids from the EPs and LPs derived from this experiment were enriched using previously described methods,16,17,28 and these lipids were subjected to quantitative lipidomics, looking specifically for C17:1 containing lipids from different lipid classes (Supporting Information Table 1). Consistent with previous lipidomics studies (Figure 1C, Supporting Information Figure 2), we found that the levels of N-17:1-ceramide were significantly elevated on LPs (Figure 3C), and there was a concomitant decrease in levels of C17:1-containing PC and PE lipids on LPs (Supporting Information Table 1, Supporting Information Figure 5, 6). These results corroborate earlier lipidomics data (Figure 1, Supporting Information Figure 2) and seem to suggest that PC and PE classes of phospholipids on EPs likely serve as the fatty acid donor for the generation of fatty acyl-CoA that is eventually converted to ceramide by the N-acylation action of CerS2.
Figure 3.
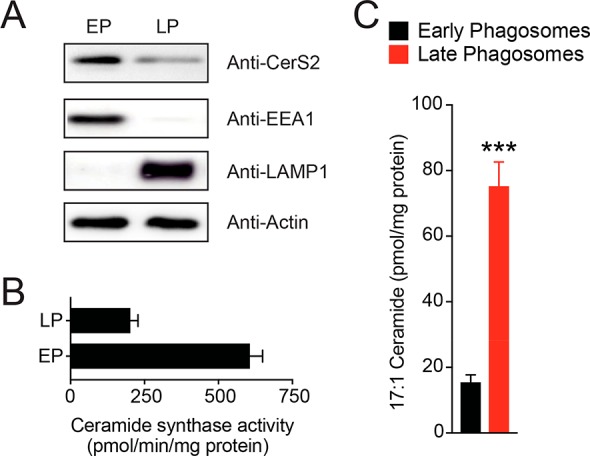
Characterization of CerS2 activity during phagosomal maturation. (A) Western blot analysis confirming the enrichment of CerS2 on EPs. As controls, EEA1 and LAMP1 were used to assess the purity of EP and LP preparations, respectively. In each case, 25 μg of lysate was loaded for all samples in this analysis, and actin was used as a loading control. The Western blot analysis was performed on eight biological replicates with reproducible results. (B) Ceramide synthase in vitro activity assay on EPs and LPs from RAW264.7 mouse macrophages, showing heightened ceramide synthase activity on EPs. Data represents mean ± SEM for three biological replicates per group. (C) Levels of C17:1 containing ceramide on EPs and LPs, following feeding of RAW264.7 mouse macrophages with 1 mM C17:1 FFA (4 h, 37 °C). Data represent mean ± SEM for five biological replicates. ***P < 0.001 for LP group versus EP group by Student’s t test.
Effects of Pharmacological Blockade of Ceramide Synthase on Phagocytosis
To validate if fumonisin was pharmacologically active, and to verify its effects of inhibition of CerS2 in RAW264.7 cells, first, we treated RAW264.7 cells in situ with fumonisin (1 or 5 μM, 4 h). We found that following fumonisin treatment, ceramide formation was potently inhibited, without the cells getting activated or undergoing apoptosis (Supporting Information Figure 7). Next, we tested whether pharmacologically disrupting CerS2 had any effect on phagosomal ceramide content. Toward this, we allowed RAW264.7 cells to phagocytose silica beads for 30 min so as to generate EPs, following which we treated cells in situ with DMSO (LP-DMSO) or 5 μM fumonisin (LP-fumonisin) for 4 h and harvested LPs at the end of this treatment using established protocols.6 As a control for this study, we included EPs. Lipids were extracted from each these phagosomal preparations and subjected to LC-MS/MS lipidomic analysis looking specifically at the concentration of phagosomal sphingolipids (Supporting Information Table 1). We were able to recapitulate changes between the EP and LP-DMSO pools as seen previously, where VLC fatty acid containing ceramides and sphingosine increased and decreased respectively on phagosomes from the LP-DMSO group compared to the EP group (Figure 4A). Thus far, no other sphingolipid changed in our study, suggesting that CerS2 is likely the major enzyme controlling the flux of sphingolipid metabolism during phagosomal maturation. Corroborating this, we found that the sphingolipid profiles for the phagosomes from the LP-fumonisin group looked near identical to those from the EP group (Figure 4A).
Figure 4.

Pharmacological blockade of CerS2 hampering phagosomal maturation. (A) Concentration of ceramides and sphingosine on LPs from RAW264.7 mouse macrophages following fumonisin treatment (LP-fumonisin, 5 μM, 4 h, 37 °C). As controls, EP and LP (LP-DMSO) were used. Data represent mean ± SEM for four biological replicates per group. **P < 0.01, ***P < 0.001 for experimental groups (LP-fumonisin and LP-DMSO) versus EP group by Student’s t test. (B) Western blot analysis confirming a defect in phagosomal maturation following inhibition of CerS2. In each case for each group, 30 μg of lysate was loaded for all samples in this analysis, and actin was also used as a loading control. The Western blot analysis was performed in triplicate with reproducible results.
Having shown that pharmacologically inhibiting CerS2 by fumonisin treatment leads to the impairment of phagosomal ceramide synthesis, we wanted to determine whether this lipid profile change had any functional effect on phagosomal maturation. We therefore looked at the levels of CerS2, and known EP and LP markers, to determine whether the pharmacological blockade of CerS2, indeed, has any effect on phagosomal maturation. We find by Western blot analysis, as seen previously, that CerS2 levels are lower on LPs (LP-DMSO group) than EPs, and the phagosomes from the LP-fumonisin group have near EP levels of CerS2 (Figure 4B). We also find that the EP protein marker, EEA1, is enriched on the phagosomal preparations from the EP group and the LP-fumonisin group but not on the LP-DMSO group (Figure 4B). Finally, we find that the LP protein markers, LAMP1 and Rab7, are enriched on the LPs (LP-DMSO group), but not on EPs. Interestingly, we find that the phagosomes from the LP-fumonisin group have significantly lesser amounts of both LAMP1 and Rab7 compared to the LP-DMSO group (Figure 4B). Taken together, these results suggest that the pharmacological blockade of CerS2 results in partial blockade or delay in phagosomal maturation.
Discussion
Our findings taken together provide compelling evidence that mature phagosomes (LPs) are enriched with ceramides, especially VLC fatty acid containing ceramides, which are biosynthesized from sphingosine and fatty acyl-CoA by the action of CerS2, a major ceramide synthase, in mammalian macrophages (Figures 1, 2). Interestingly and quite paradoxically, EPs have greater amounts and heightened activity of CerS2. It appears that this increased activity of CerS2 on EPs is an anticipatory mechanism that produces ceramides, which are eventually found enriched on LPs (Figure 3). This may stem from the possibility that ceramides are needed for maturing phagosomes, yet once enough ceramide is produced on EPs to facilitate the maturation process, elevated CerS2 activity on LPs is no longer needed. Another possibility is that cholesterol may be required for stabilizing ceramides on phagosomes. So even though EPs have more CerS2, ceramide levels may still stay low because phagosomes enrich in cholesterol content only at the LP stage.9 The regulation of CerS2 and ceramide on EPs/LPs observed here might constitute a previously unknown feedback lipid-signaling pathway in phagosomal maturation. We also show that the pharmacological blockade of CerS2 results in depleting VLC fatty acid containing ceramide levels on LPs, and thereby hampers the phagosomal maturation process (Figure 4). Additionally, from our data we speculate that the fatty acyl-CoA substrate for CerS2 is likely synthesized from PC and PE phospholipids present on EPs by unknown mechanisms (Figure 1E, Supporting Information Table 1, Supporting Information Figures 2, 3).
Phagosomes accumulate cholesterol as they mature, possibly making their membranes more rigid and making their transport within cells to lysosomes efficient.12In vitro studies from others suggest that ceramide is critical for stabilizing lipid rafts and making ordered rigid membrane microdomains.29 Our current data along with studies from others suggest that the increased ceramide on LPs is likely functioning to generate ordered lipid microdomains on maturing phagosomes, thus enabling the recruitment of appropriate proteins during the phagosome maturation process. Additionally, we believe that the membrane-associated dynein motors that drive phagosome transport toward lysosomes may be able to generate force efficiently and in a more directed manner if the underlying ceramide rich membrane is rigid and more organized.9
Projecting forward, the discovery of ceramide and CerS2 as a new player in phagosomal maturation suggests that an as-of-yet unknown sphingolipid metabolism pathway exists in mammalian phagosomal maturation (Figure 5). Unlike mammals, Dictyostelium discoideum possess only a single isoform of ceramide synthase, crsA, which shares ∼30% sequence homology to the mammalian ceramide synthase isoforms with 100% conservation in the active site Lag1P motif (Supporting Information Figure 8).20,26 Our lipidomics studies on EPs and LPs (Supporting Information Figure 1, Supporting Information Table 1) suggest that this evolutionarily conserved enzyme might serve as a nodal point for heightened ceramide levels on LPs and might, in turn, be regulating phagosomal maturation even in primitive eukaryotes. We speculate that this sphingolipid pathway has another unannotated enzyme in mammals and primitive eukaryotes, a putative fatty acyl-CoA transferase that generates fatty acyl-CoA from PC and/or PE lipids (Figure 5). Additionally, our lipidomics data also suggest that there exists a phospholipase, which prefers PUFA containing PC and/or PE phospholipids as substrates and produces free PUFAs that are found enriched on LPs (Figure 5, Supporting Information Table 1). The discovery of these enzymes would certainly help in a greater understanding of phagosomal maturation and provide new insights into the spatiotemporal regulation, cellular pathways crosstalk, and interdependence of this very important immune response. Currently, however, there are no antibodies or chemical reagents to study the ceramide lipid class and to map proteins that would interact specifically with them. Generating specific antibodies against ceramides and/or synthesizing photo-cross-linking ceramide lipid probes, like those described in the literature, would greatly advance our understanding of the protein ligands for ceramides, and link these biochemically to the process of phagosomal maturation.30,31 Finally, functional cell biological studies are required to better understand the exact role that ceramides and CerS2 play in phagosomal maturation. Our findings open up several new questions such as the origin and mechanism of CerS2 localization on phagosomes, the effect of ceramide on lipid microdomains and resultant transport of phagosomes inside cells, and the possible synergistic requirement of cholesterol and ceramide for the assembly of lipid rafts on a phagosome.9
Figure 5.
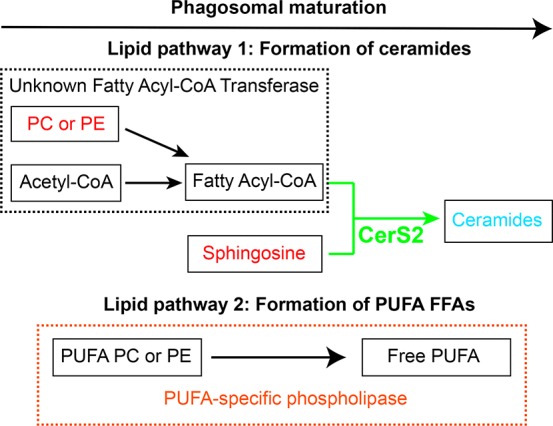
Schematic representation of putative lipid pathways in phagosomal maturation. As phagosomes mature, their ceramide content increases due to the activity of CerS2, which is found enriched on EPs. Two other cryptic activities are postulated in this scheme based on our lipidomics results. The first, an unknown fatty acyl-CoA transferase, which converts PC and/or PE to generate fatty acyl-CoA, which is eventually converted to ceramide, by the N-acylation activity of CerS2. The second is a PUFA-specific phospholipase activity, which converts PUFA containing PC and/or PE lipids into free PUFAs that are found enriched on LPs.
Methods
Materials
All chemicals, buffers, solvents, and reagents were purchased from Sigma-Aldrich unless otherwise mentioned. All MS quantitation lipid standards were purchased from Avanti Polar Lipids Inc. unless otherwise mentioned.
Phagosome Preparations from RAW264.7 Cells
The RAW264.7 mouse macrophage cell line (ATCC) was cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; HiMedia) with 10% (v/v) Fetal Bovine Serum (FBS; Thermofisher Scientific) and 1% (v/v) Penicillin-Streptomycin (MP Biomedicals) at 37 °C and 5% (v/v) CO2. Then, 10 × 10 cm culture dishes (Eppendorf) were used for phagosomal preparations using a previously established protocol.9 Briefly, 300 μL of 1-μm silica beads was washed with DMEM (3 times), pelleted by centrifugation at 500g for 5 min after each wash, and finally resuspended in 1 mL of serum free DMEM. This solution was vortexed, sonicated for 5 min, and added to 30 mL of serum-free DMEM prewarmed to 37 °C. Three mL of this solution was added to a 10 cm culture dish containing RAW264.7 cells at 80% confluency. The cells were subsequently kept at 4 °C for 5 min to synchronize the uptake of beads. Postsynchronization, the bead pulse was 15 min for EP or LP preparations. The unphagocytosed beads were removed by washing the cells with sterile Phosphate Buffer Saline (PBS; three times). For EP preparations, cells were harvested at this stage and transferred to a sterile 50 mL tube, while for LP preparations, the cells were cultured for 4 h in DMEM with 10% (v/v) FBS at 37 °C. Upon desired bead maturation, the cells were washed with sterile PBS (three times), harvested by scraping, and transferred into sterile 50 mL tubes. The harvested cells are washed again in sterile PBS (two times) and pelleted by spinning at 500g for 5 min after each wash. The cells were then washed with 25 mL of lysis buffer (250 mM sucrose, 3 mM imidazole, pH 7.4) followed by pelleting by centrifugation at 500g for 5 min. Post-washing, the cells were resuspended in 1 mL of lysis buffer containing protease inhibitor cocktail (Roche), 3 mM dithiothreitol (DTT), and 10 μg mL–1 pepstatin A. This cell suspension was lysed using a cell cracker with a 10 μm clearance (Isobiotec) by 10 passages through the syringes. This cellular lysate was overlaid on a sucrose step gradient with 5 mL each of 85% (w/v) sucrose and 60% (w/v) sucrose with 3 mM imidazole. The gradient was centrifuged at 100 000g at 4 °C for 1 h. The bead pellet was separated from the cellular debris and resuspended in 200 μL of TNE buffer (50 mM Tris, 140 mM NaCl, 5 mM EDTA, pH 7.4) and stored at −20 °C before processing for lipid extraction. For the C17:1 FFA feeding experiment, the RAW264.7 cells in 10 cm culture dish at 80% confluence were fed with 1 mM C17:1 FFA for 4 h prior to starting the aforementioned phagosomal preparations. For CerS2 inhibition studies, 5 μM fumonisin was added after the initial bead phagocytosis, post-synchronization, and the phagosomal preparations were performed as described above.
Preparation of Phagosomes from Dictyostelium discoideum
Phagosome preparations from Dictyostelium discoideum were done using established protocols.9,32 Briefly, silica beads (1 μm diameter, Polysciences Inc.) were washed in HL5 medium, and the medium was separated by centrifugation (1000g, 5 min, 4 °C). The prewashed silica beads were resuspended in 0.5 mL of Sorensen’s buffer and sonicated for 10 min. Dictyostelium discoideum AX-2 strain was grown to 6 million cells mL–1 at 22 °C, at which point the cells were pelleted by centrifugation at 900g for 5 min. After resuspending the cell pellet in Sorensen’s buffer, the prewashed silica beads were added and incubated with cells at 4 °C for 15 min with gentle mixing for synchronous bead uptake. The pulse begins by transferring the bead-cell suspension to 100 mL of HL5 medium at 22 °C and 150 rpm to initiate phagosome formation. For EPs, the pulse duration was 5 min with no chase, while LPs were pulsed for 15 min and chased for 45 min after washing off the nonphagocytosed beads. The pulse-chase cycle was quenched by the addition of 330 mL of cold Sorensen’s buffer, and the cells were pelleted by centrifugation at 900g for 5 min at 4 °C. The resulting cell pellet was resuspended in 1:1 (w/v) LB-30 buffer containing 30% (w/v) sucrose, 30 mM Tris (pH 8.0), 4 mM EGTA, protease inhibitor cocktail (Roche), 3 mM DTT, 20 μg mL–1 pepstatin A, 5 mM phenylmethyl sulfonyl fluoride, and 5 mM benzamidine hydrochloride. The cells were lysed using cell cracker (Isobiotec) with a 10 μm clearance ball and 10 passage strokes. The lysate was layered on a sucrose step gradient consisting of 4 mL each of 85% (w/v) and 65% (w/v) sucrose solution containing 30 mM Tris and 4 mM EGTA. The gradient was centrifuged at 100 000g for 1 h at 4 °C. Purified phagosomes collected at the bottom of the tube and were resuspended in 300 μL of sterile PBS by mild pipetting, and lipids were extracted from them.
Lipid Extraction and Targeted Lipid Profiling
The phagosomal lipid extractions were performed using an established protocol.16,17,28 Briefly, the phagosomal preparations were washed with sterile Dulbecco’s PBS (DPBS; three times) and transferred into a glass vial using 1 mL of DPBS. A total of 3 mL of 2:1 (v/v) chloroform (CHCl3)/methanol (MeOH) with the internal standard mix (50 pmol of each internal standard listed in Supporting Information Table 1) was added, and the mixture was vortexed. The two phases were separated by centrifugation at 2800g for 5 min. The organic phase (bottom) was removed. A total of 50 μL of formic acid was added to acidify the aqueous homogenate, and CHCl3 was added to make up a 4 mL volume. The mixture was vortexed, and separated by centrifugation at 2800g for 5 min. Both the organic extracts were pooled and dried under a stream of N2. The lipidome was solubilized in 200 μL of 2:1 (v/v) CHCl3/MeOH, and 20 μL was used for the lipidomics analysis. All the lipid species analyzed in this study were quantified using the multiple reaction monitoring high resolution (MRM-HR) scanning method (Supporting Information Table 1) on a Sciex X500R QTOF mass spectrometer (MS) fitted with an Exion-LC series UHPLC. All data were acquired and analyzed using the SciexOS software. The LC separation was achieved using a Gemini 5U C18 column (Phenomenex, 5 μm, 50 × 4.6 mm) coupled to a Gemini guard column (Phenomenex, 4 × 3 mm). The LC solvents were as follows: positive mode, buffer A, 95:5 (v/v) H2O/MeOH + 0.1% formic acid + 10 mM ammonium formate; buffer B, 60:35:5 (v/v) isopropanol (IPA)/MeOH/H2O + 0.1% (v/v) formic acid + 10 mM ammonium formate; negative mode, buffer A, 95:5 (v/v) H2O/MeOH + 0.1% (v/v) NH4OH; buffer B, 60:35:5 (v/v) IPA/MeOH/H2O + 0.1% (v/v) NH4OH. All the lipid estimations were performed using an electrospray ion (ESI) source, with following MS parameters: turbo spray ion source, medium collision gas, curtain gas = 20 L min–1, ion spray voltage = 4500 V (positive mode) or −5500 V (negative mode), at 400 °C. A typical LC-run was 55 min, with the following solvent run sequence post injection: 0.3 mL min–1 0% B for 5 min, 0.5 mL min–1 0% B for 5 min, 0.5 mL min–1 linear gradient of B from 0–100% over 25 min, 0.5 mL min–1 of 100% B for 10 min, and re-equilibration with 0.5 mL min–1 of 0% B for 10 min. A detailed list of all the species targeted in this MRM-HR study, describing the precursor ion mass and adduct, the product ion targeted, and MS voltage parameters can be found in Supporting Information Table 1. All the endogenous lipid species were quantified by measuring the area under the curve in comparison to the respective internal standard, and then normalizing to the total protein content of the phagosomal preparation. All the lipidomics data are represented as mean ± SEM of four (or more) biological replicates per group (Supporting Information Table 1).
Western Blot Analysis
All Western blots were done using established protocols6,9 with the following primary antibodies (rabbit)/anti-CerS2 (1:1000, Sigma-Aldrich, HPA027262) or anti-EEA-1 (1:2000, Cell Signaling Technology, 2411S) or anti-LAMP1 (1:2000, Abcam, ab24170) or anti-Rab7 (1:2000, Abcam, ab137029). The goat antirabbit IgG (H+L) HRP conjugated (1:10 000, Abcam, ab6789) was used a secondary antibody (1 h, 25 °C), following which the protein signal was visualized using the SuperSignal West Pico Plus Chemiluminescent substrate (Thermofisher Scientific) on a Syngene G-Box Chemi-XRQ gel documentation system.
Ceramide Synthase Substrate Assays
The substrate assay was adapted from a previously described protocol albeit with a different substrate.26 Briefly, 50 μM behenoyl-coenzyme A (C22:0-CoA) and 20 μM sphingosine were mixed by sonication and incubated at 37 °C with shaking at 750 rpm for 5 min, following which 10 μg of proteome was added to a final volume of 100 μL in DPBS, and the mixture was incubated at 37 °C with shaking at 750 rpm for 30 min. The reaction was quenched by the addition of 250 μL of 2:1 CHCl3/MeOH containing 50 pmol of N-25:0-ceramide internal standard, and the mixture was vigorously vortexed. The two phases were separated by centrifugation at 2800g for 5 min, and the organic phase (bottom) was removed. The organic extracts were dried under a stream of N2 and solubilized in 100 μL of 2:1 (v/v) CHCl3/MeOH. The LC-MS protocol was similar to a previously established protocol.33 All MS analysis was performed using an ESI in the positive ion mode for ceramide formation. All MS parameters are described in Supporting Information Table 1. Measuring the area under the peak, and normalizing it to the internal standard, quantified the product formed for the ceramide synthase assays. The enzymatic rate was corrected by subtracting the nonenzymatic rate of hydrolysis, which was obtained by using heat denatured proteome as a control. All the data are represented as mean ± SEM of at least three biological replicates.
Statistical Analysis
All data are presented as mean ± SEM of three (or more) biological replicates per group for substrate assays, and as mean ± SEM of at least four (or more) biological replicates per group for lipidomics experiments. Statistical analysis was performed using GraphPad Prism 7 (Mac OS X), and the Student’s t test (two-tailed) of this software was used to calculate statistical significance between the different study groups. A P value of <0.05 was considered statistically significant in this study.
Acknowledgments
This work was supported by grants from the Wellcome Trust DBT India Alliance (IA/I/15/2/502058 to S.S.K., IA/S/11/2500255 to R.M.), and a DST-FIST Infrastructure Development Grant to IISER Pune Biology Department. We thank A. Rajendran and D. Kelkar for technical assistance.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.8b00438.
Author Contributions
§ These authors contributed equally to this work
Author Contributions
D.P. and N.M. contributed equally to this work. S.S.K. and R.M. conceived the project. All authors performed the experiments and analyzed the data. S.S.K. wrote the paper, with inputs from all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Tauber A. I. (2003) Metchnikoff and the phagocytosis theory. Nat. Rev. Mol. Cell Biol. 4, 897–901. 10.1038/nrm1244. [DOI] [PubMed] [Google Scholar]
- Flannagan R. S.; Jaumouille V.; Grinstein S. (2012) The cell biology of phagocytosis. Annu. Rev. Pathol.: Mech. Dis. 7, 61–98. 10.1146/annurev-pathol-011811-132445. [DOI] [PubMed] [Google Scholar]
- Levin R.; Grinstein S.; Canton J. (2016) The life cycle of phagosomes: formation, maturation, and resolution. Immunol Rev. 273, 156–179. 10.1111/imr.12439. [DOI] [PubMed] [Google Scholar]
- Vieira O. V.; Botelho R. J.; Grinstein S. (2002) Phagosome maturation: aging gracefully. Biochem. J. 366, 689–704. 10.1042/bj20020691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinchen J. M.; Ravichandran K. S. (2008) Phagosome maturation: going through the acid test. Nat. Rev. Mol. Cell Biol. 9, 781–795. 10.1038/nrm2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai A. K.; Rai A.; Ramaiya A. J.; Jha R.; Mallik R. (2013) Molecular adaptations allow dynein to generate large collective forces inside cells. Cell 152, 172–182. 10.1016/j.cell.2012.11.044. [DOI] [PubMed] [Google Scholar]
- Steinberg B. E.; Grinstein S. (2008) Pathogen destruction versus intracellular survival: the role of lipids as phagosomal fate determinants. J. Clin. Invest. 118, 2002–2011. 10.1172/JCI35433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung T.; Grinstein S. (2007) Lipid signaling and the modulation of surface charge during phagocytosis. Immunol. Rev. 219, 17–36. 10.1111/j.1600-065X.2007.00546.x. [DOI] [PubMed] [Google Scholar]
- Rai A.; Pathak D.; Thakur S.; Singh S.; Dubey A. K.; Mallik R. (2016) Dynein Clusters into Lipid Microdomains on Phagosomes to Drive Rapid Transport toward Lysosomes. Cell 164, 722–734. 10.1016/j.cell.2015.12.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermine J. F.; Duclos S.; Garin J.; St-Louis F.; Rea S.; Parton R. G.; Desjardins M. (2001) Flotillin-1-enriched lipid raft domains accumulate on maturing phagosomes. J. Biol. Chem. 276, 18507–18512. 10.1074/jbc.M101113200. [DOI] [PubMed] [Google Scholar]
- Huynh K. K.; Gershenzon E.; Grinstein S. (2008) Cholesterol accumulation by macrophages impairs phagosome maturation. J. Biol. Chem. 283, 35745–35755. 10.1074/jbc.M806232200. [DOI] [PubMed] [Google Scholar]
- Pathak D.; Mallik R. (2017) Lipid - Motor Interactions: Soap Opera or Symphony?. Curr. Opin. Cell Biol. 44, 79–85. 10.1016/j.ceb.2016.09.005. [DOI] [PubMed] [Google Scholar]
- Sud M.; Fahy E.; Cotter D.; Brown A.; Dennis E. A.; Glass C. K.; Merrill A. H. Jr.; Murphy R. C.; Raetz C. R.; Russell D. W.; Subramaniam S. (2007) LMSD: LIPID MAPS structure database. Nucleic Acids Res. 35, D527–532. 10.1093/nar/gkl838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill A. H.; Dennis E. A.; McDonald J. G.; Fahy E. (2013) Lipidomics technologies at the end of the first decade and the beginning of the next. Adv. Nutr. 4, 565–567. 10.3945/an.113.004333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahy E.; Sud M.; Cotter D.; Subramaniam S. (2007) LIPID MAPS online tools for lipid research. Nucleic Acids Res. 35, W606–612. 10.1093/nar/gkm324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamat S. S.; Camara K.; Parsons W. H.; Chen D. H.; Dix M. M.; Bird T. D.; Howell A. R.; Cravatt B. F. (2015) Immunomodulatory lysophosphatidylserines are regulated by ABHD16A and ABHD12 interplay. Nat. Chem. Biol. 11, 164–171. 10.1038/nchembio.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kory N.; Grond S.; Kamat S. S.; Li Z.; Krahmer N.; Chitraju C.; Zhou P.; Frohlich F.; Semova I.; Ejsing C.; Zechner R.; Cravatt B. F.; Farese R. V. Jr.; Walther T. C. (2017) Mice lacking lipid droplet-associated hydrolase, a gene linked to human prostate cancer, have normal cholesterol ester metabolism. J. Lipid Res. 58, 226–235. 10.1194/jlr.M072538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugger B. (2014) Lipidomics: analysis of the lipid composition of cells and subcellular organelles by electrospray ionization mass spectrometry. Annu. Rev. Biochem. 83, 79–98. 10.1146/annurev-biochem-060713-035324. [DOI] [PubMed] [Google Scholar]
- Brown H. A.; Murphy R. C. (2009) Working towards an exegesis for lipids in biology. Nat. Chem. Biol. 5, 602–606. 10.1038/nchembio0909-602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy M.; Futerman A. H. (2010) Mammalian ceramide synthases. IUBMB Life 62, 347–356. 10.1002/iub.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.; Jin X.; Tsueng G.; Afrasiabi C.; Su A. I. (2016) BioGPS: building your own mash-up of gene annotations and expression profiles. Nucleic Acids Res. 44, D313–316. 10.1093/nar/gkv1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halasiddappa L. M.; Koefeler H.; Futerman A. H.; Hermetter A. (2013) Oxidized phospholipids induce ceramide accumulation in RAW 264.7 macrophages: role of ceramide synthases. PLoS One 8, e70002. 10.1371/journal.pone.0070002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laviad E. L.; Albee L.; Pankova-Kholmyansky I.; Epstein S.; Park H.; Merrill A. H. Jr.; Futerman A. H. (2008) Characterization of ceramide synthase 2: tissue distribution, substrate specificity, and inhibition by sphingosine 1-phosphate. J. Biol. Chem. 283, 5677–5684. 10.1074/jbc.M707386200. [DOI] [PubMed] [Google Scholar]
- Pewzner-Jung Y.; Park H.; Laviad E. L.; Silva L. C.; Lahiri S.; Stiban J.; Erez-Roman R.; Brugger B.; Sachsenheimer T.; Wieland F.; Prieto M.; Merrill A. H. Jr.; Futerman A. H. (2010) A critical role for ceramide synthase 2 in liver homeostasis: I. alterations in lipid metabolic pathways. J. Biol. Chem. 285, 10902–10910. 10.1074/jbc.M109.077594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill A. H. Jr.; van Echten G.; Wang E.; Sandhoff K. (1993) Fumonisin B1 inhibits sphingosine (sphinganine) N-acyltransferase and de novo sphingolipid biosynthesis in cultured neurons in situ. J. Biol. Chem. 268, 27299–27306. [PubMed] [Google Scholar]
- Spassieva S.; Seo J. G.; Jiang J. C.; Bielawski J.; Alvarez-Vasquez F.; Jazwinski S. M.; Hannun Y. A.; Obeid L. M. (2006) Necessary role for the Lag1p motif in (dihydro)ceramide synthase activity. J. Biol. Chem. 281, 33931–33938. 10.1074/jbc.M608092200. [DOI] [PubMed] [Google Scholar]
- Ogura Y.; Parsons W. H.; Kamat S. S.; Cravatt B. F. (2016) A calcium-dependent acyltransferase that produces N-acyl phosphatidylethanolamines. Nat. Chem. Biol. 12, 669–671. 10.1038/nchembio.2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J.; Lees M.; Sloane Stanley G. H. (1957) A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 226, 497–509. [PubMed] [Google Scholar]
- Megha; London E. (2004) Ceramide selectively displaces cholesterol from ordered lipid domains (rafts): implications for lipid raft structure and function. J. Biol. Chem. 279, 9997–10004. 10.1074/jbc.M309992200. [DOI] [PubMed] [Google Scholar]
- Niphakis M. J.; Lum K. M.; Cognetta A. B. 3rd; Correia B. E.; Ichu T. A.; Olucha J.; Brown S. J.; Kundu S.; Piscitelli F.; Rosen H.; Cravatt B. F. (2015) A Global Map of Lipid-Binding Proteins and Their Ligandability in Cells. Cell 161, 1668–1680. 10.1016/j.cell.2015.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulce J. J.; Cognetta A. B.; Niphakis M. J.; Tully S. E.; Cravatt B. F. (2013) Proteome-wide mapping of cholesterol-interacting proteins in mammalian cells. Nat. Methods 10, 259–264. 10.1038/nmeth.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza A., Sanghavi P., Rai A., Pathak D., and Mallik R. (2016) Isolation of Latex Bead Phagosomes from Dictyostelium for in vitro Functional Assays, Bio Protoc. 6, 10.21769/BioProtoc.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai P.; Kumar M.; Sharma G.; Barak P.; Das S.; Kamat S. S.; Mallik R. (2017) Kinesin-dependent mechanism for controlling triglyceride secretion from the liver. Proc. Natl. Acad. Sci. U. S. A. 114, 12958–12963. 10.1073/pnas.1713292114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.