

Abstract
Quercetin is a flavonoid compound that is widely present in food and drink. Quercetin-3-O-β-D-glucoside (Q3GA) is a major metabolite of quercetin. The aim of the present study was to investigate the effect of Q3GA on the pharmacokinetics of orally and intravenously administered cyclosporin A (CsA) in rats, and to assess the effect of Q3GA on drug-metabolizing enzymes (DMEs), drug transporters (DTs) and nuclear receptors (NRs). The pharmacokinetic parameters of CsA were measured following oral (10 mg/kg) and intravenous (2.5 mg/kg) administration of CsA in the presence or absence of Q3GA. The mRNA and protein expression levels of DMEs, DTs and NRs in the liver and small intestine were detected by quantitative polymerase chain reaction and western blot analysis. The results indicated that the intravenous administration of Q3GA (2.5, 5 or 10 mg/kg) for 7 consecutive days reduced the bioavailability of oral CsA. By contrast, the pharmacokinetics of the intravenous administration of CsA were not affected by Q3GA. However, the mRNA and protein expression levels of DMEs and DTs were inhibited by Q3GA. The activation of DMEs and DTs by NRs, and the interplay between DMEs and DTs, may explain these results. The present study identified a novel flavonoid-drug interaction, which may have implications for patients taking CsA and quercetin supplements or on a quercetin-containing diet.
Keywords: Q3GA, CsA, pharmacokinetics, DMEs, DTs, NRs
Introduction
Quercetin, the most widely-occurring type of flavonol, is one of the most common flavonoids in plant-derived food (1). It is widely present in vegetables (including onions and broccoli), fruits (including apples, grapes and berry crops), certain herbs, tea and wine (1). It is marketed as a dietary supplement (2). Onions are the primary source of dietary quercetin (up to 1.2 g/kg fresh weight) in numerous countries (3). Red wine and tea contain up to 45 mg/l flavonols (3). Other fruits and vegetables, including broccoli, asparagus, green pepper and tomato, are good sources of ubiquitous quercetin, particularly during the summer (1). Quercetin exhibits a variety of pharmacological effects, including anti-inflammatory, antioxidant, anticancer, antianaphylaxis and antiaging properties (1,4,5). Usually, quercetin exists in plants in the form of hydrophilic glycosides (6). Upon ingestion of vegetables and fruits, quercetin glycosides are metabolized, absorbed and rapidly hydrolyzed to generate quercetin aglycone (7). Absorbed quercetin is rapidly conjugated with glucuronic acid and/or sulfate during first-pass metabolism, and a portion of the metabolites are methylated; thus, the primary metabolites of quercetin in animal and human plasma are quercetin-3-O-β-D-glucoside (Q3GA), quercetin-3′-O-sulfate and isorhamnetin-3-O-glucuronide, whereas aglycone is generally undetectable (8). Q3GA exhibits anti-arteriosclerotic, anti-inflammatory, antiviral and antioxidant activities in vivo and in vitro (9–11). It also enhances the anti-inflammatory properties of M2a macrophages, and regulates the immune response to pro-inflammatory stimuli (12).
Quercetin modulates a number of drug-metabolizing enzymes (DMEs) and drug transporters (DTs), including cytochrome P450 (CYP) 1A1, CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4, uridine 5-diphospho-glucuronosyltransferase glucuronosyltransferases (UGTs), sulfotransferases (SULTs), glutathione-S-transferase (GSTs), P-glycoprotein (P-gp), multidrug resistance-associated protein 2 (MRP2), breast cancer resistance protein (BCRP) and organic anion transporting polypeptide 1B1 (OATP1B1) (13–17). Moreover, quercetin may activate the nuclear receptors (NRs) pregnane X receptor (PXR) and constitutive androstane receptor (CAR) (18). PXR and CAR control the expression of a largely overlapping array of target genes encoding phase I and II DMEs and DTs (19). However, only one study has been conducted regarding the effect of quercetin metabolites on DME, DT and NR expression (20). It demonstrated that quercetin metabolites downregulate the expression of CYP1A1/1A2 (20), and the regulation of the other DMEs and DTs by Q3GA remains to be investigated.
Cyclosporin (CsA) is an immunosuppressant with a narrow therapeutic window (21); it is widely used to prevent organ or tissue transplant rejection (21). CsA is a substrate of P-gp, CYP3A, MRP2, UGT1A and 2B (21–23). In addition, it is also a potent inhibitor of P-gp, CYP3A4, MRP2, BCRP, OATP1B1 and 2B (24,25). Thus, foods or drugs that are able to modulate DMEs and DTs, and/or their interplay, may alter the pharmacokinetics of CsA. The co-administration of CsA and drugs that modulate DMEs and DTs may pose serious risks. As quercetin is widely present in foods and is used as a dietary supplement, quercetin and CsA may frequently be administered simultaneously (26). The co-administration of quercetin and CsA reduces CsA-induced nephrotoxicity in male rats (27). The predominant form of quercetin in human plasma is its phase II metabolites, including Q3GA. Therefore, the effect of Q3GA on the pharmacokinetics of CsA and on DMEs, DTs and NRs require investigation.
Our previous study demonstrated that quercetin reduces the bioavailability of CsA following multiple concomitant doses in rats, and regulates the DMEs and DTs in the liver and small intestine (28). Further research is required to determine the effect of Q3GA on the pharmacokinetics of CsA and on DMEs, DTs and NRs in the liver and small intestine.
The present study aimed to assess whether multidose intravenous (i.v.) administration of Q3GA for 7 consecutive days affected the pharmacokinetics of orally and intravenously administered CsA in rats. To investigate the interaction mechanisms, reverse transcription-quantitative polymerase chain reaction (RT-qPCR) and western blot analyses were performed to evaluate the mRNA and protein expression levels of DMEs, DTs and NRs in the livers and small intestines of the rats.
Materials and methods
Materials
Q3GA was purchased from Chengdu Sino Standards Bio-Tech Co., Ltd. (Chengdu, China). CsA, CsD, verapamil (VER) and ketoconazole (KET) were purchased from Dalian Meilun Biotechnology Co., Ltd. (Dalian, China). The CsA preparation was Sandimmune® injection (50 mg/ml; Novartis International AG, Basel, Switzerland) including Cremophor® EL (polyethoxylated castor oil), and the infusion concentrate was diluted in normal saline prior to use.
Animals
A total of 54 male Sprague-Dawley rats (weight, 180–220 g; age, 8–9 weeks), were purchased from and reared in the Laboratory Animal Research Center of Tongji Medical College of Huazhong University of Science and Technology (Wuhan, China). The rats were allowed ad libitum access to food and tap water and were housed at 22±2°C with 50–60% relative humidity, with a 12-h light/dark cycle during experiments. The experiments were started following one week of acclimation. All experiments were performed with the approval of the Animal Research Ethics Committee of Tongji Medical College of Huazhong University of Science and Technology. All animal experiments were conducted in accordance with the standard ethical guidelines provided by the Laboratory Animal Research Center of Tongji Medical College of Huazhong University of Science and Technology (Wuhan, China).
Experiment I
The rats were randomly divided into eight groups with six rats in each group: (A) Control group (pretreated with phosphate buffer; i.v.; 5 ml/kg); (B) inhibition group [pretreated with KET in olive oil; intragastric (i.g.); 75 mg/kg]; (C) inhibition group (pretreated with VER in deionized water; i.g.; 4 mg/kg); (D) low-dose group (pretreated with Q3GA in phosphate buffer; i.v.; 2.5 mg/kg); (E) middle-dose group (pretreated with Q3GA in phosphate buffer; i.v.; 5 mg/kg); (F) high-dose group (pretreated with Q3GA in phosphate buffer; i.v.; 10 mg/kg); (G) control group (pretreated with phosphate buffer; i.v.; 5 ml/kg); and (H) middle-dose group (pretreated with Q3GA in phosphate buffer, i.v., 5 mg/kg). Groups A-F were used to investigate the effect of Q3GA on the pharmacokinetics of the oral administration of CsA, whereas Groups G and H were used to investigate the effect of Q3GA on the pharmacokinetics of the i.v. administration of CsA. All rats received the treatment once-a-day for 7 consecutive days. On the 7th day, the rats were treated with the vehicle (phosphate buffer) or drug, and at 30 min following this, CsA (10 mg/kg) was administered to the rats by oral gavage in the A-F groups; CsA (2.5 mg/kg) was administered to rats by i.v. injection in the G and H groups. Following administration, blood samples (0.2 ml) were collected from the rat jugular vein at 20, 40 min, 1, 1.5, 2, 4, 8, 12, 24, 36 and 48 h. The blood samples were stored at −80°C until analysis.
Experiment II
Rats were randomly divided into six groups, with three rats in each group. The groups were treated in the same manner as the A-F groups of experiment I. All rats received the treatment for 7 consecutive days. On the 7th day, 30 min following treatment, the CsA vehicle (5 ml/kg olive oil) was administered to the rats by oral gavage. Following the 7-day treatment all rats were fasted overnight prior to sacrifice. On day 8, 24 h following the final dose, the rats were sacrificed via overdose of anesthesia. The livers were rinsed with saline and blotted dry, then snap-frozen in liquid nitrogen and stored at −80°C until use. The intestines were scraped with a glass slide to collect the mucosal layers while fresh, then snap-frozen in liquid nitrogen and stored at −80°C until use.
Analytical experiments
The blood concentrations of CsA in the rats were measured with liquid chromatography-tandem mass spectrometry; CsD was used as the internal standard. CsA and CsD were analyzed on an API-4000 triple quadruple mass spectrometer (Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA) in the positive electrospray ionization mode and the multiple reactions monitoring scan type. The electrospray ionization source was operated with the ion spray voltage set at 5,500 V and ion source temperature set at 300°C. The optimized working parameters for mass detection were as follows: Declustering potential 65 V (CsA) and 62 V (CsD), collision energy 24 V (CsA) and 29 V (CsD), entrance potential 8 V (CsA) and 10 V (CsD), collision cell exit potential 15 V (CsA) and 14 V (CsD), curtain gas 30 psi, collision activated dissociation gas 8 psi, Gas1 4 psi and Gas2 5 psi. The precursor/product ion transitions were monitored at m/z 1220.0→1203.0 for CsA, and m/z 1234.1→1217.2 for CsD. The mobile phase was composed of 100% methanol:10 mM ammonium acetate containing 0.1% formic acid (90:10, v/v), and was pumped at a flow rate of 0.35 ml/min at 60°C. The calibration curve for CsA ranged from 5.00–4,000 ng/ml and the lower limit of quantitation for CsA was 5.00 ng/ml.
Pharmacokinetic analysis
The blood concentration data were analyzed by a noncompartmental method using Drug and Statistics (DAS) software version 3.2.8 (Shanghai BioGuider Medicinal Technology Co. Ltd., Shanghai, China). The peak blood concentration (Cmax) and the time to reach peak blood concentration (Tmax) of CsA were obtained directly from the concentration-time curve. The area under the blood concentration-time curve (AUC0-t) from time zero to the last measured concentration (Clast) was calculated using the linear trapezoidal rule (29). The AUC from zero to infinity (AUC0-∞) was the sum of the area of AUC0-t and AUCt-∞; AUCt-∞ was calculated by Clast/Kel, where Clast was the final blood concentration measured at time t and Kel was the slope of the end portion of the ln C-t curve. The terminal half-life (t1/2) was calculated as 0.693/Kel. The mean residence time (MRT) was calculated from area under the moment curve (AUMC)/AUC, where AUMC represents the area under the first moment vs. the time curve. The clearance (CL/F) following administration of single doses was calculated as Dose/AUC0-∞. The apparent volume of distribution (V/F) was based on the terminal elimination phase (CL/Kel).
RT-qPCR assay
RT-qPCR was used to quantify the mRNA expression levels of Cyp3a1, Cyp3a2, Ugt1a1, Sult1a1, Gst-µ1 (Gstm1) (liver only), solute carrier organic anion transporter family member (Slco)2b1 (small intestine only), Slco1b2 (liver only), multidrug resistance (Mdr)1, Bcrp, Mrp2, Pxr and Car in the small intestine and liver. Approximately 100 mg tissue was taken and ground in 1 ml of precooled TRIzol® reagent (Invitrogen; Thermo Fisher Scientific, Inc.). Total RNA was extracted from all tissues according to the manufacturer's protocol. RNA was reverse transcribed to cDNA using the PrimeScript RT reagent kit with gDNA Eraser (RR047A; Takara Biotechnology Co., Ltd., Dalian, China). qPCR was performed using SYBR® Premix Ex Taq™ (Takara Biotechnology Co., Ltd.) and a StepOne Real-Time PCR System (Thermo Fisher Scientific, Inc.). Specific primers for Cyp3a1, Cyp3a2, Ugt1a1, Sult1a1, Gst1a1, Slco2b1, Slco1b2, Mdr1, Bcrp, Mrp2, Pxr, Car and the housekeeping gene GAPDH were designed by Invitrogen (Thermo Fisher Scientific, Inc.) and are presented in Table I. The reaction procedure included a pre-denaturation at 95°C for 1 min followed by 40 cycles of 15 sec at 95°C, 20 sec at 58°C and 45 sec at 72°C, and an extension step at 72°C for 5 min. The relative mRNA levels were calculated using the 2−ΔΔCq method (30).
Table I.
Summary of the gene-specific PCR primer sequences, the length of production and the appropriate annealing temperature used in the experiments.
| Gene | Genbank accession no. | Sense primer, 5′→3′ | Antisense primer, 5′→3′ | Product size, bp | Tm, °C |
|---|---|---|---|---|---|
| GAPDH | NM_031144 | CGCTAACATCAAATGGGGTG | TTGCTGACAATCTTGAGGGAG | 201 | 58 |
| Cyp3a1 | NM_013105.2 | ACTGCATTGGCATGAGGTTTG | ATCCCGTGGCACAACCTTT | 170 | 58 |
| Cyp3a2 | NM_153312.2 | ATTCTAAGCATAAGCACCGAGTG | TGTGCTGCTGGTGGTTTCAT | 158 | 58 |
| Ugt1a1 | NM_012683.2 | ACTATTCTTGTCAAATGGCTACCC | GTTTTCCAAATCATCGGCAGT | 231 | 58 |
| Sult1a1 | NM_031834.1 | CTCTACCCGAGGAGACTGTGG | TAGCATAGTGGGCATCAAAGC | 204 | 58 |
| Gstm1 | NM_017014.1 | GACAGAGGAGGAGCGGATTC | CCACATAGGTGACCTTGTCCC | 203 | 58 |
| Slco2b1 | NM_080786.1 | TCGCTGTTGTGTCTGCTACTCAG | AACAGGGTTAAAGTCATCTGATTGG | 162 | 58 |
| Slco1b2 | NM_031650.3 | TTCGTGGTGATAAGAAGCCG | CAATTCAGGTTGGACGCTCTT | 218 | 58 |
| Mdr1 | NM_012623.2 | TCCTATGCTGCTTGTTTCCG | ATCCTGATGATGTGGGATGCT | 179 | 58 |
| Bcrp1 | NM_181381.2 | ATTGGTGCCCTTTACTTTGGTC | ACACTTGGCAAGAACCTCATAGG | 236 | 58 |
| Mrp2 | NM_012833.2 | TGTGGCAGTTGAGCGAATAAGT | AAGAGGCAGTTTGTGAGGGATG | 246 | 58 |
| Pxr | NM_052980.2 | CTTCGCCAAAGTCATATCCCA | TGTTTCCGTGTCGAACATCG | 121 | 58 |
| Car | NM_001270838.1 | GAGCCACGGGCTATCATTTC | CTCCCAGCAAACGGACAGAT | 160 | 58 |
Tm, melting temperature; Cyp, cytochrome P450; Gstm, glutathione-S-transferase-µ; Q3GA, quercetin-3-O-β-D-glucoside; Slco, solute carrier organic anion transporter family member; Sult, sulfotransferase; Car, constitutive androstane receptor; Mdr1, multidrug resistance protein 1; Mrp2; multidrug resistance protein 2; Pxr, pregnane X receptor; Bcrp, breast cancer resistance protein; Ugt, UDP glucuronosyltransferase.
Western blot analysis
Western blot analysis was performed to analyze the protein expression levels of CYP3A1, CYP3A2, UGT1A, SULT1A1, GSTM1 (liver only), OATP2B1 (small intestine only), OATP1B2 (liver only), P-gp, BCRP and MRP2, PXR and CAR in the small intestine and liver. Tissues were thoroughly homogenized in tissue protein extraction reagent with protease inhibitor (Aspen Biological Co., Ltd., Wuhan, China) added min prior to use and centrifuged at 12,000 rpm for 5 min at 4°C. The supernatant was collected, and the total protein solution was extracted. The sample protein concentration was determined using the BCA Protein Concentration Assay kit (Aspen Biological Co., Ltd.). Protein samples (40 µg) were loaded onto 8–20% SDS-PAGE gels and subjected to electrophoresis at 120 V for 90 min at room temperature. The proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Merck KGaA, Darmstadt, Germany) that had been activated with methanol. The PVDF membranes were blocked for 1 h at room temperature with 5% skim milk in Tris-buffered saline containing 0.1% Tween-20 (TBST) and incubated with diluted primary antibody at 4°C overnight. Membranes were washed three times with TBST for 5 min each and incubated for 30 min at room temperature with the secondary antibody, which was diluted with 5% skim milk (1:10,000).
The following primary antibodies were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA): Rabbit anti-CYP3A1 monoclonal antibody (1:500; cat. no. sc-53246), mouse anti-OATP1B2 polyclonal antibody (1:500; cat. no. sc-376904), rabbit anti-BCRP polyclonal antibody (1:500; cat. no. sc-25822) and rabbit anti-MRP2 polyclonal antibody (1:300; cat. no. sc-20766). Primary antibodies purchased from Abcam (Cambridge, UK) included: Rabbit anti-GAPDH monoclonal antibody (1:10,000; cat. no. ab37168), rabbit anti-P-gp monoclonal antibody (1:1,000; cat. no. sc-ab170903), rabbit anti-UGT1A1 polyclonal antibody (1:1,000; cat. no. ab194697), rabbit anti-GSTM1 polyclonal antibody (1:1,000; cat. no. ab77925) and rabbit anti-CAR polyclonal antibody (1:500; cat. no. ab62590). A mouse anti-CYP3A2 monoclonal antibody (1:1,000; cat. no. AB1276) was purchased from EMD Millipore (Billerica, MA, USA). A rabbit anti-SULT1A1 polyclonal antibody (1:1,000; cat. no. bs-6283R) was purchased from Bioss (Beijing, China). A mouse anti-PXR monoclonal antibody (1:1,000; cat. no. H00008856-B01P) was purchased from Novus Biologicals LLC (Littleton, CO, USA). Secondary antibodies, including horseradish peroxidase (HRP)-Goat anti Rabbit (1:10,000; cat. no. 074-1506) and HRP-Goat anti Mouse (1:10,000; cat. no. 074-1806), were purchased from KPL, Inc. (Gaithersburg, MD, USA). Freshly prepared enhanced chemiluminescence solution (EMD Millipore) was added dropwise to the surface of the membranes and exposed in the dark; the optical density values of the target bands were analyzed using the AlphaEaseFC software version 4.0 processing system (Protein Simple, San Jose, CA, USA). Results were normalized to GAPDH expression.
Statistical analysis
The pharmacokinetic parameters for CsA (AUC, Cmax, Tmax, CL/F, V/F and t1/2; n=6) and the mRNA and protein expression levels of DEMs, DTs and NRs (n=3) were compared between the control group and pretreatment groups with different doses of Q3GA, KET or VER. Data are expressed as the means ± standard deviation. All statistical analyses were performed via one-way analysis of variance followed by Tukey's or Dunnett's post hoc test, or unpaired Student's t-tests using GraphPad Prism 5.0 software (GraphPad Software, Inc., La Jolla, CA, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Effect of Q3GA on CsA pharmacokinetics
Mean blood concentration-time curves for CsA following oral administration at a dose of 10 mg/kg with different pretreatments are demonstrated in Fig. 1 and the corresponding pharmacokinetic parameters are presented in Table II. Compared with CsA alone (control), combined use of CsA and KET or VER increased CsA blood concentration, while CsA in combination with Q3GA decreased CsA blood concentration. Compared with the control group, in the CYP3A inhibitor (KET) pretreatment group, the Cmax of CsA was increased by 65.50% (P<0.05), and the P-gp inhibitor VER increased the Cmax of CsA by 36.30% (P>0.05), while the Cmax of CsA in the Q3GA-administration rats decreased by 33.12 (P>0.05), 33.76 (P>0.05) and 40.41% (P>0.05) in the low-, middle- and high-dose groups, respectively. In addition, in the KET pretreatment group, the AUC0-t and AUC0-∞ of CsA increased by 59.10 (P<0.05) and 54.82% respectively, while in the low-, middle- and high-dose Q3GA-administered rats, the AUC0-t of CsA decreased by 34.26, 36.32 and 47.79%, respectively, and the AUC0-∞ of CsA decreased by 34.66, 34.81 and 46.30%, respectively. The pharmacokinetic parameters of CsA demonstrated that pre-administration of Q3GA at daily dosages of 5 and 10 mg/kg for 7 consecutive days resulted in a 72.71 and 79.57% increase in the CL/F of CsA, and a 103.32 (P<0.01) and 93.19% (P<0.05) increase in the V/F of CsA, respectively. No significant differences were observed in the Cmax, AUC0-t, AUC0-∞, CL/F and V/F among the three Q3GA treatment groups. The differences in the Tmax, t1/2 and MRT0-t among all groups were not statistically significant (P>0.05).
Figure 1.
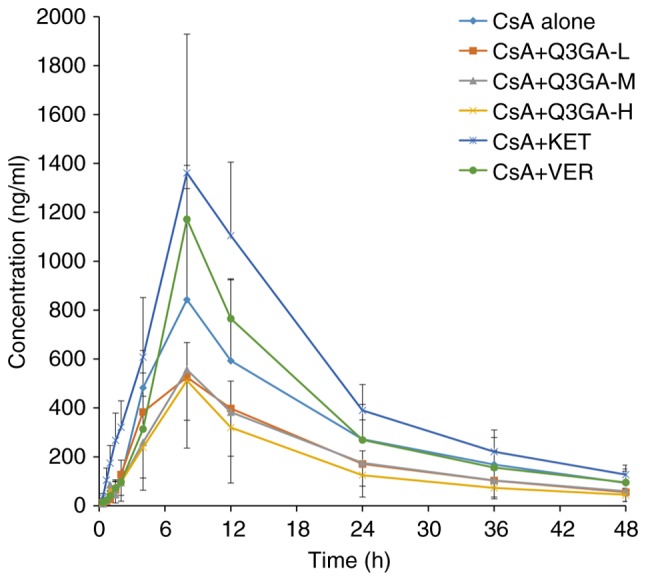
Mean blood concentration-time curves of CsA. Mean blood concentration-time curves of CsA following oral administration of CsA to the rats pretreated with phosphate buffer (control group), KET, VER and Q3GA for 7 consecutive days. Values are expressed as the mean ± standard deviation (n=6). Q3GA-L, low-dose Q3GA (2.5 mg/kg); Q3GA-M, middle-dose Q3GA (5 mg/kg); Q3GA-H, high-dose Q3GA (10 mg/kg). CsA, cyclosporin A; Q3GA, quercetin-3-O-β-D-glucoside; KET, ketoconazole; VER, verapamil.
Table II.
Principal pharmacokinetic parameters of CsA following the oral administration of CsA (10 mg/kg) to the rats with different pretreatments (mean ± standard deviation; n=6).
| Parameters | CsA alone | CsA+Q3GA-L | CsA+Q3GA-M | CsA+Q3GA-H | CsA+KET | CsA+VER |
|---|---|---|---|---|---|---|
| AUC0-t, ng·h/ml | 15621.14±8125.94 | 10269.92±2280.77 | 9947.3±6275.55 | 8155.59±2859.2 | 24853.52±6406.13a | 17679.05±3891.04 |
| AUC0-∞, ng·h/ml | 17358.61±9343.68 | 11342.33±2601.74 | 11315.61±7112.51 | 9321.12±3803.17 | 26873.83±7021.64 | 19540.26±5286.88 |
| AUMC0-t, ng·h2/ml | 267987.06±151617.53 | 169740.81±47732.93 | 170215.54±119823.64 | 130458.04±51166.97 | 402400.10±102138.59 | 288084.43±81765.85 |
| MRT0-t, h | 16.83±1.08 | 16.35±1.14 | 16.67±1.58 | 15.86±1.54 | 16.28±1.60 | 16.10±1.46 |
| VRT0-t, h2 | 132.76±6.97 | 130.62±16.21 | 132.07±13.04 | 121.76±19.29 | 119.65±16.78 | 115.83±15.51 |
| t1/2 | 13.00±1.55 | 13.47±4.07 | 15.73±2.56 | 15.38±6.01 | 11.91±2.49 | 13.73±4.65 |
| Tmax | 7.33±1.63 | 6.67±2.07 | 6.67±2.07 | 8.0±0.00 | 9.33±2.07 | 8.0±0.00 |
| VZ/F, l/kg | 12.87±4.96 | 17.56±5.13 | 26.17±12.39b | 24.87±5.40a | 6.63±1.68 | 10.27±2.69 |
| CLZ/F, l/h/kg | 0.7±0.29 | 0.93±0.24 | 1.21±0.75 | 1.26±0.56 | 0.39±0.09 | 0.54±0.15 |
| Cmax, ng/ml | 859.5±440.62 | 574.83±117.52 | 569.33±312.76 | 512.17±162.86 | 1422.5±501.00a | 1171.5±220.57 |
Rats were subjected to different pretreatments for 7 consecutive days. On the 7th day, the rats were treated with vehicle or drug; 30 min subsequently, CsA (10 mg/kg) was administered orally to the rats.
P<0.05
P<0.01 vs. CsA alone. AUC0-t, area under the blood concentration-time curve from time zero to the time of last measured concentration; AUC0-∞, area under the blood concentration-time curve from 0 h to infinity; AUMC, area under the moment curve; MRT, mean residence time; VRT, variance of the mean residence time; t1/2, terminal half-life; VZ/F, apparent volume of distribution; CLZ/F, apparent total clearance; Cmax, peak blood concentration; Tmax, time to reach Cmax, Q3GA, quercetin-3-O-β-D-glucoside; Q3GA-L, low-dose Q3GA (2.5 mg/kg); Q3GA-M, middle-dose Q3GA (5 mg/kg); Q3GA-H, high-dose Q3GA (10 mg/kg); CsA, cyclosporin A; KET, ketoconazole; VER, verapamil.
Mean blood concentration-time curves of CsA following the i.v. administration of 2.5 mg/kg CsA in the presence or absence of Q3GA are demonstrated in Fig. 2, and the corresponding pharmacokinetic parameters are presented in Table III. The blood concentration of CsA after intravenous injection of CsA was not affected by Q3GA. The differences in Cmax, AUC0-t, AUC0-∞, AUMC0-t, Tmax, t1/2, CL/F and V/F of CsA were not statistically significant between the control group and the Q3GA pretreatment groups (P>0.05).
Figure 2.
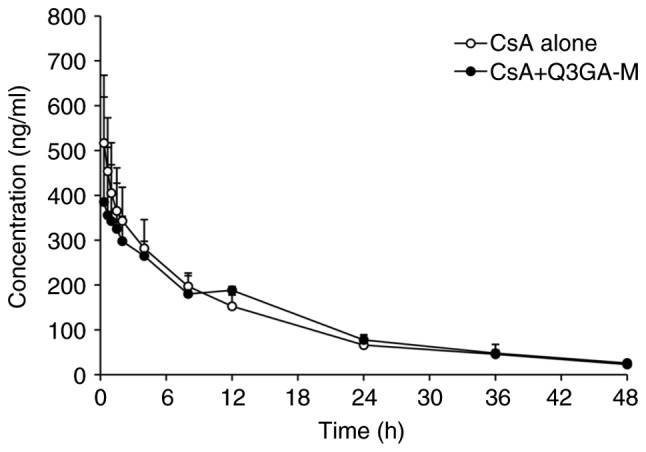
Mean blood concentration-time curves of CsA following intravenous injection of 2.5 mg/kg CsA in the presence or absence of Q3GA. Intravenous injection of 2.5 mg/kg CsA was performed on the rats in the presence or absence of Q3GA. Values are expressed as the mean ± standard deviation (n=6). Q3GA-M, middle dose Q3GA (5 mg/kg). CsA, cyclosporin A; Q3GA, quercetin-3-O-β-D-glucoside.
Table III.
Principal pharmacokinetic parameters of cyclosporin A following the intravenous administration of CsA (2.5 mg/kg) to the rats in the presence or absence of Q3GA (mean ± standard deviation; n=6).
| Pharmacokinetic parameters | CsA alone | CsA+Q3GA-M |
|---|---|---|
| AUC0-t, ng·ml−1·h | 5534.95±1053.54 | 5665.82±465.02 |
| AUC0-∞, ng·ml−1·h | 6021.50±1212.92 | 6182.46±524.10 |
| AUMC0-t, ng·h2/ml | 71130.28±20385.43 | 78787.27±7012.15 |
| MRT0-t, h | 12.69±1.74 | 13.96±1.39 |
| t1/2, h | 14.44±3.48 | 13.71±2.29 |
| Tmax, h | 0.39±0.14 | 1.305±1.39 |
| VZ/F, l/kg | 8.87±2.68 | 8.00±1.18 |
| CLZ/F, l/h/kg | 0.43±0.09 | 0.41±0.03 |
| Cmax, ng/ml | 519.67±145.09 | 396.67±185.67 |
Intravenous injection of 2.5 mg/kg CsA was performed on the rats in the presence or absence of Q3GA. AUC0-t, area under the blood concentration-time curve from time zero to the time of last measured concentration; AUC0-∞, area under the blood concentration-time curve from 0 h to infinity; AUMC, area under the moment curve; MRT, mean residence time; VRT, variance of the mean residence time; t1/2, terminal half-life; VZ/F, apparent volume of distribution; CLZ/F, apparent total clearance; Cmax, peak blood concentration; Tmax, time to reach Cmax, Q3GA-M, middle-dose Q3GA (5 mg/kg); CsA, cyclosporin A; Q3GA, quercetin-3-O-β-D-glucoside.
mRNA expression levels of DMEs, DTs and NRs in the small intestine and liver
RT-qPCR was used to quantify the mRNA expression levels of Cyp3a1, Cyp3a2, Ugt1a1, Sult1a1, Gstm1 (liver only), Slco2b1 (small intestine only), Slco1b2 (liver only), Mdr1, Bcrp, Mrp2, Pxr and Car in the small intestine and liver (Fig. 3).
Figure 3.


Effect of Q3GA on the intestinal and hepatic mRNA expression levels of Cyp3a1, Cyp3a2, Ugt1a1, Sult1a1, Gstm1, Slco2b1, Slco1b2, Mdr1, Bcrp, Mrp2, Pxr, and Car. In rats of the control and Q3GA-L, Q3GA-M, Q3GA-H, KET and VER pretreatment groups, the relative mRNA content was measured by reverse transcription-quantitative polymerase chain reaction. mRNA expression levels of (A) Cyp3a1, (B) Cyp3a2, (C) Ugt1a1, (D) Sult1a1, (E) Gstm1, (F) Slco2b1/Slco1b2, (G) Mdr1, (H) Bcrp, (I) Mrp2, (J) Pxr, and (K) Car were measured in the small intestine and liver of the rats. Values are expressed as the mean ± standard deviation (n=3). aP<0.05, bP<0.01, cP<0.001 vs. the control. dP<0.05, eP<0.01, fP<0.001 vs. the inhibitor group (KET/VER). Q3GA-L, low-dose Q3GA (2.5 mg/kg); Q3GA-M, middle-dose Q3GA (5 mg/kg); Q3GA-H, high-dose Q3GA (10 mg/kg). CsA, cyclosporin A; Q3GA, quercetin-3-O-β-D-glucoside; KET, ketoconazole; VER: Verapamil; Cyp, cytochrome P450; Gstm, glutathione-S-transferase-µ; Q3GA, quercetin-3-O-β-D-glucoside; Slco, solute carrier organic anion transporter family member; Sult, sulfotransferase; Car, constitutive androstane receptor; Mdr1, multidrug resistance protein 1; Mrp2; multidrug resistance protein 2; Pxr, pregnane X receptor; Bcrp, breast cancer resistance protein.
mRNA expression levels of Cyp3a1 and Cyp3a2
In more detail, as presented in Fig. 3A and B, compared with the control group, in the KET, low- and high-dose Q3GA groups, the mRNA expression levels of Cyp3a1 and Cyp3a2 in the liver and intestine were significantly decreased, whereas the middle-dose group demonstrated a significant decrease in mRNA expression in the liver only. The intestinal mRNA expression levels of Cyp3a1 decreased by 78.27 (P<0.001), 63.78 (P<0.001), 26.08 and 43.57% (P<0.05), in the KET, low-, middle- and high-dose Q3GA groups, respectively. In addition, the intestinal mRNA expression levels of Cyp3a2 decreased by 70.09 (P<0.001), 49.70 (P<0.001), 17.93 and 39.36% (P<0.001), in the KET, low-, middle- and high-dose Q3GA groups, respectively. Similarly, the liver mRNA expression levels of Cyp3a1 decreased by 78.64 (P<0.001), 65.14 (P<0.001), 54.94 (P<0.001) and 33.73% (P<0.05), in the KET, low-, middle- and high-dose Q3GA groups, respectively. Additionally, the liver mRNA expression levels of Cyp3a2 decreased by 87.39 (P<0.001), 64.68 (P<0.001), 26.18 (P<0.05) and 34.64% (P<0.01), in the KET, low-, middle- and high-dose Q3GA groups, respectively. Notably, the effect of Q3GA was not dose-dependent, and the low-dose group exhibited the strongest inhibitory effect on Cyp3a1 and Cyp3a2, in the small intestine and liver (Fig. 3A and B).
mRNA expression levels of Ugt1a1, Sult1a1 and Gstm1
Compared with the control group, in the low-, middle- and high-dose Q3GA groups, Ugt1a1 was decreased by 70.14 (P<0.001), 24.08 (P<0.05) and 37.83% (P<0.001), respectively, in the small intestine, and by 76.13 (P<0.001), 48.78 (P<0.01) and 41.04% (P<0.05), respectively, in the liver (Fig. 3C). Sult1a1 was significantly decreased by 64.54 (P<0.001), 33.80 (P<0.05) and 52.02% (P<0.01), respectively, in the small intestine, and by 58.06 (P<0.001), 31.94 (P<0.05) and 18.39%, respectively, in the liver (Fig. 3D). Gstm1 in the liver was increased by 248.40% (P<0.001) in the low-dose Q3GA group and increased by 85.41 and 75.09% in the middle- and high-dose Q3GA groups, respectively (Fig. 3E). Notably, the effect of Q3GA was not dose-dependent, and the low-dose Q3GA group exhibited the strongest alteration in Ugt1a1, Sult1a1 and Gstm1. These results indicated that the mRNA expression levels of Ugt1a1 and Sult1a1 were inhibited by Q3GA at a similar level in the small intestine and liver, and that the mRNA expression levels of Gstm1 were induced by Q3GA in the liver.
mRNA expression levels of Slco2b1/Slco1b2
As presented in Fig. 3F, compared with the control group, in the low-, middle- and high-dose Q3GA groups, the mRNA expression levels of Slco2b1 in the small intestine were decreased by 62.19 (P<0.001), 20.49 and 39.57% (P<0.01), respectively; whereas, the mRNA expression levels of Slco1b2 in the liver were decreased by 68.06 (P<0.001), 38.39 (P<0.01) and 53.54% (P<0.001), respectively. In addition, the effect of Q3GA was not dose-dependent, and the low-dose group exhibited the strongest inhibitory effect on Slco2b1 and Slco1b2.
mRNA expression levels of Mdr1, Bcrp and Mrp2
Compared with the control group, in the VER, low-, middle- and high- dose Q3GA groups, Mdr1 was decreased by 65.32 (P<0.001), 47.47 (P<0.001), 17.51 (P<0.001) and 33.33% (P<0.001), respectively in the small intestine, and by 84.33 (P<0.001), 67.60 (P<0.001), 45.09 (P<0.05) and 47.32% (P<0.01), respectively in the liver (Fig. 3G). In the low-, middle- and high-dose Q3GA groups, Bcrp was decreased by 63.83 (P<0.001), 27.52 (P<0.05) and 43.68% (P<0.01), respectively in the small intestine. However, in the liver, only the low-dose Q3GA group demonstrated a significant decrease in the Bcrp mRNA expression levels (56.15%; P<0.01); the middle- and high-dose Q3GA groups decreased by 29.23 and 29.23%, respectively (Fig. 3H). In the low-, middle- and high-dose Q3GA groups, Mrp2 was significantly decreased by 66.03 (P<0.001), 34.53 (P<0.01) and 28.66% (P<0.01), respectively in the small intestine, and by 68.86 (P<0.001), 32.70 (P<0.01) and 20.44% (P<0.05), respectively in the liver (Fig. 3I). In addition, the effect of Q3GA was not dose-dependent, and the low-dose Q3GA group exhibited the strongest inhibitory effect on Mdr1, Bcrp and Mrp2. These results revealed that their mRNA expression levels were inhibited by Q3GA to a similar extent in the small intestine and liver.
mRNA expression levels of Pxr and Car
Compared with the control group, in the low-, middle- and high-dose Q3GA groups, Pxr was increased by 415.18 (P<0.001), 130.03 (P<0.001) and 200.21% (P<0.001), respectively in the small intestine, however, only the low-dose Q3GA group exhibited a significant increase in the Pxr mRNA expression level (281.32%; P<0.001); the middle- and high-dose Q3GA groups increased by 84.98 and 66.30%, but these were not statistically significant (Fig. 3J). Similarly, in the low-, middle- and high-dose Q3GA groups, Car was significantly increased by 265.44 (P<0.001), 99.57 (P<0.05) and 117.54% (P<0.05), respectively in the small intestine, and by 224.76 (P<0.001), 98.73 (P<0.001) and 74.60% (P<0.01), respectively in the liver (Fig. 3K). Notably, the effect of Q3GA was not dose-dependent, and the low-dose Q3GA group exhibited the strongest induction effect on Pxr and Car. These results demonstrated that the mRNA expression levels of Pxr and Car were induced by Q3GA to a similar extent in both the small intestine and liver.
In summary, the results indicated that the mRNA expression levels of Cyp3a1, Cyp3a2, Ugt1a1, Sult1a1, Slco2b1 (small intestine only), Slco1b2 (liver only), Mdr1, Bcrp and Mrp2 were inhibited by Q3GA to a similar degree in the small intestine and liver. By contrast, the mRNA expression levels of Gstm1 (liver only), Pxr and Car were significantly induced by Q3GA to a similar degree in the small intestine and liver. In addition, the effect of Q3GA was not dose-dependent, and the low-dose group exhibited the strongest effect on DMEs, DTs and NRs.
Protein expression levels of DMEs, DTs and NRs in the small intestine and liver
Western blot analysis was performed to analyze the protein expression levels of CYP3A1, CYP3A2, UGT1A, SULT1A1, GSTM1 (liver only), OATP2B1 (small intestine only), OATP1B2 (liver only), P-gp, BCRP, MRP2, PXR and CAR in the small intestine and liver (Fig. 4). Western blots of the small intestine and liver tissue of rats are exhibited in Fig. 4A and B, respectively, and the quantification is presented in Fig. 5.
Figure 4.

Effect of Q3GA on the intestinal and hepatic protein expression levels of CYP3A1, CYP3A2, UGT1A1, SULT1A1, GSTM1, OATP2B1, OATP1B2, P-gp, BCRP, MRP2, PXR, and CAR. In rats of the control and Q3GA-L, Q3GA-M, Q3GA-H, KET, VER pretreatment groups, the relative protein contents were assessed by western blotting. Representative western blotting results in (A) liver and (B) in small intestine. Western blotting results of (Aa) UGT1A1, SULT1A1, GSTM1, OATP1B2, BCRP, MRP2, PXR and CAR, (Ab) CYP3A1/2 and (Ac) P-gp in liver. Western blotting results of (Ba) UGT1A1, SULT1A1, GSTM1, OATP2B1, BCRP, MRP2, PXR and CAR, (Bb) CYP3A1/2 and (Bc) P-gp in small intestine. Q3GA-L, low-dose Q3GA (2.5 mg/kg); Q3GA-M, middle-dose Q3GA (5 mg/kg); Q3GA-H, high-dose Q3GA (10 mg/kg). BCRP, breast cancer resistance protein; CAR, constitutive androstane receptor; CsA, cyclosporin A; CYP, cytochrome P450; GSTM, glutathione-S-transferase-µ; KET, ketoconazole; MRP, multidrug resistance-associated protein; OATP1B1, organic anion transporting polypeptide 1B1; P-gp, P-glycoprotein; PXR, pregnane X receptor; Q3GA, quercetin-3-O-β-D-glucoside; SULT, sulfotransferase; UGT, UDP glucuronosyltransferase; VER, verapamil.
Figure 5.


Quantification of the effect of Q3GA on the intestinal and hepatic protein levels, assessed by western blotting. Quantification of western blotting results revealed protein expression levels of (A) CYP3A1, (B) CYP3A2, (C) UGT1A1, (D) SULT1A1, (E) GSTM1, (F) OATP2B1/OATP1B2, (G) P-gp, (H) BCRP, (I) MRP2, (J) PXR, and (K) CAR measured in the small intestine and liver of rats. Values are expressed as the mean ± standard deviation (n=3). aP<0.05, bP<0.01, cP<0.001 vs. control; dP<0.05, eP<0.01, fP<0.001 vs. inhibitor group (KET/VER). Q3GA-L, low-dose Q3GA (2.5 mg/kg); Q3GA-M, middle-dose Q3GA (5 mg/kg); Q3GA-H, high-dose Q3GA (10 mg/kg). BCRP, breast cancer resistance protein; CAR, constitutive androstane receptor; CsA, cyclosporin A; CYP, cytochrome P450; GSTM, glutathione-S-transferase-µ; KET, ketoconazole; MRP2 multidrug resistance-associated protein 2; OATP, organic anion transporting polypeptide; P-gp, P-glycoprotein; PXR, pregnane X receptor; Q3GA, quercetin-3-O-β-D-glucoside; SULT, sulfotransferase; UGT, UDP glucuronosyltransferase; VER, verapamil.
Protein expression levels of CYP3A1 and CYP3A2
Similar to the mRNA results, in Fig. 5A and B, compared with the control group, the protein expression levels of CYP3A1 and CYP3A2 in the liver and intestine were significantly decreased in the KET, low-, middle- and high- dose Q3GA groups. The intestinal protein expression levels of Cyp3a1 decreased by 90.79 (P<0.001), 75.25 (P<0.001), 39.22 (P<0.01) and 62.95% (P<0.001), in the KET, low-, middle- and high-dose Q3GA groups, respectively. Additionally, the intestinal protein expression levels of Cyp3a2 decreased by 80.93 (P<0.001), 55.28 (P<0.001), 21.68 and 38.67% (P<0.01), in the KET, low-, middle- and high-dose Q3GA groups, respectively. Similarly, the liver protein expression levels of Cyp3a1 decreased by 77.50 (P<0.001), 57.66 (P<0.01), 43.95 (P<0.05) and 52.99% (P<0.01), in the KET, low-, middle- and high-dose Q3GA groups, respectively. Additionally, the liver protein expression levels of Cyp3a2 decreased by 60.13 (P<0.001), 54.18 (P<0.001), 33.07 (P<0.001) and 47.16% (P<0.001), in the KET, low-, middle- and high-dose Q3GA groups, respectively. Notably, the effect of Q3GA was not dose-dependent, and the low-dose group exhibited the strongest inhibitory effect on CYP3A1 and CYP3A2.
Protein expression levels of UGT1A1, SULT1A1 and GSTM1
Compared with the control group, in the low-, middle- and high-dose Q3GA groups, UGT1A1 was decreased by 76.81 (P<0.001), 36.26 (P<0.001) and 46.57% (P<0.001), respectively in the small intestine, and by 71.06 (P<0.001), 38.05 (P<0.001) and 49.63% (P<0.001), respectively in the liver (Fig. 5C). In the low-, middle- and high-dose Q3GA groups, SULT1A1 was significantly decreased by 75.44 (P<0.001), 36.33 (P<0.05) and 57.47% (P<0.001), respectively in the small intestine. In the liver, only the low-dose Q3GA group exhibited a significant decrease in the SULT1A1 protein expression levels (75.89%; P<0.01), whereas the middle- and high-dose Q3GA groups decreased by 32.43 and 34.66%, respectively, but these were not statistically significant (Fig. 5D). GSTM1 was increased by 417.29 (P<0.001) and 229.75% (P<0.05) in the low- and high-dose Q3GA groups and increased by 159.37% in the middle-dose Q3GA group, in the liver (Fig. 5E). Notably, the effect of Q3GA was not dose-dependent, and the low-dose Q3GA group exhibited the strongest effect on UGT1A1, SULT1A1 and GSTM1. These results indicated that the protein expression levels of UGT1A1 and SULT1A1 were inhibited by Q3GA to a similar level in the small intestine and liver, and the protein expression levels of GSTM1 were induced by Q3GA in the liver, similar to the results for mRNA expression.
Protein expression levels of OATP2B1/OATP1B2
As presented in Fig. 5F, compared with the control group, in the low-, middle- and high-dose Q3GA groups, the protein expression levels of OATP2B1 in the small intestine were decreased by 86.44 (P<0.001), 42.21 (P<0.01) and 51.44% (P<0.01), respectively. Additionally, the protein expression levels of OATP1B2 in the liver were decreased by 67.47 (P<0.01), 26.52 and 33.73%, respectively. In addition, the effect of Q3GA was not dose-dependent, and the low-dose group exhibited the strongest inhibitory effect on OATP2B1 and OATP1B2.
Protein expression levels of P-gp, BCRP and MRP2
Compared with the control group, in the VER, low-, middle- and high-dose Q3GA groups, P-gp was decreased by 85.02 (P<0.001), 65.31 (P<0.001), 30.22 (P<0.01) and 41.70% (P<0.001), respectively in the small intestine, and by 84.54 (P<0.001), 67.93 (P<0.001), 39.92 (P<0.01) and 47.26% (P<0.001), respectively in the liver (Fig. 5G). In the low-, middle- and high-dose Q3GA groups, BCRP was decreased by 70.61 (P<0.001), 32.93 (P<0.05) and 36.44% (P<0.01), respectively, in the small intestine and by 69.78 (P<0.001), 27.73 (P<0.05) and 30.86% (P<0.05), respectively, in the liver (Fig. 5H). In the low-, middle- and high-dose Q3GA groups, MRP2 was decreased by 75.95 (P<0.001), 42.60 (P<0.001) and 50.84% (P<0.001), respectively in the small intestine. In the liver, only the low-dose Q3GA group had a significant decrease in the MRP2 protein expression levels (70.70%; P<0.001), whereas the middle- and high-dose Q3GA groups decreased by 26.09 and 30.71%, and these were not statistically significant (Fig. 5I). In addition, the effect of Q3GA was not dose-dependent, and the low-dose Q3GA group exhibited the strongest inhibitory effect on P-gp, BCRP and MRP2. These results were similar to those obtained for mRNA; the protein expression levels of P-gp, BCRP and MRP2 were inhibited by Q3GA to a similar extent in the small intestine and liver.
Protein expression levels of PXR and CAR
Compared with the control group, in the low-, middle- and high-dose Q3GA groups, PXR was increased by 258.85 (P<0.001), 81.66 (P<0.05) and 147.68% (P<0.01) in the small intestine. In the liver, only the low-dose Q3GA group exhibited a significant increase in the PXR protein expression levels (182.93%; P<0.01), whereas the middle- and high-dose Q3GA groups increased by 69.71 and 79.76%, respectively, but these were not statistically significant (Fig. 5J). Similarly, in the low-, middle- and high-dose Q3GA groups, CAR was increased by 235.46 (P<0.001), 78.59 (P<0.01) and 91.07% (P<0.01) in the small intestine; only the low-dose Q3GA group exhibited a significant increase of the CAR protein expression levels (456.37%; P<0.01) in the liver, compared with 208.32 and 211.76% in the middle- and high-dose Q3GA groups (Fig. 5K). Notably, the effect of Q3GA was not dose-dependent and the low-dose Q3GA group exhibited the strongest induction effect on PXR and CAR. These results demonstrated that the protein expression levels of PXR and CAR were induced by Q3GA to a similar degree in the small intestine and liver.
In summary, the results indicated that the protein expression levels of CYP3A1, CYP3A2, UGT1A1, SULT1A1, OATP2B1 (small intestine only), OATP1B2 (liver only), P-gp, BCRP and MRP2 were inhibited by Q3GA treatment to a similar degree in the small intestine and liver. By contrast, the protein expression of GSTM1 (liver only), PXR and CAR was significantly promoted by Q3GA to a similar degree in the small intestine and liver. In addition, the effect of Q3GA was not dose-dependent, and the low-dose Q3GA group exhibited the strongest effect on DMEs, DTs and NRs.
Discussion
In recent years, research on herbal medicine-drug interactions has received considerable attention (31–33). These interactions may increase the risk to patients, particularly for drugs with a narrow therapeutic index, including CsA (34). A number of studies on the interaction between herbs, their pharmacological components and CsA content have been conducted. For example, schisandra extract, polyphyllin I, ginkgolic acid, mulberry, quercetin and rutin influence the pharmacokinetics of CsA (30–33,35,36). Quercetin exhibits various effects on the pharmacokinetics of CsA, and it has been demonstrated to either increase or decrease the bioavailability of CsA (26,33,37). Thus, the interaction mechanisms between quercetin and CsA remain to be completely elucidated. The predominant form of quercetin in human plasma is its phase II metabolites. Therefore, the interactions and mechanisms of quercetin and its metabolites with drugs require further investigation. The present study reported on the effects of the quercetin metabolite Q3GA on the pharmacokinetics of CsA in rats, and the regulation of DMEs, DTs and NRs.
The results of the present study demonstrated that the i.v. administration of Q3GA for 7 consecutive days decreased the Cmax of CsA (10 mg/kg), which suggests that Q3GA decreased the oral bioavailability of CsA. In addition, CsA was orally administered to rats following treatment with Q3GA for 1 week and the AUC values for CsA decreased. Pre-administration with Q3GA for 7 consecutive days resulted in an increase in the CL/F and in the V/F of CsA. No significant differences in the Cmax, AUC0-t, AUC0-∞, CL/F and V/F among the three Q3GA treatment groups were observed. The differences in the Tmax, t1/2 and MRT0-t among all groups were also not statistically significant. Therefore, the Q3GA co-administration exerted a marked effect on the pharmacokinetics of CsA. These results were consistent with our previous report that the oral administration of quercetin for 7 consecutive days reduced the bioavailability of CsA in rats (28). Yu et al (33) and Hsiu et al (37) reported the same results. By contrast, Choi et al (26) reported that quercetin increases the bioavailability of CsA in healthy volunteers. They demonstrated that the co-administration of quercetin (5 mg/kg) and CsA increases the AUC of CsA by 18%. Furthermore, the administration of CsA 30 min and 3 days following quercetin administration increased the AUC of CsA by 36 and 47%, respectively. These contrasting results may be due to the differences in the subjects, administration method and doses. In the present study, the effect of Q3GA was not dose-dependent, and the low-dose group exhibited the strongest effect on DMEs, DTs and NRs. This suggested that the dose reached a saturation point, as the dose increment did not improve the response. In addition, no significant difference was observed in the corresponding pharmacokinetic parameters of CsA following the i.v. administration of 2.5 mg/kg CsA in the presence or absence of Q3GA. This result indicated that Q3GA may decrease the oral bioavailability of CsA by influencing absorption and first-pass metabolism in the intestine.
Furthermore, the results of the present study demonstrated that the mRNA and protein expression levels of CYP3A1, CYP3A2, UGT1A1, SULT1A1, OATP2B1 (small intestine only), OATP1B2 (liver only), P-gp, BCRP and MRP2 were significantly inhibited by Q3GA to a similar degree in the small intestine and liver. By contrast, the mRNA and protein expression levels of GSTM1 (liver only), PXR and CAR were significantly increased by Q3GA to a similar degree in the small intestine and liver. Q3GA exerted a similar effect on the mRNA and protein expression levels. These results were consistent with our previous report about the regulation of DMEs and DTs by quercetin (28).
CsA is a substrate of CYP3A, P-gp, MRP2, and UGT1A and 2B (21–23). These results indicated that Q3GA inhibited CYP3A4, P-gp, MRP2 and UGT1A1. Such inhibition may potentially improve the oral bioavailability of CsA. However, the in vivo results demonstrated that Q3GA significantly decreased the oral bioavailability of CsA. This finding indicated that the effect of Q3GA on CsA may not be attributed to its regulation of DMEs or DTs alone. Flavonoids are able to regulate DMEs and DTs, possibly requiring the involvement of various NRs (38). A previous study identified that the effect of flavonoids on the regulation of CYP expression is mediated by PXR (39). Tangeretin, a flavonoid compound, activates PXR and thus modulates P-gp (40). However, the expression of DMEs and DTs, including CYP2B6, CYP3A4, UGT1A1, MDR1 and MRP2, are induced by the terpenoids of EGb 761 through the selective activation of PXR, CAR and aryl hydrocarbon receptor, and not by the flavonoids of EGb 761 (18). The association with NRs, the regulation of DMEs and DTs by quercetin and its metabolites, and the molecular mechanisms of such regulation remain to be studied.
CYP3A4, CYP2C9, CYP2C19, CYP2B6, UGT1A1, GST, SULT2A1, MDR1, MRP2, OATPA and other DMEs and DTs are activated by PXR and/or CAR (41). The present study demonstrated that Q3GA significantly induced PXR and CAR protein expression; however, this finding was not consistent with the recent report of Lau et al (42), which indicated that quercetin activates mouse and human PXR, and not rat PXR. Q3GA strongly activates PXR and CAR, and PXR and CAR induce DMEs and DTs. However, Q3GA inhibits DMEs and DTs. These contradicting effects may lead to a decrease in CsA bioavailability through Q3GA treatment. Comprehensively investigating the mechanisms of quercetin and its metabolites will contribute to the understanding of the role of quercetin in herbal medicine-drug interactions.
The complex enzyme-transporter interplay may be used to explain the interaction of Q3GA and CsA. Drug efflux by intestinal P-gp reduces the bioavailability of a number of CYP3A4 substrates (43). Benet et al (44) revealed the dynamic interplay between CYP3A and P-gp in the intestine and liver where P-gp efflux transport can enhance or block the metabolism of CYP3A4. When P-gp was inhibited, it appeared that the degree of metabolism of the double CYP3A and P-gp substrates was enhanced (44). Intestinal P-gp increases CYP3A metabolism by pumping the substrate back into the intestine, thereby avoiding the saturation of the metabolic enzyme (45). Based on the extension of this idea of interplay between CYP3A and P-gp, interactions between UGTs/SULTs and BCRP/MRP have been identified in the study of the metabolism and clearance of flavonoids in animal and cellular models (38,46). The interplay between phase II enzymes and efflux transporters results in the widespread metabolism and low bioavailability of flavonoids (47). The modeling of the complex interplay between DMEs and DTs is at an early stage, and the basic mechanism of this interaction requires full elucidation.
The present experiments demonstrated the effect of Q3GA on DMEs, DTs and NRs in vivo, although not in vitro. In vitro studies may allow for the identification of the mechanisms involved in the DME-DT interplay, and in the regulation of DMEs, DTs and NRs by Q3GA. Certain in vitro studies have demonstrated the effect of Q3GA on CYP1A1/2, MRP1, MRP2 and SULT (20,48,49). Thus, the effect of Q3GA on other DMEs, DTs and NRs requires further in vitro study.
In conclusion, the present study investigated the effects of the quercetin metabolite Q3GA on the pharmacokinetics of CsA in rats. Q3GA reduced the bioavailability of CsA, which was in contrast to the inhibitory effect of Q3GA on the mRNA and protein expression levels of DMEs and DTs in the liver and intestine. The activation of DMEs and DTs by PXR and CAR in the liver and intestine, and the interplay between DMEs and DTs may explain this result, although this requires further investigation. The present study identified a novel flavonoid-drug interaction and may have implications for patients who simultaneously receive CsA and quercetin/Q3GA-containing dietary/herbal supplements.
Acknowledgements
Not applicable.
Glossary
Abbreviations
- AUC
area under the curve
- BCRP
breast cancer resistance protein
- CAR
constitutive androstane receptor
- CL/F
clearance
- Cmax
the peak blood concentration
- CsA
cyclosporin A
- CsD
cyclosporin D
- CYP
cytochrome P450
- DMEs
drug-metabolizing enzymes
- DTs
drug transporters
- GSTs
glutathione-S-transferase
- KET
ketoconazole
- MRP2
multidrug resistance-associated protein 2
- MDR1
multidrug resistance protein 1
- MRT
the mean residence time
- NRs
nuclear receptors
- OATP1B1
organic anion transporting polypeptide 1B1
- P-gp
P-glycoprotein
- PXR
pregnane X receptor
- Q3GA
quercetin-3-O-β-D-glucoside
- RT-qPCR
reverse transcription-quantitative polymerase chain reaction
- SULTs
sulfotransferases
- t1/2
the terminal half-life
- Tmax
time to reach peak blood concentration
- UGTs
UDP glucuronosyltransferases
- VER
verapamil
- V/F
the apparent volume of distribution
Funding
The present study was supported by National Natural Science Foundation of China (grant nos. 81503161 and 81273591) and the Fundamental Research Funds for the Central Universities (China; grant nos. 2014YGYL003 and 2016YXZD050). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors' contributions
TY, YL, YZ, JW and SS designed all the experiments. TY, YL and XH performed animal experiments and RT-qPCR assays. RZ and JW performed western blotting. Statistical analysis was performed by CY and JZ. TY, YL, JW and SS wrote the manuscript. All authors reviewed the manuscript.
Ethics approval and consent to participate
All rat experiments were approved by the Animal Research Ethics Committee of Tongji Medical College of Huazhong University of Science and Technology (Wuhan, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Mlcek J, Jurikova T, Skrovankova S, Sochor J. Quercetin and its anti-allergic immune response. Molecules. 2016;21 doi: 10.3390/molecules21050623. pii: E623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miclaus MO, Filip X, Filip C, Martin FA, Grosu IG. Highly sensitive solid forms discrimination on the whole tablet of the active ingredients in quercetin dietary supplements by NMR crystallography approaches. J Pharm Biomed Anal. 2016;124:274–280. doi: 10.1016/j.jpba.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 3.Manach C, Scalbert A, Morand C, Rémésy C, Jiménez L. Polyphenols: Food sources and bioavailability. Am J Clin Nutr. 2004;79:727–747. doi: 10.1093/ajcn/79.5.727. [DOI] [PubMed] [Google Scholar]
- 4.Cai X, Fang Z, Dou J, Yu A, Zhai G. Bioavailability of quercetin: Problems and promises. Curr Med Chem. 2013;20:2572–2582. doi: 10.2174/09298673113209990120. [DOI] [PubMed] [Google Scholar]
- 5.Russo M, Spagnuolo C, Tedesco I, Bilotto S, Russo GL. The flavonoid quercetin in disease prevention and therapy: Facts and fancies. Biochem Pharmacol. 2012;83:6–15. doi: 10.1016/j.bcp.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 6.Yang F, Song L, Wang H, Wang J, Xu Z, Xing N. Quercetin in prostate cancer: Chemotherapeutic and chemopreventive effects, mechanisms and clinical application potential (Review) Oncol Rep. 2015;33:2659–2668. doi: 10.3892/or.2015.3886. [DOI] [PubMed] [Google Scholar]
- 7.Kawabata K, Mukai R, Ishisaka A. Quercetin and related polyphenols: New insights and implications for their bioactivity and bioavailability. Food Funct. 2015;6:1399–1417. doi: 10.1039/C4FO01178C. [DOI] [PubMed] [Google Scholar]
- 8.Galindo P, Rodriguez-Gómez I, González-Manzano S, Dueñas M, Jiménez R, Menéndez C, Vargas F, Tamargo J, Santos-Buelga C, Pérez-Vizcaíno F, Duarte J. Glucuronidated quercetin lowers blood pressure in spontaneously hypertensive rats via deconjugation. PLoS One. 2012;7:e32673. doi: 10.1371/journal.pone.0032673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ishizawa K, Yoshizumi M, Kawai Y, Terao J, Kihira Y, Ikeda Y, Tomita S, Minakuchi K, Tsuchiya K, Tamaki T. Pharmacology in health food: Metabolism of quercetin in vivo and its protective effect against arteriosclerosis. J Pharmacol Sci. 2011;115:466–470. doi: 10.1254/jphs.10R38FM. [DOI] [PubMed] [Google Scholar]
- 10.Fan D, Zhou X, Zhao C, Chen H, Zhao Y, Gong X. Anti-inflammatory, antiviral and quantitative study of quercetin-3-O-β-D-glucuronide in polygonum perfoliatum L. Fitoterapia. 2011;82:805–810. doi: 10.1016/j.fitote.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 11.Yokoyama A, Sakakibara H, Crozier A, Kawai Y, Matsui A, Terao J, Kumazawa S, Shimoi K. Quercetin metabolites and protection against peroxynitrite-induced oxidative hepatic injury in rats. Free Radic Res. 2009;43:913–921. doi: 10.1080/10715760903137010. [DOI] [PubMed] [Google Scholar]
- 12.Derlindati E, Dall'Asta M, Ardigò D, Brighenti F, Zavaroni I, Crozier A, Del Rio D. Quercetin-3-O-glucuronide affects the gene expression profile of M1 and M2a human macrophages exhibiting anti-inflammatory effects. Food Funct. 2012;3:1144–1152. doi: 10.1039/c2fo30127j. [DOI] [PubMed] [Google Scholar]
- 13.Arinç E, Yilmaz D, Bozcaarmutlu A. Mechanism of inhibition of CYP1A1 and glutathione S-transferase activities in fish liver by quercetin, resveratrol, naringenin, hesperidin, and rutin. Nutr Cancer. 2015;67:137–144. doi: 10.1080/01635581.2015.965335. [DOI] [PubMed] [Google Scholar]
- 14.Rastogi H, Jana S. Evaluation of inhibitory effects of caffeic acid and quercetin on human liver cytochrome p450 activities. Phytother Res. 2014;28:1873–1878. doi: 10.1002/ptr.5220. [DOI] [PubMed] [Google Scholar]
- 15.Stachel N, Skopp G. Formation and inhibition of ethyl glucuronide and ethyl sulfate. Forensic Sci Int. 2016;265:61–64. doi: 10.1016/j.forsciint.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 16.Li Y, Revalde J, Paxton JW. The effects of dietary and herbal phytochemicals on drug transporters. Adv Drug Deliv Rev. 2017;116:45–62. doi: 10.1016/j.addr.2016.09.004. [DOI] [PubMed] [Google Scholar]
- 17.Shi M, Liu Y, Bian JL, Jin M, Gui CS. The interactions between natural products and OATP1B1. Yao Xue Xue Bao. 2015;50:848–853. (In Chinese) [PubMed] [Google Scholar]
- 18.Li L, Stanton JD, Tolson AH, Luo Y, Wang H. Bioactive terpenoids and flavonoids from Ginkgo biloba extract induce the expression of hepatic drug-metabolizing enzymes through pregnane X receptor, constitutive androstane receptor, and aryl hydrocarbon receptor-mediated pathways. Pharm Res. 2009;26:872–882. doi: 10.1007/s11095-008-9788-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tolson AH, Wang H. Regulation of drug-metabolizing enzymes by xenobiotic receptors: PXR and CAR. Adv Drug Deliv Rev. 2010;62:1238–1249. doi: 10.1016/j.addr.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang YZ, Lin HC, Chan ST, Yeh SL. Effects of quercetin metabolites on the enhancing effect of β-carotene on DNA damage and cytochrome P1A1/2 expression in benzo[a]pyrene-exposed A549 cells. Food Chem. 2012;133:445–450. doi: 10.1016/j.foodchem.2012.01.060. [DOI] [PubMed] [Google Scholar]
- 21.Yu CP, Lin HJ, Lin SP, Shia CS, Chang PH, Hou YC, Hsieh YW. Rhubarb decreased the systemic exposure of cyclosporine, a probe substrate of P-glycoprotein and CYP 3A. Xenobiotica. 2016;46:677–682. doi: 10.3109/00498254.2015.1117159. [DOI] [PubMed] [Google Scholar]
- 22.Dupuis R, Yuen A, Innocenti F. The influence of UGT polymorphisms as biomarkers in solid organ transplantation. Clin Chim Acta. 2012;413:1318–1325. doi: 10.1016/j.cca.2012.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kato R, Nishide M, Kozu C, Iwamoto A, Urashima K, Suzuki K, Ijiri Y, Hayashi T, Tanaka K. Is cyclosporine a transport inhibited by pravastatin via multidrug resistant protein 2? Eur J Clin Pharmacol. 2010;66:153–158. doi: 10.1007/s00228-009-0740-6. [DOI] [PubMed] [Google Scholar]
- 24.Ouwerkerk-Mahadevan S, Snoeys J, Peeters M, Beumont-Mauviel M, Simion A. Drug-drug interactions with the NS3/4A protease inhibitor simeprevir. Clin Pharmacokinet. 2016;55:197–208. doi: 10.1007/s40262-015-0314-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu J, Tjandra M, Becker C, Bednarczyk D, Capparelli M, Elling R, Hanna I, Fujimoto R, Furegati M, Karur S, et al. Potent nonimmunosuppressive cyclophilin inhibitors with improved pharmaceutical properties and decreased transporter inhibition. J Med Chem. 2014;57:8503–8516. doi: 10.1021/jm500862r. [DOI] [PubMed] [Google Scholar]
- 26.Choi JS, Choi BC, Choi KE. Effect of quercetin on the pharmacokinetics of oral cyclosporine. Am J Health Syst Pharm. 2004;61:2406–2409. doi: 10.1093/ajhp/61.22.2406. [DOI] [PubMed] [Google Scholar]
- 27.Zal F, Mostafavi-Pour Z, Vessal M. Comparison of the effects of vitamin E and/or quercetin in attenuating chronic cyclosporine A-induced nephrotoxicity in male rats. Clin Exp Pharmacol Physiol. 2007;34:720–724. doi: 10.1111/j.1440-1681.2007.04623.x. [DOI] [PubMed] [Google Scholar]
- 28.Liu Y, Luo X, Yang C, Yang T, Zhou J, Shi S. Impact of quercetin-induced changes in drug-metabolizing enzyme and transporter expression on the pharmacokinetics of cyclosporine in rats. Mol Med Rep. 2016;14:3073–3085. doi: 10.3892/mmr.2016.5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chiou WL. Critical evaluation of the potential error in pharmacokinetic studies of using the linear trapezoidal rule method for the calculation of the area under the plasma level-time curve. J Pharmacokinet Biopharm. 1978;6:539–546. doi: 10.1007/BF01062108. [DOI] [PubMed] [Google Scholar]
- 30.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 31.Lai Q, Wei J, Mahmoodurrahman M, Zhang C, Quan S, Li T, Yu Y. Pharmacokinetic and nephroprotective benefits of using Schisandra chinensis extracts in a cyclosporine A-based immune-suppressive regime. Drug Des Devel Ther. 2015;9:4997–5018. doi: 10.2147/DDDT.S89876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu H, Zhu SC, Shakya S, Mao Q, Ding CH, Long MH, Li SL. Study on the pharmacokinetics profiles of polyphyllin I and its bioavailability enhancement through co-administration with P-glycoprotein inhibitors by LC-MS/MS method. J Pharm Biomed Anal. 2015;107:119–124. doi: 10.1016/j.jpba.2014.12.002. [DOI] [PubMed] [Google Scholar]
- 33.Yu CP, Wu PP, Hou YC, Lin SP, Tsai SY, Chen CT, Chao PD. Quercetin and rutin reduced the bioavailability of cyclosporine from Neoral, an immunosuppressant, through activating P-glycoprotein and CYP 3A4. J Agric Food Chem. 2011;59:4644–4648. doi: 10.1021/jf104786t. [DOI] [PubMed] [Google Scholar]
- 34.Shi S, Klotz U. Drug interactions with herbal medicines. Clin Pharmacokinet. 2012;51:77–104. doi: 10.2165/11597910-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 35.Li L, Yao QQ, Xu SY, Hu HH, Shen Q, Tian Y, Pan LY, Zhou H, Jiang HD, Lu C, et al. Cyclosporin A affects the bioavailability of ginkgolic acids via inhibition of P-gp and BCRP. Eur J Pharm Biopharm. 2014;88:759–767. doi: 10.1016/j.ejpb.2014.06.012. [DOI] [PubMed] [Google Scholar]
- 36.Hsu PW, Shia CS, Lin SP, Chao PD, Juang SH, Hou YC. Potential risk of mulberry-drug interaction: Modulation on P-glycoprotein and cytochrome P450 3A. J Agric Food Chem. 2013;61:4464–4469. doi: 10.1021/jf3052384. [DOI] [PubMed] [Google Scholar]
- 37.Hsiu SL, Hou YC, Wang YH, Tsao CW, Su SF, Chao PD. Quercetin significantly decreased cyclosporin oral bioavailability in pigs and rats. Life Sci. 2002;72:227–235. doi: 10.1016/S0024-3205(02)02235-X. [DOI] [PubMed] [Google Scholar]
- 38.Jiang W, Hu M. Mutual interactions between flavonoids and enzymatic and transporter elements responsible for flavonoid disposition via phase II metabolic pathways. RSC Adv. 2012;2:7948–7963. doi: 10.1039/c2ra01369j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dong H, Lin W, Wu J, Chen T. Flavonoids activate pregnane × receptor-mediated CYP3A4 gene expression by inhibiting cyclin-dependent kinases in HepG2 liver carcinoma cells. BMC Biochem. 2010;11:23. doi: 10.1186/1471-2091-11-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Satsu H, Hiura Y, Mochizuki K, Hamada M, Shimizu M. Activation of pregnane X receptor and induction of MDR1 by dietary phytochemicals. J Agric Food Chem. 2008;56:5366–5373. doi: 10.1021/jf073350e. [DOI] [PubMed] [Google Scholar]
- 41.Urquhart BL, Tirona RG, Kim RB. Nuclear receptors and the regulation of drug-metabolizing enzymes and drug transporters: Implications for interindividual variability in response to drugs. J Clin Pharmacol. 2007;47:566–578. doi: 10.1177/0091270007299930. [DOI] [PubMed] [Google Scholar]
- 42.Lau AJ, Chang TK. 3-Hydroxyflavone and structural analogues differentially activate pregnane X receptor: Implication for inflammatory bowel disease. Pharmacol Res. 2015;100:64–72. doi: 10.1016/j.phrs.2015.07.031. [DOI] [PubMed] [Google Scholar]
- 43.Benet LZ, Cummins CL, Wu CY. Transporter-enzyme interactions: Implications for predicting drug-drug interactions from in vitro data. Curr Drug Metab. 2003;4:393–398. doi: 10.2174/1389200033489389. [DOI] [PubMed] [Google Scholar]
- 44.Benet LZ, Cummins CL, Wu CY. Unmasking the dynamic interplay between efflux transporters and metabolic enzymes. Int J Pharm. 2004;277:3–9. doi: 10.1016/j.ijpharm.2002.12.002. [DOI] [PubMed] [Google Scholar]
- 45.Shi S, Li Y. Interplay of drug-metabolizing enzymes and transporters in drug absorption and disposition. Curr Drug Metab. 2014;15:915–941. doi: 10.2174/1389200216666150401110610. [DOI] [PubMed] [Google Scholar]
- 46.Li Y, Zhou J, Ramsden D, Taub ME, O'Brien D, Xu J, Busacca CA, Gonnella N, Tweedie DJ. Enzyme-transporter interplay in the formation and clearance of abundant metabolites of faldaprevir found in excreta but not in circulation. Drug Metab Dispos. 2014;42:384–393. doi: 10.1124/dmd.113.055863. [DOI] [PubMed] [Google Scholar]
- 47.Jiang W, Xu B, Wu B, Yu R, Hu M. UDP-glucuronosyltransferase (UGT) 1A9-overexpressing HeLa cells is an appropriate tool to delineate the kinetic interplay between breast cancer resistance protein (BRCP) and UGT and to rapidly identify the glucuronide substrates of BCRP. Drug Metab Dispos. 2012;40:336–345. doi: 10.1124/dmd.111.041467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Zanden JJ, van der Woude H, Vaessen J, Usta M, Wortelboer HM, Cnubben NH, Rietjens IM. The effect of quercetin phase II metabolism on its MRP1 and MRP2 inhibiting potential. Biochem Pharmacol. 2007;74:345–351. doi: 10.1016/j.bcp.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 49.Wong CC, Williamson G. Inhibition of hydroxycinnamic acid sulfation by flavonoids and their conjugated metabolites. Biofactors. 2013;39:644–651. doi: 10.1002/biof.1127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.