

The presence of volatile phenols is considered a major organoleptic defect of several fermented alcoholic beverages. The biosynthesis of these compounds has been mainly associated with Brettanomyces/Dekkera yeasts. However, the potential importance of lactic acid bacteria in volatile phenol spoilage is emphasized by reports describing a faster ethylphenol production by these bacteria than by yeasts. The genetic identification of the bacterial vinylphenol reductase involved in volatile phenol production provides new insights into the role of lactic acid bacteria in the production of these off flavors. The development of a molecular method for the detection of ethylphenol-producing bacteria could be helpful to design strategies to reduce the bacterial production of vinylphenols in fermented foods.
KEYWORDS: aroma, cider, ethylguaiacol, ethylphenol, lactic acid bacteria, off flavors, phenolic compounds, spoilage, volatile phenols, wine
ABSTRACT
Ethylphenols are strong odorants produced by microbial activity that are described as off flavors in several foods. Lactobacillus plantarum is a lactic acid bacterial species able to produce ethylphenols by the reduction of vinylphenols during the metabolism of hydroxycinnamic acids. However, the reductase involved has not been yet uncovered. In this study, the involvement in vinylphenol reduction of a gene encoding a putative reductase (lp_3125) was confirmed by the absence of reduction activity in the Δlp_3125 knockout mutant. The protein encoded by lp_3125, VprA, was recombinantly produced in Escherichia coli. VprA was assayed against vinylphenols (4-vinylphenol, 4-vinylcatechol, and 4-vinylguaiacol), and all were reduced to their corresponding ethylphenols (4-ethylphenol, 4-ethylcatechol, and 4-ethylguaiacol). PCR and high-performance liquid chromatography (HPLC) detection methods revealed that the VprA reductase is not widely distributed among the lactic acid bacteria studied and that only the bacteria possessing the vprA gene were able to produce ethylphenol from vinylphenol. However, all the species belonging to the L. plantarum group were ethylphenol producers. The identification of the L. plantarum VprA protein involved in hydroxycinnamate degradation completes the route of degradation of these compounds in lactic acid bacteria.
IMPORTANCE The presence of volatile phenols is considered a major organoleptic defect of several fermented alcoholic beverages. The biosynthesis of these compounds has been mainly associated with Brettanomyces/Dekkera yeasts. However, the potential importance of lactic acid bacteria in volatile phenol spoilage is emphasized by reports describing a faster ethylphenol production by these bacteria than by yeasts. The genetic identification of the bacterial vinylphenol reductase involved in volatile phenol production provides new insights into the role of lactic acid bacteria in the production of these off flavors. The development of a molecular method for the detection of ethylphenol-producing bacteria could be helpful to design strategies to reduce the bacterial production of vinylphenols in fermented foods.
INTRODUCTION
One of the main organoleptic problems occurring during the elaboration of many fermented alcoholic beverages is the biosynthesis of volatile phenols (1). Although these aromatic compounds are essential for the overall flavor perception of some types of beers and wines, they become undesirable when their concentrations exceed certain limits (1). Volatile phenols (mainly 4-ethylphenol, 4-vinylphenol, 4-ethylguaiacol, and 4-vinylguaiacol) have very low perception thresholds, and they can thus have a major impact on the aroma of wine, beer, or cider (2, 3). These compounds are associated with animal, leather, medicinal, and “horse sweat” odors. Winemakers consider the presence of these compounds in wine a key concern in the control of wine quality and a serious economic problem (4).
Volatile phenols have been found to be mainly produced from hydroxycinnamates, the major phenols in grape juice and the major class of phenolics in white wine (5), by microbial activity. Volatile phenols are originated by the sequential action of two enzymes on hydroxycinnamic acids, mainly p-coumaric and ferulic acids. First, a hydroxycinnamate decarboxylase enzyme decarboxylates these acids into their vinyl derivatives, and they are then reduced to ethyl derivatives by a reductase enzyme (4).
The biosynthesis of volatile phenols has been studied mainly in wines. Here, the main microorganism associated with volatile phenol production is Dekkera/Brettanomyces yeast (4). A hydroxycinnamate decarboxylase from Brettanomyces was purified and characterized (6, 7). A partial amino acid sequence of this decarboxylase revealed the lack of similarity with previously sequenced phenolic acid decarboxylases (PDCs) from other yeasts or bacteria (8). The vinyl reductase enzyme that catalyzes the reduction step is found in a small number of yeasts (4). Although Godoy et al. (2008) (7) and Tchobanov et al. (2008) (9) isolated a potential vinylphenol reductase from Dekkera bruxellensis, the complete sequence of the enzyme has been only recently known (10). While the protein showed a high similarity to superoxide dismutase SDO1 of Saccharomyces cerevisiae, a deeper bioinformatic examination revealed that the enzyme is a dehydrogenase/reductase protein that hosts domains required to bind NAD(P)H. The biological functionality of this protein as the one responsible for the production of off-flavors in D. bruxellensis has been recently demonstrated (11).
Although lactic acid bacteria have been shown to produce low levels of volatile phenols in wines (12), in synthetic media simulating cider conditions, the production of ethylphenols by these bacteria was faster than production by Brettanomyces/Dekkera, the microorganism traditionally associated with volatile phenol defects in wine (13). This result emphasizes the potential importance of lactic acid bacteria in volatile phenol spoilage. Several studies have reported the ability of lactic acid bacteria to produce volatile phenols (1, 3, 14–22). Similarly to yeasts, in some bacterial species, such as Lactobacillus plantarum, p-coumaric, caffeic, and ferulic acids are decarboxylated to their corresponding vinyl derivatives and subsequently reduced to ethyl derivatives. L. plantarum has been shown to synthesize an inducible PDC, which decarboxylates these hydroxycinnamic acids into their vinyl derivatives. L. plantarum PDC enzyme was identified and biochemically characterized (23, 24). As other lactic acid bacteria were also able to produce vinylphenols, a pdc-based PCR method was designed to identify bacteria potentially producing these volatile phenols (16). However, in L. plantarum, as well as in other bacterial species, the reductase involved in the production of ethylphenols remains unknown. In this work, the L. plantarum gene involved in ethylphenol formation has been identified and the corresponding reductase protein characterized.
RESULTS AND DISCUSSION
Identification of Lp_3125 as the enzyme responsible for vinylphenol reduction in L. plantarum WCFS1.
Numerous studies demonstrated that L. plantarum strains could produce volatile phenols from hydroxycinnamic acids (14, 15, 17, 20, 22, 25–27). The production of ethylphenols from hydroxycinnamic acids involved two consecutive steps. The first step is a decarboxylation carried out by the PDC (Lp_3665) enzyme that transforms hydroxycinnamic acids into their vinyl derivatives, and the second step involves the subsequent reduction of these latter compounds into the corresponding ethyl derivatives. As far as we know, this reductase enzyme remains unknown. A previous study demonstrated that L. plantarum fully converts p-coumaric acid into 4-ethylphenol when grown in MRS broth supplemented with this hydroxycinnamic acid (20). The presence of p-coumaric acid increased the expression of PDC (23) and might also induce the reductase involved in the second step of ethylphenol formation. As expected, the L. plantarum transcriptomic response to p-coumaric acid revealed the induction of the pdc gene (112-fold), but also of the genes encoding hydroxycinnamate reductase (hcrA and hcrB; 15.3 and 19.7-fold, respectively). In this regard, we have recently described the involvement of HcrAB in the reduction of hydroxycinnamic acids into substituted phenylpropionic acid (28). Since the reaction catalyzed by HcrAB also implies the reduction of a carbon-carbon double bond, the possible involvement of HcrAB in vinylphenol reduction needs to be eliminated. To this end, the HrcA and HcrB knockout mutants (28) were grown in the presence of 4-vinylphenol and, as shown in Fig. 1, both mutants were able to reduce this compound, demonstrating that neither of the two proteins was involved in vinylphenol reduction.
FIG 1.

Effect of disruption of hcrA (lp_1424), hcrB (lp_1425), and lp_3125 in L. plantarum WCFS1 on the reduction of an hydroxycinnamic acid (m-coumaric acid) and 4-vinylphenol. HPLC chromatograms of L. plantarum cultures incubated in 1.5 mM m-coumaric acid or 4-vinylphenol are shown for L. plantarum WCFS1 (wild type [wt]), L. plantarum WCFS1(pUCE191-hcrA) (ΔhcrA mutant), L. plantarum WCFS1(pUCE191-hcrB) (ΔhcrB mutant), and L. plantarum WCFS1(pUCE191-lp_3125) (Δlp_3125 mutant). Results for uninoculated medium are also shown (control). The m-coumaric acid (mCA), 4-vinylphenol (VP), 3-(3-hydroxyphenyl) propionic acid (3-HPPA), and 4-ethylphenol (EP) detected are indicated. Chromatograms were recorded at 280 nm. mAU, milli-absorbance units.
A more detailed examination of the global transcriptomic response of L. plantarum to p-coumaric acid revealed a 4-fold induction of the lp_3125 gene, which putatively encoded the flavoprotein subunit of a fumarate reductase (29). In order to know whether the lp_3125 gene codes for the reductase involved in vinylphenol reduction, a lp_3125 knockout mutant was constructed by an insertion-duplication strategy. This mutant strain was grown in MRS media containing vinylphenol (1.5 mM) for 10 days. After incubation, the phenolic compounds present in the supernatant were extracted twice with ethyl acetate and analyzed by high-performance liquid chromatography (HPLC) (Fig. 1). Unlike HcrAB mutants, which reduced 4-vinylphenol to 4-ethylphenol but were unable to reduce m-coumaric acid to 3-(3-hydroxyphenyl) propionic acid (3-HPPA), the lp_3125 mutant was unable to reduce vinylphenol, while its ability to reduce hydroxycinnamic acid was not affected (Fig. 1). The disruption of the lp_3125 gene demonstrated its involvement in the reduction of vinylphenol, despite the Lp_3125 protein being annotated as a fumarate reductase. This result illustrates that assignments of function based on genomic data must be verified by experimental data generated by assays of the isolated, purified enzyme or recombinantly produced enzyme (30).
Organization of the genes required for vinylphenol reductase activity in L. plantarum WCFS1.
Once the involvement of lp_3125 (vprA, vinyl phenol reductase) in ethylphenol formation was confirmed, the genetic organization of the region surrounding vprA was analyzed. The lp_3123 and lp_3124 open reading frames (ORFs) are divergently transcribed to the gene pair vprA and lp_3127. The lp_3123 gene encodes a putative NADH pyrophosphatase of the Nudix family, and lp_3124 encodes a LysR family transcriptional regulator. Downstream of vprA, lp_3127 putatively encodes a mucus-binding protein containing the LPXTG motif for the cell wall anchor (Fig. 2A).
FIG 2.
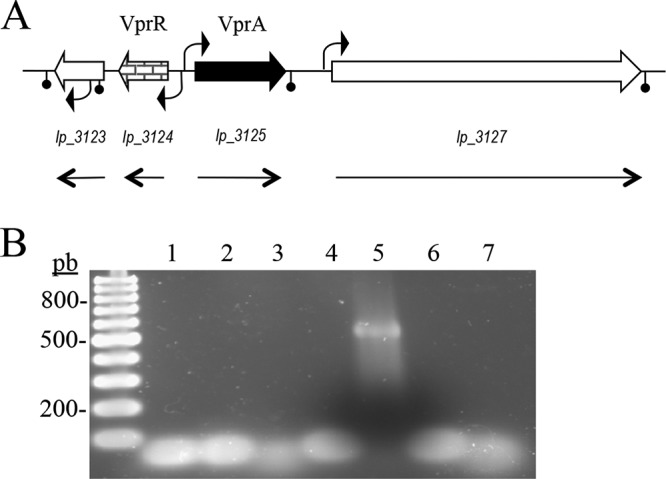
Genetic organization of the L. plantarum WCFS1 chromosomal region containing the vinylphenol reductase encoding genes. (A) (NCBI accession number NC_004567, positions 2788661 to 2798829). Arrows indicate genes. The shaded genes encode genes putatively involved in vinylphenol reductase (vpr) activity. The location of putative promoters and transcription terminators are also indicated. The size and direction of the transcripts revealed by reverse transcription are also shown. (B) Transcriptional analysis by RT-PCR of the L. plantarum WCFS1 genome in the vinylphenol reductase locus. RT-PCR amplification was performed with primers designed to amplify internal gene regions or intergenic regions, as follows: lp_3123 (primers 1810 and 1811, 617 bp) (1), lp_3123-vprR (1684 and 1685, 613 bp) (2), vprR (1686 and 1687, 596 bp) (3), vprR-vprA (1688 and 1689, 800 bp) (4), vprA (891 and 892, 472 bp) (5), hcrB-hcrC (1385 and 1052, 778 bp) (6), and hcrC (1051 and 1952, 384 bp) (7). Left lane, 100-bp molecular size ladder. Numbers indicate some of the molecular sizes.
The RNA isolated from a L. plantarum WCFS1 culture grown in MRS medium containing 1.5 mM 4-vinylphenol was used to determine the transcriptional profile of the VprA region by reverse transcription PCR (RT-PCR). Oligonucleotides were designed to amplify the four genes (lp_3123 to lp_3127), as well as the regions spanning their gene junctions (Table 1). As shown in Fig. 2B, only the vprA gene yielded a PCR amplicon of the expected size, indicating that vprA is transcribed as a monocistronic mRNA. This result is supported by the in silico identification of a Rho-independent transcriptional terminator downstream vprA (ΔG, −10.5 kcal/mol).
TABLE 1.
Primers used in this study
| Primer | Sequence (5′→3′)a | Amplified fragment/cloning strategy |
|---|---|---|
| 891 | GGGGTACCGGTGTAATGATTCAAAGTG | The 472-bp PCR vprA internal fragment was double digested with KpnI/XbaI and cloned into a KpnI/XbaI doubly digested pUCE191 plasmid, generating pUCE191-vprA |
| 892 | GCTCTAGATCCCGTCCTGAGCCATTTTG | |
| 965 | ATGACGTTAGCAAAACATGA | Amplification with oligonucleotide 1233 to corroborate the correct insertion of pUCE191-vprA into the L. plantarum WCFS1 chromosome |
| 1233 | AGCGGATAACAATTTCACACAGGA | 24-mer reverse sequencing primer (−48) for pUCE19/pUCE191 |
| 1470 | CATGCCTGGAACCTGTGTTG | Amplification of a 62-bp vprA fragment for real-time quantitative PCR |
| 1471 | TGCCGACCGGAATTGC | Amplification with oligonucleotide 1690 of an 804-bp vprA-lp_3127 intergenic region |
| 1516 | TAACTTTAAGAAGGAGATATACATATGACGTTAGCAAAACATGATTCAT | The 1.5-kb vprA gene amplified with oligonucleotides 1516 and 1517 was introduced into vector pURI3-Cter by using an LICb strategy |
| 1517 | GCTATTAATGATGATGATGATGATGATGACTAACGATTGACTGCTGGTG | |
| 1633 | CAGCGGTACCTATTACCGGCCTTA | The 308-bp PCR vprR internal fragment was doubly digested with KpnI/XbaI and cloned into a KpnI/XbaI doubly digested pUCE191 plasmid, generating pUCE191-vprR |
| 1634 | GTTGAGCTTTCTAGACTGTAATAT | |
| 1635 | ATGGATCTTGACCGGCTACAGA | Amplification with oligonucleotide 1233 to corroborate the correct insertion of pUCE191-vprR into the L. plantarum WCFS1 chromosome |
| 1657 | GAYVTNGTNGTNGTNGG | Degenerate oligonucleotides coding for the VprA conserved motifs D(V/L/I)VVVG (1657) and GLYAAG (1658); they are used to amplify a 1.3-kb internal fragment of vprA in lactic acid bacteria |
| 1658 | CCNGCNGCRTANAGNCC | |
| 1684 | CAACGGTATAGTTATCATTCGC | Amplification of a 613-bp lp_3123-vprR intergenic region |
| 1685 | TGTTAGTTGCGTTGAACCAAGC | |
| 1686 | TGCTCATAGTCTTGAATAAACG | Amplification of a 596-bp vprR internal fragment |
| 1687 | GAATATCGTTCACAGCGGACAG | |
| 1688 | CAGTAGGAACATTAAATTGACG | Amplification of an 800-bp vprR-vrpA intergenic region |
| 1689 | TCGCCGGCACCCATTGCTTGTACG | |
| 1690 | ACTGGTTGTAGTACTGCCAGTTGC | Amplification with oligonucleotide 1470 of an 804-bp vprA-lp_3127 intergenic region |
| 1719 | GCTGCGCAACAAGGCTATG | Amplification of a 54-bp vprR fragment for real-time quantitative PCR |
| 1720 | CCGGGACCGGTTTGATTT | |
| 1810 | GCCACGTTCTCATTAAGGTCCGC | Amplification of a 617-bp lp_3123 internal fragment |
| 1811 | CTTAATTGGTCGTCAACAACAGG | |
| 1812 | CATTGATGTCAGCAATTGGCTAGC | Amplification of a 573-bp lp_3127 internal fragment |
| 1813 | CACTAGCTGCCATCTTAGCACCAC |
R = G or A; Y = C or T; V = A, C or G; and N = G, A, C or T. Engineered restriction sites are underlined; nucleotides pairing the vector sequence are italicized.
LIC, ligation-independent cloning.
Lp_3124 is annotated as a LysR-type transcriptional regulator. LysR-type transcriptional regulators comprise the largest family of prokaryotic regulatory proteins identified and are frequently associated with degradation pathways of aromatic compounds (31). In general, a gene encoding this type of regulator lies upstream of its target-regulated gene and is transcribed in the opposite direction (32, 33). In view of this, the involvement of lp_3124 in vinylphenol reductase activity demands further studies. An L. plantarum WCFS1 lp_3124 knockout mutant was constructed by plasmid insertion-duplication and was grown in the presence of 4-vinylphenol. The HPLC analysis of the culture supernatants revealed that, compared to the wild-type strain, the disruption of lp_3124 (vprR) (as well as the vprA gene) avoided the reduction of vinylphenol in L. plantarum (see Fig. S1 in the supplemental material). This result indicates that, most probably, the VprR (Lp_3124) transcriptional regulator acts as an activator, as its absence avoids the production of VprA. Most of the LysR-type transcriptional regulators involved in aromatic degradation pathways act as transcriptional activators for their target metabolic genes in the presence of a chemical inducer, which is usually a pathway intermediate. LysR transcriptional regulators repress their own expression, and both autorepression and activation of the catabolic promoter are exerted from the same binding site. Relatively few data exist on autorepression mechanisms, since most studies on this type of regulators have focused on the mechanism of target gene activation (31).
Reductase activity of purified VprA protein.
As explained before, the disruption of the vprA gene of L. plantarum WCFS1 renders a variant which is unable to reduce vinylphenol to ethylphenol. In order to know whether the VprA protein is the only protein involved in the catalytic activity, its encoding gene was cloned into the expression vector pURI3-Cter by a restriction enzyme- and ligation-free cloning strategy described previously (34). Oligonucleotides 1516 and 1517 were used to amplify the 1.5-kb vprA gene (Table 1). Escherichia coli DH10B cells were transformed, and the recombinant plasmids were isolated. Those containing the correct insert were used for transformation of E. coli BL21(DE3) cells. Cell extracts were used to detect the presence of overproduced proteins by SDS-PAGE analysis. Control cells containing the pURI3-Cter vector plasmid did not show protein overexpression; in addition, in the presence of 4-vinylphenol, no reductase activity was observed in this control extract (Fig. 3B). However, an overproduced protein with an apparent molecular mass of around 50 kDa was observed in cells harboring the pURI3-Cter-VprA plasmid (Fig. 3A). Since the cloning strategy yielded a His-tagged protein variant, L. plantarum VprA protein could be purified by using an immobilized metal affinity chromatography (IMAC) resin (Fig. 3A). Purified VprA protein showed the characteristic yellow color indicative of the binding of a flavin cofactor to oxidoreductases. VprA protein in a reaction mixture containing NADH or NADPH (15 mM) was able to reduce 4-vinylphenol to 4-ethylphenol (Fig. 3B). Although activity was observed with both cofactors, 4-vinylphenol was fully reduced only in the presence of NADH.
FIG 3.
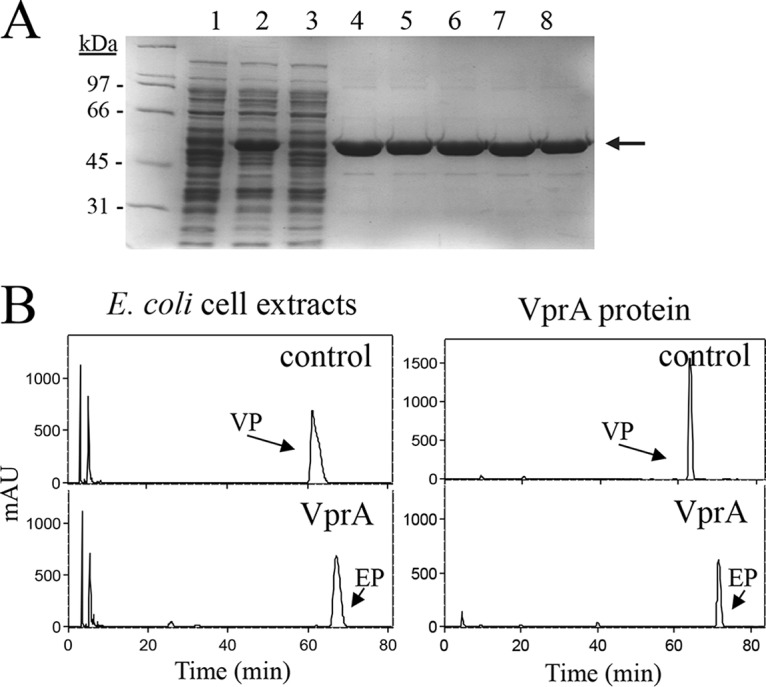
Purification and enzymatic activity of E. coli extracts expressing L. plantarum vprA and recombinant VprA protein. (A) SDS-PAGE analysis of the expression and purification of the His6-tagged VprA. Data represent results of analysis of soluble cell extracts of IPTG-induced E. coli BL21(DE3)(pURI3-Cter) (lane 1) or E. coli BL21(DE3)(pURI3-Cter-VprA) (lane 2), flowthrough from the affinity resin (lane 3), or fractions eluted after His affinity resin (lanes 4 to 8). The 10% gel was stained with Coomassie blue. Molecular mass markers are located on the left (SDS-PAGE standards; Bio-Rad). (B) HPLC chromatograms showing vinylphenol reductase activity of soluble cell extracts of IPTG-induced E. coli BL21(DE3)(pURI3-Cter) (control) or E. coli BL21(DE3)(pURI3-Cter-VprA) (VprA) incubated in 1.5 mM 4-vinylphenol and 15 mM NADH. HPLC chromatograms also showed the reductase activity of purified His6-VprA protein (500 μg) (VprA) or the reaction mix without VprA protein (control). The 4-vinylphenol (VP) and 4-ethylphenol (EP) detected are indicated. Chromatograms were recorded at 280 nm.
The reductase activity shown by VprA confirms that this protein is responsible for the vinylphenol reductase activity exhibited by L. plantarum cells. Therefore, this is a description of a vinylphenol reductase enzyme in bacteria. In the yeast Dekkera bruxellensis, a protein exhibiting vinylphenol reductase activity was recently described (10, 11). The Dekkera protein was annotated as a Cu/Zn superoxide dismutase, and it was indeed found to possess both vinylphenol reductase and superoxide dismutase activities (10). This protein presented cofactor-binding structural features that are absent in superoxide dismutases from related microorganisms that do not display vinylphenol reductase activity. This Dekkera protein belongs to the short-chain dehydrogenase/reductase (SDR) family, a functionally diverse family of NAD(P)H-dependent oxidoreductases that have a single domain with a structurally conserved Rossmann fold, an NAD(P)H-binding region, and a structurally diverse C-terminal region. The most conserved feature in this family is a structural motif characterized by a highly variable Gly-rich sequence pattern (GXGXXG), critical for accommodation and binding of the pyrophosphate portion of the nucleotide cofactor. In this sequence, the first two glycines participate in NAD(P) binding, and the third one facilitates close packing of the Rossmann fold. This Rossmann fold region is conserved in D. bruxellensis wine yeasts, although nucleotide polymorphism of the gene is present and, apparently, it is not directly linked to the production of volatile phenols (11).
A domain analysis of the VprA protein at the NCBI site revealed the presence of an oxidoreductase conserved domain (COG3573). Like the D. bruxellensis protein, VprA showed similarity to NAD(P)H-binding proteins possessing the Rossmann fold. When the vinyl reductase from D. bruxellensis (154 amino acid residues) was aligned to the 493-amino-acid VprA protein from L. plantarum, a 28.5% sequence identity was found in the C-terminal overlapping region (see Fig. S2A in the supplemental material). A sequence identity of 29.5% was found for VprA and the recently described L. plantarum hydroxycinnamate reductase HcrB protein (812 amino acid residues) along the 493-amino-acid overlapping region (Fig. S2B). L. plantarum HcrB also possesses an oxidoreductase conserved domain exhibiting similarity to NAD(P)H-binding proteins possessing the Rossmann fold. HcrB and VprA are annotated as flavoproteins, and both purified proteins exhibited the characteristic yellow color indicative of the binding of a flavin cofactor. The presence of the flavin adenine dinucleotide (FAD) cofactor was identified by mass spectrometry analysis in both proteins (see Fig. S3 in the supplemental material).
This work identifies the L. plantarum VprA flavoprotein NADH oxidoreductase as a bacterial enzyme possessing vinylphenol reductase activity.
VprA and PDC exhibited the same substrate range.
The production of ethylphenols from hydroxycinnamic acids involved two consecutive steps. First, a decarboxylation is carried out by the PDC (Lp_3665) enzyme, which transforms hydroxycinnamic acids into their vinyl derivatives, and subsequently, reduction of the vinyl derivatives to ethyl derivatives. It was previously described that PDC decarboxylase is able to decarboxylate only the hydroxycinnamic acids p-coumaric, caffeic, and ferulic acids to their vinyl derivatives (35). In order to know if VprA is able to reduce all the vinyl derivatives produced by PDC action, 4-vinylphenol and 4-vinylguaiacol were used as potential substrates (Fig. 4). As shown in Fig. 4, VprA was able to fully reduce both vinylphenols, producing 4-ethylphenol and 4-ethylguaiacol, respectively. These two compounds were traditionally identified as the main volatile phenols responsible for off flavors in beer and red wines (2) and consequently have received most of the research attention. However, another volatile phenol, 4-ethylcatechol, has been recently described as contributor to horsey flavor in wines (36) or ciders (13). With the aim to determine if VprA is able to produce this volatile phenol, we first produced its 4-vinylphenol precursor, which is not commercially available, by using recombinant E. coli cells harboring a plasmid expressing the L. plantarum PDC. E. coli cell cultures were grown in the presence of caffeic acid, and, after incubation, the phenolic compounds present in the supernatant were extracted with ethyl acetate and analyzed by HPLC. As shown in Fig. S4 in the supplemental material, E. coli cells fully decarboxylated caffeic acid into 4-vinylcatechol. The identification of vinylcatechol was carried out by comparing the retention time and the spectra of the compound previously identified as 4-vinylcatechol by liquid chromatography-diode array detection (LC-DAD/electrospray ionization-mass spectrometry) (ESI-MS). The vinylcatechol produced by E. coli was used as the substrate for VprA activity. Figure 4 shows that VprA partially reduced vinylcatechol to ethylcatechol. The production of ethylphenol, ethylcatechol, and ethylguaiacol from reduction of their corresponding vinylphenols has been previously described in L. plantarum strains (35). This result confirms that the two enzymatic activities involved in volatile phenol production in L. plantarum shared a common transformation pathway as the compounds produced by decarboxylase action are substrates for reductase activity. This concomitant action is also supported by gene expression analysis. Reverse transcription-PCR (RT-PCR) assays revealed that vprA expression is induced by its substrate, 4-vinylphenol, as well as by the substrate of the PDC decarboxylase enzyme, p-coumaric acid (see Fig. S5 in the supplemental material).
FIG 4.

Reductase activity of VprA on several vinylphenols. HPLC chromatograms represent E. coli BL21(DE3)(pURI3-Cter) (control) or E. coli BL21(DE3)(pURI3-Cter-vprA) (VprA) cell extracts incubated at 37°C during 16 h. The vinylphenols assayed were vinyl catechol (VC), 4-vinylphenol (VP), and vinylguaiacol (VG). The corresponding ethyl derivatives such as ethyl catechol (EC), 4-ethylphenol (EP), and ethylguaiacol (EG) detected are indicated. Chromatograms were recorded at 280 nm.
L. plantarum VprA is a bacterial protein able to reduce vinylphenol and vinylcatechol, to ethylphenol and ethylcatechol, respectively.
Ability among lactic acid bacteria to produce volatile phenols.
As VprA and PDC are enzymes acting on the same transformation pathway, it is tempting to speculate about the copresence of both enzymatic activities in the same bacterial strains. However, previous studies demonstrated that the ability to decarboxylate hydroxycinnamic acids to produce vinylphenols is more widely distributed among lactic acid bacteria than the ability to reduce them to ethylphenols (16). Although Lactobacillus brevis and Pediococcus pentosaceus strains were able to produce vinylphenols, only L. plantarum strains produced ethylphenols (16). A pdc PCR assay was described for the molecular screening of wine lactic acid bacteria producing vinylphenols. L. plantarum, L. brevis, and P. pentosaceus strains produced a positive response in the pdc PCR assay, whereas Oenococcus oeni, Lactobacillus hilgardii, and Leuconostoc mesenteroides strains did not produce the expected PCR product. Moreover, the strains that gave a positive pdc PCR response produce vinylphenols, whereas strains that did not produce a PCR amplicon did not produce them (16).
In order to design a detection method for lactic acid bacteria producing ethylphenols, proteins similar to VprA were searched for in databases. Among lactic acid bacteria, only strains from the Lactobacillus plantarum group (L. plantarum, Lactobacillus paraplantarum, Lactobacillus pentosus, and Lactobacillus fabifermentans), Lactobacillus collinoides, Lactobacillus paracollinoides, Lactobacillus bifermentans, Lactobacillus similis, and Lactobacillus rossiae species possessed a protein similar to L. plantarum VprA (see Fig. S6 in the supplemental material). The degree of identity among these proteins ranged from 40.71% (L. rossiae and L. fabifermentans proteins) to 89.94% (L. paraplantarum and L. pentosus proteins). The protein most similar to L. plantarum VprA was the protein from L. pentosus (89.25% identity) and the least similar was that from L. collinoides (42.32%). Most probably, these proteins could possess vinylphenol reductase activity since, apart from the L. plantarum group, the L. rossiae (21) and L. collinoides (15, 17) strains have been described as ethylphenol producers. The multiple-sequence alignment of these proteins revealed the existence of conserved amino acid motifs. Two conserved domains were selected to design the degenerate oligonucleotides 1657 and 1658, which amplify a 1.3-kb vprA fragment. DNA extracted from 39 lactic acid bacterial strains was used as the template in PCRs, using these degenerate oligonucleotides to detect the presence of vprA-like genes. Figure 5A shows that only the strains belonging to the L. plantarum group produced a PCR-positive response. None of the strains belonging to other lactic acid bacteria species produced the expected PCR fragment. So far, these results seem to indicate that the molecular screening for the presence of a vprA gene copy could result in an adequate method to detect the potential production of ethylphenols. To ascertain this finding, these strains were grown in culture media containing 4-vinylphenol, and their supernatants were analyzed for the production of 4-ethylphenol (Fig. 5B). HPLC results confirmed the PCR results (Table 1). Only bacteria from the L. plantarum group were able to reduce the vinylphenol present in the media. Strains belonging to the genera Lactobacillus (Lactobacillus fermentum, L. brevis, Lactobacillus sakei, and Lactobacillus fructivorans), Enterococcus (Enterococcus durans, Enterococcus faecalis, Enterococcus faecium, Enterococcus casseliflavus, Enterococcus gallinarum, and Enterococcus hirae), and to the Leuconostoc citreum and Streptococcus gallolyticus species did not amplify the expected fragment and did not reduce vinylphenol. Most of these results are in agreement with those reported previously in relation to the inability to produce ethylphenols by L. brevis (14, 15, 19, 22, 37, 38), L. fermentum (14), L. paracollinoides (39, 40), or L. fructivorans (15) strains. Strains from bacterial species not included in this study were also revealed as ethylphenol nonproducers; among these bacteria are Lactobacillus (Lactobacillus paracasei, Lactobacillus casei, Lactobacillus mali, Lactobacillus confusus, Lactobacillus curvatus, Lactobacillus hilgardii, and Lactobacillus reuteri) (14, 15, 21), Pediococcus (Pediococcus acidilactici, Pediococcus parvulus, Pediococcus pentosaceus, and Pediococcus damnosus) (15, 17), and Leuconostoc (Leuconostoc mesenteroides) (17, 21) strains. From all these studies, it could be envisaged that the ability to reduce vinylphenol is an enzymatic activity that is scarcely present among lactic acid bacteria, being less common than the ability to carry out the decarboxylation of hydroxycinnamic acid, the first step of the transformation of these acids.
FIG 5.

Vinylphenol reductase activity in lactic acid bacteria. (A) PCR amplification of the VprA gene. Chromosomal DNA from several lactic acid bacteria was used for PCR amplification with oligonucleotides 1657 and 1658 to amplify 1.3 kb of the vprA gene. PCR products were subjected to gel electrophoresis and stained with GelRed. Left lane, λ/EcoT14I (TaKaRa) molecular size marker. Numbers indicate some of the molecular sizes. (B) HPLC chromatograms of supernatants from lactic acid bacteria grown during 10 days at 30°C in MRS media supplemented with 1.5 mM 4-vinylphenol. The 4-vinylphenol (VP) and 4-ethylphenol (EP) detected are indicated. Chromatograms were recorded at 280 nm. The strains assayed were Enterococcus casseliflavus DSM 20680 (1), E. durans DSM 20633 (2), E. faecalis DSM 20478 (3), E. faecium CECT 4102 (4), E. gallinarum DSM 24841 (5), E. hirae DSM 20160 (6), Lactobacillus brevis CECT 5354 (7), L. fermentum CECT 4007 (8), L. fructivorans CECT 4785 (9), L. paraplantarum DSM 10641 (10), L. plantarum subsp. argentoratensis DSM 16365 (11), L. plantarum subsp. plantarum ATCC 14917 (CECT 748) (12), L. plantarum DSM 10492 (13), L. pentosus DSM 16366 (14), L. sakei subsp. carnosus DSM 15831 (15), Leuconostoc citreum CECT 4025 (16), and Streptococcus gallolyticus UCN34 (17).
In this work, the description of the L. plantarum vinylphenol reductase enzyme identifies the final step of the hydroxycinnamic acid metabolism in lactic acid bacteria (Fig. 6). In plant food substrates, a high proportion of hydroxycinnamic acids are found esterified. The biochemical pathway for the degradation of cell wall hydroxycinnamates starts with the action of esterases, which releases free hydroxycinnamic acids from the naturally esterified forms. In particular, feruloyl esterases are the enzymes involved in the release of hydroxycinnamic acids from plant cell walls. A feruloyl esterase (Lp_0796) has been identified as the esterase responsible for the esterase activity observed on L. plantarum WCFS1 cell extracts (41). Moreover, some L. plantarum strains possess an additional feruloyl esterase (Est_1092) that confers this activity to cell cultures (42). After esterase action, some hydroxycinnamic acids are subsequently metabolized by L. plantarum strains. In culture media, the main transformation of hydroxycinnamic acids is their decarboxylation (Fig. 6). The phenolic acid decarboxylase enzyme (PAD), encoded by the lp_3665 gene, is able to decarboxylate only p-coumaric, caffeic, and ferulic acids (23, 24). Only acids with a para hydroxyl group with respect to the unsaturated side chain and with a substitution of –H, –OH, or –OCH3 in the meta position were decarboxylated. The decarboxylation of p-coumaric, caffeic, and ferulic acids originates their 4-vinyl derivatives (4-vinylphenol, 4-vinylcatechol, and 4-vinylguaiacol, respectively), which are considered food additives and are approved as flavoring agents. Subsequently, L. plantarum strains are able to reduce these vinyl derivatives into their corresponding ethyl derivatives (4-ethylphenol, 4-ethylcatechol, and 4-ethylguaiacol) by the action of the VprA protein (encoded by lp_3125) described in this work (Fig. 6). These compounds are considered the most important flavor components of fermented soy sauce (43), and, on the other hand, are considered off flavor and responsible for sensorial wine and cider alterations (2).
FIG 6.

Schematic representation of hydroxycinnamic acid metabolism in L. plantarum WCFS1. When R1 is –OH and R2 is –H, the represented compounds are p-coumaric acid (B), vinylphenol (C), ethylphenol (D), and phloretic acid (E). The esters (A) are methyl p-coumarate or ethyl coumarate when R3 is –OCH3 or –OCH2CH3, respectively. When R1 is –OH and R2 is –OH, the represented compounds are caffeic acid (B), vinylcatechol (C), ethylcatechol (D), and hydrocaffeic acid (E). The esters (A) are methyl caffeate or ethyl caffeate when R3 is –OCH3 or –OCH2CH3, respectively. When R1 is –OH and R2 is –OCH3, the compounds are ferulic acid (B), vinylguaiacol (C), ethylguaiacol (D), and hydroferulic acid (E). The esters (A) are methyl ferulate or ethyl ferulate when R3 is –OCH3 or –OCH2CH3, respectively. When R1 is –H and R2 is –OH, only the reduction reaction is carried out, and the represented compounds are m-coumaric acid (B) and 3-(3-hydroxyphenyl) propionic acid (E).
In addition, L. plantarum possesses an alternative pathway to transform hydroxycinnamic acids (Fig. 6). Some of these acids could be reduced to their corresponding substituted phenylpropionic acids by the action of recently described HcrAB proteins (encoded by lp_1424 and lp_1425 genes) (28). m-Coumaric, o-coumaric, p-coumaric, caffeic, ferulic, and sinapic acids are also reduced by HcrAB to originate 3-(3-hydroxyphenyl) propionic, melilotic, phloretic, hydrocaffeic, hydroferulic, and hydrosinapic acids, respectively (28). These reduced acids were not further degraded by L. plantarum strains. The biotransformation pathways described in this work are the metabolic strategies followed by L. plantarum to tolerate the hostile environment generated by the presence of hydroxycinnamic acids.
MATERIALS AND METHODS
Strains and growth conditions.
In this study, 26 L. plantarum strains were analyzed. L. plantarum WCFS1, used through this study, was kindly provided by Michiel Kleerebezem (NIZO Food Research, The Netherlands) and L. plantarum NC8 by L. Axelsson (Norwegian Institute of Food, Fisheries and Aquaculture Research, Norway). Several strains were purchased from the Spanish Type Culture Collection (CECT), namely L. plantarum CECT 220 (ATCC 8014), CECT 221 (ATCC 14431), CECT 223, CECT 224, CECT 749 (ATCC 10241), CECT 4645 (NCFB 1193), and the type strain L. plantarum subsp. plantarum CECT 748T (ATCC 14917, DSMZ 20174), or from the German Collection of Microorganisms and Cell Cultures (DSMZ) (L. plantarum DSM 1055, DSM 2648, DSM 10492, DSM 13273, DSM 20246, and the type strain L. plantarum subsp. argentoratensis DSM 16365T). L. plantarum RM28, RM31, RM35, RM38, RM39, RM40, RM41, RM71, RM72, and RM73 were isolated from must grape or wine from different wine-producing areas of Spain over the period from 1998 to 2001 (44). Strains from other lactic acid bacterial species were also purchased from the DSMZ or CECT collections, Lactobacillus paraplantarum (DSM 10641 [ATCC 10776] and DSM 10667T), Lactobacillus pentosus (DSM 16366, DSM 20314, and DSM 20199), Enterococcus casseliflavus (DSM 20680), E. durans (DSM 20633), E. faecalis (DSM 20478), E. faecium (CECT 4102 and DSM 20477), E. gallinarum (DSM 24841), E. hirae (DSM 20160), Lactobacillus brevis (CECT 5354, CECT 216, and CECT 4121), L. fermentum (CECT 4007), L. fructivorans (CECT 4785), L. sakei (DSM 15831), and Leuconostoc citreum (CECT 4025 and CECT 4700). Philipe Glaser (Institut Pasteur, France) kindly provided Streptococcus gallolyticus subsp. gallolyticus strain UCN34 (CIP 110142).
Escherichia coli DH10B was used for DNA manipulations. E. coli BL21(DE3) was used for expression in the pURI3-Cter vector (34). E. coli strains were cultured in Luria-Bertani (LB) medium at 37°C and 140 rpm. When required, ampicillin was added to the medium at concentrations of 100 μg/ml.
MRS broth was used to routinely grow lactic acid bacteria. To assay vinylphenol reductase activity, MRS media was supplemented with vinylphenols (1.5 mM final concentration). Where appropriate, erythromycin was added to the culture medium at 10 μg/ml and lincomycin was added at 100 μg/ml. The inoculated MRS media were incubated at 30°C in the dark, without shaking, for 7 days.
Disruption of L. plantarum vpr genes.
Insertion-duplication mutagenesis was employed to knock out vpr genes. Internal fragments from the vprR (lp_3124) and vprA (lp_3125) genes were cloned into the pUCE191 suicide vector (45) by using primers described in Table 1. pUCE191-derivative plasmids were used to transform L. plantarum WCFS1 competent cells by electroporation (46). L. plantarum transformants were selected by plating with 100 μg/ml lincomycin and 10 μg/ml erythromycin. PCR analysis was used to check the correct insertion of the donor pUCE191-derivative plasmid into the L. plantarum WCFS1 chromosome (Table 1).
RNA isolation, reverse transcription-PCR, and quantitative PCR.
L. plantarum WCFS1 MRS cultures were grown to an optical density at 600 nm (OD600) of 0.8 to 0.9 and then supplemented with 4-vinylphenol or p-coumaric acid at 1.5 mM final concentration. After 10 min of incubation, the cultures were immediately processed for RNA extraction, and, after DNase I treatment, the DNA-free RNA was retrotranscribed (28). The cDNA obtained in the presence of 4-vinylphenol was used for RT-PCR experiments. The lp_3123, vprR (lp_3124), vprA (lp_3125), and lp_3127 genes as well as the lp_3123-vprR, vprR-vprA, and vprA-lp_3127 intergenic regions were analyzed by PCR. PCR amplifications were performed as previously described (28).
Quantitative gene expression analyses were performed in an AbiPrism 7500 fast real-time PCR system, using the L. plantarum ldh gene as the endogenous gene (47) and growth in the absence of hydroxycinnamic derivative as the growth condition calibrator. Results were analyzed using the comparative threshold cycle (CT) method (also named the 2ΔΔCT method) (28).
Production and purification of L. plantarum VprA.
The vprA gene from L. plantarum WCFS1 was amplified by PCR using the 1516 and 1517 primer pair. The purified PCR fragment was inserted into the pURI3-Cter vector using a restriction enzyme- and ligation-free cloning strategy described previously (34). The vectors produce recombinant proteins having a six-histidine affinity tag in their C termini. E. coli DH10B cells were transformed, recombinant plasmids were isolated, and plasmids containing the correct insert were identified by restriction enzyme analysis, verified by DNA sequencing, and transformed into E. coli BL21(DE3) cells for expression.
E. coli BL21(DE3) was transformed with the pURI3-Cter-vprA recombinant plasmid. E. coli cells were grown at 22°C in LB medium containing 1 M d-sorbitol, 2.5 mM glycine betaine, and 0.25 mM isopropyl-β-d-thiogalactopyranoside (IPTG) to prevent formation of inclusion bodies (48). The cells were disrupted by French press lysis, and the insoluble fraction of the lysate was removed by centrifugation at 47,000 × g for 30 min at 4°C. The filtered supernatant containing VprA protein was analyzed for reductase activity by adding 1.5 mM 4-vinylphenol and incubating at 37°C during 16 h. VprA protein was purified for affinity chromatography in a Talon Superflow resin (Clontech), and the purity of the enzyme was determined by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) in Tris-glycine buffer.
Vinylphenol reductase activity assay.
Fractions containing the His6-tagged proteins were pooled and analyzed for vinylphenol reductase activity. Eluted purified VprA protein (500 μg) was incubated at 37°C for 18 h in the presence of a vinylphenol (4-vinylphenol, and 4-vinylguaiacol) (1.5 mM) and 15 mM NAD(P)H. To test vinylcatechol reductase activity, vinylcatechol was biotechnologically produced from caffeic acid using recombinant L. plantarum PDC protein (24). The phenolic compounds present in the reaction were extracted by ethyl acetate and analyzed by HPLC with diode array detection (DAD) as described previously (24).
PCR detection of hydroxycinnamate reductase activity.
Genes encoding VprA proteins were amplified by PCR using chromosomal DNA from several lactic acid bacterial strains. Amplifications were performed by using degenerate primers 1657 and 1658. The reactions were performed using 30 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 1 min, and extension at 72°C for 30 s. The expected size of the amplicon was 1.3 kb.
Sequence data analyses.
A homology search with finished and unfinished microbial genome databases was performed with the BLAST algorithm at the National Center for Biotechnology Information served (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Multiple alignments were made using the Clustal Omega program (http://www.ebi.ac.uk/Tools/msa/clustalo/) on the European Bioinformatics Institute (EBI) site, after retrieval of sequences from the GenBank and Swiss-Prot databases. Computer promoter predictions were carried out at the Berkeley Drosophila Genome Project website (http://fruitfly.org/seq_tools/promoter.html), and predicted transcription terminators were analyzed at the ARNold site (http://rna.igmors.u-psud.fr/toolbox/arnold/index.php).
Statistical analysis.
A two-tailed Student's t test performed using GraphPad InStat version 3.0 (GraphPad Software, San Diego, CA) was used to determine the differences between means. The data are representative means of at least three independent experiments.
Supplementary Material
ACKNOWLEDGMENTS
We thank J. M. Mancheño for the identification of the flavin cofactor by mass spectrometry and for critical reading of the manuscript. We are grateful to M. V. Santamaría and J. M. Barcenilla for their assistance.
This work ws supported by grant AGL2014-52911-R (AEI/FEDER, UE) (MINEICO). L.S. is the recipient of an FPI Fellowship from the MINEICO.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01064-18.
REFERENCES
- 1.Tempère S, Cuzange E, Schaaper MH, de Lescar R, de Revel G, Sicard G. 2014. “Brett character” in wine? Is there a consensus among professional assessors? A perceptual and conceptual approach. Food Qual Pref 34:29–36. [Google Scholar]
- 2.Chatonnet P, Dubordieu D, Boidron JN, Pons M. 1992. The origin of ethylphenols in wines. J Sci Food Agric 60:165–178. doi: 10.1002/jsfa.2740600205. [DOI] [Google Scholar]
- 3.Buron N, Coton M, Legendre P, Ledauphin J, Kientz-Bouchart V, Guichard H, Barillier D, Coton E. 2012. Implications of Lactobacillus collinoides and Brettanomyces/Dekkera anomala in phenolic off-flavour defects of ciders. Int J Food Microbiol 153:159–165. doi: 10.1016/j.ijfoodmicro.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 4.Suarez R, Suarez-Lepe JA, Morata A, Calderón F. 2007. The production of ethylphenols in wine by yeasts of the genera Brettanomyces and Dekkera. A review. Food Chem 102:10–21. doi: 10.1016/j.foodchem.2006.03.030. [DOI] [Google Scholar]
- 5.Shahidi F, Naczk M. 2003. Phenolics in food and nutraceuticals. CRC Press, London, United Kingdom. [Google Scholar]
- 6.Edlin DAN, Narbad A, Gasson MJ, Dickinson JR, Lloyd D. 1998. Purification and characterization of hydroxycinnamate decarboxylase from Brettanomyces anomalus. Enzyme Microb Tech 22:232–239. doi: 10.1016/S0141-0229(97)00169-5. [DOI] [Google Scholar]
- 7.Godoy L, Martínez C, Carrasco N, Ganga MA. 2008. Purification and characterization of a p-coumarate decarboxylase and a vinylphenolreductase from Brettanomyces bruxellensis. Int J Food Microbiol 127:6–11. doi: 10.1016/j.ijfoodmicro.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 8.Harris V, Ford CM, Jiranek V, Grbin PR. 2009. Survey of enzyme activity responsable for phenolic off-flavour production by Dekkera and Brettanomyces yeast. Appl Microbiol Biotechnol 81:1117–1127. doi: 10.1007/s00253-008-1708-7. [DOI] [PubMed] [Google Scholar]
- 9.Tchobanov I, Gal L, Guilloux-Benatier M, Remize F, Nardi T, Guzzo J, Serpaggi V, Alexandre H. 2008. Partial vinylphenol reductase purification and characterization from Brettanomyces bruxellensis. FEMS Microbiol Lett 284:213–217. doi: 10.1111/j.1574-6968.2008.01192.x. [DOI] [PubMed] [Google Scholar]
- 10.Granato TM, Romano D, Vigentini I, Foschino RC, Monti D, Mamone G, Ferranti P, Nitride C, Iametti S, Bonomi F, Molinari F. 2015. New insights on the features of the vinyl phenol reductase from the wine-spoilage yeast Dekkera/Brettanomyces bruxellensis. Ann Microbiol 65:321–329. [Google Scholar]
- 11.Romano D, Valdetara F, Zambelli P, Galafassi S, De Vitis V, Molinari F, Compagno C, Foschino R, Vigentini I. 2017. Cloning the putative gene of vinyl phenol reductase of Dekkera bruxellensis in Saccharomyces cerevisiae. Food Microb 63:92–100. doi: 10.1016/j.fm.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 12.Rayne S, Eggers NJ. 2007. 4-Ethylphenol and 4-ethylguaiacol in wines: estimating non-microbial sourced contributions and toxicological considerations. J Environ Sci Health B 42:887–897. doi: 10.1080/03601230701623365. [DOI] [PubMed] [Google Scholar]
- 13.Buron N, Coton M, Desmarais C, Ledauphin J, Guichard H, Barillier D, Coton E. 2011. Screening of representative cider yeasts and bacteria for volatile phenol-production ability. Food Microbiol 28:1243–1251. doi: 10.1016/j.fm.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 14.van Beek S, Priest FG. 2000. Decarboxylation of substituted cinnamic acids by lactic acid bacteria isolated during malt whisky fermentation. Appl Environ Microbiol 66:5322–5328. doi: 10.1128/AEM.66.12.5322-5328.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Couto JA, Campos FM, Figueiredo AR, Hogg TA. 2006. Ability of lactic acid bacteria to produce volatile phenols. Am J Enol Vitic 57:166–171. [Google Scholar]
- 16.de las Rivas B, Rodríguez H, Curiel JA, Landete JN, Muñoz R. 2009. Molecular screening of wine lactic acid bacteria degrading hydroxycinnamic acids. J Agric Food Chem 57:490–494. doi: 10.1021/jf803016p. [DOI] [PubMed] [Google Scholar]
- 17.Silva I, Campos FM, Hogg T, Couto JA. 2011. Wine phenolic compounds influence the production of volatile phenols by wine-related lactic acid bacteria. J Appl Microbiol 111:360–370. doi: 10.1111/j.1365-2672.2011.05052.x. [DOI] [PubMed] [Google Scholar]
- 18.Silva I, Campos FM, Hogg T, Couto JA. 2011. Factors influencing the production of volatile phenols by wine lactic acid bacteria. Int J Food Microbiol 145:471–475. doi: 10.1016/j.ijfoodmicro.2011.01.029. [DOI] [PubMed] [Google Scholar]
- 19.Buron N, Guichard H, Coton E, Ledauphin J, Barillier D. 2011. Evidence of 4-ethylcatechol as one of the main phenolic off-flavour markers in French ciders. Food Chem 125:542–548. doi: 10.1016/j.foodchem.2010.09.046. [DOI] [Google Scholar]
- 20.Fras P, Campos FM, Hogg T, Couto JA. 2014. Production of volatile phenols by Lactobacillus plantarum in wine conditions. Biotechnol Lett 36:281–285. doi: 10.1007/s10529-013-1351-y. [DOI] [PubMed] [Google Scholar]
- 21.Filannino P, Gobbetti M, de Angelis M, di Cagno R. 2014. Hydroxycinnamic acids used as external acceptos of electrons: an energetic advantage for strictly heterofermentative lactic acid bacteria. Appl Environ Microbiol 80:7574–7582. doi: 10.1128/AEM.02413-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Filannino P, Bai Y, di Cagno R, Gobbetti M. 2015. Metabolism of phenolic compounds by Lactobacillus spp. during fermentation of cherry juice and broccoli puree. Food Microbiol 46:272–279. doi: 10.1016/j.fm.2014.08.018. [DOI] [PubMed] [Google Scholar]
- 23.Cavin JF, Barthelmebs L, Guzzo J, van Breeumen J, Samyn R, Travers JF, Diviès C. 1997. Purification and characterization of an inducible p-coumaric acid decarboxylase from Lactobacillus plantarum. FEMS Microbiol Lett 147:291–295. doi: 10.1111/j.1574-6968.1997.tb10256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodríguez H, Landete JM, Curiel JA, de las Rivas B, Mancheño JM, Muñoz R. 2008. Characterization of the p-coumaric acid decarboxylase from Lactobacillus plantarum CECT 748T. J Agric Food Chem 56:3068–3072. doi: 10.1021/jf703779s. [DOI] [PubMed] [Google Scholar]
- 25.Chatonnet P, Dubordieu D, Biodron JN. 1995. The influence of Brettanomyces/Dekkera sp. yeasts and lactic acid bacteria on the ethylphenol content of red wines. Am J Enol Vitic 46:463–468. [Google Scholar]
- 26.Chatonnet P, Viala C, Dubordieu D. 1997. Influence of polyphenolic components of red wines on the microbial synthesis of volatile phenols. Am J Enol Vitic 48:443–448. [Google Scholar]
- 27.Barthelmebs L, Diviès C, Cavin J-F. 2000. Knockout of the p-coumarate decarboxylase gene from Lactobacillus plantarum reveals the existence of two other inducible enzymatic activities involved in phenolic acid metabolism. Appl Environ Microbiol 66:3368–3375. doi: 10.1128/AEM.66.8.3368-3375.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Santamaría L, Reverón I, López de Felipe F, de las Rivas B, Muñoz R. 2018. Unravelling the reduction pathway as alternative metabolic route to hydroxycinnamate decarboxylation in Lactobacillus plantarum. Appl Environ Microbiol 84. doi: 10.1128/AEM.01123-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reverón I, de las Rivas B, Muñoz R, López de Felipe F. 2012. Genome-wide transcriptomic responses of a human isolate of Lactobacillus plantarum exposed to p-coumaric acid stress. Mol Nutr Food Res 56:1848–1859. doi: 10.1002/mnfr.201200384. [DOI] [PubMed] [Google Scholar]
- 30.White RH. 2006. The difficult road from sequence to function. J Bacteriol 188:3431–3432. doi: 10.1128/JB.188.10.3431-3432.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tropel D, van der Meer JR. 2004. Bacterial transcriptional regulators for degradation pathways of aromatic compounds. Microbiol Mol Biol Rev 68:474–500. doi: 10.1128/MMBR.68.3.474-500.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maddocks SE, Oyston CF. 2008. Structure and fuction of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology 154:3609–3623. doi: 10.1099/mic.0.2008/022772-0. [DOI] [PubMed] [Google Scholar]
- 33.Pandee SG, Pagliari FA, Gardner CL, Wrench A, Narvel R, Gonzalez CF, Lorca GL. 2011. Lactobacillus brevis responds to flavonoids through KaeR, a LysR-type of transcriptional regulator. Mol Microbiol 81:1623–1639. doi: 10.1111/j.1365-2958.2011.07796.x. [DOI] [PubMed] [Google Scholar]
- 34.Curiel JA, de las Rivas B, Mancheño JM, Muñoz R. 2011. The pURI family of expression vectors: a versatile set of ligation independent cloning plasmids for producing recombinant His-fusion proteins. Protein Expr Purif 76:44–53. doi: 10.1016/j.pep.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 35.Rodríguez H, Landete JM, de las Rivas B, Muñoz R. 2008. Metabolism of food phenolic acids by Lactobacillus plantarum CECT 748T. Food Chem 107:1393–1398. doi: 10.1016/j.foodchem.2007.09.067. [DOI] [Google Scholar]
- 36.Diako C, Vixie B, Weller KM, Dycus DA, Ross CF. 2017. Determination of 4-ethylcatechol in a Merlot wine using sensory evaluation and the electronic tongue. Int J Food Sci Technol. 52:2489–2496. [Google Scholar]
- 37.Cavin JF, Andioc V, Etievant PX, Divies C. 1993. Ability of wine lactic acid bacteria to metabolize phenol carboxylic acids. Am J Enol Vitic 44:76–80. [Google Scholar]
- 38.Curiel JA, Rodríguez H, Landete JM, de las Rivas B, Muñoz R. 2010. Ability of Lactobacillus brevis strains to degrade food phenolic acids. Food Chem 120:225–229. doi: 10.1016/j.foodchem.2009.10.012. [DOI] [Google Scholar]
- 39.Whitting GC, Carr JG. 1959. Metabolism of cinnamic acid and hydroxycinnamic acids by Lactobacillus pastorianus var. quinicus. Nature 4696(Suppl 18):1427–1428. doi: 10.1038/1841427a0. [DOI] [PubMed] [Google Scholar]
- 40.Ehrmann MA, Vogel RF. 2005. Taxonomic note “Lactobacillus pastorianus” (Van Laer, 1892) a former synonym for Lactobacillus paracollinoides. Syst Appl Microbiol 28:54–56. doi: 10.1016/j.syapm.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 41.Esteban-Torres M, Reverón I, Mancheño JM, de las Rivas B, Muñoz R. 2013. Characterization of a feruloyl esterase from Lactobacillus plantarum. Appl Environ Microbiol 79:5130–5136. doi: 10.1128/AEM.01523-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Esteban-Torres M, Landete JM, Reverón I, Santamaría L, de las Rivas B, Muñoz R. 2015. A Lactobacillus plantarum esterase active on a broad range of phenolic esters. Appl Environ Microbiol 81:3235–3242. doi: 10.1128/AEM.00323-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yokosuka Y. 1986. Soy sauce biochemistry. Adv Food Res 30:196–220. [DOI] [PubMed] [Google Scholar]
- 44.Moreno-Arribas MV, Polo MC, Jorganes F, Muñoz R. 2003. Screening of biogenic amine production by lactic acid bacteria isolated from grape must and wine. Int J Food Microbiol 84:117–123. doi: 10.1016/S0168-1605(02)00391-4. [DOI] [PubMed] [Google Scholar]
- 45.Arrecubieta C, García E, López R. 1995. Sequence and transcriptional analysis of a DNA region involved in the production of capsular polysaccharide in Streptococcus pneumoniae type 3. Gene 167:1–7. doi: 10.1016/0378-1119(95)00657-5. [DOI] [PubMed] [Google Scholar]
- 46.Aukrust T, Blom H. 1992. Transformation of Lactobacillus strains used in meat and vegetable fermentations. Food Res Int 25:253–261. doi: 10.1016/0963-9969(92)90121-K. [DOI] [Google Scholar]
- 47.Fiocco D, Crisetti E, Capozzi V, Spano G. 2008. Validation of an internal control gene to apply reverse transcription quantitative PCR to study heat, cold and ethanol stresses in Lactobacillus plantarum. World J Microbiol Biotechnol 24:899–902. doi: 10.1007/s11274-007-9556-7. [DOI] [Google Scholar]
- 48.Ackerley DF, González CF, Park CH, Blake R, Keyhan M, Martin A. 2004. Chromate-reducing properties of soluble flavoproteins from Pseudomonas putida and Escherichia coli. Appl Environ Microbiol 70:873–882. doi: 10.1128/AEM.70.2.873-882.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.