

Abstract
Molecularly imprinted polymers (MIPs) with selective affinity for protein biomarkers could find extensive utility as environmentally robust, cost-efficient biomaterials for diagnostic and therapeutic applications. In order to develop recognitive, synthetic biomaterials for prohibitively expensive protein biomarkers, we have developed a molecular imprinting technique that utilizes structurally similar, analogue proteins. Hydrogel microparticles synthesized by molecular imprinting with trypsin, lysozyme, and cytochrome c possessed an increased affinity for alternate high isoelectric point biomarkers both in isolation and plasma-mimicking adsorption conditions. Imprinted and non-imprinted P(MAA-co-AAm-co-DEAEMA) microgels containing PMAO-PEGMA functionalized polycaprolactone nanoparticles were net-anionic, polydisperse, and irregularly shaped. MIPs and control non-imprinted polymers (NIPs) exhibited regions of Freundlich and BET isotherm adsorption behavior in a range of non-competitive protein solutions, where MIPs exhibited enhanced adsorption capacity in the Freundlich isotherm regions. In a competitive condition, imprinting with analogue templates (trypsin, lysozyme) increased the adsorption capacity of microgels for cytochrome c by 162% and 219%, respectively, as compared to a 122% increase provided by traditional bulk imprinting with cytochrome c. Our results suggest that molecular imprinting with analogue protein templates is a viable synthetic strategy for enhancing hydrogel-biomarker affinity and promoting specific protein adsorption behavior in biological fluids.
Keywords: protein adsorption, molecular imprinting, biomimetics, intelligent hydrogels
INTRODUCTION
In recent years, molecularly imprinted polymers (MIPs) have been suggested as a synthetic alternative to antibodies and antibody-conjugates for medical applications including diagnostic biosensing,1 targeted drug delivery,2 regenerative medicine.3 Compared to antibodies, synthetic polymers are robust to environmental conditions, highly reproducible, and inexpensive.4 Therefore, the development of a synthetic MIP system with specificity for disease biomarkers can make a significant impact in the medical field, particularly in global regions where advanced healthcare infrastructure is lacking.5
MIPs are crosslinked networks formed in the presence of a molecular template, which in this case is a protein. A critical aspect of MIP purification is template extraction with a series of solvents to leave void nanocavities that subsequently recognize the template in biological fluids.6 These extraction conditions are traditionally denaturing, which prevents the collection and recycling of protein templates. Consequently, it is not cost-effective to fabricate MIP systems via traditional synthesis regimes for expensive protein markers with extensive biological relevance, such as membrane bound proteins, cytokines, or growth factors.7
Research efforts toward the development of intelligent MIP networks for diagnostic and therapeutic applications have largely employed two approaches. The first and most common approach is to synthesize, optimize, and characterize different MIPs with model templates.8 This approach has led to numerous advancements in sophisticated MIP polymerizations, including but not limited to bulk and surface imprinting through free radical and living polymerizations9 in nanoparticles,10 microparticles,11 and films.12 However, these systems are not designed to recognize expensive non-model biomarkers with great therapeutic relevance. The second approach entails epitope imprinting, where a peptide fragment is utilized as an imprinting template.10,13 These epitopes are fabricated through peptide synthesis methods, and the resultant MIPs possess elevated affinity for the native protein through epitope recognition. This approach has the upside of enabling MIPs for diverse protein biomarkers, but does not create nanocavities with whole-protein geometry.
Our approach was motivated by advancements in small molecule imprinting for chromatography applications. MIP sorbents were prepared with a small molecule template that shared key characteristics with the molecule that the researchers sought to eventually extract and collect.14 These MIP materials subsequently possessed elevated affinity for the target molecule, enabling its separation from complex solutions.
We hypothesized that MIPs formed in the presence of rationally selected, low-cost analogue proteins with similar geometries, molecular weights, and isoelectric points to biomarkers of interest would possess similar affinity for the biomarker of interest as compared to MIPs formed by traditional bulk imprinting methods. We demonstrated the feasibility of this approach, detailed herein, through the synthesis of MIPs for three proteins (Table I), which served as a model library of high isoelectric point, low molecular weight biomarkers. High isoelectric point proteins currently administered for therapeutic purposes include, but are not limited to, erythropoietin, calcitonin, interferon-β, vascular endothelial growth factor (VEGF), and bone morphogenic protein-2 (BMP-2). Increasing a material’s affinity for one of these therapeutic proteins through the inclusion of a low-cost analogue template would dramatically improve its utility as a component of a biosensor,1 controlled release system,2 or engineered tissue construct.3
TABLE I.
Physiochemical Properties, and Labeling Convention for Protein Templates
| Protein | Trypsin | Lysozyme | Cytochrome c |
|---|---|---|---|
| MW (kDa) | 24.0 | 14.3 | 12.4 |
| pI | 10.1 | 11.35 | 10.5 |
| Geometry | Globular | Globular | Globular |
| Polymer Label | TMIP | LMIP | CMIP |
MATERIALS AND METHODS
Materials
All reagents and solvents were purchased from Sigma Aldrich or Fisher Scientific and used as received. Bovine serum albumin, fibrinogen, gamma globulins, hemoglobin, cytochrome c, and trypsin, as well as lysozyme and trypsin inhibitor type II from chicken egg white were purchased as lyophilized powders from Sigma Aldrich and used as received. Poly(maleic anhydride-alt–1-octadecene)-g-poly (ethylene glycol) methacrylate (PMAO-PEGMA) was prepared as described by Culver et al.15 Ultrapure water (18.2 MΩ) was obtained from a Barnstead GenPure purification system from Thermo Scientific.
Instrumentation and analysis
Dynamic light scattering and zeta potential measurements were obtained with a ZetaSizer Nano-ZS (Malvern). TEM imaging was conducted on a FEI Tecnai Transmission Electron Microscope operating at 80 kV. FTIR spectra were acquired on a Nicolet is10 FT-IR spectrometer, and analyzed in OMNIC software (Thermo Scientific). A Hanna Instruments HI 902 potentiometric titrator was utilized for autotitration of microparticle suspensions. All absorbance and fluorescence measurements in protein quantification assays were performed with a Cytation 3 Cell Imaging Multi-Mode Plate Reader (Bio Tek Instruments). Data and statistical analyses were performed using MATLAB (Mathworks) or PRISM (GraphPad).
Fabrication of PMAO-PEGMA functionalized polycaprolactone nanoparticles
Polycaprolactone (PCL) (Mn~10,000) (10 mg/mL) in acetone was added dropwise at 500 μL/min to a rapidly stirring (1000 rpm) solution of PMAO-PEGMA (5 mg/mL) in ultrapure water using a Harvard Apparatus syringe pump until a final mass ratio of 4:5 (PCL:PMAO-PEGMA) was achieved. PCL nanoparticles (PCL-NPs), stabilized with PMAO-PEGMA, formed spontaneously by nanoprecipitation. Solvent exchange was achieved by rotary evaporation of acetone and dialysis against ultrapure water (frequent water changes) for 72 h.15
Synthesis of imprinted and non-imprinted microgels
All values in parentheses represent final concentration in the pre-polymer solution. Methacrylic acid (MAA) (12.5 mM), (diethylamino)ethyl methacrylate (DEAEMA) (12.5 mM), acrylamide (AAm) (22.5 mM), and methylenebisacrylamide (MBA) (2.5 mM) were dissolved in 0.1 × phosphate buffered saline (PBS) and sonicated for 10 min to ensure a uniform solution. Following sonication, 45 mg of PCL-NPs, 25 mg of template (cytochrome c, lysozyme, or trypsin), and 15 μL of N,N,N′,N′-tetramethylethylenediamine (TEMED) were added to the monomer solution (final volume = 45 mL).
Following 30 min of nitrogen purging to remove dissolved oxygen, each pre-polymer solution was allowed to mix at room temperature for 30 min to allow template-monomer self-assembly. An initiator solution of ammonium persulfate (APS) (5 mL, 10 mg/mL) in 0.1× PBS was purged with nitrogen for 10 min and injected into each pre-polymer solution immediately following self-assembly. Reactions were allowed to proceed under rapid stirring (500 rpm) at room temperature for 16 h (Fig. 1). Non-imprinted microgels (NIPs) were prepared in parallel to MIPs using the same synthesis protocol, excluding the protein template. Following polymerization, 2.9 g of sodium chloride was added to each polymer solution and MIPs and NIPs were collected by centrifugation at 3200 g for 10 min. MIPs and NIPs were suspended in 6 mL of ultrapure water and transferred to microcentrifuge tubes. Template removal was achieved by four purification cycles, where each cycle consisted of resuspension, sonication, pelleting, and supernatant removal in 10 vol% acetic acid, followed by 1× PBS, and then ultrapure water. To enhance template removal from the microparticles, two of the four acetic acid washes were allowed to mix overnight with constant agitation (300 rpm). Following the completion of all four cycles, 12 washes in total, microparticle suspensions were dialyzed against ultrapure water for 72 h (frequent water changes) for solvent exchange, and were lyophilized.
FIGURE 1.
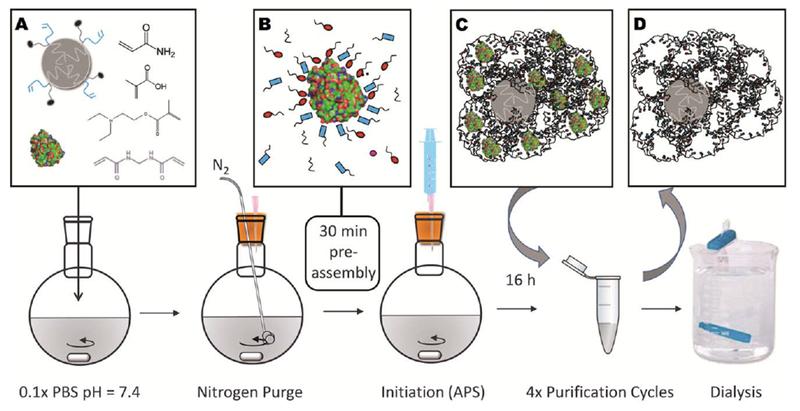
(a) PCL-NP, template protein, and functional monomers were dissolved in phosphate buffer and were (b) allowed to self-assemble for 30 min to form complementary hydrostatic and electrostatic interactions. In the graphic, the red ovals and blue rectangles represent the anionic, cationic, or otherwise functional moieties, which are contributed by each monomer. (c) Polymerized microparticles contained entrapped template, which was extracted with four purification cycles using 10% acetic acid, PBS, and ultrapure water, (d) leaving MIPs with void nanocavities. Dialysis was utilized for solvent exchange into water prior to lyophilization.
Analysis of bulk microgel properties
For DLS and zeta-potential measurements, MIPs and NIPs were suspended in ultrapure water at a final concentration of 0.5 mg/mL. Values presented are the average of three measurements. For TEM, MIPs and NIPs were suspended in 0.1× PBS at 0.5 mg/mL, cast on a carbon-coated grid, and stained with uranyl acetate. Representative images are presented, to illustrate PCL-NP incorporation, as well as microparticle dispersity and morphology.
Characterization of microgel composition
Following lyophilization, FTIR spectra were obtained for the dried powders from 4000 to 680 cm−1. Absorbance spectra presented are the average of 128 scans, with the background subtracted, and are normalized to the carbonyl peak appearing at 1732 cm−1. For composition analysis by potentiometric titration, dried microgels were suspended in 5 mM KCl, adjusted to pH = 11 and allowed to equilibrate overnight. Microgels were titrated with 0.01 N HCl until an equilibrium pH of 3.25 was reached. The normality of the HCl titrant was established by titration with a standardized NaOH buffer. For analysis, the moles of titrant were normalized to the polymer dry weight. The relative incorporation of functional monomers was quantified as the mole ratio of titrant required to pass through the equivalence point for each monomer in the polymer (pH = 8.5 DEAEMA, pH = 4.8 MAA), plus or minus one pH unit. Remaining template entrapped in MIP microgels was quantified using a Micro BCA colorimetric assay, where the NIP absorbance was subtracted from each MIP signal as polymer background and this corrected signal was compared to a template standard.
Assessment of non-competitive protein adsorption
MIPs and NIPs were suspended in 0.1× PBS, and adjusted to pH = 7.4 immediately prior to each adsorption experiment. Each formulation of MIPs and NIPs at a concentration of 0.3 mg/mL were incubated individually with lysozyme,16 trypsin,17 cytochrome c,18 trypsin inhibitor type II,19 BSA,18 and hemoglobin20 (Table II) in pH = 7.4, 0.1× PBS at initial concentrations from 0.25 to 2 mg/mL. Following one-hour incubation with constant orbital mixing, microparticles and adsorbed proteins were separated from free molecules by centrifugation at 15,000 g for 7 min. Free protein was quantified by direct absorbance, relative to a standard of known concentration, in 0.1× PBS at 280 nm (trypsin, lysozyme), 405 nm (cytochrome c, hemoglobin), and by a Micro BCA colorimetric assay (trypsin inhibitor type II, BSA). The equilibrium adsorption capacity (Q) was calculated using equation 1, where Co and Ce are the initial and equilibrium protein concentrations (varied), V is the solution volume (0.6 mL), and m is the MIP or NIP mass (0.00018 g). Each adsorption assay was performed in triplicate.
TABLE II.
Physiochemical Properties of Model Proteins Studied in Noncompetitive and Competitive Adsorption Assays
| Protein | Lysozyme | Trypsin | Cytochrome c | Hemoglobin | Trypsin Inhibitor | BSA |
|---|---|---|---|---|---|---|
| MW (kDa) | 14.3 | 24.0 | 12.4 | 60.2 | 28.0 | 66.5 |
| pI | 11.35 | 10.1 | 10.5 | 6.8 | 4.9 | 4.7 |
| (1) |
Evaluation of competitive protein adsorption
Two competitive adsorption experiments were performed. To enable their detection and quantification in competitive conditions, trypsin and trypsin inhibitor type II were labeled with fluorescein isothiocyanate (FITC). Each protein (10 mg/mL) in 0.1M bicarbonate buffer was mixed with FITC (10 mg/mL in DMF) at a 10:1 volume ratio (protein: FITC). The reaction was quenched after 1 h with 100 μL of 1.5 M hydroxylamine, and labeled protein was purified by dialysis against 1× PBS at 4°C for 72 h, followed by ultrapure water for 24 h.
The purpose of the first competitive assay was to assess the adsorption of proteins of various isoelectric points in the presence of a high concentration of competing proteins. A “serum buffer” consisting of 40 mg/mL cocktail of BSA (61%), gamma globulins (35%) and fibrinogen (4%) in pH = 7.4, 0.1× PBS was chosen to mimic the protein distribution in human blood plasma.21 All values in parenthesis signify the percent of total protein by mass. Cytochrome c, hemoglobin, and FITC-trypsin inhibitor were chosen as model high, neutral, and low isoelectric point proteins for adsorption, and were incubated with MIPs or NIPs at an initial concentration of 0.25 mg/mL in serum buffer for 1 h. Supernatants were collected as described above, and free cytochrome c or hemoglobin was quantified by absorbance at 405 nm, while FITC-trypsin inhibitor was quantified by absorbance at 490 nm.
The second competitive assay determined the relative adsorption of trypsin, lysozyme, and cytochrome c to MIPs and NIPs when incubated simultaneously. Lysozyme, cytochrome c, and FITC-trypsin (1:1:1 mass ratio, 0.75 mg/mL total protein) were incubated with MIPs or NIPs in pH = 7.4, 0.1× PBS. Following one-hour incubation, the absorption spectrum of the supernatant from 260–600 nm was measured, and the equilibrium concentrations of lysozyme, cytochrome c, and fluorescent trypsin were determined from the deconvolution of the supernatant absorbance spectrum, relative to protein standards. In addition to the calculation of adsorption capacity, imprinting factor (IF) and selectivity factor (a) were also calculated by Eqs. 2 and 3:
| (2) |
| (3) |
IF calculations demonstrate the relative increase in adsorption capacity for a given protein imparted by molecular imprinting in a competitive or non-competitive condition, whereas the selectivity factor reveals the relative adsorption of one protein over another in a competitive state.
RESULTS
Microgel synthesis
Microgels containing PCL nanoparticles were successfully synthesized as previously described.22 PMAO-PEGMA functionalized PCL nanoparticles can encapsulate a therapeutic agent or fluorophore,15 and were synthetically incorporated to make the microgel system readily translatable for theranostic applications. The polymerization was conducted in 0.1× PBS to provide ionic strength and buffering capacity to protect the templates’ natural tertiary structure. 0.1× PBS was selected specifically, as opposed to a buffer with greater ionic strength, in order to prevent shielding of template-monomer electrostatic interactions.23 Monomer concentration was kept intentionally low (45 mM) to mitigate protein denaturation, which could occur in concentrated monomer solutions24. Purification cycles, which followed polymerization, successfully extracted templates from MIPs, as monitored through absorbance measurement of each wash buffer. A stable condition was reached by the end of the fourth purification cycle, as no additional template was eluted in consecutive washes. However, it should be noted that some template remained entrapped in the microgels, which accounted for 7.3%, 8.1%, and 10.7% of the TMIP, LMIP, and CMIP dry weight, respectively (Table III). Microgels were irregularly shaped [Fig. 2(a-c)], negatively charged [Fig. 2(d)], and highly polydisperse [Fig. 2(e), Table III]. The majority of formed microparticles, comprising 97.9% or more of the DLS intensity measurement for both MIPs and NIPs, were <2 μm in swollen diameter, while all formulations also had a small population of larger microparticles [Fig. 2(e)]. The inclusion, or identity of, an imprinting template did not significantly alter the microparticles’ size, morphology, polydispersity, or zeta potential.
TABLE III.
Summary of MIP and NIP Macroscopic Properties and Composition Analysis
| Formulation: | Dh (z-avg, DLS) (μm) | PDI | ζ-potential (mV) | Remaining Template (% of total Dry Weight) | Approximate DEAEMA/MAA Ratio (titration) |
|---|---|---|---|---|---|
| NIP | 1.868 | 0.758±0.026 | −44.2±5.5 | - | 0.629 |
| TMIP | 1.766 | 0.548±0.047 | −47.0±8.3 | 7.3±3.9 | 0.942 |
| LMIP | 4.042 | 0.851±0.135 | −45.3±6.7 | 8.1±1.2 | 0.934 |
| CMIP | 3.609 | 0.692±0.131 | −46.2±11.6 | 10.7±2.2 | 0.731 |
FIGURE 2.
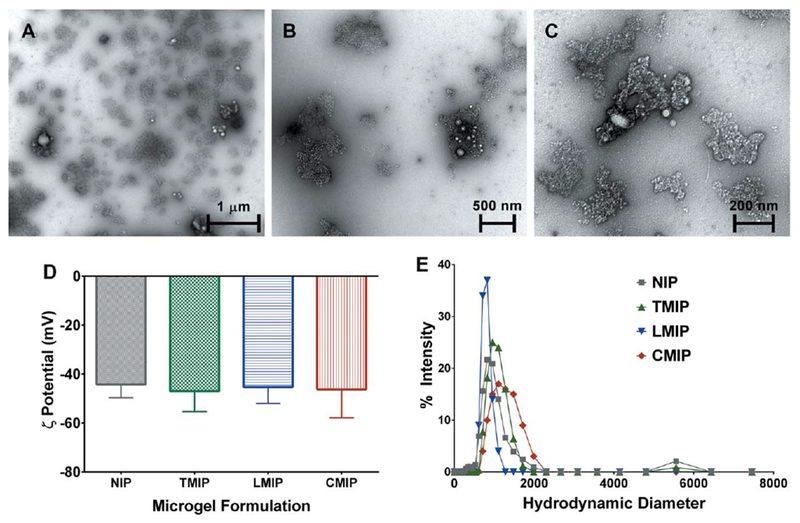
(a-c) Representative TEM images for imprinted and non-imprinted microgels. Imaging revealed that microparticles were highly polydisperse, irregularly shaped, and contained a varying number of PCL-NPs. LMIP shown, at increasing magnification to illustrate (a) the general distribution of microparticles, (b) typical microgel morphology, and (c) PCL-NP incorporation. No morphological differences were observed between MIPs and NIPs. (d) Zeta potential and (e) hydrodynamic diameter measurements were obtained at a microparticle concentration of 0.5 mg/mL in ultrapure water. The imprinting template identity did not significantly alter the microparticle z-average diameter or zeta potential. Quantitative metrics are presented in Table III. n = 3, presented as (d) zeta potential ± zeta deviation, and (e) average intensity.
Microparticle composition
The FTIR spectra shown here [Fig. 3(a)] were obtained following the completion of purification, dialysis, and lyophilization, and were normalized to the absorbance magnitude at 1732 cm−1, which received contributions from the carbonyls of MAA, DEAEMA, PMAO-PEGMA, and PCL. This was chosen as the standard for normalization as this summative quantity should be consistent between formulations. All formulations possessed significant and similar IR absorbance at 755 cm−1 and 3000–2800 cm−1, which can be attributed to the inclusion of PCL and PMAO-PEGMA respectively, which are contributed by functionalized PCL-NP. Similar absorbance spectra also emerged from 1340–1020 cm−1, which received contributions from the esters present in the functionalized PCL-NP, MAA and DEAEMA. There is a visual difference between imprinted and non-imprinted formulations in the peak between 1690–1650 cm−1, which is caused by the stretch of the amide carbonyl contributed by AAm. Purification conditions, which included extended incubations in 10 vol % acetic acid, likely caused the hydrolysis of some pendant amides in the polymerized MIP or NIP.25 Consequently, either the inclusion of an imprinting template enhanced AAm incorporation into microparticles, with trypsin and lysozyme templates offering greater enhancement than cytochrome c, or entrapped protein from imprinting protected some amides from hydrolysis during purification [Fig. 3(a)]. Another contributor to imprinted, but not non-imprinted formulations’ absorbance in the 1690–1650 cm−1 range is also the remaining entrapped protein (Table III).
FIGURE 3.
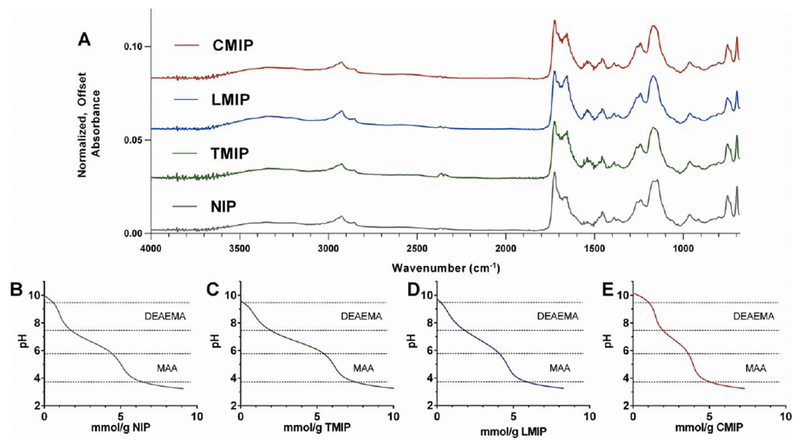
(a) FTIR spectra were obtained for each MIP or NIP formulation and normalized to the absorption peak at 1732 cm−1, which is proportional to the carbonyl content, which should be constant between formulations. Titration of (b) NIP, (c) TMIP, (d) LMIP and (e) CMIP revealed two unique equivalence points, validating the presence of MAA and DEAEMA in each formulation. Quantitative titration analysis is presented in Table III.
Titration of microparticle suspensions revealed two distinct equivalence points contributed by DEAEMA at pH = 8.5 and MAA at pH = 4.8 [Fig. 3(b-e)]. These equivalence points were present at the same pH for all formulations, but the volume of titrant required to pass through equivalence varied slightly between formulations (Table III). It should be noted that physiological pH (7.4) falls within an ionized range for both MAA and DEAEMA. The existence of both equivalence points validated the presence of both DEAEMA and MAA in the purified microparticles; however, it is challenging to draw quantitative conclusions about MAA incorporation specifically due to the overlapping equivalence points of the carboxylic acid moieties contributed by PMAO-PEGMA, MAA, and any hydrolyzed AAm.
Non-competitive protein adsorption
All MIPs and NIPs bound significant quantities of all tested high isoelectric point proteins (lysozyme, cytochrome c, trypsin) [Fig. 4(a-c)], as well as hemoglobin [Fig. 4(d)], which possesses an isoelectric point of 6.8.20 Both low isoelectric point proteins (BSA, trypsin inhibitor type II) were adsorbed in minimal or undetectable quantities [Fig. 4(e,f)]. This adsorption behavior is likely a result of favorable electrostatic interactions between the net-anionic MIPs and NIPs and net-cationic high isoelectric point proteins. As hemoglobin possesses numerous charged residues and a minimal formal charge at pH = 7.4, it did not experience nearly the extent of electrostatic repulsion exerted by MIPs and NIPs on BSA and trypsin inhibitor.
FIGURE 4.
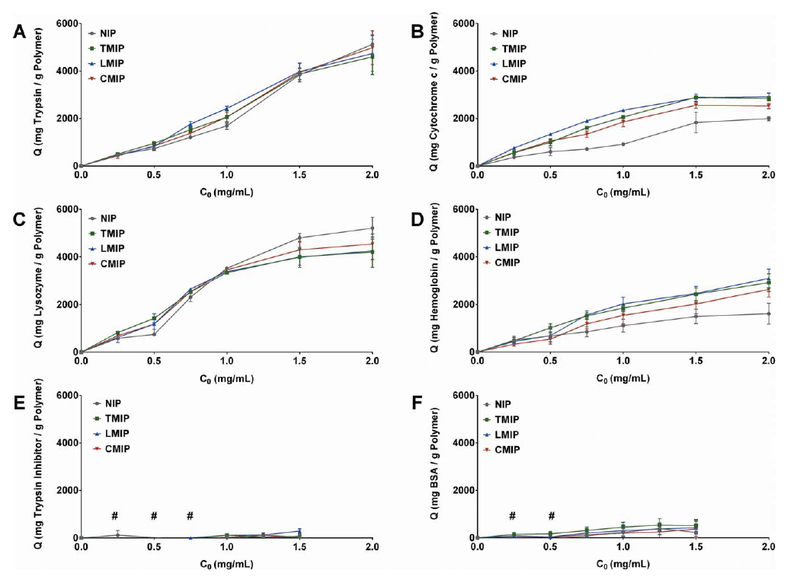
Concentration-dependent protein adsorption to MIP, TMIP, LMIP, and CMIP, separated by protein: (a) trypsin, (b) cytochrome c, (c) lysozyme, (d) hemoglobin, (e) trypsin inhibitor type II, (f) BSA. n = 3, presented as average ± SEM, with connecting lines for visualization. (#) indicates adsorption quantities below assay detection.
For initial protein concentrations less than or equal to 0.75 mg/mL, corresponding to a 2.5-fold mass excess of protein relative to polymer, MIPs adsorbed more trypsin, lysozyme, cytochrome c, and hemoglobin than NIPs irrespective of template identity (Fig. 5). As the initial concentration of protein was increased beyond the 2.5-fold excess, MIPs continued to adsorb more cytochrome c and hemoglobin than NIPs, while trypsin and lysozyme were adsorbed comparably by both MIPs and NIPs (Table IV). The ability of MIPs to demonstrate an elevated adsorption capacity for cytochrome c and hemoglobin at these greater solution concentrations is most likely a result of their generally reduced adsorption when compared to trypsin or lysozyme, preventing polymer saturation with protein at the solution concentrations up to 2 mg/mL.
FIGURE 5.
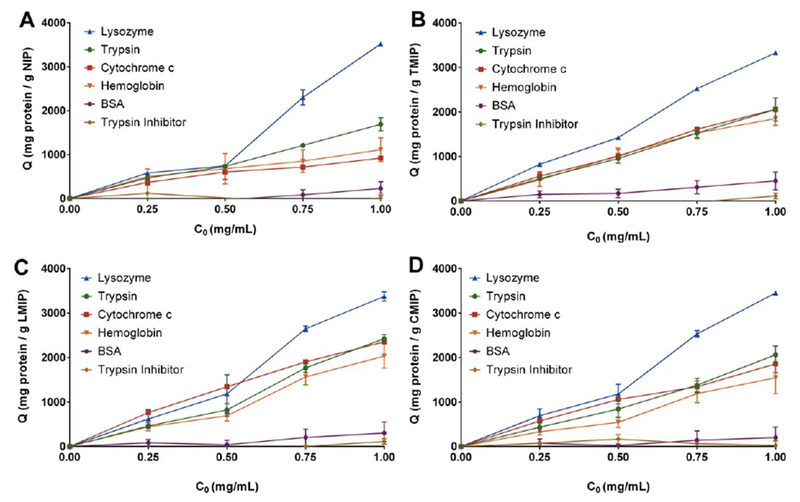
Non-competitive protein adsorption behavior of (a) NIP, (b) TMIP, (c) LMIP, and (d) CMIP. While all MIP formulations exhibited different adsorption behavior for cytochrome c and hemoglobin than NIP, no significant difference was observed between the TMIP, LMIP, and CMIP. n = 3, presented as average ± SEM, with connecting lines for visualization.
TABLE IV.
Statistical Comparison of MIP and NIP Noncompetitive Adsorption Behavior
| Formulation Comparison | Trypsin | Lysozyme | Cytochrome c | Hemoglobin | Trypsin Inhibitor | BSA |
|---|---|---|---|---|---|---|
| NIP vs TMIP | ns | ns | * | * | ns | ns |
| NIP vs. LMIP | ns | ns | * | * | ns | ns |
| NIP vs. CMIP | ns | ns | * | * | ns | ns |
| TMIP vs. LMIP | ns | ns | ns | ns | ns | ns |
| TMIP vs CMIP | ns | ns | ns | ns | ns | ns |
| LMIP vs. CMIP | ns | ns | ns | ns | ns | ns |
Statistically significant at α = 0.05 level. ns = no significant difference. 2-way ANOVA with multiple comparisons.
Protein adsorption in competitive and crowded environments
In a protein-rich serum buffer comprised of 40 mg/mL bovine serum albumin, gamma globulins, and fibrinogen; cytochrome c was adsorbed in significantly greater capacity by MIPs than NIPs, with a 162%, 219%, and 122% increase in adsorption for TMIPs, LMIPs, and CMIPs, respectively [Fig. 6(a), Table V]. This imprinting factor was much greater than that exhibited for hemoglobin for each formulation. A minimal quantity of trypsin inhibitor was adsorbed by each polymer irrespective of imprinting. It should be noted that this crowded protein buffer, containing plasma-mimicking protein ratios contained only low isoelectric point (fibrinogen pI = 4.4, albumin pI = 4.7) and neutral isoelectric point (gamma globulin pI = 7.2) proteins. Therefore, while these abundant proteins compete with high isoelectric point proteins, such as cytochrome c, for adsorption to MIPs and NIPs they would not directly compete for electrostatic interactions.
FIGURE 6.
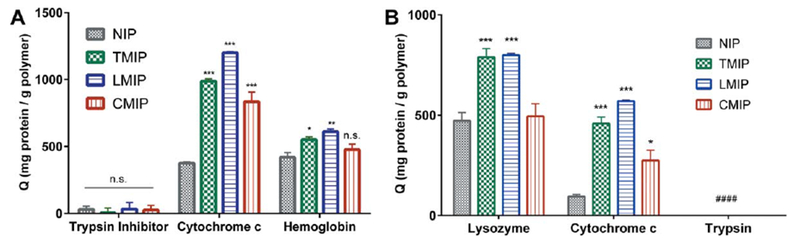
Competitive adsorption of (a) trypsin inhibitor, cytochrome c, or hemoglobin to MIPs and NIPs in serum buffer and (b) lysozyme, cytochrome c, and fluorescent trypsin when incubated simultaneously in 0.1× PBS. MIPs adsorbed significantly more (a) cytochrome c and (b) cytochrome c and lysozyme, than their non-imprinted controls. n = 4–5, presented as average ± SEM. *p< 0.05, **p< 0.01, ***p< 0.001 compared to NIP 2-way ANOVA with multiple comparisons. n.s. = no statistical significance, # = below assay detection.
TABLE V.
Summary of Imprinting Factor and Selectivity Factor Values Obtained from Competitive Adsorption Analysis
| Imprinting Factor (IF) |
Lysozyme Selectivity Factor (α) |
|||
|---|---|---|---|---|
| Formulation | Cytochrome c | Hemoglobin | Trypsin inhibitor | Qlysozyme/ Qcytochrome c |
| NIP | – | – | – | 4.99±0.41 |
| TMIP | 2.62±0.09 | 1.31±0.12 | 0.12±1.25 | 1.72±0.09 |
| LMIP | 3.19±0.08 | 1.46±0.13 | 1.12±1.85 | 1.40±0.01 |
| CMIP | 2.22±0.20 | 1.14±0.14 | 0.87±1.34 | 1.80±0.23 |
To directly address the question of protein adsorption behavior in the presence of multiple cationic proteins, trypsin, lysozyme, and cytochrome c were co-incubated with MIPs and NIPs in 0.1× PBS. Lysozyme adsorbed to all formulations in the greatest capacity, with TMIPs and LMIPs adsorbing more lysozyme than NIPs. Simultaneously, MIPs adsorbed more cytochrome c than NIPs [Fig. 6(b)], and no trypsin adsorption was observed by any formulation.
DISCUSSION
Molecular imprinting and adsorption behavior
While imprinting did not significantly alter each microgel’s macroscopic properties, it caused small differences in the relative incorporation and supramolecular structure of functional monomers within the resulting PCL-NP containing hydrogel networks. Through our MIP polymerization protocol, the resultant polymers have a bulk distribution of heterogeneous, high-affinity sites for protein adsorption. Consequently, while these sites are available, MIPs exhibit an increased affinity, revealed through adsorption capacity, for lysozyme, trypsin, cytochrome c, and hemoglobin. This behavior closely follows the assumptions of the Freundlich adsorption isotherm,26 which is an empirical relation that describes solute adsorption behavior to surfaces which contain heterogeneous binding sites. When polymers saturate with protein at a greater mass adsorption, which appears to exist between 2500 and 3000 mg protein/g polymer, subsequent protein adsorption will occur in a layered manner. At this point, the adsorption behavior fails to satisfy the assumptions of the Freundlich model, and more closely follows Brunauer-Emmet-Teller (BET) isotherm,27 which describes multilayered adsorption behavior to a solid surface, where each layer exhibits ideal behavior. In the BET adsorption regime for this system, such a significant quantity of protein has been adsorbed on the MIP or NIP surface that the surface being presented to additional protein molecules is analogous between formulations. This is substantially different from the case of protein-void polymer, where MIPs and NIPs present unique heterogeneous adsorption sites.
Insights for productive analogue imprinting
The elevated affinity of MIPs for high isoelectric point proteins, irrespective of template identity (lysozyme, cytochrome c, trypsin) can be explained mechanistically in several ways. By altering the chemical makeup of the final polymer, in terms of acrylamide and functional monomer content, molecular imprinting imparted elevated affinity for select proteins. Additionally, as it was noted that template extraction was maximized by purification methods but was still incomplete, a small quantity of protein and peptide fragments remained in MIPs but not NIPs. This remaining protein content would interact favorably with certain, but not all proteins when presented in solution and could explain the differential affinity imparted by protein imprinting. Alternatively, molecular imprinting could alter the arrangement and orientation of polymerized monomers within the final network. The orientation of complementary anionic monomer moieties relative to void nanocavities during imprinting should result in favorable interactions with each high isoelectric point protein, template or non-template. Synergistically, the protein templates acted as a porogen, which under optimal conditions enabled the diffusion of free proteins into the polymer bulk for favorable adsorption. Any combination of these theorized mechanisms could have contributed to, and ultimately explain the protein selectivity exhibited by MIPs.
The increased polymer affinity for high isoelectric point proteins imparted by molecular imprinting did not differ between TMIPs, LMIPs and CMIPs in non-competitive conditions. While there were statistically significant differences in the adsorption behavior of cytochrome c and hemoglobin to each MIP formulation as compared to NIPs, there were no significant differences in the adsorption behavior of any protein between MIP formulations. This result is in agreement with the preceding analysis of physiochemical properties by FTIR, DLS, and zeta potential measurement, which demonstrated that while molecular imprinting alters the composition of the final MIP relative to NIP, the differences between TMIPs, LMIPs, and CMIPs were minimal.
The inability of MIPs and NIPs to adsorb trypsin in competitive solution could be a result of trypsin’s larger size presenting a diffusion limitation in competitive solution, or an artifact of FITC-labeling. We can be confident that the increased adsorption of cytochrome c and lysozyme in competitive experiments falls within the Freundlich-isotherm regime and is due to affinity interactions with adsorption sites, as the adsorbed protein falls below the 2500–3000 mg/g threshold identified in non-competitive experiments. The polymer, irrespective of imprinting, has the greatest affinity for lysozyme, as evidenced by each formulation possessing the greatest adsorption capacity for lysozyme in competitive solution. The increased adsorption of cytochrome c by MIPs, as compared to NIPs, is attributable to the increased affinity provided by lysozyme, trypsin, or cytochrome c imprinting. The decrease in lysozyme selectivity factor as a result of imprinting demonstrates that, while the MIPs possessed the greatest affinity for lysozyme, imprinting lysozyme, cytochrome c, or trypsin impacted cytochrome c affinity to a greater extent than lysozyme affinity. This trend was consistent irrespective of the high isoelectric point template identity (trypsin, lysozyme, cytochrome c).
Summary
We have demonstrated that it is possible to utilize molecular imprinting techniques with rationally selected protein templates to augment the affinity of microgels for desired non-templates. Molecular imprinting of trypsin, lysozyme, or cytochrome c significantly increased high isoelectric point protein-polymer affinity, evidenced through the quantity of adsorbed protein at equilibrium, but there was no evidence to suggest that the identity of the high isoelectric point template affected the magnitude of this affinity benefit. We believe that, in the future, this strategy can be employed in parallel with traditional bulk and epitope imprinting approaches to develop biomaterials with engineered affinity for costly protein biomarkers for applications in disease diagnosis and therapy.
ACKNOWEDGMENTS
We would like to acknowledge Ms. Heidi Culver and Dr. Julia Vela-Ramirez for many helpful discussions during experiment planning, Dr. Dwight Romanovicz and the Institute for Cell and Molecular Biology (ICMB) at UT Austin for TEM technical support, and Ms. Clair LaVaye for administrative support.
Contract grant sponsor: The Cockrell Family Regents Chair, the UT-Portugal Collaborative Research Program (CO-LAB), and UT Austin Undergraduate Research Fellowships (to JXZ, ASI, and JG)
Contract grant sponsor: NSF Graduate Research Fellowships (JRC and DSS)
REFERENCES
- 1.Shimizu KD, Stephenson CJ. Molecularly imprinted polymer sensor arrays. Curr Opin Chem Biol 2010;14:743–750. ). [DOI] [PubMed] [Google Scholar]
- 2.Byrne ME, Hilt JZ, Peppas NA. Recognitive biomimetic networks with moiety imprinting for intelligent drug delivery. J Biomed Mater Res A 2008;84:137–147. [DOI] [PubMed] [Google Scholar]
- 3.Neves MI, Wechsler ME, Gomes ME, Reis RL, Granja PL, Peppas NA. Molecularly imprinted intelligent scaffolds for tissue engineering applications. Tissue Eng 2017;23:27–43. [DOI] [PubMed] [Google Scholar]
- 4.Chen L, Xu S, Li J. Recent advances in molecular imprinting technology: current status, challenges and highlighted applications. Chem Soc Rev 2011;40:2922–2942. [DOI] [PubMed] [Google Scholar]
- 5.Shephard GS, Gelderblom WCA. Rapid testing and regulating for mycotoxin concerns: a perspective from developing countries. World Mycotoxin J 2014;7:431–437. [Google Scholar]
- 6.Verheyen E, Schillemans JP, van Wijk M, Demeniex MA, Hennink WE, van Nostrum CF. Challenges for the effective molecular imprinting of proteins. Biomaterials 2011;32:3008–3020. [DOI] [PubMed] [Google Scholar]
- 7.Peppas NA, Clegg JR. The challenge to improve the response of biomaterials to the physiological environment. Regen Biomater 2016;3:67–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kryscio DR, Peppas NA. Critical review and perspective of macromolecularly imprinted polymers. Acta Biomater 2012;8:461–473. [DOI] [PubMed] [Google Scholar]
- 9.Beyazit S, Bui BTS, Haupt K, Gonzato C. Molecularly imprinted polymer nanomaterials and nanocomposites by controlled/living radical polymerization. Prog Polym Sci 2016;62:1–21. [Google Scholar]
- 10.Hoshino Y, Kodama T, Okahata Y, Shea KJ. Peptide imprinted polymer nanoparticles: a plastic antibody. J Am Chem Soc 2008; 130:15242–15243. [DOI] [PubMed] [Google Scholar]
- 11.Oral E, Peppas NA. Hydrophilic molecularly imprinted poly (hydroxyethyl-methacrylate) polymers. J Biomed Mater Res A 2006;78:205–210. [DOI] [PubMed] [Google Scholar]
- 12.Byrne ME, Oral E, Hilt JZ, Peppas NA. Networks for recognition of biomolecules: Molecular imprinting and micropatterning poly (ethylene glycol)-containing films. Polym Adv Technol 2002;13: 798–816. [Google Scholar]
- 13.Zhang Y, Deng C, Liu S, Wu J, Zhangbao C, Li C, Lu W. Active targeting of tumors through conformational epitope imprinting. Angew Chem Int Ed 2015;54:5157–5160. [DOI] [PubMed] [Google Scholar]
- 14.Wang XJ, Xu ZL, Feng JL, Bing NC, Yang ZG. Molecularly imprinted membranes for the recognition of lovastatin acid in aqueous medium by a template analogue imprinting strategy. J Membr Sci 2008;313:97–105. [Google Scholar]
- 15.Culver HR, Steichen SD, Herrera-Alonso M, Peppas NAA. Versatile route to colloidal stability and surface functionalization of hydrophobic nanomaterials. Langmuir 2016;32:5629–5636. [DOI] [PubMed] [Google Scholar]
- 16.Wetter LR, Deutsch HF. Immunological studies on egg white proteins IV. Immunochemical and physical studies of lysozyme. J Biol Chem 1951;192:237–242. [PubMed] [Google Scholar]
- 17.Walsh KA. Trypsinogens and trypsins of various species. Methods Enzymol 1970;19:41–63. [Google Scholar]
- 18.Malamud D, Drysdale JW. Isoelectric points of proteins: a table. Anal Biochem 1978;86:620–647. [DOI] [PubMed] [Google Scholar]
- 19.Melamed MD. Electrophoretic properties of ovomucoid. Biochem J 1967;103:805–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Conway-Jacobs A, Lewin LM. Isoelectric focusing in acrylamide gels: use of amphoteric dyes as internal markers for determination of isoelectric points. Anal Biochem 1971;43:394–400. [DOI] [PubMed] [Google Scholar]
- 21.Anderson L, Anderson NG. High resolution two-dimensional electrophoresis of human plasma proteins. Proc Natl Acad Sci 1977; 74:5421–5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Culver HR, Steichen SD, Peppas NA. A closer look at the impact of molecular imprinting on adsorption capcity and selectivity for protein templates. Biomacromolecules 2016;17:4045–4053. [DOI] [PubMed] [Google Scholar]
- 23.Koetting MC, Peppas NA. pH-Responsive poly (itaconic acid-co-N-vinylpyrrolidone) hydrogels with reduced ionic strength loading solutions offer improved oral delivery potential for high isoelectric point-exhibiting therapeutic proteins. Int J Pharm 2014;471: 83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kryscio DR, Fleming MQ, Peppas NA. Protein conformational studies for macromolecularly imprinted polymers. Macromol Bio-sci 2012;12:1137–1144. [DOI] [PubMed] [Google Scholar]
- 25.Moens J, Smets G. Alkaline and acid hydrolysis of polyvinylamides. J Polym Sci 1957;23:931–948. [Google Scholar]
- 26.Umpleby RJ, Baxter SC, Chen Y, Shah RN, Shimizu KD. Characterization of molecularly imprinted polymers with the Langmuir-Freundlich isotherm. Anal Chem 2001;73:4584–4591. [DOI] [PubMed] [Google Scholar]
- 27.Latour RA. The langmuir isotherm: A commonly applied but misleading approach for the analysis of protein adsorption behavior. J Biomed Mater Res A 2015;103:949–958. [DOI] [PubMed] [Google Scholar]