

ABSTRACT
Chaperone-mediated autophagy (CMA), a form of selective autophagy, maintains cellular proteostasis in response to diverse stress conditions. Whether and how endoplasmic reticulum (ER) stress triggers CMA remains elusive. In our recent study, we demonstrate that various types of ER stress activate the CMA pathway via an EIF2AK3/PERK-MAP2K4/MKK4-MAPK14/p38-dependent manner. We term this process ERICA for ER stress-induced chaperone-mediated autophagy. This pathway is activated in response to stress associated with Parkinson disease and is required for the viability of the SNc dopaminergic neurons in an animal model of Parkinson disease.
KEYWORDS: CMA, ER stress, p38 MAPK, Parkinson's disease, PERK
The ER and lysosomes are 2 of the most critical subcellular organelles involved in sensing and regulating stress response. The ER depends upon a specialized environment to modify, fold, and assemble newly-synthesized proteins targeted for secretion or membrane compartments. Disturbance of the ER environment interferes with the ability of the ER to properly process proteins and causes the unfolded protein response (UPR). Various physiological and pathological conditions and several pharmacological reagents can trigger the UPR. HSPA5/Bip is a key protein that responds to the calcium level of the ER and is involved in the regulation of 3 ER-resident transmembrane proteins, EIF2AK3/PERK (eukaryotic translation initiation factor 2 alpha kinase 3), ATF6 (activating transcription factor 6), and ERN1/IRE1α (endoplasmic reticulum [ER] to nucleus signaling 1). These ER sensors induce an adaptive mechanism to restore ER homeostasis and maintain cellular survival. Among adaptive responses, autophagy triggered by ER stress can address the accumulation of unfolded proteins. However, although both ER stress and CMA occur under several conditions, it is completely unclear whether ER stress and CMA are functionally linked. In our recent study, we demonstrate that ER stress is coupled to CMA.
The UPR and CMA are implicated in disposing of proteins in response to stress. Thus, we wondered about the possibility that these 2 key protein quality control processes may be functionally related. In dopaminergic-like SN4741 cells, 4 classical ER stressors—the Ca2+pump inhibitor thapsigargin, the N-glycosylation suppresser tunicamycin, the reducing agent 2-mercaptoethanol, and the ER-Golgi protein transport inhibitor brefeldin A—and 1 nonclassical ER stressor—6-hydroxydopamine—can dramatically activate CMA at their effective concentrations to trigger the UPR. This activation is determined and confirmed by CMA substrate decrease and CMA binding and uptake assays. The CMA activity is largely controlled by 2 key regulators, the rate-limiting lysosomal membrane receptor LAMP2A, and the key chaperone protein HSPA8, and we determined their levels following ER stress. Treatment of SN4741 cells with ER stressors significantly increased the level of LAMP2A but not HSPA8 in lysosomes. Analysis of the sub-lysosomal fractions showed that ER stressors markedly increased the LAMP2A level and oligomerization in the lysosomal membrane.
We addressed the underlying molecular mechanism(s) by which ER stress induces LAMP2A activation. MAPK14/p38 is a key kinase activated by many cellular stress conditions including ER stress. In our system, ER stressors can activate MAPK14, whereas inhibition of MAPK14 can block the increase of LAMP2A in lysosomes and the activation of CMA by ER stressors. Furthermore, overexpression of MAPK14 can mimic ER-induced CMA activation. Interestingly, ER stressors cause a biphasic activation of MAPK14 with the first peak being at 5 to 15 min and the second one being at 1 to 3 h after ER stressor challenge. Consistently, a fraction of activated MAPK14 is localized to the lysosomes following 2 h of ER stress, and inhibition of the second phase of MAPK14 activation abolishes ER stress-induced CMA. We knocked down the UPR transducers EIF2AK3/PERK, ERN1/IRE1α, and ATF6 and determined that EIF2AK3/PERK, but not ERN1/IRE1α or ATF6, is required for signaling ER stress to activate CMA. Knockdown of MAP2K4/MKK4 but not the classical MAPK14 upstream activator MAP2K3/MKK3-MAP2K6/MKK6 and MAP3K5/ASK1 prevents ER stress-induced phosphorylation and activation of lysosomal MAPK14 and CMA. Because MAPK14 is required for ER stress-induced regulation of LAMP2A and CMA, we confirmed the possibility that MAPK14 directly phosphorylates and regulates LAMP2A. First, we carried out immunoprecipitation and established that the endogenous MAPK14 and endogenous LAMP2A associate with each other. Second, high MAPK14 activity leads to robust phosphorylation of LAMP2A at Thr, but not Ser, residues. Third, the purified MAPK14 directly phosphorylates LAMP2A mainly at threonines at position 211 and 213. Mutation of threonines 211 and 213 completely abolishes phosphorylation of LAMP2A by MAPK14 in vitro and blocks LAMP2A phosphorylation and CMA activity induced by ER stressors in vivo.
We further demonstrate that prolonged and severe ER stress induces apoptosis, whereas inhibition of CMA exacerbates the loss of cellular viability under such stress, indicating that CMA following initial ER stress is protective. The cell protective activity of ERICA has the following characteristics: 1) the survival signaling pathway involves EIF2AK3/PERK-MAPK24/MKK4-MAPK14/p38-p-LAMP2A; 2) it involves only the second phase of lysosomal MAPK14; and 3) the activation of ERICA inhibits both DDIT3/CHOP and CASP3 (caspase 3) activity. More importantly, we established the engagement of the ERICA pathway in mouse brain following intraperitoneal injection of the ER stressor tunicamycin. Analysis showed that ER stress increases the level of LAMP2A, and HSPA5/Bip and reduces the level of MEF2D in the striatum. Analysis of the lysosomes purified from the striatum showed that ER stress induces higher levels of phosphorylated MAPK14 and phosphorylated LAMP2A, whereas SB203580 reverses the ER stress-induced increase in total and phosphorylated LAMP2A and CMA activity. Furthermore, the neurotoxin 6-hydroxydopamine causes a significant loss of TH (tyrosine hydroxylase)-positive dopaminergic neurons in the SNc. The loss of dopaminergic neuronal viability was significantly exacerbated by uncoupling ER stress and CMA with co-administration of SB203580.
In summary, ER stressors trigger CMA through an EIF2AK3/PERK-dependent activation and recruitment of MAP2K4/MKK4 to lysosomes, activating MAPK14 at lysosomes (Figure 1). Lysosomal MAPK14 directly phosphorylates LAMP2A and causes its membrane accumulation and active conformational change. CMA activation protects cells against ER stress-induced death. Neurotoxins associated with Parkinson disease engage the ER-MAPK14-CMA pathway in the mouse brain and uncoupling it results in a greater loss of SNc dopaminergic neurons. Engaging ERICA is functionally required for maintaining cellular protein homeostasis and protecting cells from the initial stress. Given that ER stress has been observed under many pathological conditions, it is possible that ERICA is involved or even plays a critical role in maintaining proteostasis in many tissues in many diseases. It is reasonable to postulate that ER-associated protein degradation (ERAD) and ERICA function to restore intra- and extra-ER proteostasis, respectively, under ER stress conditions. Whether ERICA may help dispose of unfolded proteins retrotransported out of the ER remains an interesting possibility and should be clarified.
Figure 1.
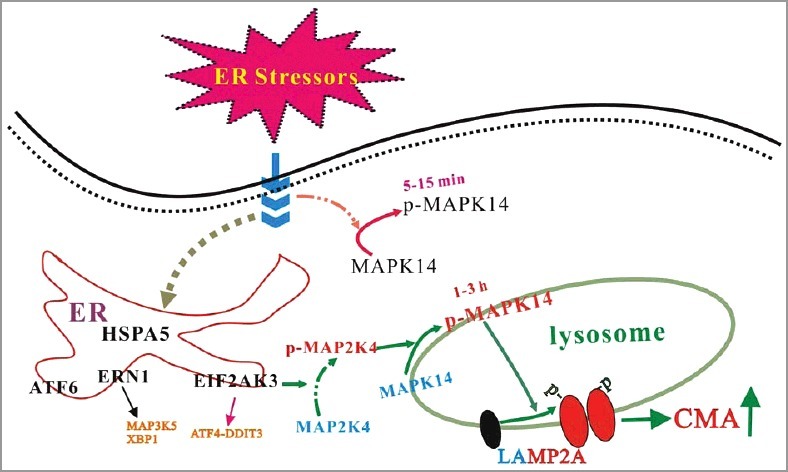
MAPK14 links ER stress to CMA. Multiple ER stressors lead to an EIF2AK3/PERK-dependent activation and recruitment of MAP2K4/MKK4 to the lysosomes, activating a lysosomal pool of MAPK14. Lysosomal MAPK14 directly phosphorylates the CMA receptor LAMP2A at T211 and T213. This dual phosphorylation modification constitutes a key regulatory event in modulating the LAMP2A level and oligomerization on the lysosomal membrane.
Funding Statement
This work was partially supported by grants from NIH [grant number NS079858], [grant number AG023695], and [grant number NS095269] to Z. Mao and National Natural Science Foundation of China [grant number 31371400] and [grant number 31671060] to Q. Yang. National Institute of Neurological Disorders and Stroke [grant number NS079858].
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.