

ABSTRACT
The biogenesis of the proton pump V-ATPase commences with the assembly of the proton pore sector V0 in the endoplasmic reticulum (ER). This process occurs under the control of a group of assembly factors whose mutations have recently been shown to cause glycosylation disorders with overlapping phenotypes in humans. Using whole exome sequencing, we demonstrate that mutations of the accessory V-ATPase subunit ATP6AP2 cause a similar disease characterized by hepatosteatosis, lipid abnormalities, immunodeficiency and cognitive impairment. ATP6AP2 interacts with members of the V0 assembly complex, and its ER localization is crucial for V-ATPase activity. Moreover, ATP6AP2 mutations can cause developmental defects and steatotic phenotypes when introduced into Drosophila. Altogether, our data suggest that these phenotypes are the result of a pathogenetic cascade that includes impaired V-ATPase assembly, defective lysosomal acidification, reduced MTOR signaling and autophagic misregulation.
KEYWORDS: acidification, autophagy, Drosophila, endoplasmic reticulum, human genetics, lysosomes, MTORC1, (pro)renin receptor, V-ATPase
The vacuolar-type H+-translocating ATPase (V-ATPase) is a multisubunit proton pump essential for the acidification of membraneous compartments. It consists of a V0 sector, forming the proton pore in the lipid bilayer, and a cytosolic-facing V1 sector, which is the ATP hydrolysis domain. Both sectors are pre-assembled separately before uniting as a functional holoenzyme in the Golgi. Altogether, there are at least 13 subunits and 2 accessory subunits. The first accessory subunit, ATP6AP1, has structural and functional homology with Voa1 in yeast. Together with 4 other proteins, Voa1 functions as a chaperone for the assembly of the V0 sector in the endoplasmic reticulum. Mutations in the genes encoding 3 of these assembly factors, including ATP6AP1, were recently identified to cause congenital disorders of glycosylation (CDG) in humans. CDGs form a genetically and clinically heterogeneous group of diseases with aberrant protein glycosylation as a hallmark. The glycosylation defects caused by the assembly factor deficiencies are thought to be the result of impaired Golgi acidification, with liver disease as the main phenotype observed in all 3 diseases.
Prior to our study, the only diseases associated with ATP6AP2, the second accessory protein, were X-linked Parkinsonism and epilepsy caused by exon skipping mutations. ATP6AP2 was first discovered as a binding partner to the V-ATPase complex in bovine membrane preparations. Accordingly, genetic manipulation of ATP6AP2 in cell culture and mouse models mainly results in lysosomal phenotypes. Using Drosophila, we and others could also show that ATP6AP2 functions in signaling pathways such as MTOR, NOTCH, and WNT signaling that require endosome-lysosomal acidification. In addition, it has been suggested that ATP6AP2 is a receptor for (pro)REN/renin, but this function remains very controversial.
ATP6AP2 is a type I transmembrane protein that can be cleaved in the Golgi apparatus into a lumenal or extracellular N-terminal fragment and a membrane-bound C-terminal fragment, harboring an ER retrieval motif (see Figure 1). Using whole exome sequencing, we found 2 missense mutations in the N-terminal domain of ATP6AP2 in patients with a glycosylation disorder highly similar to the deficiencies found in other assembly factors, particularly ATP6AP1 deficiency. Patients with ATP6AP2 mutations exhibit hepatosteatosis, immunodeficiency and psychomotor impairment. To validate the pathogenicity of the mutations, we used several experimental approaches. Hypoglycosylation of serum proteins, as found in the patients, was recapitulated in mice with temporal downregulation of ATP6AP2 in the liver. Expression of constructs harboring the 2 mutations in HEK293T cells show reduced protein levels. Interestingly, the turnover can be blocked by disturbing ER-associated degradation, indicating that degradation occurs upon misfolding in the ER. Using co-immunoprecipitaton and yeast two-hybrid assays, we could further show that both missense mutations lead to the reduced binding of ATP6AP2 to the assembly factor ATP6AP1. By contrast, a Parkinson-associated exon skipping mutation in ATP6AP2 does not impair the interaction with ATP6AP1, suggesting that this mutation has milder or different effects on protein function.
Figure 1.
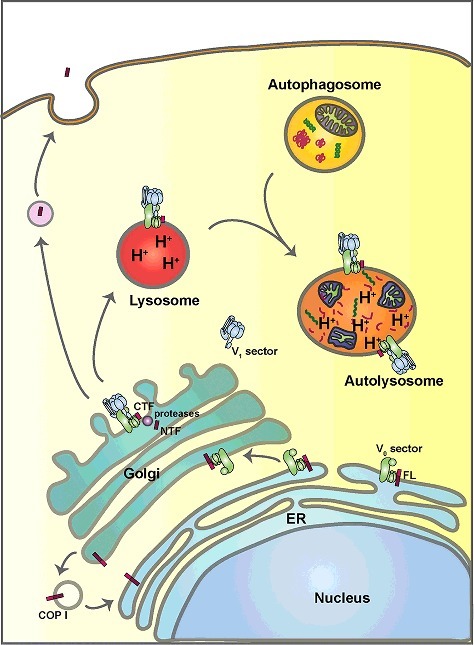
Schematic diagram of the putative intracellular dynamics of ATP6AP2 and the V-ATPase. ATP6AP2 is synthesized as a type I transmembrane protein in the endoplasmic reticulum (ER), where it contributes to the assembly of the V0 sector of the V-ATPase. Upon assembly, ATP6AP2 and the V-ATPase traffic together to the Golgi, where the V1 sector is added giving rise to a functional holoenzyme. ATP6AP2 proteins not undergoing retrograde transport to the ER are cleaved into 2 fragments, the amino-terminal fragment (NTF) and the carboxyl-terminal fragment (CTF). While the NTF can be secreted, the CTF follows the V-ATPase to the lysosome. The presence of the V-ATPase in lysosomes and autolysosomes ensures low intralumenal pH, which is essential for lysosomal hydrolase activity and the degradation of autophagic cargos.
The phenotypic consequences of the mutations were assessed using a Drosophila model that our lab has previously used to study structure-function relationships of the ATP6AP2 gene. The null mutant of ATP6AP2 shows early developmental lethality that can be partially rescued by the expression of a full-length transgene with the endogenous promoter. A similar ability to rescue is shown by a construct with mutations in the cleavage site, suggesting that an essential function is carried out by the full-length protein. By contrast, a deletion of the C-terminal ER retrieval motif causes developmental arrest in the late pupal stage. The strongest developmental defects, however, are found upon introducing the conserved patient mutation L98S into the rescue constructs. Animals carrying this mutation (ATP6AP2L98S ) already show defects in the transition from larval to pupal stage. They also show weaker expression of ATP6AP2 in agreement with the cell culture findings.
To better understand the reason for the developmental arrest, we turned to the main organs affected in the patients, the central nervous system and the liver. In the brain of the fly, we found severe effects on the morphology of the optic lobes, which constitute a major part of the brain. These defects could be linked to a premature differentiation of neuroblasts, possibly indicating a Notch-dependent phenotype. To study the liver involvement, we used the fat body, which performs both liver-like and adipose functions in flies. ATP6AP2L98S animals not only show an increased load of triglycerides in whole animal samples, but also have fat body cells with larger fat storage organelles, so-called lipid droplets. A similar phenotype is found for the knockdown of ATP6AP2, suggesting that L98S leads to reduced ATP6AP2 function.
Because the phenotype is very similar to the liver steatosis in the patient, we next sought to understand the mechanism underlying the increased lipid storage. In agreement with the proposed role of ATP6AP2 in V-ATPase assembly, we obtained the following results:
An increase in lipid droplet size was found for the knockdown of other V-ATPase subunits and assembly factors.
Lysosomes in ATP6AP2L98S fat body cells show reduced acidification.
The number and size of autophagosomes marked by Atg8a are increased in ATP6AP2-deficient cells; electron microscopy reveals the presence of enlarged autolysomes with undegraded content, including lipid droplets.
Ref(2)p/p62 and lipidated Atg8a/LC3 is increased in ATP6AP2L98S animals.
Tor signaling is reduced in ATP6AP2L98S animals, consistent with the proposed role of the V-ATPase in MTORC1 activation at the lysosomal membrane.
Altogether, the results suggested that there are 2 problems related to autophagy: first, Tor/MTOR downregulation leads to an induction of autophagy; and second, due to the degradation block, clearance cannot take place in the autolysosomes. As a result, enlarged autolysosomes accumulate undegraded material, giving rise to the steatotic phenotype in the fat body cells and, most likely, in the hepatocytes of the patients.
Thus, we were able to link mutations in ATP6AP2 with both glycosylation and autophagic defects. We also propose a novel role for this protein in V-ATPase assembly in the ER, which may be the basis for all proposed ATP6AP2 functions. Future work will be needed to elucidate the precise mechanism by which ATP6AP2 cooperates with the other assembly factors to facilitate not only the assembly but also the transport of the V0 sector to the Golgi apparatus.
Funding Statement
Funding in the Simons lab is supported by Fondation Bettencourt-Schüller, Cystinosis Research Foundation, ATIP-Avenir and l'Agence Nationale de la Recherche (ANR) (NEPHROFLY) grants [grant number ACHN-0013].
Disclosure of potential conflicts of interest
There are no potential conflict of interest relevant to this article.