

ABSTRACT
The primary cilia are evolutionarily conserved microtubule-based cellular organelles that perceive metabolic status and thus link the sensory system to cellular signaling pathways. Therefore, ciliogenesis is thought to be tightly linked to autophagy, which is also regulated by nutrient-sensing transcription factors, such as PPARA (peroxisome proliferator activated receptor alpha) and NR1H4/FXR (nuclear receptor subfamily 1, group H, member 4). However, the relationship between these factors and ciliogenesis has not been clearly demonstrated. Here, we present direct evidence for the involvement of macroautophagic/autophagic regulators in controlling ciliogenesis. We showed that activation of PPARA facilitated ciliogenesis independently of cellular nutritional states. Importantly, PPARA-induced ciliogenesis was mediated by controlling autophagy, since either pharmacological or genetic inactivation of autophagy significantly repressed ciliogenesis. Moreover, we showed that pharmacological activator of autophagy, rapamycin, recovered repressed ciliogenesis in ppara−/− cells. Conversely, activation of NR1H4 repressed cilia formation, while knockdown of NR1H4 enhanced ciliogenesis by inducing autophagy. The reciprocal activities of PPARA and NR1H4 in regulating ciliogenesis were highlighted in a condition where de-repressed ciliogenesis by NR1H4 knockdown was further enhanced by PPARA activation. The in vivo roles of PPARA and NR1H4 in regulating ciliogenesis were examined in greater detail in ppara−/− mice. In response to starvation, ciliogenesis was facilitated in wild-type mice via enhanced autophagy in kidney, while ppara−/− mice displayed impaired autophagy and kidney damage resembling ciliopathy. Furthermore, an NR1H4 agonist exacerbated kidney damage associated with starvation in ppara−/− mice. These findings indicate a previously unknown role for PPARA and NR1H4 in regulating the autophagy-ciliogenesis axis in vivo.
KEYWORDS: autophagy, ciliogenesis, kidney, NR1H4/FXR, PPARA
Abbreviations
- A549
human lung adenocarcinoma epithelial cell
- ACTB
actin, beta
- AQP2
aquaporin 2
- ARL13B
ADP-ribosylation factor-like 13B
- Ac-TUBA
acetylated tubulin, alpha
- ATG
autophagy related
- BNIP3
BCL2/adenovirus E1B interacting protein 3
- BUN
blood urea nitrogen
- CQ
chloroquine
- DAPI
4’,6-diamidino-2-phenylindole
- DBA
Dolichos biflorus agglutinin
- GFP
green fluorescent protein
- HK2
human proximal tubule epithelial cell
- H&E
hematoxylin and eosin
- IFT
intraflagellar transport
- 3-MA
3-methyladenine
- MAP1LC3/LC3
microtubule-associated protein 1 light chain 3
- MEF
mouse embryonic fibroblast
- NR1H4/FXR
nuclear receptor subfamily 1, group H, member 4
- OFD1
oral-facial-digital syndrome 1
- PKD
polycystic kidney disease
- PPARA
peroxisome proliferator activated receptor alpha
- RPE1
human retinal pigmented epithelial cell
- SESN2
sestrin 2
- SQSTM1/p62
(sequestosome 1)
- SMO
smoothened
- siRNA
small interfering RNA
- TUBG
tubulin, gamma 1
- ULK1
unc-51 like kinase 1
Introduction
Primary cilia are dynamic microtubule-based organelles that protrude from the cell surface of plasma membrane in various cell types. They act as sensory receptors and play a critical role in sensing environmental changes and transducing extracellular signals into different cellular pathways [1,2]. The importance of primary cilia in the human body is emphasized by the existence of numerous primary cilia-related congenital disorders known as ciliopathies [3,4]. Mutations in genes important for cilia structure and function are often associated with developmental defects, retinal degeneration, obesity, mental retardation, and cystic kidney disease [5,6]. Primary cilia contain the axoneme, a microtubule-based structure, whose formation is initiated by nucleation from the basal body, which originates from the mother centriole of the centrosome [7,8]. Ciliogenesis is tightly regulated by coordinated action of polarized vesicle trafficking and intraflagellar transport (IFT) that results in ciliary membrane biogenesis, extension of microtubule axoneme as well as maintenance of primary cilia [4,9].
Ciliogenesis is tightly governed by the extracellular environment and nutrient availability [10,11]. In cultured cells, serum starvation is a widely used protocol for promoting primary cilia formation [11,12]. In addition, it is well established that nutrient deprivation induces autophagy, a catabolic pathway by which cytosolic components and organelles are broken down inside lysosomes [13–15]. Autophagy generally functions to protect cells in response to various cellular stresses, including nutrient depletion, subcellular organelle damage, oxidative stress, and intracellular pathogens [16]. Recent studies have indicated that crosstalk exists between the processes of cilia formation and macroautophagy/autophagy. For example, autophagic machinery is located at ciliary structures, such as the axoneme and the basal body, to induce autophagosome formation [17]. In addition, hedgehog signaling regulates autophagy through primary cilia, while autophagy-dependent removal of OFD1 (oral-facial-digital syndrome 1) from centriolar satellites promotes ciliogenesis [18]. Although autophagy is clearly associated with ciliogenesis, the precise roles of major factors involved in autophagy and their impact on ciliogenesis require further investigation.
Nutrient metabolism and cellular homeostasis are tightly regulated by various regulatory systems including specific transcription factors [19,20]. It has previously been shown that the mechanisms for regulating autophagy take place in the cytoplasm and are controlled by various proteins in coordination with lysosomes. However, recent studies showed that both PPARA (peroxisome proliferator activated receptor alpha) and NR1H4/FXR (nuclear receptor subfamily 1, group H, member 4) regulate autophagy by controlling transcription of genes involved in autophagic pathways [21,22]. PPARA is a member of the ligand-activated nuclear hormone receptor family and plays an important role in fatty acid oxidation to maintain energy production and lipid utilization in response to starvation in various tissues, such as the liver, kidney, and heart [23,24]. NR1H4, another nuclear hormone receptor, is involved in metabolic regulation mediated by bile acids in a postprandial state [25,26]. In the present study, we investigated the potential roles of PPARA and NR1H4 activity that may regulate primary cilia formation in association with their activities in modulating autophagy pathway. We demonstrate that reciprocal activity of PPARA and NR1H4 in autophagy plays a critical role in regulating ciliogenesis in various cell lines. Furthermore, it is involved in the maintenance of kidney function in vivo by regulating its activity in response to different states of nutrition.
Results
The PPARA ligand promotes ciliogenesis in mammalian cells
To examine the role of PPARA on ciliogenesis, human retinal pigment epithelial cells that stably express GFP-tagged SMO (smoothened) (RPE1-SMO-GFP) were treated with PPARA ligands. SMO is a transmembrane protein that accumulates in the ciliary membrane during ciliogenesis. Cells were treated with 0.5 μΜ GW 7647 or 10 μΜ Wy 14643 for 24 h in normal or serum-starvation conditions. The basal body was detected with anti-TUBG (tubulin, gamma 1) antibody, and cilia formation by detection of the RPE1-SMO-GFP signal with a fluorescence microscope. As shown in Figure 1A, GW 7647 treatment increased primary cilia formation in normal and serum-free media. The RPE1-SMO-GFP signal corresponding to cilia changed from a punctate form, representing the basal body, to an elongated form. In addition to increased ciliogenesis, GW 7647 induced ciliary elongation compared to the non-treated control. To examine whether Wy 14643, another PPARA ligand, has a similar effect on cilia formation, RPE1-SMO-GFP cells were treated with Wy 14643. As shown in Figure S1, ciliary length and formation were significantly increased in normal and serum starvation conditions. Also, we examined the effect of PPARA ligand on ciliogenesis in RPE1 cells, which have not expressed the GFP-tagged SMO. After incubation with GW 7647, cells were immunostained with antibodies recognizing ciliary markers such as Ac-TUBA (acetylated tubulin, alpha) and ARL13B (ADP-ribosylation factor-like 13B). In Figure 1B, ciliary formation was significantly increased by PPARA ligand in normal and serum starvation conditions in RPE1 cells. These data suggest that activation of PPARA increases cilia formation in RPE1 cells. To confirm the stimulatory effect of PPARA activation on ciliogenesis in other mammalian cell lines, A549 (human lung adenocarcinoma epithelial cell) and HK2 (human proximal tubule epithelial cell) lines were treated with GW 7647, then immunostained with Ac-TUBA and ARL13B antibodies. As shown in Figure 1C, GW 7647 treatment induced ciliogenesis and increased ciliary length in A549 cells in normal and serum starvation conditions. A similar effect of GW 7647 on cilia formation was observed in HK2 cells (Figure 1D). In Figure S2, PPARA protein is expressed in all the cell line that tested here for cilia formation. These data indicate that activation of PPARA promotes cilia formation, which may be a general feature of mammalian cells.
Figure 1.
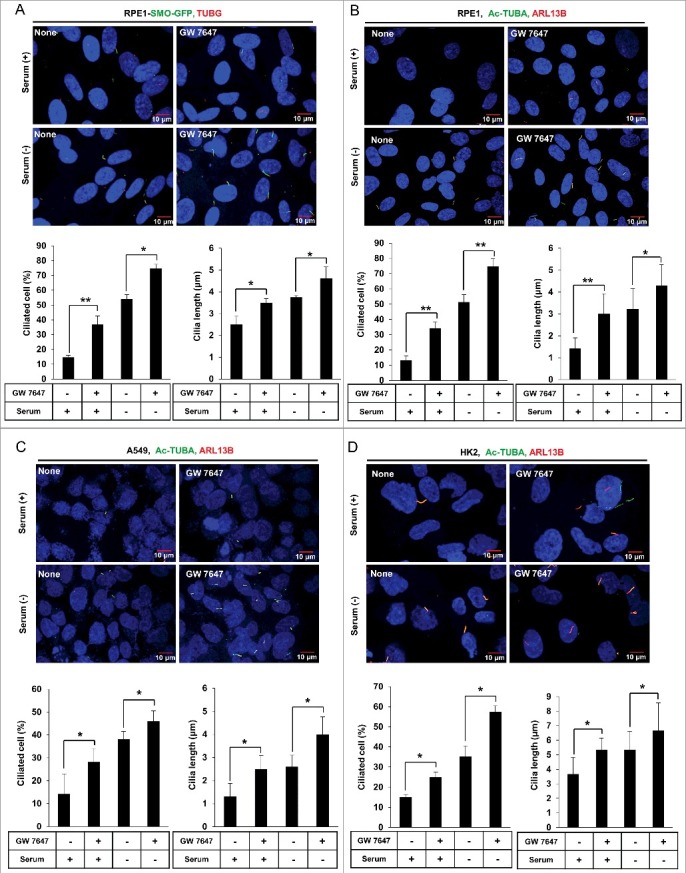
The PPARA ligand promotes ciliogenesis in mammalian cells. (A) RPE1-SMO-GFP cells were treated with 0.5 μΜ GW 7647 as indicated. After incubation for 24 h, cells were immunostained with anti-TUBG Alexa Fluor 568-conjugated antibody and observed using fluorescence microscopy. Merged signal showed the primary cilium. The number of ciliated cells and the length of cilia were quantified as described in the materials and methods section. (B, C, and D) RPE1, A549, and HK2 cells were treated with 0.5 μΜ GW 7647 as indicated. After 24 h, cells were immunostained with anti-Ac-TUBA Alexa Fluor 488-conjugated antibody and anti-ARL13B Alexa Fluor 568-conjugated antibody. The cells were observed using fluorescence microscopy. The primary cilium was showed by merged signal. The number of ciliated cells and the ciliary length of the cells were quantified as described in the materials and method section. All data shown represent mean ± s. d. percentage of cells with primary cilia or length of cilia for 200 cells per well in triplicate samples. *P < 0.05, **P < 0.01 and ***P < 0.001, one-way ANOVA, as compared to that of cells incubated in normal or serum-free media only.
The PPARA ligand induces expression of autophagic genes and upregulates autophagy in mammalian cells
Because PPARA is supposed to regulate autophagy by controlling expression of genes related to autophagy, we examined whether treatment of GW 7647, a PPARA ligand, can induce autophagy in these cell lines. To this end, the conversion of MAP1LC3/LC3-I (microtubule-associated protein 1 light chain 3) to LC3-II, which is conjugated to phosphatidylethanolamine, was analyzed. As shown in Figure 2A, GW 7647 treatment increased LC3-II protein levels in RPE1 cells in normal and serum-free media for 24 h, compared to untreated controls. LC3-II was increased in GW 7647-treated cells in both normal and serum-free conditions. The PPARA-induced increase in LC3-II levels was also observed in HK2 and A549 cells. To confirm whether the autophagy flux is also induced by PPARA ligand, we measured the level of SQSTM1/p62 (sequestosome 1), which is a receptor protein that can be degraded through autophagy [27]. Our result revealed that SQSTM1 degradation was increased by GW 7647 treatment in both normal and serum-free conditions in Figure 2A. Next, the treatment of chloroquine, an inhibitor of autophagy, induced the further accumulation of LC3-II and prevented the degradation of SQSTM1 protein shown in Figure S3A. To further elucidate the effect of the PPARA ligand on autophagy, we measured autophagosome formation in RPE1-GFP-LC3 cells. As shown in Figure S3B, GW 7647 treatment induced the formation of GFP-LC3 punctate structures in both normal and serum-free conditions. Next, we examined the effect of PPARA ligand on the autophagic flux by using RPE1 cells expressing RFP-GFP-LC3. As shown in Figure 2B, the number of red puncta, which means RFP-GFP-LC3 was delivered in autolysosome, were induced by treatment of GW 7647. Also, the ratio of red puncta to total puncta was increased, strongly suggesting that PPARA ligand increases the formation of autolysosomes due to the increased autophagic flux. These results indicate that activation of PPARA induces autophagy.
Figure 2.
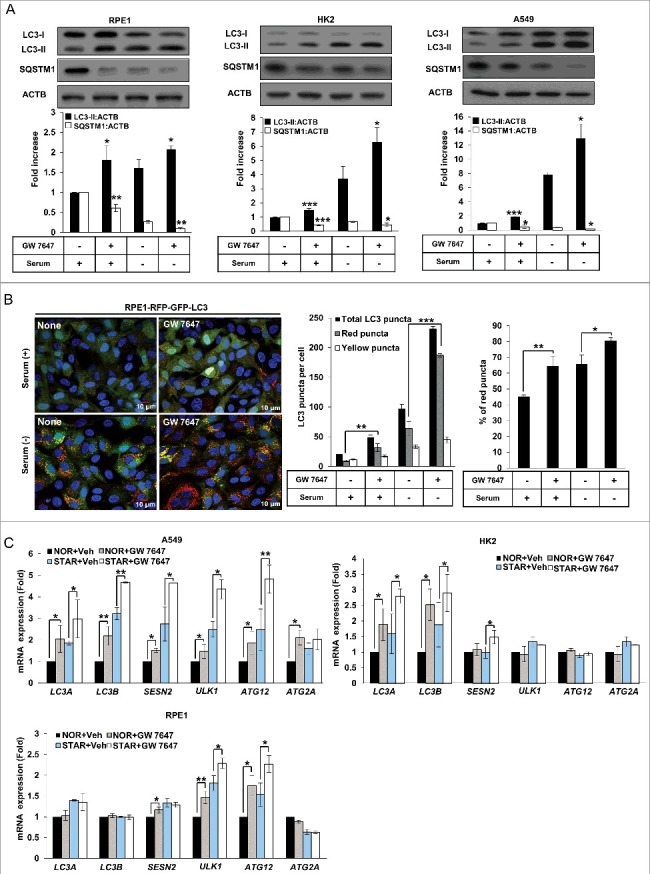
The PPARA ligand upregulates autophagy in mammalian cells. (A) RPE1, HK2, and A549 cells were treated with 0.5 μM GW 7647 as indicated. After 24 h, whole extracts were separated by SDS-PAGE and western blot analysis was conducted with antibodies against LC3, SQSTM1, and ACTB. (B) RPE1-RFP-GFP-LC3 cells were treated with 0.5 μΜ GW 7647 as indicated. After incubation for 24 h, cells were fixed with 4% paraformaldehyde, and mounted with DAPI. The number of red LC3 puncta and yellow LC3 puncta per cell in each condition was quantified. Total puncta are the sum of the number of red and yellow puncta. Percentage of the red puncta is from the total LC3 puncta. Data shown represent mean ± s. d. number of red and yellow puncta for 50 cells per well. (C) RPE1, HK2, or A549 cells were treated with vehicle (Veh) or 0.5 μM GW 7647 as indicated. After incubation for 12 h, total RNAs were extracted, and expression of LC3A, LC3B, SESN2, ULK1, ATG12, and ATG2A was analyzed with quantitative real-time polymerase chain reaction (qPCR). NOR: normal; STAR: serum starvation. All the experiments were performed 3 times. *P < 0.05, **P < 0.01 and ***P < 0.001, one-way ANOVA, compared to cells incubated in normal or serum-free media only.
Since PPARA acts as a transcription factor, the transcriptional effect of autophagic genes mediated by the PPARA ligand was measured. As shown in Figure 2C, LC3A, LC3B, SESN2 (sestrin 2), ULK1 (unc-51 like autophagy activating kinase 1), ATG2A (autophagy related 2A), and ATG12 mRNA was increased by GW 7647 in A549 cells in both normal and serum-free conditions. A similar increase in LC3A, LC3B, and SESN2 mRNA was also observed as an effect of the PPARA ligand in HK2 cells (Figure 2C). ULK1 and ATG12 mRNA were induced by PPARA ligand in RPE1 cells (Figure 2C). These results indicate that PPARA activation facilitates autophagy by inducing expression of autophagic genes.
The PPARA ligand promotes ciliogenesis via the process of autophagy in mammalian cells
Since PPARA-induced autophagy and ciliogenesis appeared to be closely related, we examined whether PPARA-induced autophagy is required for cilia formation. As shown in Figure 3A, treatment with chloroquine prevented the PPARA-induced cilia formation in RPE1 cells in a dose-dependent manner in serum-free conditions. Chloroquine itself also blocked the ciliogenesis in RPE1 cells in a dose-dependent manner in serum-starved condition. A similar result was observed with 3-methyladenine (3-MA) treatment (Figure 3B). To confirm the relationship between cilia formation and PPARA-induced autophagy, we examined the effect of autophagy inhibition in atg7 knockout mouse embryonic fibroblasts (MEFs). Atg7+/+ MEFs and atg7−/− MEFs were treated with GW 7647, and cilia formation and LC3 protein conversion were measured. In Figure 3C, GW 7647 treatment induced cilia formation in wild-type (WT) MEFs in both normal and serum-free medium. By contrast, this phenomenon was completely blocked in atg7−/− MEFs. In addition, LC3 protein conversion was increased in WT MEFs, but not in atg7−/− MEFs (Figure 3D). These data suggest that PPARA ligand-induced cilia formation is dependent on activation of autophagy.
Figure 3.
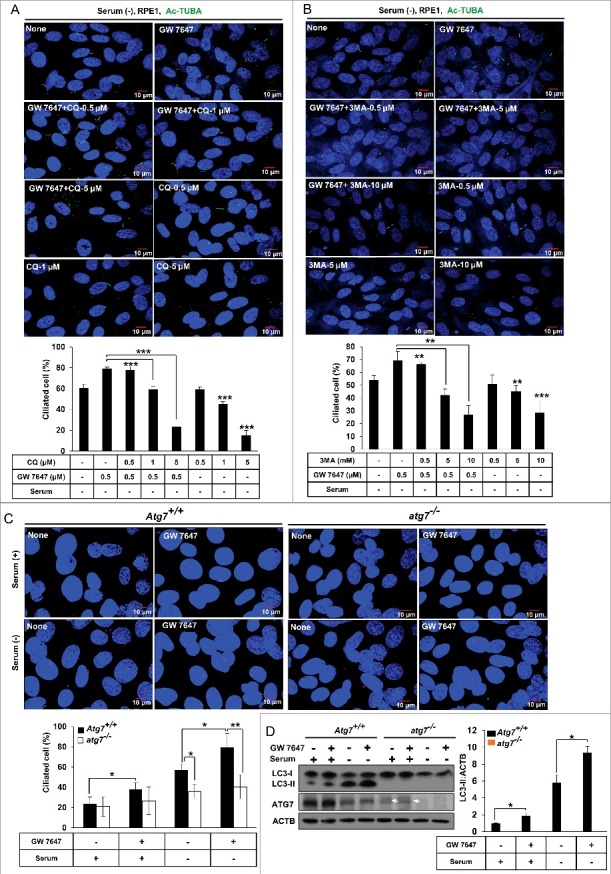
The PPARA ligand promotes ciliogenesis by activating autophagy in mammalian cells. (A and B) RPE1 cells were treated with 0.5 μΜ GW 7647, CQ and 3-MA as indicated. After 24 h starvation, cells were fixed with 4% paraformaldehyde and immunostained with anti-Ac-TUBA Alexa Fluor 488-conjugated antibody. The cells were observed using fluorescence microscopy. Data shown represent mean ± s. d. percentage of cells with primary cilia, for 200 cells per well in triplicate samples. (C) MEFs with indicated genotype were treated with 0.5 μΜ GW 7647 for 24 h, and immunostained with anti-ARL13B Alexa Fluor 568-conjugated antibody. The cells were observed using fluorescence microscopy. Data shown represent mean ± s. d. percentage of cells with primary cilia, for 200 cells per well in triplicate samples. (D) MEFs with indicated genotype were treated with 0.5 μM GW 7647 as indicated. After 24 h, cells were lysed and immunoblotted with antibodies against LC3, ATG7, and ACTB in triplicate samples. All the experiments were performed 3 times. Arrows show nonspecific bands. *P < 0.05, **P < 0.01 and ***P < 0.001, one-way ANOVA, compared to cells incubated in normal or serum-free media, or GW 7647 only.
Ciliogenesis requires PPARA-mediated activation of autophagy
To confirm whether PPARA is required for ciliogenesis, Ppara+/+ MEFs and ppara−/− MEFs were isolated from Ppara+/+ and ppara−/− mouse embryos. The cells were immunostained with ARL13B antibodies, a ciliary marker, and analyzed using fluorescence microscopy. As shown in Figure 4A, treatment with GW 7647 in WT MEFs increased the number of ciliated cells in normal and serum-free conditions compared to untreated control WT MEFs. However, ppara−/− MEFs showed reduced cilia formation in both normal and serum-free conditions, compared to WT MEFs. In addition, PPARA ligand-induced LC3 protein conversion was diminished in ppara−/− MEFs, but not in WT MEFs (Figure 4B). Also, the level of SQSTM1 protein was not reduced by GW 7647 treatment in ppara−/− MEFs compared to in WT MEFs in Figure 4B. The treatment with autophagy inhibitor CQ prevents the reduction of SQSTM1 protein by PPARA ligand in WT MEFs. To further examine whether the defect of cilia formation in ppara−/− MEFs is recovered by activation of autophagy, we treated cells with rapamycin, which is an mTORC1 inhibitor and activates autophagy. As shown in Figure 4C, treatment with rapamycin induced cilia formation in ppara−/− MEFs. Also, the level of SQSTM1 protein was reduced by rapamycin treatment in ppara−/− MEFs, as compared with no treatment in Figure 4D. The recovery effect of ciliogenesis and autophagy was blocked by ATG7 depletion in ppara−/− MEFs as shown in Figure S4A and B, indicating that autophagy was required for rescuing ciliogensis by rapamycin in ppara−/− MEFs. These results suggested that PPARA may play a key role in cilia formation by activation of autophagy in mammalian cells.
Figure 4.
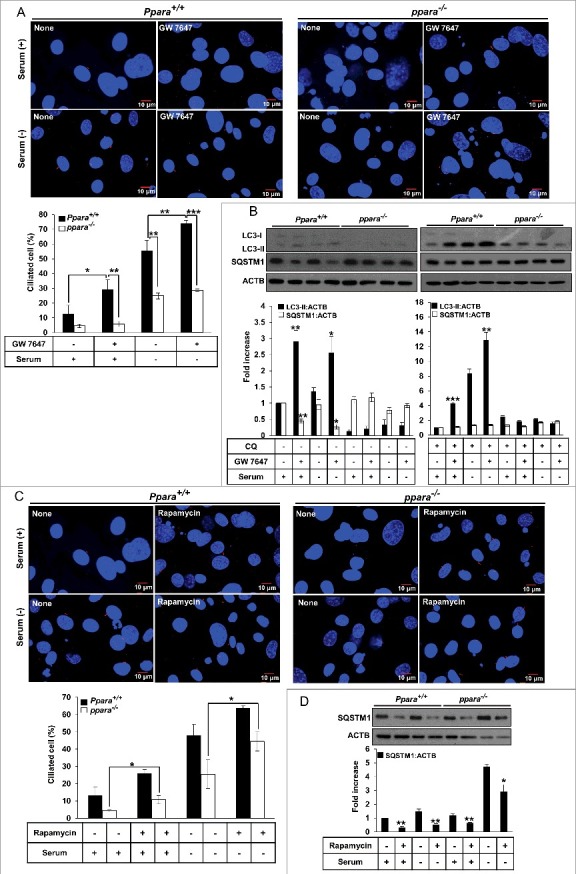
PPARA requires autophagy for ligand-induced ciliogenesis. (A) MEFs with indicated genotypes were treated with 0.5 μΜ GW 7647 for 24 h, cells were immunostained with anti-ARL13B Alexa Fluor 568-conjugated antibody. The cells were observed using fluorescence microscopy. Data shown represent mean ± s. d. percentage of cells with primary cilia, for 200 cells per well in triplicate samples. (B) MEFs with indicated genotypes were treated with 0.5 μM GW 7647 for 24 hrs or 5 μM CQ for CQ for 12 h as indicated. After incubation, cells were lysed and immunoblotted with antibodies against LC3, SQSTM1, and ACTB. (C) MEFs with indicated genotypes were treated with 1 μΜ rapamycin for 24 h, cells were immunostained with anti-ARL13B Alexa Fluor 568-conjugated antibody. Data shown represent mean ± s. d. percentage of cells with primary cilia, for 200 cells per well in triplicate samples. (D) MEFs with indicated genotypes were treated with 1 μM Rapamycin as indicated. After 24 h, cells were lysed and immunoblotted with antibodies against SQSTM1, and ACTB. All the experiments were performed 3 times. *P < 0.05, **P < 0.01 and ***P < 0.001, one-way ANOVA, compared to that of cells incubated in normal, serum-free media or GW 7647 only.
Ciliogenesis is reciprocally controlled by PPARA and NR1H4
Recent studies have demonstrated that NR1H4 binds promoter regions and suppresses transcription of autophagy genes, resulting in repression of autophagy [21,22]. Since our data showed that cilia formation was induced by PPARA-induced autophagy, we hypothesized that NR1H4 might suppress cilia formation by negatively regulating autophagy. To examine whether NR1H4 represses cilia formation, cells were treated with GW 4064, an NR1H4 ligand, in serum-free medium, a favorable condition for cilia formation. As shown in Figure 5A, treatment with GW 4064 reduced cilia formation in RPE1-SMO-GFP expressing cells in a dose-dependent manner. Similarly, cilia formation in HK2 cells was inhibited by GW 4064 treatment during starvation as shown in Figure 5B. To confirm the effect of NR1H4-mediated suppression of cilia formation, we performed small interfering RNA (siRNA)-mediated knockdown experiments to deplete NR1H4 in HK2 cells in normal medium, an unfavorable condition for cilia formation. Indeed, cilia formation was induced in NR1H4-knockdown HK2 cells in normal medium (Figure 5C). Immunoblotting confirmed that our siRNA against NR1H4 efficiently depleted NR1H4 expression in RPE1 and HK2 cells (Figure S5). We reasoned that NR1H4 and PPARA reciprocally regulate ciliogenesis, and therefore tested a condition where ciliogenesis can be maximally achieved in normal medium. As shown in Figure 5C, a combination of NR1H4 siRNA and GW 7647 further enhanced cilia formation in NR1H4-knockdown HK2 cells compared to NR1H4 siRNA or GW 7647 alone.
Figure 5.
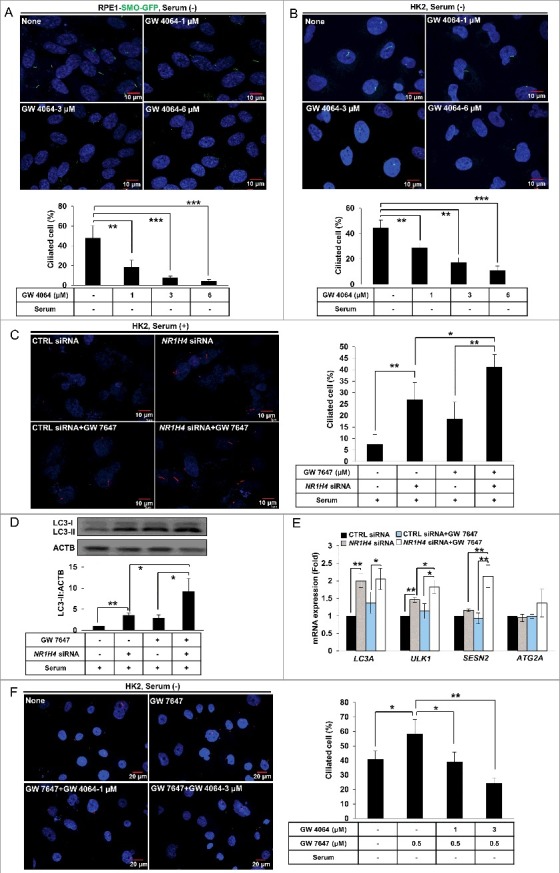
PPARA and NR1H4 reciprocally control ciliogenesis. (A and B) RPE1-SMO-GFP or HK2 cells were treated with 1, 3, or 6 μΜ GW 4064 for 24 h, as indicated, and immunostained with anti-ARL13B Alexa Fluor 488-conjugated antibody. The cells were observed under fluorescence microscope. Data shown represent mean ± s. d. percentage of cells with primary cilia for 200 cells per well in triplicate samples. (C) HK2 cells were transfected with scrambled control or NR1H4 siRNAs. After 48 h, cells were treated with 0.5 μΜ GW 7647 as indicated, and immunostained with anti-ARL13B Alexa Fluor 568-conjugated antibody. Data shown represent mean ± s. d. percentage of cells with primary cilia for 200 cells per well in triplicate samples. (D and E) HK2 cells were transfected with control or NR1H4 siRNAs for 48 h followed by treatment with 0.5 μΜ GW 7647 for 12 or 24 h. Total RNA or protein was extracted and analyzed by quantitative real-time polymerase chain reaction (qPCR) or immunoblotted, respectively. (F) HK2 cells were pretreated with 0.5 μΜ GW 7647 for 30 min, treated with different doses of GW 4064, and ciliated cells were visualized by immunofluorescence using anti-ARL13B Alexa Fluor 568-conjugated antibody. Data shown represent mean ± s. d. percentage of cells with primary cilia for 200 cells per well in triplicate samples. All the experiments were performed 3 times. *P < 0.05, **P < 0.01 and ***P < 0.001, one-way ANOVA, compared to that of cells in normal, serum-free media or GW 7647 only. CTRL; scrambled control.
Since PPARA induces ciliogenesis by activating autophagy, we examined the combined effect of NR1H4 repression and PPARA activation on autophagy. As shown in Figure 5D, the level of LC3-II was significantly increased by NR1H4 knockdown, which was further enhanced by the PPARA ligand. Furthermore, mRNA of autophagic genes, such as LC3A, ULK1, and SESN2, was increased by NR1H4 knockdown in HK2 cells, and expression of ULK1 and SESN2 was further induced by the PPARA ligand as shown in Figure 5E. These results strongly suggest that reciprocal activity of NR1H4 and PPARA controls ciliogenesis by regulating expression of autophagic genes. Next, we performed the qPCR with RPE1, HK2, and A549 cells with GW 4064 to measure the transcription of autophagy genes in Figure S6. The results showed the induced transcription of autophagy genes by starvation was suppressed by treatment of GW 4064 in RPE1, HK2, and A549 cells. In Figure S7, CQ treatment inhibited ciliogenesis induced by NR1H4 depletion, indicating depletion of NR1H4 enhances ciliogenesis by activation of autophagy. We performed a competition assay to test whether activation of NR1H4 resulting from treatment with its ligand, GW 4064, can repress PPARA-induced ciliogenesis. In particular, HK2 cells were treated with different concentrations of GW 4064 in the presence of a PPARA ligand in a serum-free condition. We found that GW 4064 suppressed PPARA ligand-induced cilia formation in a dose-dependent manner (Figure 5F). Therefore, these results suggested that NR1H4 activity can repress PPARA–induced ciliogenesis through regulating autophagy in mammalian cells.
PPARA deficiency induces kidney damage and loss of cilia via regulation of autophagy
Dysfunction of primary cilia is associated with a various pathophysiologies and can contribute to problems in many organs including kidney and eye [3,4]. Recent studies suggest that polycystic kidney disease (PKD) is caused by the abnormal functioning of primary cilia [28–31]. In addition, autophagy has been suggested to play an important role in protection against kidney injury [32,33]. To investigate the effect of PPARA on kidney function and cilia formation, Ppara+/+ and ppara−/− mice were subjected to 48 h starvation, a condition for upregulating PPARA activity in kidney [34]. Following starvation, blood urea nitrogen (BUN) and serum creatinine, biochemical markers for kidney damage, were measured. As shown in Figure 6A, starvation increased the levels of BUN (97 ± 8.271 mg/dl) and creatinine (0.36 ± 0.027 mg/dl) in ppara−/− mice, compared to those of BUN (29.64 ± 2.06 mg/dl) and creatinine (0.25 ± 0.04 mg/dl) in wild-type (WT) mice. Next, kidney sections were stained with hematoxylin and eosin (H&E) for histological examination. As shown in Figure 6B, starved ppara−/− mice showed typical features of kidney damage, such as tubular dilation and vacuole formation, while starved WT mice did not. To confirm the kidney damage induced by starvation in ppara−/− mice, the collecting ducts were stained with rhodamine-Dolichos biflorus agglutinin (rhodamine-DBA). In Figure 6C, the lumen of collecting ducts is shown to be dilated in starved ppara−/− mice compared to that in starved or fed WT mice. These results suggest that PPARA deficiency induces tubular dilation and kidney abnormality during starvation. To verify that PPARA deficiency affects the formation of cilia, the kidney sections were immunostained with an antibody against ARL13B. As shown in Figure 6D, starvation stimulated cilia formation in the tubular lumen of WT mice, whereas the number of cilia in ppara−/− mice was reduced in dilated tubules. The lack of ciliogenesis due to PPARA deficiency seemed to be closely linked with impaired autophagy in ppara−/− mice upon starvation. In contrast, cilia length is not statistically different in all conditions in Figure S8.
Figure 6.
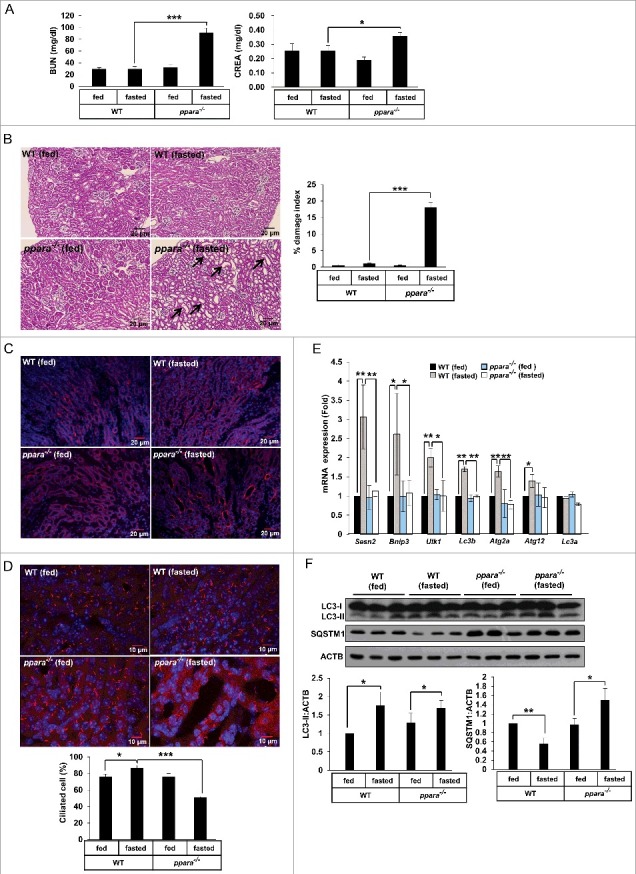
PPARA deficiency induces kidney damage and loss of cilia through impaired autophagy. (A) Blood samples from chow-fed wild-type (WT) mice, 48 h fasted WT mice, chow-fed ppara−/− mice, and 48 h fasted ppara−/− mice (n = 6) were prepared and serum levels of creatinine (CREA) and blood urea nitrogen (BUN) were determined. (B) Kidneys from chow-fed WT mice, 48 h fasted WT mice, chow-fed ppara−/− mice group, and 48 h fasted ppara−/− mice (n = 6) were removed and embedded in paraffin, and 5 μm sections were prepared. Kidney specimens were stained with hematoxylin and eosin (H&E). Damaged indices were calculated based on representative renal sections as described in Materials and Methods. (C) Kidney specimens (n = 6) were stained with Dolichos biflorus agglutinin (DBA, red). (D) Kidney specimens (n = 6) were immunostained with anti-ARL13B and Alexa Fluor 568-conjugated antibody as indicated. Data shown represent mean ± s. d. percentage of cells with primary cilia for 1000 cells per kidney in 6 samples. (E and F) Total RNAs or proteins were extracted from kidneys (n = 6) and analyzed by quantitative real-time polymerase chain reaction (qPCR) or immunoblotted as indicated. Black arrows, damaged area. *P < 0.05, **P < 0.01 and ***P < 0.001, one-way ANOVA, as compared to those from WT fasted group or WT fed group.
We found that starvation induced the expression of Sesn2, Bnip3 (BCL2/Adenovirus E1B Interacting Protein 3), Ulk1, Lc3b, Atg2a and Atg12 in WT mice, but not in ppara−/− mice (Figure 6E). In addition, the level of SQSTM1 protein was decreased in WT mice during starvation, while it accumulated in ppara−/− mice during starvation (Figure 6F). To examine whether PPARA is critical to form autophagic vesicles in kidney during starvation, we performed transmission electron microscopy. As shown in Figure S9, the formation of autophagic vesicles is increased in kidney sections from WT fasted mice, but not KO fasted mice. To further examine whether the defect of cilia formation in ppara−/− mice is recovered by activation of autophagy, mice were injected with rapamycin. As shown in Figure S10, rapamycin ameliorated the kidney damage by measuring the levels of BUN and creatinine in ppara−/− mice during fasting. Also, ciliogenesis in the damaged area and autophagy were restored by rapamycin in fasted ppara−/− mice (Figure S11 and S12). These results strongly suggest that PPARA deficiency causes impaired autophagy and ciliogenesis during starvation, which may result in tubular dilation and vacuolar formation in kidney.
Activation of NR1H4 stimulates kidney damage and loss of cilia in kidney
Since an NR1H4 ligand inhibits cilia formation in various cell lines, we investigated whether NR1H4 activation could lead to inhibition of cilia formation and kidney damage in mice. After injection of GW 4064, an NR1H4 ligand, in WT and ppara−/− mice, BUN, and serum creatinine, biochemical markers for kidney damage, were measured. As shown in Figure 7A, we did not find an increase in BUN or creatinine levels in fed or starved WT mice in the presence or absence of GW 4064. However, we found that the serum levels of BUN (74.1 ± 24.0 mg/dl) and creatinine (0.28 ± 0.03 mg/dl) were significantly increased in starved ppara−/− mice in the absence of GW 4064, compared to those of BUN (24.98 ± 3.67 mg/dl) and creatinine (0.17 ± 0.032 mg/dl) in fasted WT mice in the absence of GW 4064, confirming the requirement of PPARA in kidney function. Moreover, we found that GW 4064 further increased the serum levels of BUN (113.18 ± 21.0 mg/dl) and creatinine (0.45 ± 0.08 mg/dl) in starved ppara−/− mice, compared to those in fasted ppara−/− mice that did not receive an injection of GW 4064. This indicated an additive effect of NR1H4 in PPARA deficiency during starvation. To correlate between increased levels of BUN and creatinine and kidney histology, kidney sections were stained with H&E. As shown in Figure 7B, starved ppara−/− mice displayed typical features of kidney damage, such as tubular dilation and vacuole formation, when compared to starved WT mice. However, we could not find any difference by histological examination between DMSO-injected and GW 4064-injected kidneys of starved ppara−/− mice, in spite of a significant increase in the levels of BUN and creatinine in GW 4064-injected, starved ppara−/− mice.
Figure 7.
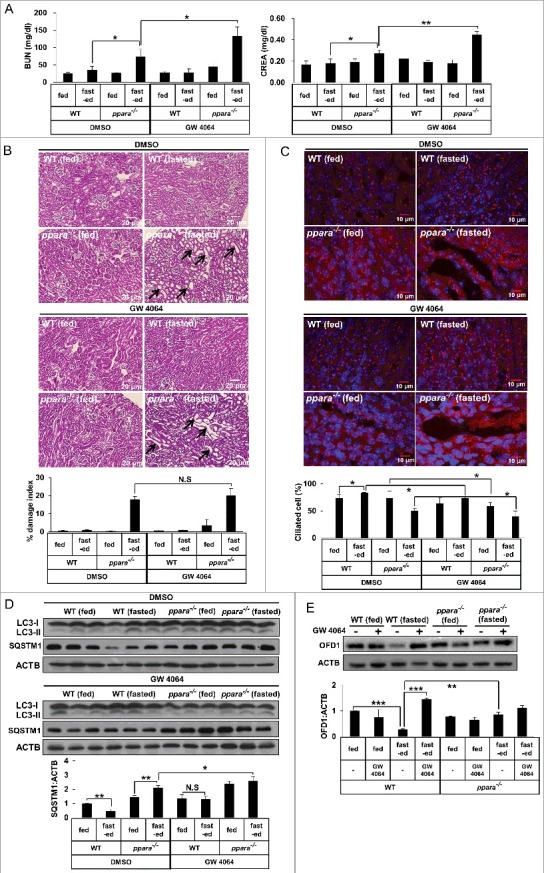
Activation of NR1H4 stimulates kidney damage and loss of cilia formation in kidney. (A) Blood samples from chow-fed wild-type (WT) mice, 48-h fasted WT mice, chow-fed ppara−/− mice, and 48-h fasted ppara−/− mice (n = 6) injected with DMSO or GW 4064 were collected and serum creatinine (CREA) and blood urea nitrogen (BUN) were analyzed. (B) Kidneys from chow-fed wild-type (WT) mice, 48-h fasted WT mice, chow-fed ppara−/− mice, and 48-h fasted ppara−/− mice (n = 6) were injected with DMSO or GW 4064. Kidney specimens were stained with hematoxylin and eosin (H&E). Damaged indices were calculated based on representative renal sections as described in Materials and Methods. (C) Kidney specimens were immunostained with anti-ARL13B and Alexa Fluor 568-conjugated antibody, as indicated. Data shown represent mean ± s. d. percentage of cells with primary cilia for 1000 cells per kidney in 6 samples. (D and E) Total protein was extracted from kidneys (N = 6) and immunoblotted with antibodies against LC3, SQSTM1, OFD1, or ACTB. Black arrows; damage area. *P < 0.05, **P < 0.01 and ***P < 0.001, one-way ANOVA, compared to WT fasted or ppara−/− group treated with DMSO. NS, non-significant. [Graphic Note: 1Homozygous recessive mutants are spelled with all lower case letters, whereas heterozygotes have the first letter capitalized. Thus, in Figure 3 Atg7+/+ and in Figure 4 Ppara+/+; 2Figure 7 [Labels within micrographs presented in B are almost impossible to read. Try changing to white font.; 3Figure S10 has the same problem as Figure 7.
To confirm that activation of NR1H4 prevents cilia formation, the number of cilia was measured by staining the kidney sections with anti-ARL13B antibody. Consistent with the result obtained in vitro, starvation induced cilia formation in WT mice, but not in ppara−/− mice (Figure 7C). Moreover, GW 4064 efficiently suppressed starvation-induced cilia formation in WT mice during starvation and further decreased cilia number in ppara−/− mice at the dilated tubules as compared to DMSO-injection group (Figure 7C). Next, to further examine the effect of NR1H4 activation on autophagy in kidney, clearance of SQSTM1 protein was measured. As shown in Figure 7D, the amount of SQSTM1 protein was reduced in DMSO-injected WT mice during starvation, although it was not reduced by administration of NR1H4 in WT and ppara−/− mice during starvation, indicating that autophagy in the kidney is blocked by the activation of NR1H4 during starvation. OFD1 was proposed to act as a negative regulator of ciliogenesis [18]. In Figure 7E, the amount of OFD1 protein in the kidney of WT mice was significantly reduced in starved mice compared to that in fed mice. In contrast, we found that GW 4064 prevented the starvation-induced OFD1 reduction in wild type mice. Moreover, OFD1 was accumulated in starved ppara−/− mice compared to WT starved mice. These data indicated that NR1H4 can reduce ciliogenesis by inhibiting autophagy, resulting in kidney damage during starvation.
Discussion
Primary cilia are specialized organelles that sense environmental conditions, and its functional abnormalities are emphasized by congenital disorders related to ciliary structure and function, known as ciliopathies [3,35]. Since nutrient availability is a common trigger for both primary cilia formation and autophagy activation, recent studies have shown that ciliogenesis is closely linked to autophagy. For instance, autophagy is required for the degradation of negative regulators of ciliogenesis such as OFD1 [18], while primary cilia are involved in autophagosome formation [17]. Furthermore, several studies have suggested that autophagy activation induces ciliogenesis [18,36,37]. However, the relationship between cilia formation and nutrient sensing proteins whose activities are controlled by fasting or feeding remain poorly understood. In this study, we demonstrated newly recognized roles of PPARA and NR1H4 in ciliogenesis through controlling autophagy in vitro and in vivo. PPARA and NR1H4 act as nutrient-sensing receptors. PPARA is known to become activated during starvation by binding a variety of fatty acids as a ligand, and upregulates fatty acid oxidation [38,39]. On the other hand, NR1H4 is thought to be activated by bile acids in a normal, fed condition and suppresses autophagy by downregulating expression of genes related to autophagy [21,22]. We showed that activation of PPARA induced ciliogenesis by activating autophagy, which was confirmed by the use of chemical inhibitors or genetic inactivation of autophagy. In addition, ppara−/− MEFs did not respond to treatment with a PPARA ligand, and ciliogenesis was not induced. Furthermore, our data demonstrated that rapamycin restored the cilia formation in ppara−/− MEFs through activating autophagy. This result strongly suggested that PPARA depletion suppresses the ciliogenesis through blocking autophagy activation. Conversely, we found that NR1H4 negatively regulates ciliogenesis. In particular, treatment with an NR1H4 ligand suppressed ciliogenesis in RPE1 and HK2 cells, while knockdown of NR1H4 induced ciliogenesis even in medium containing serum, which is known to be an unfavorable condition for cilia formation. Furthermore, knockdown of NR1H4 induced expression of autophagy genes and enhanced the effect of PPARA-induced ciliogenesis. These results suggested that PPARA and NR1H4 regulate ciliogenesis during fasting and feeding, respectively, via a mechanism that controls expression of autophagy genes.
In the kidney, mutations in cilia-related genes have been known to cause severe defects in kidney function [40,41]. PKD is a well-known genetic disorder derived from abnormalities of primary cilia formation and function. Renal cilia protrude from tubular epithelial cells into tubule lumen and respond to fluid movement, which stimulates intracellular calcium signaling, indicating the importance of primary cilia in kidney [3,42]. Several reports also suggested that defective autophagy may be involved in kidney damage, such as acute kidney injury or PKD [43,44]. PPARA is highly expressed in the kidney and plays an important role in regulating metabolic reactions governing energy production and expenditure [45]. Using ppara−/− mice, we showed that PPARA is required for maintaining kidney function and stimulating primary cilia formation in kidney during starvation, which is dependent on activation of autophagy. PPARA has been reported to protect against renal dysfunction caused by hypertension or acute kidney injury [46,47]. Based on our study, nutrient deprivation, a potentially acute stress, also induces kidney damage due to dysregulation of autophagy and ciliogenesis as shown in ppara−/− mice kidney. In agreement with the reported renoprotective effect of PPARA, we found cystically dilated tubular structures in the kidneys of ppara−/− mice during starvation. Consistent with the effect of rapamycin in vitro, our results showed that the autophagy activator rescues cilia defect and renal damage in ppara−/− mice kidney. Although the detail mechanisms are required for further investigation, we speculated that this tubular dilation might impair normal kidney function resulting in renal cysts similar to those observed in PKD [48,49]. Since an increase in cell proliferation and activation of mTOR are necessary for cyst formation in PKD [50–52], it is conceivable that depletion of PPARA may stimulate the cell cycle, leading to tubular dilation via inhibition of autophagy [53].
NR1H4 is highly expressed in kidney, and controls renal lipid metabolism and water reabsorption in renal tubules [54–56]. We showed that NR1H4 acts as negative regulator for autophagy to prevent ciliogenesis. In kidney, treatment with an NR1H4 agonist prevents autophagy and ciliogenesis, which may impair kidney function as evidenced by the increase of BUN and creatinine in serum and dilated tubules during starvation. Several reports showed that cysts originate from collecting ducts where AQP2 (aquaporin 2) is expressed [57,58]. Since NR1H4 regulates expression of AQP2 and the lack of cilia is proposed to dysregulate AQP2 location in collecting ducts [59], NR1H4 activity might be responsible for the AQP2 abnormality that leads to kidney damage through controlling autophagy and ciliogenesis. Taken together, our results indicate that PPARA and NR1H4 reciprocally control ciliogenesis through regulating autophagy, which may be critical for ciliary function, and mediating the appropriate response to nutrient availability in the kidney.
Materials and methods
Antibodies and reagents
Antibodies against the following proteins were used: ARL13B (Proteintech Group, 17711-1-AP), ATG7 (Cell Signaling Technology, 2631S), ACTB (Santa Cruz Biotechnology, sc-47778), SQSTM1 (Abnova, H00008878-M01), OFD1 (Abcam, ab122960), LC3 (Sigma-Aldrich, L8918), acetylated TUBA (Sigma-Aldrich, T7451), TUBG (Sigma-Aldrich, T5326). Secondary antibodies for immunoblotting were from Bethyl Laboratories (goat anti-mouse 31430, goat anti-rabbit 31460) and for immunofluorescent staining (goat anti-mouse Alexa Fluor 488 A-11001, goat anti-rabbit Alexa Fluor 568 A-11011) were from ThermoFisher. Rhodamine-DBA (RL-1032) was purchased from Vector Laboratories. Rapamycin (S1039) was purchased from Selleck Chemicals. GW 7647 (10008613) and Wy 14643 (70730) was bought from Cayman Chemical; GW 4064 (2473) was bought from Tocris Bioscience. Dimethyl sulfoxide (DMSO, D2650), CQ (C6628) and 3-MA (M9281) were bought from Sigma-Aldrich. To detect cell nuclei, samples were mounted with the Prolong Gold anti-fade reagent containing DAPI (4’-6-diamidino-2-phenylindole) (Sigma-Aldrich, P36931).
Cell culture and drug treatment
htRPE1, htRPE1-SMO-GFP (a gift from Joon Kim, KAIST, Korea), htRPE1-RFP-GFP-LC3, and MEFs were cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM; Gibco, 11965092) supplemented with 10% fetal bovine serum (FBS; Gibco, 16000044) and 100 IU/ml penicillin and 100 µg/ml streptomycin at 37°C and 5% CO2 in a humidified atmosphere. Ppara+/+ and ppara−/− mouse embryonic fibroblasts (MEFs) were isolated from Ppara+/+ and ppara−/− mouse embryos according to a previously described protocol [60]. Atg7+/+ and atg7−/− MEFs were a gift from Inhe Lee, Ewha University, Korea. HK2 and A549 were cultured in RPMI-1640 (Sigma, R6504) supplemented with 10% FBS, 100 IU/ml penicillin, and 100 µg/ml streptomycin. All cell lines were tested for mycoplasma contamination and validated as negative. For the treatment of cells, GW 7647 (0.5 μM), GW 4064 (1 μM, 3 μM, or 6 μM), or rapamycin (1 μM) was added to the medium for 24 h. The chemicals 3-MA (5 mM or 10 mM) or CQ (1 μM or 5 μM) were added for 12 h. For the treatment of cells in qPCR, GW 7647 (0.5 μM) was added into DMEM or RPMI-1640 media for 12 h.
htRPE1-RFP-GFP-LC3
To generate stable cell lines expressing ptfLC3 (Addgene, 21074; deposited by Tamotsu Yoshimori), RPE1 cells were transfected with 2 μg of RFP-GFP-LC3 plasmids using X-tremeGENE HP DNA transfection reagent (Sigma-Aldrich, 6366244001) [61]. Twenty-four h after transfection, cells were switched to a medium supplemented with 700 mg/ml G418 (Sigma-Aldrich, A1720) to select neomycin-resistant cells. Fresh medium was added every 2 to 3 d until colonies were formed at about 15 d. Individual colonies were isolated with cloning cylinders and the expression of RFP-GFP-LC3 was assessed under a fluorescence microscope.
Primary cilia biogenesis assay in vitro
To ensure that cultures reached the same level of confluence in normal medium and serum-free medium, different quantities of cells were seeded into 12-well dishes (105 for normal medium and 2 × 105 for serum free medium). For MEF and RPE1 cells, just after they reached confluence on the coverslips in either fresh normal medium (10% FBS, DMEM) or serum-starved medium (0.5% FBS, DMEM), they were treated as indicated for 24 h. A549 cells were treated for 24 h after reaching overconfluence in both conditions. HK2 cells were treated for 12 h at subconfluent levels. Cells were then fixed with 4% paraformaldehyde (Electron Microscopy Sciences, 15714) for immunofluorescent staining. For quantification of the number of ciliated cells, Z-stack sections were acquired by Olympus FluoView 1000 confocal laser scanning system (Shinjuku-ku, Tokyo, Japan), and 200 cells were counted per well in triplicate samples. Cilia length was measured using ImageJ (NIH, Bethesda, MD) after thresholding of the images.
Immunofluorescent staining in vitro
Cells were treated according to the protocol described above with or without serum starvation or treatment with compounds. Cells were fixed with 4% paraformaldehyde for 20 min or cold methanol for 5 min at -20°C. Cells were then washed 3 times with phosphate-buffered saline (PBS; Gibco, 10010023) and blocked with blocking buffer (2.5% bovine serum albumin [BSA; bioWORLD, 22070008], 0.1% Triton X-100 [Sigma-Aldrich, T8787] in PBS) at room temperature for 1 h. Cells were incubated with primary antibodies at 4°C overnight, washed 5 times with PBS buffer and then incubated with the appropriate secondary antibodies conjugated to Alexa Fluor 488 or Alexa Fluor 568 for 1 h at room temperature. DNA was stained with DAPI. Slides were examined using an Olympus FluoView 1000 confocal laser scanning system.
Western blot analysis
Proteins from cells or tissues were extracted with RIPA buffer as previously described [62]. In brief, kidney tissues were isolated from mice. Cells (3 × 106) were scraped off culture plates and centrifuged at 1000 × g for 5 min at 4°C. Cultured cells or kidney tissue were homogenized with buffer (10 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate (Sigma-Aldrich, D6750), 1 mM ethylenediaminetetraacetic acid [EDTA], and 1 × protease inhibitor cocktail [GenDEPOT, P3100-001]) and centrifuged at 13,000 × g for 10 min at 4°C. The supernatant was heated at 96°C for 10 min then separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were electroblotted onto PVDF membranes (EMD Millipore, IPFL00010). The membranes were incubated in TBST blocking buffer (5% nonfat dry milk [Lab Scientific, M0841] in TBST (20 mM Tris-HCl, pH 7.6, 150 mM NaCl, 0.1% Tween 20 [Sigma-Aldrich, P1379])) for 1 h, followed by the primary antibody and the secondary horseradish peroxidase (HRP)-conjugated antibody (Bethyl Laboratories, goat anti-mouse 31430, goat anti-rabbit 31460). Quantification was performed by densitometry and analyzed using Gel-Pro Analyzer software.
Small interfering RNA (siRNA) transfection
Pre-designed mouse NR1H4 (Santa Cruz Biotechnology, sc-38848) and scrambled control siRNAs (Santa Cruz Biotechnology, sc-37007) were transfected into cells. Transfections were carried out using Lipofectamine RNAiMAX (Invitrogen, 13778-150) according to the manufacturer's protocol. After siRNA transfection for 48 h, cells were treated with 0.5 μM GW 7647 for 18 h. Autophagy was analyzed, followed by expression analysis using quantitative real-time polymerase chain reaction (qPCR) and western blotting.
Quantitative real time polymerase chain reaction (qPCR)
Total RNA was extracted from cultured cells or kidney tissue using TRIzol reagent (Invitrogen, 15596026). A reverse transcription kit (Roche, 04379012001) was used to transcribe cDNA for qPCR analysis using LightCycler with FastStart Essential DNA Green Master (Roche, 06402712001). The human primer sequences (forward and reverse, respectively) were as follows: intracellular control 36B4, 5′-TGCATCAGTACCCCATTCTATCA-3′, 5′-AAGGTGTAATCCGTCTCCACAGA-3′; LC3A, 5′-CATGAGCGAGTTGGTCAAGA-3′, 5′-CCATGCTGTGCTGGTTCA-3′; LC3B, 5′-CGCACCTTCGAACAAAGAG-3′, 5′-CTCACCCTTGTATCGTTCTATTATCA-3′; SESN2, 5′-CGCTTTGAGCTGGAGAAGTC-3′, 5′-TCCACAAAGCACAGCATGTC-3′; ULK1, 5′-CAGACGACTTCGTCATGGTC-3′, 5′-AGCTCCCACTGCACATCAG-3′; ATG2A, 5′-CCTCGCCCTTCTCCTCTAAG-3′, 5′-TCTGGAACGTCCTCATCTCC-3′; ATG12, 5′-TCTTCCGCTGCAGTTTCC-3′, 5′-TGTCTCCCACAGCCTTTAGC-3′. The mouse primer sequences (forward and reverse) were as follows: intracellular control Rplp0/36B4, 5′-CACTGGTCTAGGACCCGAGAAG-3′, 5′-GGTGCCTCTGGAGATTTTCG-3′; Atg12, 5′-GTCTGTCTATGCCTCCAA-3′, 5′-TCCACTTCCTCAATGCTA-3′; Atg2a, 5′-CACTCTACGCCACTACAT-3′, 5′-ATCCAGCACATCCAAGAA-3′; Bnip3, 5′- TGGACGAAGTAGCTCCAAGAG-3′, 5′-TCCTCAGACAGAGTGCTGTTTT-3′; Lc3a, 5′-GACCAGCACCCCAGTAAGAT-3′, 5′-TGGGACCAGAAACTTGGTCT-3′; Lc3b, 5′-CCCCACCAAGATCCCAGT-3′, 5′-CGCTCATGTTCACGTGGT-3′; Sesn2, 5′-CAGCGCTTTCATTCCAGTG-3′, 5′-GGGTGTAGACCCATCACCAC-3′.
Animal and drug treatments
Experiments were conducted with 8-wk-old to 10-wk-old wild-type (WT) and ppara−/− male mice [63]. Mice were permitted free access to water and standard mouse chow. All experimental protocols were approved by the Committee for Ethics in Animal Experiments of the Wonkwang University (WKU15-104) and carried out under the Guidelines for Animal Experiments. ppara−/− mice were divided into 4 groups after 48 h fasting: wild-type fed group (WT-fed, n = 6), wild-type fasted group (WT-fasted, n = 6), ppara−/− fed group (n = 6), ppara−/− fasted group (n = 6). For the PPARA and NR1H4 competition in vivo, the experimental mice were divided into 8 groups: vehicle injection in wild-type fed group (WT-fed, n = 6), wild-type fasted group (WT-fasted, n = 6), ppara−/− fed group (n = 6), and ppara−/− fasted group (n = 6). GW 4064 injection in wild-type fed group (WT-Fed, n = 6), wild-type fasted group (WT-fasted, n = 6), ppara−/− fed group (n = 6), and ppara−/− fasted group (n = 6). Vehicle (DMSO) and GW 4064 (40 mg/kg body weight) were administered via intraperitoneal injection 4 times during 2 d fasting. Then mice were sacrificed to collect blood and kidneys. Collected kidney tissues were immediately frozen in liquid nitrogen for molecular analysis.
Histology
Kidneys were perfused and fixed as previously described [64]. Kidneys were perfused via the left ventricle with 100 ml PBS, followed fixing by 4% paraformaldehyde in PBS. After perfusion, the kidneys were excised and placed in 4% paraformaldehyde for 4 h at 4°C. Then, kidneys were dehydrated with an alcohol series. Paraffin-embedded tissue samples were then sliced into 20-μm sections using a Leica RM2255 Fully Automated Rotary microtome (Heidelberger, Nußloch, Germany). Paraffin-embedded tissue sections were deparaffinized with xylene, rehydrated with 100, 90, 80, and 70% ethanol, and then washed with PBS for 10 min. Paraffin sections were stained with hematoxylin and eosin (H&E) staining for morphological evaluation. H&E staining was conducted in accordance with the manufacturer's protocols (Vector Laboratories, H-3502). Images were collected with Olympus light IX71 microscope (Shinjuku-ku, Tokyo). Each experimental animal group consisted of more than 6 mice.
Immunofluorescent staining in vivo
Prepared sections were incubated in PBS containing 0.1% sodium dodecyl sulfate (SDS; Sigma-Aldrich, 436143) for 5 min and washed for 10 min in PBS. To reveal the antigen epitope, the sections were boiled in 10 mM sodium citrate buffer (10 mM Tri-sodium citrate in PBS, pH 6.0) for 10 min using a microwave, cooled for 20 min at room temperature, and then washed 3 times with PBS for 5 min each. The sections were blocked with blocking buffer (PBS containing 1% BSA) for 30 min at room temperature and then incubated with ARL13B overnight at 4°C. After incubation, the sections were washed 3 times in PBS for 5 min each, incubated with Alexa Fluor 568-conjugated secondary antibodies for 60 min at room temperature, and washed 3 times with PBS for 5 min each. To detect the cell nuclei, the sections were mounted with the Prolong Gold anti-fade reagent containing DAPI and observed using an Olympus FluoView 1000 confocal laser scanning system. Each experimental animal group consisted of 6 mice. For quantification of the number of ciliated cells, Z-stack sections were acquired and 1000 cells were counted per section in kidney samples.
DBA staining
Kidney sections were processed for DBA staining as described above. Sections were stained with Rhodamine-DBA overnight at 4°C, and washed 3 times with PBS for 5 min each. To detect the cell nuclei, the sections were mounted with the Prolong Gold anti-fade reagent containing DAPI.
Damage index calculations
The images of kidney section were obtained from H&E staining. A grid was placed over the H&E stained images, and the damage index was calculated as the percentage of grid intersection points that bisected damage or nondamaged areas [51].
Supplementary Material
Funding Statement
This work was supported by National Research Foundation of Korea (NRF) grants funded by the Korean government [grant number 2014M3A9D8034463], [grant number 2011–0030130], [grant number 2015R1A2A1A15054674], and [grant number 2017R1A2B2008840] and by GIST Research Institute (GRI) grant funded by GIST in 2017.
Disclosure of potential conflicts of interest
The authors have no conflict of interest to declare.
Acknowledgements
ppara-/- mice were a generous gift from Frank J. Gonzalez. This work was supported by National Research Foundation of Korea (NRF) grants funded by the Korean government (No. 2014M3A9D8034463, 2011–0030130, 2015R1A2A1A15054674, and 2017R1A2B2008840) and by GIST Research Institute (GRI) grant funded by GIST in 2017.
References
- [1].Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010;11:331–344. doi: 10.1038/nrg2774. PMID:20395968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Pedersen LB, Veland IR, Schrøder JM, et al. Assembly of primary cilia. Dev Dyn. 2008;237:1993–2006. doi: 10.1002/dvdy.21521. PMID:18393310 [DOI] [PubMed] [Google Scholar]
- [3].Fliegauf M, Benzing T, Omran H. When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol. 2007;8:880–893. doi: 10.1038/nrm2278. PMID:17955020 [DOI] [PubMed] [Google Scholar]
- [4].Gerdes JM, Davis EE, Katsanis N. The vertebrate primary cilium in development, homeostasis, and disease. Cell. 2009;137:32–45. doi: 10.1016/j.cell.2009.03.023. PMID:19345185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lancaster MA, Gleeson JG. The primary cilium as a cellular signaling center: lessons from disease. Curr Opin Genet Dev. 2009;19:220–229. doi: 10.1016/j.gde.2009.04.008. PMID:19477114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Oh EC, Katsanis N. Cilia in vertebrate development and disease, Development. 2012;139:443–448. doi: 10.1242/dev.050054. PMID:22223675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Nigg EA, Stearns T. The centrosome cycle: Centriole biogenesis, duplication and inherent asymmetries. Nat Cell Biol. 2011;13:1154–1160. doi: 10.1038/ncb2345. PMID:21968988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kobayashi T, Dynlacht BD. Regulating the transition from centriole to basal body. J Cell Biol. 2011;193:435–444. doi: 10.1083/jcb.201101005. PMID:21536747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ishikawa H, Marshall WF. Ciliogenesis: building the cell's antenna. Nat Rev Mol Cell Biol. 2011;12:222–234. doi: 10.1038/nrm3085. PMID:21427764 [DOI] [PubMed] [Google Scholar]
- [10].Kim J, Lee JE, Heynen-Genel S, et al. Functional genomic screen for modulators of ciliogenesis and cilium length. Nature. 2010;464:1048–1051. doi: 10.1038/nature08895. PMID:20393563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kim J, Jo H, Hong H, et al. Actin remodelling factors control ciliogenesis by regulating YAP/TAZ activity and vesicle trafficking. Nat Commun. 2015;6:6781. doi: 10.1038/ncomms7781. PMID:25849865 [DOI] [PubMed] [Google Scholar]
- [12].Santos N, Reiter JF. Building it up and taking it down: the regulation of vertebrate ciliogenesis. Dev Dyn. 2008;237:1972–1981. doi: 10.1002/dvdy.21540. PMID:18435467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. PMID:18191218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. PMID:22078875 [DOI] [PubMed] [Google Scholar]
- [15].Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell Metab. 2011;13:495–504. doi: 10.1016/j.cmet.2011.04.004. PMID:21531332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157:65–75. doi: 10.1016/j.cell.2014.02.049. PMID:24679527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Pampliega O, Orhon I, Patel B, et al. Functional interaction between autophagy and ciliogenesis. Nature. 2013;502:194–200. doi: 10.1038/nature12639. PMID:24089209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tang Z, Lin MG, Stowe TR, et al. Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature. 2013;502:254–257. doi: 10.1038/nature12606. PMID:24089205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bookout AL, Jeong Y, Downes M, et al. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. 2006;126:789–799. doi: 10.1016/j.cell.2006.06.049. PMID:16923397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hong SH, Ahmadian M, Yu RT, et al. Nuclear receptors and metabolism: from feast to famine. Diabetologia. 2014;57:860–867. doi: 10.1007/s00125-014-3209-9. PMID:24619218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lee JM, Wagner M, Xiao R, et al. Nutrient-sensing nuclear receptors coordinate autophagy. Nature. 2014;516:112–115. PMID:25383539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Seok S, Fu T, Choi SE, et al. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature. 2014;516:108–111. PMID:25383523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–650. doi: 10.1038/347645a0. PMID:2129546 [DOI] [PubMed] [Google Scholar]
- [24].Mandard S, Müller M, Kersten S. Peroxisome proliferator-activated receptor alpha target genes. Cell Mol Life Sci. 2004;61:393–416. doi: 10.1007/s00018-003-3216-3. PMID:14999402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Calkin AC, Tontonoz P. Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat Rev Mol Cell Biol. 2012;13:213–224. doi: 10.1038/nrm3312. PMID:22414897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gadaleta RM, Cariello M, Sabbà C, et al. Tissue-specific actions of FXR in metabolism and cancer. Biochim Biophys Acta. 2015;1851:30–39. doi: 10.1016/j.bbalip.2014.08.005. PMID:25139561 [DOI] [PubMed] [Google Scholar]
- [27].Komatsu M, Waguri S, Koike M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. PMID:18083104 [DOI] [PubMed] [Google Scholar]
- [28].Pazour GJ, Dickert BL, Vucica Y, et al. Chlamydomonas. IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Biol. 2000;151:709–718. doi: 10.1083/jcb.151.3.709. PMID:11062270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhang Q, Taulman PD, Yoder BK. Cystic kidney diseases: all roads lead to the cilium. Physiology (Bethesda). 2004;19:225–230. PMID:15304637 [DOI] [PubMed] [Google Scholar]
- [30].Patel V, Li L, Cobo-Stark P, et al. Acute kidney injury and aberrant planar cell polarity induce cyst formation in mice lacking renal cilia. Hum Mol Genet. 2008;17:1578–1590. doi: 10.1093/hmg/ddn045. PMID:18263895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lin F, Satlin LM. Polycystic kidney disease: the cilium as a common pathway in cystogenesis. Curr Opin Pediatr. 2004;16:171–176. doi: 10.1097/00008480-200404000-00010. PMID:15021197 [DOI] [PubMed] [Google Scholar]
- [32].Huber TB, Edelstein CL, Hartleben B, et al. Emerging role of autophagy in kidney function, diseases and aging. Autophagy. 2012;8:1009–1031. doi: 10.4161/auto.19821. PMID:22692002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Jiang M, Wei Q, Dong G, et al. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012;82:1271–1283. doi: 10.1038/ki.2012.261. PMID:22854643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sugden MC, Bulmer K, Gibbons GF, et al. Role of peroxisome proliferator-activated receptor-alpha in the mechanism underlying changes in renal pyruvate dehydrogenase kinase isoform 4 protein expression in starvation and after refeeding. Arch Biochem Biophys. 2001;395:246–252. doi: 10.1006/abbi.2001.2586. PMID:11697863 [DOI] [PubMed] [Google Scholar]
- [35].Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med. 2011;364:1533–1543. doi: 10.1056/NEJMra1010172. PMID:21506742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang S, Livingston MJ, Su Y, et al. Reciprocal regulation of cilia and autophagy via the MTOR and proteasome pathways. Autophagy. 2015;11:607–616. doi: 10.1080/15548627.2015.1023983. PMID:25906314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Shin JH, Kim PS, Kim ES, et al. BIX-01294-induced autophagy regulates elongation of primary cilia. Biochem Biophys Res Commun. 2015;460:428–433. doi: 10.1016/j.bbrc.2015.03.050. PMID:25796328 [DOI] [PubMed] [Google Scholar]
- [38].Kliewer SA, Sundseth SS, Jones SA, et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc Natl Acad Sci USA. 1997;94:4318–4323. doi: 10.1073/pnas.94.9.4318. PMID:9113987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Keller H, Dreyer C, Medin J, et al. Fatty acids and retinoids control lipid metabolism through activation of peroxisome proliferator-activated receptor-retinoid X receptor heterodimers. Proc Natl Acad Sci USA. 1993;90:2160–2164. doi: 10.1073/pnas.90.6.2160. PMID:8384714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Jonassen JA, San Agustin J, Follit JA, et al. Deletion of IFT20 in the mouse kidney causes misorientation of the mitotic spindle and cystic kidney disease. J Cell Biol. 2008;183:377–384. doi: 10.1083/jcb.200808137. PMID:18981227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lin F, Hiesberger T, Cordes K, et al. Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci USA. 2003;100:5286–5291. doi: 10.1073/pnas.0836980100. PMID:12672950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ko JY, Park JH. Mouse models of polycystic kidney disease induced by defects of ciliary proteins. BMB Rep. 2013;46:73–79. doi: 10.5483/BMBRep.2013.46.2.022. PMID:23433108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Takiar V, Nishio S, Seo-Mayer P, et al. Activating AMP-activated protein kinase (AMPK) slows renal cystogenesis. Proc Natl Acad Sci USA. 2011;108:2462–2467. doi: 10.1073/pnas.1011498108. PMID:21262823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ravichandran K, Edelstein CL. Polycystic kidney disease: a case of suppressed autophagy? Semin Nephrol. 2014;34:27–33. doi: 10.1016/j.semnephrol.2013.11.005. PMID:24485027 [DOI] [PubMed] [Google Scholar]
- [45].Park CW, Zhang Y, Zhang X, et al. PPARalpha agonist fenofibrate improves diabetic nephropathy in db/db mice. Kidney Int. 2006;69:1511–1517. doi: 10.1038/sj.ki.5000209. PMID:16672921 [DOI] [PubMed] [Google Scholar]
- [46].Portilla D, Dai G, Peters JM, et al. Etomoxir-induced PPAR alpha-modulated enzymes protect during acute renal failure. Am J Physiol Ren Physiol. 2000;278:667–675. doi: 10.1152/ajprenal.2000.278.4.F667. [DOI] [PubMed] [Google Scholar]
- [47].Li S, Nagothu KK, Desai V, et al. Transgenic expression of proximal tubule peroxisome proliferator-activated receptor-alpha in mice confers protection during acute kidney injury. Kidney Int. 2009;76:1049–1062. doi: 10.1038/ki.2009.330. PMID:19710628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yoder BK. Role of primary cilia in the pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2007;18:1381–1388. doi: 10.1681/ASN.2006111215. PMID:17429051 [DOI] [PubMed] [Google Scholar]
- [49].Zhou J. Polycystins and primary cilia: primers for cell cycle progression. Annu Rev Physiol. 2009;71:83–113. doi: 10.1146/annurev.physiol.70.113006.100621. PMID:19572811 [DOI] [PubMed] [Google Scholar]
- [50].Huber TB, Walz G, Kuehn EW. mTOR and rapamycin in the kidney: signaling and therapeutic implications beyond immunosuppression. Kidney Int. 2011;79:502–511. doi: 10.1038/ki.2010.457. PMID:21085109 [DOI] [PubMed] [Google Scholar]
- [51].Shillingford JM, Murcia NS, Larson CH, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci USA. 2006;103:5466–5471. doi: 10.1073/pnas.0509694103. PMID:16567633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Boehlke C, Kotsis F, Patel V, et al. Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat Cell Biol. 2010;12:1115–1122. doi: 10.1038/ncb2117. PMID:20972424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Gizard F, Amant C, Barbier O, et al. PPAR alpha inhibits vascular smooth muscle cell proliferation underlying intimal hyperplasia by inducing the tumor suppressor p16INK4a. J Clin Invest. 2005;115:3228–3238. doi: 10.1172/JCI22756. PMID:16239970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sinal CJ, Tohkin M, Miyata M, et al. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731–744. doi: 10.1016/S0092-8674(00)00062-3. PMID:11030617 [DOI] [PubMed] [Google Scholar]
- [55].Zhang X, Huang S, Gao M, et al. Farnesoid X receptor (FXR) gene deficiency impairs urine concentration in mice. Proc Natl Acad Sci USA. 2014;111:2277–2282. doi: 10.1073/pnas.1323977111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wang XX, Jiang T, Shen Y, et al. Diabetic nephropathy is accelerated by farnesoid X receptor deficiency and inhibited by farnesoid X receptor activation in a type 1 diabetes model. Diabetes. 2010;59:2916–2927. doi: 10.2337/db10-0019. PMID:20699418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Shibazaki S, Yu Z, Nishio S, et al. Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific inactivation of Pkd1. Hum Mol Genet. 2008;17:1505–1516. doi: 10.1093/hmg/ddn039. PMID:18263604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Lin F, Hiesberger T, Cordes K, et al. Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci USA. 2003;100:5286–5291. doi: 10.1073/pnas.0836980100. PMID:12672950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Saigusa T, Reichert R, Guare J, et al. Collecting duct cells that lack normal cilia have mislocalized vasopressin-2 receptors. Am J Physiol Renal Physiol. 2012;302:F801–F808. doi: 10.1152/ajprenal.00253.2011. PMID:22205228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Conner DA. Mouse embryo fibroblast (MEF) feeder cell preparation. Curr Protocols Mol Biol. 2001;23:23.2 [DOI] [PubMed] [Google Scholar]
- [61].Jin A, Lee JN, Kim MS, et al. 2,2'-dipyridyl induces pexophagy. Biochem Biophys Res Commun. 2016;469:941–947. doi: 10.1016/j.bbrc.2015.12.098. PMID:26721431 [DOI] [PubMed] [Google Scholar]
- [62].Lee JN, Kim SG, Lim JY, et al. 3-Aminotriazole protects from CoCl2-induced ototoxicity by inhibiting the generation of reactive oxygen species and proinflammatory cytokines in mice. Arch Toxicol. 2016;90:781–791. doi: 10.1007/s00204-015-1506-9. PMID:25820916 [DOI] [PubMed] [Google Scholar]
- [63].Lee SS, Pineau T, Drago J, et al. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol. 1995;15:3012–3022. doi: 10.1128/MCB.15.6.3012. PMID:7539101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Gage GJ, Kipke DR, Shain W. Whole animal perfusion fixation for rodents. J Vis Exp. 2012;65:pii: 3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.