

Abstract
An approach to synthesize the pentacyclic framework of the polyol diterpenoid ryanodol is reported. The ABC tricycle was constructed by a Comediated Pauson-Khand reaction, and both radical and anionic cyclization pathways were developed to form the E-ring. In addition, a reaction sequence involving SeO2-mediated enone oxidation and hydroxyl-directed oxy-Michael addition was developed to introduce the A-ring oxidation. The feasibility of forming the bridging D-ring by an oxidative dearomatization was established.
Ryanodine (l) is a diterpenoid natural product that was isolated from the insecticidal shrub Ryania speciose Vahli n 1958 (Figure 1).1 Investigations of ryanodine’s insecticidal properties ultimately led to the identification of the ryanodine receptors (RyRs), high conductance intracellular calcium ion channels that are involved in biological signaling pathways such as excitation-contraction coupling.2,3 Ryanodine binds to the open state of RyRs with high affinity and is widely used as a probe molecule to interrogate the functional state of RyRs.4 Hydrolysis of the C3-ester of ryanodine provides ryanodol,1b,5 a compound that retains affinity for insect RyRs but exhibits significantly lower affinity for the mammalian receptors. In addition to ryanodine, a series of related ryanoids with variations in the peripheral oxidation have been isolated over the past several decades (e.g. 3, 4, 5).6 Whereas the pharmacology of ryanodine has been extensively investigated,7 far less is known about the biological activities of these minor ryanoid natural products. As part of a program aimed at the development of new ryanoid probes of RyR function, we initiated a synthetic effort to prepare ryanodol, ryanodine, and related ryanoid congeners.
Figure 1.

Representative examples of ryanoid natural products.
Although our long-term objective was to prepare ryanodine (l), initial studies focused on the hydrolysis product, ryanodol (2), to validate our synthetic strategy. Structural analysis of ryanodol identified the central bridging D and E rings as opportunities for strategic disconnections (Scheme 1). Prior work by Deslongchamps and coworkers had established that 2 can be prepared in two steps from anhydroryanodol (6),8 establishing the viability of late-stage E-ring formation. In 2016, we reported a 13-step synthesis of anhydroryanodol from (S)-pulegone (7)9 and subsequently demonstrated that a similar approach furnishes (+)-ryanodine (1) in 18 steps.10,11
Scheme 1.

Retrosynthetic analysis of ryanodol (2).
Our successful syntheses of 1 and 2 were heavily guided by a first-generation approach to 2, wherein we envisioned late-stage formation of the D-ring by an oxidative dearomatization of phenol 8 (Scheme 1). Phenol 8 was expected to be accessible by a radical or anionic cyclization of 9a or 9b, either of which could be prepared from the product of a Pauson-Khand reaction12 of enyne 10. Enyne 10 could be prepared from arene 11. An appealing aspect of this approach is that it would enable divergent, late-stage oxidation of the C-ring, which could provide access to additional ryanoids such as ga-rajonone (3), 2,3-didehydrocinnzeylanone (4), or 9-hydroxy-10-epi-ryanodine (5, Figure 1). In this report, we disclose the chemistry developed as part of our first-generation approach to ryanodol (2). While this approach was not ultimately used to prepare 1 and 2, these studies established that the carbocyclic framework of ryanodol can be prepared by an oxidative dearomatization. In addition, these studies identified the use of a Pauson-Khand reaction to prepare the ABC tricycle, and a sequence for A-ring functionalization involving SeO2-mediated cyclopentenone oxidation and isopropenyl cross-coupling, tactics that were incorporated into the successful syntheses of 1 and 2.9,10
The first objective of our preliminary investigations was to validate the feasibility of preparing the pentacyclic ryanodol core by an oxidative dearomatization. To this end, phenol 1213 was protected as the methyl ether and the iodide was chemoselectively cross-coupled with n-butyl vinyl ether via a Pd-catalyzed Heck reaction,14 which upon acidic workup delivered ketone 13 (Scheme 2). Sonogashira coupling of the bromide with propyne furnished alkyne 14 in 77% yield.15 Subsequent 1,2-addition of vinylmagnesium bromide to the methyl ketone afforded the tertiary alcohol (10, Scheme 1), which underwent a Co-mediated Pauson-Khand cyclization and acid-catalyzed dehydration to afford tricyclic enone 15.16 This efficient, six-step protocol readily establishes the ABC tricycle present in the ryanoids, and has been executed on multigram scale.
Scheme 2.

Synthesis of the ryanodol ABC tricycle.
At this stage, benzyl ether 16 was also prepared to enable exploratory studies wherein the phenol could be revealed under mild conditions. Both the methyl (15) and benzyl (16) ethers were advanced to the corresponding cylcopentenones 20 and 21. This two-step sequence introduces the first of two all-carbon quaternary centers via chemoselective epoxidation of the more electron rich alkene, followed by Lewis acid-catalyzed epoxide opening with silyl ketene acetal 19.17
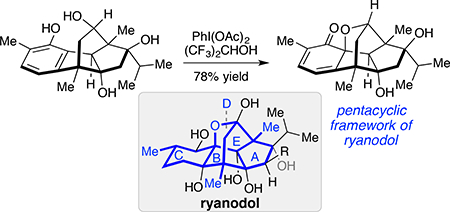
Initial studies focused on E-ring formation from methyl ether 20. 1,2-Addition of isopropyl magnesium chloride to 20 in the presence of CeCl3 2LiCl delivered the isopropyl group from the α-face (Scheme 3).18 Hydrolysis of the methyl ester was followed by conversion of the acid to the acyl selenide 23. Treatment of 23 with (TMS)3SiH and AIBN resulted in acyl radical cyclization to provide tetracyclic ketone 24 in moderate yield. Notably, the isomeric product resulting from radical addition to C12 was not observed; this product is likely disfavored due to the strain associated with a transfused 5–5 ring system.19 Tetracycle 24 was advanced to an oxidative cyclization substrate, 25, by NaBH(OAc)3 reduction of the C15 ketone followed by nucleophilic demethylation of the phenol. The stereoselectivity of the NaBH(OAc)3 reduction was confirmed by NOESY.20 Treatment of phenol 25 with Phl(OAc)2 led to clean formation of the desired ortho-oxidative dearomatization product 26 bearing the complete pentacyclic ryanoid carbon skeleton. Hydroxyl-directed epoxidation with m-CPBA followed by hydrogenolysis of the allylic epoxide afforded 27. Pentacycle 27 has the fully functionalized C ring of the natural ryanoid 2,3-didehydro-cinzeylanone (4), but lacks the C12 and C15 alcohols. Nonetheless, these studies validated the feasibility of an oxidative dearomatization approach to prepare the ryanoid framework.
Scheme 3.

Oxidative dearomatization to form the ryanoid carbon skeleton.
Recognizing that it would be difficult to introduce the C12 and C15 alcohols through existing C-H oxidation technologies, we investigated a second approach in which the hydroxyl group at C12 would be introduced prior to oxidative dearomatization (Scheme 4). Riley oxidation of enone 21 under microwave irradiation provided diketone 28.21 We were pleased to find that condensation of the tertiary alcohol of 28 with nBuB(OH)2 enabled intramolecular oxy-Michael addition; in situ trapping of the resulting enolate with Tf2O afforded triflate 29 in a one-pot procedure.22 The reactive enediketone of 28 is required for the oxy-Michael addition, as enone 21 did not undergo the corresponding reaction. Suzuki cross-coupling of vinyltriflate 29, followed by cleavage of the labile cyclic boronate provided diol 32.
Scheme 4.

Synthesis of Tetracycle 34
With access to the fully functionalized ABC tricycle, we turned our attention to the installation of a functional group (FG) precursor that could ultimately furnish the required hydroxyl at C15 (Scheme 4). To this end, methyl ester 32 was saponified and converted to acyl selenide 33, which upon exposure to Bu3SnH and AIBN at 80 °C underwent intramolecular acyl radical cyclization to provide tetracycle 34. Unfortunately, efforts to convert diketone 34 to tertiary alcohols, such as 35, using a variety of nucleophiles (TMSCN, vinyl Grignard, ethynyl Grignard, etc.) were unsuccessful due to competitive addition to the C3 ketone, or incorrect diastereoselectivity at C15. Given the propensity of nucleophiles to approach the C15 ketone from the sterically less encumbered trajectory (over the arene), we envisioned that dihydroxylation of a tetracycle bearing an C15 exo-methylene would provide a tertiary alcohol with the required configuration (e.g. 35, FG = CH2OH). The required C15 hydroxyl group could be revealed via a Baeyer-Villiger protocol.
To this end, exo-olefin 42 was prepared by first adding allenyl stannane 36 to epoxide 18 to give tertiary alcohol 37 as a single diastereomer in 53% yield (Scheme 5).23 The alkyne of 37 was elaborated to vinyl iodide 38 by regioselective stannylalumination/protonolysis with Bu3SnAlEt2/CuCN and subsequent iododestannylation.24 Hydroxyl-directed epoxidation of 38, followed by protection of the tertiary alcohol with TESCl furnished 40. Treatment of 40 with nBuLi in THF at 0 °C provided the the desired 1,2 addition product 41 in 65% yield, which underwent a facile semi-pinacol rearrangement in the presence of TMSOTf and 2,6-lutidine to give tetracycle 42 with the critical vicinal yn-diol at the AB ring fusion. Unfortunately, dihydroxylation of the exo-olefin of 42 proceeded with high diastereoselectivity for the incorrect diol stereoisomer, 43 (confirmed by NOESY data). Efforts to oxidize this terminal alkene by the Prévost reaction,25 Woodward reaction,26 or Sharpless dihydrox-ylation27 failed to produce the desired diol.
Scheme 5.

Synthesis of Diol 43.
These studies established the viability of preparing the ryanodol carbon framework by an oxidative dearomatization approach. Although this strategy did not successfully provide access to the natural product targets, it established several key chemistries that were incorporated into the completed route.
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. Michael Takase and Larry Henling (both of Caltech) for X-ray data collection and Ms. Julie Hofstra (Caltech) for X-ray data refinement. Dr. Scott Virgil and the Caltech Center for Catalysis and Chemical Synthesis are gratefully acknowledged for access to analytical equipment. Fellowship support was provided by the Shenzhen UVChemTech Inc. (C.X.), and the NIH (A. H., 5T32GM007616–37, 1F31GM120821). S.E.R. is a Heritage Medical Research Institute Investigator. Financial support from the NIH (NIGMS RGM097582–01, R35GM118191–01), Eli Lilly, and Novartis is gratefully acknowledged.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures, characterization data (1H and 13C NMR, HRMS, FTIR) for all new compounds (pdf)
REFERENCES
- (1).(a) Rogers EF; Koniuszy FR; Shavel J; Folkers K J. Am. Chem. Soc 1948, 70, 3086–3088. [DOI] [PubMed] [Google Scholar]; (b) Wiesner K; Valenta Z; Findlay JA Tetrahedron Lett. 1967, 8, 221. [DOI] [PubMed] [Google Scholar]
- (2).(a) Pessah IN; Waterhouse AL; Casida JE Biochem. Biophys. Res. Commun 1985, 128, 449–456. [DOI] [PubMed] [Google Scholar]; (b) Inui M; Saito A; Fleischer S J. Biol. Chem 1987, 262, 15637–15642. [PubMed] [Google Scholar]
- (3).(a) Ryanodine Receptors: Structure, Function and Dysfunction in Clinical Disease. Wehrens XHT; Marks AR, Eds.; Springer: 2005. [Google Scholar]; (b) Lanner JT Ryanodine Receptor Physiology and Its Role in Disease In Calcium Signaling, Islam MS, Ed.; Advances in Experimental Medicine and Biology; Springer: 2012. [DOI] [PubMed] [Google Scholar]
- (4).(a) Meissner G J Biol. Chem 1986, 261, 6300–6306. [PubMed] [Google Scholar]; (b)Meissner G; El-Hashem A Mol. Cell. Biochem 1992, 114, 119–123. [DOI] [PubMed] [Google Scholar]
- (5).Srivastava SN; Przybylska M Can. J. Chem 1968, 46, 795–797. [Google Scholar]
- (6).Garajonone and 2,3-didehydro-cinnzeylanone: (a) Fraga BM; Terrero D; Gutierrez C; Gonzalez-Coloma A Phytochemistry 2001, 56, 315 9-Hydroxy-10-epi-ryanodine: [DOI] [PubMed] [Google Scholar]; (b) Hübner H; Vierling W; Brandt W; Reiter M; Achenbach H Phytochemistry 2001, 57, 285. [DOI] [PubMed] [Google Scholar]
- (7).Sutko JL; Airey JA; Welch W; Ruest L Pharmacol. Rev 1997, 49, 53–98. [PubMed] [Google Scholar]
- (8).(a) Deslongchamps P; Bélanger A; Berney DJF; Borschberg HJ;Brousseau R; Doutheau A; Durand R; Katayama H; Lapalme R; Leturc DM; Liao CC; MacLachlan FN; Maffrand JP; Marazza F; Martino R; Moreau C; Ruest L; Saint-Laurent L; Saintonge R; Soucy P Can. J Chem 1979, 57, 3348–3354. [Google Scholar]; (b) Deslongchamps P; Bélanger A; Berney DJF; Borschberg HJ;Brousseau R; Doutheau A; Durand R; Katayama H; Lapalme R; Leturc DM; Liao CC; MacLachlan FN; Maffrand JP; Marazza F; Martino R; Moreau C; Ruest L; Saint-Laurent L; Saintonge R; Soucy P Can. J Chem 1990, 68, 115–126. [Google Scholar]; (c) Deslongchamps P; Bélanger A; Berney DJF; Borschberg HJ;Brousseau R; Doutheau A; Durand R; Katayama H; Lapalme R; Leturc DM; Liao CC; MacLachlan FN; Maffrand JP; Marazza F; Martino R; Moreau C; Ruest L; Saint-Laurent L; Saintonge R; Soucy P Can. J Chem 1990, 68, 127–152. [Google Scholar]; (d) Deslongchamps P; Bélanger A; Berney DJF; Borschberg HJ;Brousseau R; Doutheau A; Durand R; Katayama H; Lapalme R; Leturc DM; Liao CC; MacLachlan FN; Maffrand JP; Marazza F; Martino R; Moreau C; Ruest L; Saint-Laurent L; Saintonge R; Soucy P Can. J Chem 1990, 68, 153–185. [Google Scholar]; (e) Deslongchamps P; Bélanger A; Berney DJF; Borschberg HJ;Brousseau R; Doutheau A; Durand R; Katayama H; Lapalme R; Leturc DM; Liao CC; MacLachlan FN; Maffrand JP; Marazza F; Martino R; Moreau C; Ruest L; Saint-Laurent L; Saintonge R; Soucy P Can. J Chem 1990, 68, 186–192. [Google Scholar]
- (9).Chuang KV; Xu C; Reisman SE Science 2016, 353, 912–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Xu C; Han A; Virgil SC; Reisman SE ACS Cent. Sei 2017, 3, 278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).In addition, Inoue and coworkers have reported syntheses of ryanodol and ryanodine: (a) Nagatomo M; Koshimizu M; Masuda K; Tabuchi T; Urabe D; Inoue M J. Am. Chem. Soc 2014, 136, 5916–5919. [DOI] [PubMed] [Google Scholar]; (b) Nagatomo M; Hagiwara K; Masuda K; Koshimizu M; Kawamata T; Matsui Y; Urabe D; Inoue M Chem. Eur. J 2016,22, 222–229. [DOI] [PubMed] [Google Scholar]; (c) Masuda K; Koshimizu M; Nagatomo M; Inoue M Chem. Eur. J 2016, 22, 230–236. [DOI] [PubMed] [Google Scholar]
- (12).(a) Khand IU; Knox GR; Pauson PL; Watts WE; Forman MI J. Chem. Soc. Perkin Trans. I 1973, 1973, 977–981. [Google Scholar]; (b) Chung YK; Lee BY; Jeong N; Hudecek M; Pauson PL Organometallics, 1993, 12, 220–223 (1993). [Google Scholar]; (c) Shambayani S; Crowe WE; Schreiber SL Tetrahedron Lett. 1990, 31, 5289–5292. [Google Scholar]
- (13).Prepared in one step from commercially available 11 using the following method: Fujisaki S; Eguchi H; Omura A; Okamoto A; Nishida A Bull. Chem. Soc. Jpn 1993, 66, 1576–1579. [Google Scholar]
- (14).Hyder Z; Ruan J; Xiao J Chem. Eur. J 2008, 14, 5555. [DOI] [PubMed] [Google Scholar]
- (15).Hundertmark T; Littke AF; Buchwald SL; Fu GC Org. Lett 2000, 2, 1729–1731. [DOI] [PubMed] [Google Scholar]
- (16).Chung YK; Lee BY; Jeong N; Hudecek M; Pauson PL Organometallics 1993, 12, 220. [Google Scholar]
- (17).Silyl ketene acetal 19 was prepared according to: Wenzel AG; Jacobsen EN J. Am. Chem. Soc 2002, 124, 12964–12965. [DOI] [PubMed] [Google Scholar]
- (18).Krasovskiy A; Kopp F; Knochel P Angew. Chem. Int. Ed 2006, 45, 497–500. [DOI] [PubMed] [Google Scholar]
- (19).It is possible that 24 arises by initial 5-exo-trig cyclization followed by Dowd-Beckwith-type rearrangement to give the more thermodynamically favored product. (a) Dowd P; Choi SC J. Am. Chem. Soc 1987, 109, 3493–3494. [Google Scholar]; (b) Beckwith ALJ; O’Shea DM; Westwood SW J. Am. Chem. Soc 1988, 110, 2565–2575. [Google Scholar]
- (20). See Supporting Information.
- (21). Under the reaction conditions the methyl ester was partially hydrolyzed; treatment of the resulting mixture with TMSCHN2 re-esterifies.
- (22).Li DR; Murugan A; Falck JR J. Am. Chem. Soc 2008, 130, 46–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).The allenyl stannane 36 was prepared by the procedure reported in: Suzuki I; Kiyokawa K; Yasuda M; Baba A Org. Lett 2013, 15, 1728. [DOI] [PubMed] [Google Scholar]
- (24).Corminboeuf O; Overman LE; Pennington LD J. Am. Chem. Soc 2003, 125, 6650–6652. [DOI] [PubMed] [Google Scholar]
- (25).(a) Prévost C Comptes Rendus Acad. Sei 1933, 196, 1129–1131. [Google Scholar]; (b) Rodriguez J; Dulcère JP Synthesis 1993, 1177–1205. [Google Scholar]
- (26).Woodward RB; Brutcher FV Jr. J. Am. Chem. Soc 1958, 80, 209–211. [Google Scholar]
- (27).(a) Jacobsen EN; Marko I; Mungall WS; Schroeder G; Sharpless KB J. Am. Chem. Soc 1988, 110, 1968–1970. [Google Scholar]; (b) Kolb HC; VanNieuwenhze MS; Sharpless KB Chem. Rev 1994, 94, 2483–2547. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.