

Abstract
Protein N-glycosylation is essential for mammalian cell survival and is well-known to be involved in many biological processes. Aberrant glycosylation is directly related to human disease including cancer and infectious diseases. Global analysis of protein N-glycosylation will allow a better understanding of protein functions and cellular activities. Mass spectrometry (MS)-based proteomics provides a unique opportunity to site-specifically characterize protein glycosylation on a large scale. Due to the complexity of biological samples, effective enrichment methods are critical prior to MS analysis. Here, we compared two lectin-independent methods to enrich glycopeptides for the global analysis of protein N-glycosylation by MS. The first boronic acid-based enrichment (BA) method benefits from the universal and reversible interactions between boronic acid and sugars; the other method utilizes metabolic labeling and click chemistry (MC) to incorporate a chemical handle into glycoproteins for future affinity enrichment. We comprehensively compared the performance of the two methods in the identification and quantification of glycoproteins in statin-treated liver cells. Based on the current results, the BA method is more universal in enriching glycopeptides, while with the MC method, cell surface glycoproteins were highly enriched, and the quantification results appear to be more dynamic because only the newly-synthesized glycoproteins were analyzed. In addition, we normalized the glycosylation site ratios by the corresponding parent protein ratios to reflect the real modification changes. In combination with MS-based proteomics, effective enrichment methods will vertically advance protein glycosylation research.
Keywords: Glycoproteomics, Boronic acid-based enrichment, Metabolic labeling, Click chemistry, LC-MS/MS
Graphical Abstract
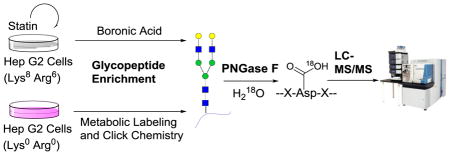
1. Introduction
Protein glycosylation is critical in determining protein folding, trafficking, stability and activity [1, 2]. Among multiple types of protein glycosylation, N- and O-linked glycosylation are the two major types [3, 4]. N-linked glycosylation occurs on the side chain of the asparagine residue and often has an N-X-S/T/C (X stands for any amino acid residues other than proline) [5, 6], while O-linked glycosylation is on the side chains of serine and threonine residues [7–9]. N-glycosylation typically begins with the synthesis of the dolichol-linked precursor oligosaccharide (GlcNAc2Man9Glc3), followed by en bloc transfer of the precursor oligosaccharide to newly synthesized peptides in the endoplasmic reticulum (ER) [10, 11]. Due to its importance in biological systems [12–16], N-glycosylation has also brought extensive attention for its role in human disease, such as Alzheimer’s disease (AD), cancer, and infectious diseases [13, 17, 18].
With the development of mass spectrometry (MS) instrumentation and computation techniques, current MS-based proteomics is very powerful in analyzing protein modifications, including glycosylation, in complex biological samples [19–32]. Due to the low abundance of many glycoproteins, sub-stoichiometry of protein glycosylation, and the complexity of biological samples, it is imperative to enrich glycoproteins prior to MS analysis [28, 33–37]. Conventional lectin-based enrichment methods have been extensively used [38, 39]. However, due to the binding specificity of lectin, no single or several types of lectin can cover all glycoproteins that have highly diverse glycans in human cells.
In recent years, several very elegant methods have been developed and tremendously advanced the glycoproteomics field [6, 28, 29, 40–45]. In this work, we systematically compared two lectin-independent chemical methods to enrich and analyze glycoproteins in human cells: one based on boronic acid and cis-diol interactions [33, 46–48] and the other benefited from metabolic labeling and click reaction [49–51]. For the first method, we utilized the universal and reversible interactions between boronic acid and sugar molecules. Boronic acid and cis-diols can form reversible covalent bonds in basic solutions, and conversely, the bonds are prone to cleavage under acidic conditions. The reversible nature of this bond ensures that glycopeptides can be effectively released after capturing. The second method takes advantage of the endogenous glycoprotein synthesis machinery to incorporate a chemical handle into glycans for further click chemistry and biotin avidin-based glycopeptide enrichment. An unnatural sugar analog containing an azide group was employed to feed cells in order to generate the chemical handle mentioned above. Comparing to BA, we reasoned that MC has the advantage of better reflecting the dynamic changes in cells since only the newly-synthesized glycoproteins are labeled by the sugar analog, while BA may be more universal for glycoprotein enrichment.
In this work, we designed an experiment to comprehensively compare the identification and quantification of glycoproteins with these two methods. We analyzed the glycoproteome changes in statin-treated liver cells using these two methods. Statins are a group of cholesterol-lowering drugs that target 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMGCR), which is the rate-limiting enzyme of the mevalonate pathway. Upon inhibition of HMGCR, the synthesis of many intermediate and end products in this pathway was affected, which induced many well-known pleiotropic effects of statins. Dolichol is one of the end products and is involved in protein N-glycosylation, functioning as the membrane anchor for precursor oligosaccharides formation. Therefore, we expected that protein N-glycosylation was attenuated in statin-treated cells. Using liver cells (HepG2) as a biological model, we systematically evaluated the performance of these two methods and explained the underlying mechanisms for the differences observed. The current work may provide useful information for future selection of enrichment methods to study the cell glycoproteome under different circumstances.
2. Experimental
2.1. Cell Culture and metabolic labeling
HepG2 (C3A) cells (from American type culture collection (ATCC)) were grown in “heavy” and “light” SILAC (stable isotope labeling with amino acids in cell culture) Dulbecco’s modified eagle’s medium (DMEM) (Sigma-Aldrich) for five generations before treatment with a statin. The medium also contained 1000 mg/L glucose and 10% dialyzed fetal bovine serum (diFBS) (Corning). “Heavy” and “light” SILAC media were freshly prepared by adding 0.146 g/L 13C615N2 L-lysine (Lys-8) and 0.84 g/L 13C6 L-arginine (Arg-6) (Cambridge Isotope Lab) or the corresponding non-labeled L-lysine (Lys-0) and L-arginine (Arg-0). When cells reached about 70% confluency, we switched to SILAC media without diFBS and added 15 μM atorvastatin to the heavy group. Meanwhile, dimethyl sulfoxide (DMSO) was used to treat the light group as a vehicle control. For the MC experiments, 100 μM tetra-acetylated N-azidoacetylgalactosamine (Ac4GalNAz) (Click Chemistry Tools) was added into both heavy and light cells at the statin or mock treatment time. Cells were then maintained in a humidified incubator at 37 °C and 5.0% CO2 for 24 h.
2.2. Cell lysis, click reaction, and protein digestion
Cells were washed twice with phosphate buffered saline (PBS), harvested by scraping in PBS, and pelleted by centrifugation at 500 g for 3 min. Two trial runs using about 2% of total cells were conducted to calibrate the heavy and light cell ratios in the BA and MC experiments. For the real experiments, heavy and light cells were mixed based on the protein ratio of 1:1 according to the results from the trial runs. The cell pellets were lysed through end-over-end rotation at 4 °C for 45 minutes in a lysis buffer (50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH=7.6, 150 mM NaCl, 0.5% sodium deoxycholate (SDC), 25 units/mL benzonase and 1 tablet/10 mL protease inhibitor (EDTA-free, Roche)). Lysates were centrifuged, and the resulting supernatant was transferred to new tubes. For the MC experiment, the supernatant was reacted with 100 μM dibenzocyclooctyne (DBCO)-sulfo-biotin to have the metabolically labeled glycoproteins tagged with biotin through the specific click reaction between the azido group and DBCO [52–54]. Proteins were subjected to disulfide reduction with 5 mM 1,4-dithiothreitol (DTT) (56 °C, 25 min) and alkylation with 14 mM iodoacetamide (RT, 20 min in the dark). Detergent was removed by the methanol-chloroform protein precipitation method. The purified proteins were digested with 10 ng/μL Lys-C (Wako) in 50 mM HEPES pH 8.2, 1.6 M urea, 5% ACN at 31 °C for 16 h, and 10 ng/uL trypsin (Promega) at 37 °C for 4 h.
2.3. Glycopeptide separation, enrichment and deglycosylation
Digestion mixtures were acidified by addition of trifluoroacetic acid (TFA) to a final concentration of 0.1%, clarified by centrifugation, desalted using tC18 SepPak cartridge (Waters), and lyophilized.
For the BA experiment, purified peptides were dried and enriched with boronic acid-conjugated magnetic beads (Fig. 1a). Briefly, beads were washed three times with 100 mM ammonium acetate. Peptides were dissolved in the same buffer and mixed with the beads. The mixture was incubated in a shaking incubator at room temperature for an hour, and the beads were washed with the buffer mentioned above to remove non-glycopeptides. Finally, the beads were eluted with ACN:H2O:TFA = 49:50:1 with shaking. The elution was lyophilized and purified with tC18 SepPak cartridge, dried overnight, and treated with four units of peptide-N-glycosidase F (PNGase F, Sigma-Aldrich) in 80 μL buffer containing 50 mM NH4HCO3 in heavy oxygen water (H218O) at 37 °C for 3 h [55]. The reaction was quenched by addition of 1% TFA to pH~2, desalted, and dried. The glycopeptides were fractionated by high pH reversed-phase high-performance liquid chromatography (HPLC) into 10 fractions with a 40-min gradient of 5–55% ACN in 10 mM ammonium acetate (pH=10). The fractions were dried and further purified with the stage-tip method.
Figure 1.

Experimental schemes of the (a) BA and (b) MC experiments. In the BA experiment, cells were treated with 15 μM atorvastatin for 24 hours before harvest. After cell lysis, proteins were reduced, alkylated, and digested. Enrichment was performed at the peptide level with boronic acid-conjugated magnetic beads. Enriched glycopeptides were deglycosylated, fractionated, and subjected to LC-MS/MS analysis. In the MC experiment, 100 μM Ac4GalNAz was added into both heavy and light cells, and heavy cells were treated with atorvastatin. After cell lysis, the lysate was incubated with 100 μMDBCO-sulfo-biotin, followed by protein reduction, alkylation, and digestion. Enrichment was performed at the peptide level with NeutrAvidin agarose beads.
For the MC experiment, purified and dried peptides were enriched with NeutrAvidin beads (Thermo) at 37 °C for 30 min (Fig. 1b). The samples were transferred to spin columns and washed according to manufacturer’s protocol. Peptides were eluted from the beads by 3-min incubations with 300 μL of 8 M guanidine-HCL (pH=1.5) at 56 °C three times. Eluates were combined, desalted using tC18 SepPak cartridge, and lyophilized overnight. Dried peptides were deglycosylated as described in the BA experiment and quenched using the same method. Subsequently, we also attempted to fractionate the glycopeptide sample using HPLC, but the results were not ideal because the enriched sample amount in the MC experiment was much lower than the sample amount in the BA experiment since only the newly-synthesized and metabolically labeled glycopeptides were enriched. Finally, we fractionated the deglycosylated peptides during the stage-tip step, and the sample was separated into 3 fractions using the elution buffer with 20%, 50% and 80% ACN, respectively, containing 1% HOAc.
2.4. LC-MS/MS analyses
Purified and dried peptide samples were dissolved in 10 μL solution of 5% ACN and 4% formic acid (FA) each, and 4 μL of the resulting solutions were loaded onto a microcapillary column packed with C18 beads (Magic C18AQ, 3 μm, 200 Å, 100 μm x 16 cm, Michrom Bioresources) by Dionex WPS-3000TPLRS autosampler (UltiMate 3000 thermostatted Rapid Separation Pulled Loop Wellplate Sampler). Peptides were separated by reversed-phase chromatography using UltiMate 3000 binary pump with a 110 min gradient with increasing concentration of ACN (in 0.125% FA). Peptides were detected with a data-dependent Top20 method [56, 57] in a hybrid dual-cell quadrupole linear ion trap – Orbitrap mass spectrometer (LTQ Orbitrap Elite, Thermo Scientific, with XCalibur 3.0.63 software). For each cycle, one full MS scan (resolution: 60,000) in the Orbitrap at 106 AGC target was followed by up to 20 MS/MS in the LTQ for the most intense ions. The selected ions were excluded from further analysis for 90 seconds. Ions with singly or unassigned charge were not sequenced. Maximum ion accumulation times were 1000 ms for each full MS scan and 50 ms for MS/MS scans.
2.5. Database searches and data filtering
Raw data files from the mass spectrometer were converted into mzXML format, and precursor ion mass measurements were refined by checking the monoisotopic peak assignments [58]. All spectra were searched using the SEQUEST algorithm (version 28) [59] and matched against a database encompassing sequences of all proteins in the UniProt Human (Homo sapiens) database containing common contaminants. Each protein sequence was listed in both forward and reverse orders to control the false discovery rate (FDR) of glycopeptide identifications. We performed the database search using the following parameters: 10 ppm precursor mass tolerance; 1.0 Da product ion mass tolerance; fully digested with trypsin; up to two missed cleavages; variable modifications: oxidation of methionine (+15.9949), O18 tag of asparagine (+2.9883), heavy lysine (+8.0142), and heavy arginine (+6.0201); fixed modifications: carbamidomethylation of cysteine (+57.0214).
The target-decoy method was employed to estimate and control FDRs at the glycopeptide levels [60, 61]. Through linear discriminant analysis (LDA), which is similar to other methods reported in the literature [62–64], several parameters (such as XCorr, ΔCn, precursor mass error, and charge state) were used to distinguish correct and incorrect peptide identifications [58]. After scoring, peptides shorter than six amino acid residues were removed, and the dataset was restricted to glycopeptides when determining FDRs. Glycopeptide FDRs were filtered to <1 % based on the number of decoy sequences in the final data set.
2.6. Glycopeptide quantification and glycosylation site localization
For peptide quantification, we required an S/N value larger than 3 for both heavy and light species. If the S/N value of one member of a heavy and light pair was less than 3, the partner was required to be greater than 5. A probabilistic algorithm was used to localize N-glycosylation sites and to estimate the assignment confidence [65, 66]. A ModScore was calculated for each glycosylation site, and sites with a ModScore >13 (P<0.05) were considered to be confidently localized.
3. Results and discussion
3.1. Examples of glycopeptide identification
The enriched glycopeptides were treated with PNGase F in heavy oxygen water to remove N-glycans and to generate a common tag. When this enzymatic reaction occurs in heavy oxygen water, the converted Asp from the glycosylation site contains heavy oxygen, which creates a mass shift of +2.9883 Da for MS analysis. In this case, heavy oxygen on Asp enabled us to distinguish authentic N-glycosylation sites from those caused by spontaneous asparagine deamidation, which may happen in vivo and during sample preparation. It could also occur during PNGase F treatment, which may result in false positive identifications of protein N-glycosylation sites. To minimize false positive identifications, we ran the reaction for 3 h, during which the effect of deamidation was nearly negligible [51].
Two examples of N-glycopeptide identifications are shown in Fig. 2. Formerly glycosylated peptide YHYN*GTLLDGTLFDSSYSR@ (*-N-glycosylation site, @-heavy arginine) was confidently identified with XCorr of 5.9 from the BA experiment (Fig. 2a). This peptide is from protein FKBP9, one of the peptidyl-prolyl cis-trans isomerases (PPIases) that accelerates the folding of proteins during protein synthesis. The other formerly glycosylated peptide SSCGKEN*TSDPSLVIAFGR shown in Fig. 2b is from protein LAMP1- lysosome-associated membrane glycoprotein 1, which has the major function of presenting carbohydrate ligands to selectins. This peptide was identified in the MC experiment with an even higher XCorr of 6.2 and mass accuracy of -0.23 PPM. The site N84 is confidently identified to be glycosylated with ModScore=1,000, and the score of 1,000 means that only one possible glycosylation site exists on the identified glycopeptide.
Figure 2.

Tandem mass spectra of (a) the formerly glycosylated peptide YHYN*GTLLDGTLFDSSYSR@ (*-N-glycosylation site, @-heavy arginine) from protein FKBP9 identified in the BA experiment, and (b) another formerly glycosylated peptide SSCGKEN*TSDPSLVIAFGR from protein LAMP1 identified in the MC experiment.
In this work, a total of 2,641 unique formerly glycosylated peptides were identified with the boronic acid-based enrichment method and 1,493 with the method combining metabolic labeling with click chemistry. These results indicated that the boronic acid enrichment method is more universal.
3.2. N-glycosylation sites identified with the two lectin-independent enrichment methods
Boronic acids and sugars can form reversible covalent bonds. Based on this universal reaction, our lab has employed this chemical enrichment method to analyze the yeast glycoproteome [33]. The results demonstrated that this method can be used to effectively enrich glycopeptides from digested whole cell lysates. The potential pitfall of this reaction is that the interactions are relatively weak, which could affect the enrichment of glycopeptides from low-abundance glycoproteins.
Metabolic labeling can be employed to label proteins and/or modified proteins, and the labeled proteins and modified proteins may bind to fluorophore for visualization or be selectively enriched for further analysis [49, 67]. In this study, we incorporated an azide-containing sugar analog (Ac4GalNAz) into glycans in glycoproteins, and this azide group was used as a chemical handle to tag a biotin molecule onto the metabolically labeled glycans through click chemistry [68, 69]. Tagging glycoproteins with biotin allowed further glycopeptide enrichment through the strong interaction between biotin on labeled peptides and NeutrAvidin beads after cell lysis and digestion. The detailed experimental procedure is shown in Fig. 1b. Stringent wash was employed to remove non-glycopeptides. Compared to BA, MC has more steps, and the enrichment is largely dependent on metabolic labeling and click reaction efficiency. Since only sugar analog-labeled peptides were enriched with the MC method, it can largely minimize sample complexity, which is an advantage when investigating cellular responses to drug treatment.
For glycosylation site identification, in addition to running the PNGase F treatment for three hours and filtering glycopeptides with <1% FDR, we also applied another criterion: all glycosylation sites must have the consensus motif of NXS/T/C [6] (X stands for a random amino acid other than proline). We confidently identified 1,045 N-glycosylation sites on 432 proteins with the boronic acid-based enrichment method, and the sites are listed in Table S1. Using the enrichment method combined with metabolic labeling and click chemistry, 685 N-glycosylation sites were identified on 351 proteins (Table S2). 418 common sites were identified in both experiments (Fig. 3a). Many proteins contain one glycosylation site while some proteins are highly glycosylated. For instance, LRP1, prolow-density lipoprotein receptor-related protein 1 is a large protein with molecular weight 504,606 Da and 4,454 amino acid residues. We identified very similar number of glycosylation sites on this protein through the two experiments: 21 sites in BA and 20 in MC. As expected, the overlap at the protein level was higher, and 259 common glycoproteins were identified from the two experiments (Fig. 3b).
Figure 3.

Comparison of glycosylation sites (a) and glycoproteins (b) identified using the two enrichment methods.
3.3. Protein clustering based on molecular function
We clustered the glycoproteins identified exclusively in either BA or MC experiment according to the molecular function analysis using the Database for Annotation, Visualization and Integrated Discovery (DAVID) [70] (Fig. 4). Interestingly, we found that the most enriched categories for glycoproteins identified in the BA experiment are intracellular enzyme activity-related, such as hydrolase and transferase activities. However, among glycoproteins identified in the MC experiment, the top enriched categories are binding, receptor, and molecular transducer activities. These activities are known to occur prominently on the cell surface. The differences may be attributed to the following reasons. The boronic acid-based enrichment method is universal, which may unbiasedly enrich cell surface and intracellular glycoproteins. However, the enrichment method based on metabolic labeling and click chemistry is very dependent on the metabolic labeling efficiency, and the latter relies on the endogenous glycan synthesis machinery. In this work, we used Ac4GalNAz to feed the cells and labeled the glycans containing GalNAc or GlcNAc. Normally, cell surface glycoproteins have mature glycan structures in order to be transported to the plasma membrane and/or be secreted, thus these glycans are more likely to have GalNAc moieties that can be substituted by GalNAz. Although all N-Glycans have GlcNAc, GalNAz must convert into GlcNAz before labeling. Therefore, the labeling of GlcNAz may not be as efficient as GalNAz over a relatively short labeling period. For intracellular glycoproteins, especially those still in the ER and Golgi, because their glycan structures are likely immature, the chance of being labeled would be lower than those on the cell surface. Overall, BA is a more global and universal method, while MC has better performance on the identification of glycoproteins located on the cell surface.
Figure 4.

Clustering of the glycoproteins identified only in the (a) BA or (b) MC experiment based on molecular function.
3.4. Quantification of cell glycoproteome changes in statin-treated cells
Statins are a family of popular drugs for lowering cholesterol, but they may affect protein N-glycosylation because the inhibition of HMGCR by statins also prevents the synthesis of other products in the mevalonate pathway, including ubiquinone, dolichol, and farnesyl-pyrophosphate (farnesyl-PP) [71]. Dolichol is essential to protein N-glycosylation in the form of dolichyl phosphate (Dol-P), which serves as the carrier in pyrophosphate-linked oligosaccharide assembly as well as acting as the acceptor in the synthesis of the sugar donors Dol-P-Man and Dol-P-Glc from GDP-Man and UDP-Glc, respectively. Thus, protein N-glycosylation is expected to be impacted while the dolichol synthesis is hindered by the statin treatment. Perturbation of protein N-glycosylation by statins may contribute to the well-known “pleiotropic effects” of statins [71, 72]. Systematic and quantitative investigation of protein N-glycosylation changes by statins will provide insight into the molecular mechanisms of the pleiotropic effects and allow patients to benefit further from the drug.
Statin is a relatively mild drug, and patients typically take it for months or years [73]. Here, we used it to treat cells only for one day, and the drug indirectly affected N-glycosylation; in addition, dolichol in cells was not depleted. Therefore, we did not expect that N-glycosylation would be dramatically influenced over a short period of the treatment.
An example of the full MS and elution profiles of the heavy and light versions of glycopeptide WSFSN*GTSWR are shown in Fig. 5a and b. Based on the areas under the curves from both elution profiles, we were able to accurately quantify the ratio of the glycopeptide as 2.17. This peptide is from protein NEU1 (sialidase-1), which catalyzes the removal of sialic acid moieties from glycoproteins and glycolipids. The protein abundance was up-regulated by 2.07 fold under the drug treatment.
Figure 5.

(a) An example of the full MS of the heavy (WSFSN*GTSWR@) and light (WSFSN*GTSWR) formerly glycosylated peptides with the same sequence; (b) the elution profiles of the two peptides; (c) comparison of unique glycopeptides quantified in the two experiments; (d) comparison of the glycosylation sites quantified from the two experiments.
We quantified a total of 1,247 unique glycopeptides from BA and 682 glycopeptides from MC with an overlap of 376 peptides (Fig. 5c). As anticipated, majority of the quantified peptides were not regulated when two-fold change was used as a threshold. With the BA method, 59 glycopeptides were down-regulated while 93 were up-regulated before normalization. With the MC method, 24 glycopeptides were down-regulated while 62 were up-regulated.
3.5. Glycosylation site quantification and normalization by their corresponding parent protein abundance changes
For the N-glycosylation site quantification, besides the identification criteria discussed above, all glycopeptides must be singly glycosylated with the site ModScore > 13. With the site localization confidence, the quantitation can be site-specific. Although 1,045 glycosylation sites were identified from BA, only 719 sites (Table S3) were quantified; while in MC, the combination of MC and SILAC led to the confident quantitation of 584 glycosylated sites (Table S4) out of 685 unique glycosylation sites identified. 330 sites were quantified in both experiments (Fig. 5d). Relatively low overlap was expected because the principles of two enrichment methods are different. The powerful MS-based proteomics can allow us to site-specifically quantify protein glycosylation changes.
When performing quantitative study of protein modifications, we need to pay attention to the abundance changes of their parent proteins. For instance, if a protein is dramatically up-regulated in treated cells while the stoichiometry of the modification sites from this protein are largely unaffected or even down-regulated, we could still profile these sites to be up-regulated because site down-regulation cannot cancel out the effect of protein up-regulation. As shown in Fig. 6a, for example, if we assume that two out of three copies of a certain protein are N-glycosylated, then it will result in 66.7% glycosylation rate. After the drug treatment, although four copies of this protein are glycosylated, the glycosylation percentage is significantly lowered. This is due to protein expression up-regulation in the treated cells. Therefore, we normalized the raw site ratios by the corresponding parent protein ratios we obtained previously [50] to provide more quantitative information. This normalization strategy was previously applied for phosphorylation analysis in the literature [74].
Figure 6.

(a) An illustration of glycosylation site and glycoprotein abundance changes; glycosylation site regulation distributions before and after normalization using corresponding protein ratios in the (b) BA and (c) MC experiments.
The site ratio distributions in the BA and MC experiments before and after normalization are shown in Fig. 6b and c. The whole series were shifted towards the down-regulation side after normalization. We listed a few quantified sites as examples in Table 1; two-fold was set as the threshold for defining a site to be regulated. All the listed sites have raw ratios larger than 2. However, their protein ratios are also larger than 2, which demonstrated that these proteins were up-regulated in the statin-treated cells. For instance, the first site in the list is from protein HMGCR, the rate-liming enzyme in the mevalonate pathway and the direct target of statins. Since the function of this protein was inhibited by the statin, this protein expression was up-regulated dramatically in the statin-treated cells, and the protein ratio for HMGCR increased by 15.4 fold. Without normalization by the protein ratio, N-glycosylation site 281 on HMGCR was up-regulated by 25.9 fold, while the normalized ratio was 1.7. After normalization, six out of seven N-glycosylation sites were determined to be not regulated.
Table 1.
Some example N-glycosylation sites quantified in the BA experiment.
| Gene Symbol | PPM | XCorr | Peptide | Site | Mod Score | Site ratio | Protein ratio | Site ratio normalized |
|---|---|---|---|---|---|---|---|---|
| HMGCR | −0.43 | 4.24 | WIADPSPQN* STADTSK# | 281 | 1000 | 25.89 | 15.35 | 1.69 |
| KLB | −0.28 | 2.17 | FALDWASVLPTGN* LSAVNR@ | 611 | 31.94 | 14.24 | 2.02 | 7.05 |
| SLCO4C1 | −1.06 | 1.93 | VYYN* CSCIER | 544 | 1000 | 5.39 | 4.18 | 1.29 |
| KLB | −1.18 | 2.32 | MGQN* VSLNLR | 391 | 59.30 | 2.88 | 2.02 | 1.43 |
| A1BG | 0.50 | 3.50 | EGDHEFLEVPEAQEDVEATFPVHQPGN* YSCSYR@ | 179 | 1000 | 2.75 | 3.05 | 0.90 |
| NEU1 | 0.06 | 1.43 | VN* LTLR@ | 343 | 1000 | 2.47 | 2.07 | 1.19 |
| 0.30 | 1.77 | WSFSN* GTSWRK | 352 | 1000 | 2.17 | 2.07 | 1.05 |
-glycosylation site,
-heavy lysine,
-heavy arginine
Among 640 normalized sites from the BA experiment (Table S5), 22 were up-regulated, and 35 were down-regulated. In contrast, 30 sites were up-regulated, and 50 were down-regulated among 518 normalized sites from the MC experiment. Although we quantified fewer sites from the MC experiment, more sites were down-regulated. This phenomenon is in very good agreement with the expectation that the results from the MC experiment may be more dynamic because it only enriches the newly-synthesized glycoproteins during the statin treatment.
4. Conclusions
Protein glycosylation alternation is often a hallmark of human disease. In-depth analysis of glycoprotein changes may aid in a better understanding of glycoprotein functions and lead to the discovery of disease biomarkers and drug targets. Modern MS-based proteomics is very powerful in globally analyzing protein modifications, but it is pivotal to enrich modified proteins in complex biological samples prior to MS analysis. Lectin-based enrichment methods have been used extensively to enrich glycopeptides. However, the binding specificity of lectin prevents high coverage of glycopeptides. Here, we evaluated two lectin-independent chemical enrichment methods (namely, BA and MC) for global analysis of protein N-glycosylation. BA is based on the reversible interactions between boronic acids and hydroxyl groups on glycans; MC utilizes the endogenous glycan synthesis pathways in human cells to incorporate a sugar analog with a chemically functional, but biologically inert group, into the glycan structure, followed by biorthogonal reactions and affinity enrichment. BA is more universal and helped identify a greater number of glycosylation sites, whereas MC has better performance on cell surface glycoprotein identification. Furthermore, the quantitative results from the MC experiment were more dynamic because it enriched the newly synthesized glycoproteins under the drug treatment. For the quantification of protein modification, normalization using the parent protein ratios can provide more quantitative information regarding the protein expression and modification changes. Because of the high abundance of proteins and sugars in human cells, the interactions between proteins and sugars are ubiquitous. Global analysis of protein glycosylation will dramatically facilitate glycoscience research in the biological and biomedical fields.
Supplementary Material
Highlights.
Two lectin-independent glycopeptide enrichment methods were comprehensively compared for global analysis of glycoproteins by mass spectrometry.
The experimental results demonstrated that boronic acid-based enrichment (BA) method was more universal for glycopeptide enrichment, and a greater number of unique glycopeptides and glycosylation sites were identified and quantified in statin-treated liver cells.
The method combining metabolic labeling with click chemistry (MC) had better performance on the identification of cell surface glycoproteins, and the quantification results were more dynamic because only the newly-synthesized glycoproteins during the statin treatment were analyzed.
The glycosylation site ratios were further normalized by the corresponding parent protein ratios to extract more quantitative information about protein expression and modification changes.
Acknowledgments
This work was supported by the National Science Foundation (CAREER Award, CHE-1454501), and the National Institutes of Health (R01GM118803). R. W. was also supported by Blanchard Assistant Professorship and an ASMS Research Award.
Footnotes
This paper is dedicated to Professor Terry McMahon on the occasion of his 70th birthday.
Table S1. Protein N-glycosylation sites identified in BA experiment; Table S2. Protein N-glycosylation sites identified in MC experiment; Table S3. N-glycosylation sites quantified in BA experiment; Table S4. N-glycosylation sites quantified in MC experiment; Table S5. Protein N-glycosylation sites quantified and normalized in BA experiment; Table S6, Protein N-glycosylation sites quantified and normalized in MC experiment.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ryan MC, Notterpek L, Tobler AR, Liu N, Shooter EM. Role of the peripheral myelin protein 22 N-linked glycan in oligomer stability. J Neurochem. 2000;75:1465–1474. doi: 10.1046/j.1471-4159.2000.0751465.x. [DOI] [PubMed] [Google Scholar]
- 2.Varki A. Nothing in glycobiology makes sense, except in the light of evolution. Cell. 2006;126:841–845. doi: 10.1016/j.cell.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 3.Konrad RJ, Kudlow JE. The role of O-linked protein glycosylation in beta-cell dysfunction (review) Int J Mol Med. 2002;10:535–539. [PubMed] [Google Scholar]
- 4.Spiro RG. Protein glycosylation: nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology. 2002;12:43r–56r. doi: 10.1093/glycob/12.4.43r. [DOI] [PubMed] [Google Scholar]
- 5.Schwarz F, Aebi M. Mechanisms and principles of N-linked protein glycosylation. Curr Opin Struc Biol. 2011;21:576–582. doi: 10.1016/j.sbi.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 6.Zielinska DF, Gnad F, Wisniewski JR, Mann M. Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell. 2010;141:897–907. doi: 10.1016/j.cell.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 7.Goto M. Protein O-glycosylation in fungi: Diverse structures and multiple functions. Biosci Biotech Bioch. 2007;71:1415–1427. doi: 10.1271/bbb.70080. [DOI] [PubMed] [Google Scholar]
- 8.Kamemura K, Hart GW. Dynamic interplay between O-glycosylation and O-phosphorylation of nucleocytoplasmic proteins: A new paradigm for metabolic control of signal transcluction and transcription. Prog Nucleic Acid Re. 2003;73:107–136. doi: 10.1016/s0079-6603(03)01004-3. [DOI] [PubMed] [Google Scholar]
- 9.Steentoft C, Vakhrushev SY, Vester-Christensen MB, Schjoldager K, Kong Y, Bennett EP, Mandel U, Wandall H, Levery SB, Clausen H. Mining the O-glycoproteome using zinc-finger nuclease-glycoengineered SimpleCell lines. Nat Methods. 2011;8:977–982. doi: 10.1038/nmeth.1731. [DOI] [PubMed] [Google Scholar]
- 10.Breitling J, Aebi M. N-linked protein glycosylation in the endoplasmic reticulum. Csh Perspect Biol. 2013;5 doi: 10.1101/cshperspect.a013359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruiz-Canada C, Kelleher DJ, Gilmore R. Cotranslational and posttranslational N-glycosylation of polypeptides by distinct mammalian OST isoforms. Cell. 2009;136:272–283. doi: 10.1016/j.cell.2008.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ibraghimovbeskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular-matrix. Nature. 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- 13.Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell. 2006;126:855–867. doi: 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 14.Roth J. Protein N-glycosylation along the secretory pathway: Relationship to organelle topography and function, protein quality control, and cell interactions. Chem Rev. 2002;102:285–303. doi: 10.1021/cr000423j. [DOI] [PubMed] [Google Scholar]
- 15.Neelamegham S, Mahal LK. Multi-level regulation of cellular glycosylation: from genes to transcript to enzyme to structure. Curr Opin Struc Biol. 2016;40:145–152. doi: 10.1016/j.sbi.2016.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kurcon T, Liu ZY, Paradkar AV, Vaiana CA, Koppolu S, Agrawal P, Mahal LK. miRNA proxy approach reveals hidden functions of glycosylation. Proc Natl Acad Sci U S A. 2015;112:7327–7332. doi: 10.1073/pnas.1502076112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ju TZ, Otto VI, Cummings RD. The Tn antigen-structural simplicity and biological complexity. Angew Chem-Int Edit. 2011;50:1770–1791. doi: 10.1002/anie.201002313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilgunn S, Conroy PJ, Saldova R, Rudd PM, O’Kennedy RJ. Aberrant PSA glycosylation - a sweet predictor of prostate cancer. Nat Rev Urol. 2013;10:99–107. doi: 10.1038/nrurol.2012.258. [DOI] [PubMed] [Google Scholar]
- 19.Harvey DJ. Proteomic analysis of glycosylation: structural determination of N- and O-linked glycans by mass spectrometry. Expert Rev Proteomic. 2005;2:87–101. doi: 10.1586/14789450.2.1.87. [DOI] [PubMed] [Google Scholar]
- 20.Morelle W, Canis K, Chirat F, Faid V, Michalski JC. The use of mass spectrometry for the proteomic analysis of glycosylation. Proteomics. 2006;6:3993–4015. doi: 10.1002/pmic.200600129. [DOI] [PubMed] [Google Scholar]
- 21.Xu SL, Medzihradszky KF, Wang ZY, Burlingame AL, Chalkley RJ. N-glycopeptide profiling in Arabidopsis Inflorescence. Mol Cell Proteomics. 2016;15:2048–2054. doi: 10.1074/mcp.M115.056101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang Y, Liu F, Franc V, Halim LA, Schellekens H, Heck AJR. Hybrid mass spectrometry approaches in glycoprotein analysis and their usage in scoring biosimilarity. Nat Commun. 2016;7:10. doi: 10.1038/ncomms13397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang XS, Yuan ZF, Fan J, Karch KR, Ball LE, Denu JM, Garcia BA. A novel quantitative mass spectrometry platform for determining protein O-GlcNAcylation dynamics. Mol Cell Proteomics. 2016;15:2462–2475. doi: 10.1074/mcp.O115.049627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang N, Goonatilleke E, Park D, Song T, Fan GR, Lebrilla CB. Quantitation of site-specific glycosylation in manufactured recombinant monoclonal antibody drugs. Anal Chem. 2016;88:7091–7100. doi: 10.1021/acs.analchem.6b00963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zacharias LG, Hartmann AK, Song EH, Zhao JF, Zhu R, Mirzaei P, Mechref Y. HILIC and ERLIC enrichment of glycopeptides derived from breast and brain cancer cells. J Proteome Res. 2016;15:3624–3634. doi: 10.1021/acs.jproteome.6b00429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu ZK, Desaire H. Carbohydrates on proteins: Site-specific glycosylation analysis by mass spectrometry. In: Cooks RG, Pemberton JE, editors. Annual Review of Analytical Chemistry. Vol. 8. Annual Reviews; Palo Alto: 2015. pp. 463–483. [DOI] [PubMed] [Google Scholar]
- 27.Ma C, Qu JY, Meisner J, Zhao XY, Li X, Wu ZG, Zhu HL, Yu ZK, Li L, Guo YX, Song J, Wang PG. Convenient and Precise Strategy for Mapping N-Glycosylation Sites Using Microwave-Assisted Acid Hydrolysis and Characteristic Ions Recognition. Anal Chem. 2015;87:7833–7839. doi: 10.1021/acs.analchem.5b02177. [DOI] [PubMed] [Google Scholar]
- 28.Woo CM, Iavarone AT, Spiciarich DR, Palaniappan KK, Bertozzi CR. Isotope-targeted glycoproteomics (IsoTaG): a mass-independent platform for intact N- and O-glycopeptide discovery and analysis. Nat Methods. 2015;12:561–567. doi: 10.1038/nmeth.3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun SS, Shah P, Eshghi ST, Yang WM, Trikannad N, Yang S, Chen LJ, Aiyetan P, Hoti N, Zhang Z, Chan DW, Zhang H. Comprehensive analysis of protein glycosylation by solid-phase extraction of N-linked glycans and glycosite-containing peptides. Nat Biotechnol. 2016;34:84–88. doi: 10.1038/nbt.3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chandler KB, Costello CE. Glycomics and glycoproteomics of membrane proteins and cell-surface receptors: Present trends and future opportunities. Electrophoresis. 2016;37:1407–1419. doi: 10.1002/elps.201500552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khatri K, Klein JA, Zaia J. Use of an informed search space maximizes confidence of site-specific assignment of glycoprotein glycosylation. Anal Bioanal Chem. 2017;409:607–618. doi: 10.1007/s00216-016-9970-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiao HP, Wu RH. Quantitative investigation of human cell surface N-glycoprotein dynamics. Chemical Science. 2017;8:268–277. doi: 10.1039/c6sc01814a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen WX, Smeekens JM, Wu RH. A universal chemical enrichment method for mapping the yeast N-glycoproteome by mass spectrometry (MS) Mol Cell Proteomics. 2014;13:1563–1572. doi: 10.1074/mcp.M113.036251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Madera M, Mechref Y, Novotny MV. Combining lectin microcolumns with high-resolution separation techniques for enrichment of glycoproteins and glycopeptides. Anal Chem. 2005;77:4081–4090. doi: 10.1021/ac050222l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng JN, Xiao HP, Wu RH. Specific identification of glycoproteins with the Tn antigen in human cells. Angew Chem Int Ed. 2017 doi: 10.1002/anie.201702191. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiao HP, Wu RH. Global and site-specific analysis revealing unexpected and extensive protein S-GlcNAcylation in human cells. Anal Chem. 2017;89:3656–3663. doi: 10.1021/acs.analchem.6b05064. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Y, Yu M, Zhang C, Wang YL, Di Y, Wang CC, Lu HJ. Highly specific enrichment of N-glycoproteome through a nonreductive amination reaction using Fe3O4@SiO2-aniline nanoparticles. Chem Commun. 2015;51:5982–5985. doi: 10.1039/c4cc10285a. [DOI] [PubMed] [Google Scholar]
- 38.Dong LP, Feng S, Li SS, Song PP, Wang JD. Preparation of concanavalin A-chelating magnetic nanoparticles for selective enrichment of glycoproteins. Anal Chem. 2015;87:6849–6853. doi: 10.1021/acs.analchem.5b01184. [DOI] [PubMed] [Google Scholar]
- 39.Calvano CD, Zambonin CG, Jensen ON. Assessment of lectin and HILIC based enrichment protocols for characterization of serum glycoproteins by mass spectrometry. J Proteomics. 2008;71:304–317. doi: 10.1016/j.jprot.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 40.Zhang H, Li XJ, Martin DB, Aebersold R. Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat Biotechnol. 2003;21:660–666. doi: 10.1038/nbt827. [DOI] [PubMed] [Google Scholar]
- 41.Wollscheid B, Bausch-Fluck D, Henderson C, O’Brien R, Bibel M, Schiess R, Aebersold R, Watts JD. Mass-spectrometric identification and relative quantification of N-linked cell surface glycoproteins. Nat Biotechnol. 2009;27:378–386. doi: 10.1038/nbt.1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeng Y, Ramya TNC, Dirksen A, Dawson PE, Paulson JC. High-efficiency labeling of sialylated glycoproteins on living cells. Nat Methods. 2009;6:207–209. doi: 10.1038/nmeth.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Palaniappan KK, Bertozzi CR. Chemical glycoproteomics. Chem Rev. 2016;116:14277–14306. doi: 10.1021/acs.chemrev.6b00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gaunitz S, Nagy G, Pohl NLB, Noyotny MV. Recent advances in the analysis of complex glycoproteins. Anal Chem. 2017;89:389–413. doi: 10.1021/acs.analchem.6b04343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thaysen-Andersen M, Packer NH, Schulz BL. Maturing glycoproteomics technologies provide unique structural insights into the N-glycoproteome and its regulation in health and disease. Mol Cell Proteomics. 2016;15:1773–1790. doi: 10.1074/mcp.O115.057638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang LJ, Xu YW, Yao HL, Xie LQ, Yao J, Lu HJ, Yang PY. Boronic acid functionalized core-satellite composite nanoparticles for advanced enrichment of glycopeptides and glycoproteins. Chem-Eur J. 2009;15:10158–10166. doi: 10.1002/chem.200901347. [DOI] [PubMed] [Google Scholar]
- 47.Xu GB, Zhang W, Wei LM, Lu HJ, Yang PY. Boronic acid-functionalized detonation nanodiamond for specific enrichment of glycopeptides in glycoproteome analysis. Analyst. 2013;138:1876–1885. doi: 10.1039/c3an36623e. [DOI] [PubMed] [Google Scholar]
- 48.Xiao HP, Smeekens JM, Wu RH. Quantification of tunicamycin-induced protein expression and N-glycosylation changes in yeast. Analyst. 2016;141:3737–3745. doi: 10.1039/c6an00144k. [DOI] [PubMed] [Google Scholar]
- 49.Chen WX, Smeekens JM, Wu RH. Systematic and site-specific analysis of N-sialoglycosylated proteins on the cell surface by integrating click chemistry and MS-based proteomics. Chemical Science. 2015;6:4681–4689. doi: 10.1039/c5sc01124h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xiao HP, Chen WX, Tang GX, Smeekens JM, Wu RH. Systematic investigation of cellular response and pleiotropic effects in atorvastatin-treated liver cells by ms-based proteomics. J Proteome Res. 2015;14:1600–1611. doi: 10.1021/pr501277g. [DOI] [PubMed] [Google Scholar]
- 51.Xiao HP, Tang GX, Wu RH. Site-specific quantification of surface N-glycoproteins in statin-treated liver cells. Anal Chem. 2016;88:3324–3332. doi: 10.1021/acs.analchem.5b04871. [DOI] [PubMed] [Google Scholar]
- 52.Hong V, Steinmetz NF, Manchester M, Finn MG. Labeling live cells by copper-catalyzed alkyne-azide click chemistry. Bioconjugate Chem. 2010;21:1912–1916. doi: 10.1021/bc100272z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shelbourne M, Chen X, Brown T, El-Sagheer AH. Fast copper-free click DNA ligation by the ring-strain promoted alkyne-azide cycloaddition reaction. Chem Commun. 2011;47:6257–6259. doi: 10.1039/c1cc10743g. [DOI] [PubMed] [Google Scholar]
- 54.Debets MF, van Berkel SS, Schoffelen S, Rutjes F, van Hest JCM, van Delft FL. Aza-dibenzocyclooctynes for fast and efficient enzyme PEGylation via copper-free (3+2) cycloaddition. Chem Commun. 2010;46:97–99. doi: 10.1039/b917797c. [DOI] [PubMed] [Google Scholar]
- 55.Kaji H, Saito H, Yamauchi Y, Shinkawa T, Taoka M, Hirabayashi J, Kasai K, Takahashi N, Isobe T. Lectin affinity capture, isotope-coded tagging and mass spectrometry to identify N-linked glycoproteins. Nat Biotechnol. 2003;21:667–672. doi: 10.1038/nbt829. [DOI] [PubMed] [Google Scholar]
- 56.Chen WX, Smeekens JM, Wu RH. Comprehensive analysis of protein N-glycosylation sites by combining chemical deglycosylation with LC-MS. J Proteome Res. 2014;13:1466–1473. doi: 10.1021/pr401000c. [DOI] [PubMed] [Google Scholar]
- 57.Wu RH, Haas W, Dephoure N, Huttlin EL, Zhai B, Sowa ME, Gygi SP. A large-scale method to measure absolute protein phosphorylation stoichiometries. Nat Methods. 2011;8:677–683. doi: 10.1038/nmeth.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausolei SA, Villen J, Haas W, Sowa ME, Gygi SP. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass-spectral data of peptides with amino-acid-sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 60.Peng JM, Elias JE, Thoreen CC, Licklider LJ, Gygi SP. Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: The yeast proteome. J Proteome Res. 2003;2:43–50. doi: 10.1021/pr025556v. [DOI] [PubMed] [Google Scholar]
- 61.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 62.Du X, Callister SJ, Manes NP, Adkins JN, Alexandridis RA, Zeng X, Roh JH, Smith WE, Donohue TJ, Kaplan S, Smith RD, Lipton MS. A computational strategy to analyze label-free temporal bottom-up proteomics data. J Proteome Res. 2008;7:2595–2604. doi: 10.1021/pr0704837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kall L, Canterbury JD, Weston J, Noble WS, MacCoss MJ. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods. 2007;4:923–925. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- 64.Zhang JY, Ma J, Dou L, Wu SF, Qian XH, Xie HW, Zhu YP, He FC. Bayesian nonparametric model for the validation of peptide identification in shotgun proteomics. Mol Cell Proteomics. 2009;8:547–557. doi: 10.1074/mcp.M700558-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol. 2006;24:1285–1292. doi: 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
- 66.Chen WX, Smeekens JM, Wu RH. Systematic study of the dynamics and half-lives of newly synthesized proteins in human cells. Chemical Science. 2016;7:1393–1400. doi: 10.1039/c5sc03826j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hang HC, Yu C, Kato DL, Bertozzi CR. A metabolic labeling approach toward proteomic analysis of mucin-type O-linked glycosylation. Proc Natl Acad Sci U S A. 2003;100:14846–14851. doi: 10.1073/pnas.2335201100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McKay CS, Finn MG. Click chemistry in complex mixtures: Bioorthogonal bioconjugation. Chem Biol. 2014;21:1075–1101. doi: 10.1016/j.chembiol.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Copper-free click chemistry for dynamic in vivo imaging. Proc Natl Acad Sci U S A. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 71.Liao JK, Laufs U. Annual Review of Pharmacology and Toxicology. Annual Reviews; Palo Alto: 2005. Pleiotropic effects of statins; pp. 89–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Forbes K, Shah VK, Siddals K, Gibson JM, Aplin JD, Westwood M. Statins inhibit insulin-like growth factor action in first trimester placenta by altering insulin-like growth factor 1 receptor glycosylation. Mol Hum Reprod. 2015;21:105–114. doi: 10.1093/molehr/gau093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, Kirby A, Sourjina T, Peto R, Collins R, Simes J, Collaborators CTT. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–1278. doi: 10.1016/S0140-6736(05)67394-1. [DOI] [PubMed] [Google Scholar]
- 74.Wu RH, Dephoure N, Haas W, Huttlin EL, Zhai B, Sowa ME, Gygi SP. Correct interpretation of comprehensive phosphorylation dynamics requires normalization by protein expression changes. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M1111.009654. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.