

Abstract
Objective:
This study aims to isolate the active anticancer compound from ethyl acetate fraction extracted from the roots of Calotropis gigantea and to determine the operating mechanism of the isolates towards WiDr colon cancer cells.
Methods:
the isolation was conducted by using bioassay guided isolation approach method. The cytotoxic potential was determined by using MTT method. The chemical structure was identified by using UPLCMS/MS and NMR-1H spectroscopy. The cell cycle arrest and apoptosis induction were determined by flow cytometry method. The expression of caspase-8 was determined by immunocytochemistry method.
Results:
The results showed that the active compounds are obtained calotroposid A compound which is glycosides terpenoids. Calotroposide A is capable of inhibiting the growth of WiDr colon cancer cells at IC50 17.23µg/ml. Cell apoptosis induction took place and was indicated by cell apoptosis increase, S and G2/M accumulation and by caspase-8 expression.
Conclusion:
Calotroposide A induces anticancer activity against WiDr colon cancer cells by means of apoptosis induction mechanism through extrinsic pathway with increased expression of caspase-8.
Keywords: Calotropis gigantea, calotroposid A, apoptosis, WiDr cell line, cell cycle arrest
Introduction
Calotropis gigantea (C.gigantea) is a plant that grows wild and is widespread in Indonesia. For generations, this plant has been traditionally used by the people of Indonesia to cure rashes, scabies, boils, cough, trachoma, constipation (the leaves), asthma, nausea, stomach pain (the flowers), syphilis, viper bites (the roots), toothache, swelling, ear inflammation, intestinal worms infestation and dysentery (Radjakmanugunsudarso 1968). Scientific evidence on anticancer activity happening in this plant has been reported to include calotropon compound of the roots that induce cytotoxic activity against leukemia cell K562 and gastric cancer cells 7901 (Wang et al., 2008). The methanol extract and chloroform fraction of its flowers induce antitumor activity on mice to ht ascites carcinoma (Habib et al., 2010). The methanol extract (ME) and chloroform fraction of the roots of C. gigantea is capable of inhibiting the growth of ascites carcinoma by 43.90% (20mg ME/kg) and 57.07% (40mg CF/kg) (Habib and Karim, 2011). It has been reported that the cytotoxic potential of the leaves’ cardenolide compound can inhibit the growth of breast cancer cell MCF-7, skin cancer cells KB, and lung cancer cells NCL-H18 (Seeka and Sutthivaiyakit 2010). The dichloromethane cytotoxic extracted from the leaves is potential to fight breast cancer cells MCF-7 and MDA-MB-231, Hela cells, colon cancer cells HT-29, ovarian cancer cells Skov-3 and also liver cancer cells Hep-G2 (Wong et al., 2011) .
In previous studies, it has also been reported that the ethanol extracted from C. gigantea leaves was capable of inhibiting the in vivo growth of fibrosarcoma at the dose of 100 and 150 mg/kg with an increased expression mechanism of caspase-3 (Muti et al. 2016). C.gigantea root extract contained higher anticancer activity than its leaves and flowers (Mutiah et al., 2016). The ethyl acetate fraction of the leaves (IC50 41.79 μg/ml) and its dichloromethane fraction (IC50 40.57μg/ml) had higher cytotoxic activity than those of butanol fraction (IC50 737.74 μg/ml) and water (IC50 8493 μg/ml) (Mutiah et al., 2017).
In a preliminary test on the fractionated ethanol extract of the roots of calotropis gigantea, the ethyl acetate fraction (IC50 0.063 μg/ml) showed higher cytotoxic activity than that of dichloromethane fraction (IC50 0.367 μg /ml), butanol (IC50 12:18 g/ml) and water (IC50 8493μg/ml). Based on these data, it can be seen that the ethyl acetate fraction of C. gigantea’s roots performed a higher anticancer activity than the other fractions. It was presumed that the ethyl acetate fraction contained in the active compound was responsible for the anticancer activity on the roots of C. gigantea. Therefore, it is important to conduct further research on the ethyl acetate fraction on C gigantea’s roots, which is mainly related to the active compounds comprised in these fractions and the operating mechanism of the active compound.
In this study, an isolation on anticancer compound from ethyl acetate fraction of the roots of C. gigantea had been conducted which was active towards WiDr colon cancer cells. The isolation was conducted by using bioassay guided isolation approach method. The isolation was conducted based on anticancer activities, ranging from fractions, sub-fractions and isolates. Then, the chemical structures of the active isolates were determined. This research is important because there are still many active anticancer compounds from the roots of C. gigantea which are left undiscovered. In addition, the approach taken by previous researchers have not been based on bioassay guided isolation. This step is important to take as it determines the compounds responsible for anticancer activity (lead compounds) contained in the roots of C. gigantea.
Materials and Methods
Experiment Method
Materials, equipment and reagents
Materials and reagents
The research materials: The plant employed in this study was C. gigantea taken from Malang, East Java. The plant parts used were the roots. The roots were dried in an oven at a temperature of 40oC. The dried simplicia was then ground into powder and put in a dry brown bottle.
Materials for the extraction, fractionation, and isolation of the active compound of C. gigantea roots: The solvent used for ethanol extraction on the maceration stage was technical ethanol, while the one used in fractionation stage was distilled water, dichloromethane p.a (Merck), ethyl acetate p.a (Merck), and butanol p.a (Merck). The materials for sub-fractionation and isolation were n-hexane p.a, (Merck) ethyl acetate p.a (Merck), chloroform p.a (Merck), ethanol p.a (Merck), acetonitrile p.a (Merck), Methanol pro HPLC (JT Backer), pro HPLC acetonitrile (JT Backer), TLC plates of silica gel 60 F254 (Merck 0.25 mm), silica gel 60 for chromatography column (Merck 0.063 to 0.200 mm), and last silica gel 60G (Merck).
Materials for cell culture: The cancer cells used in this study were colon cancer cell lines. The colon cancer cells used were WiDr cells obtained from the Cancer Chemoprevention Research Centre (CCRC) of Faculty of Pharmacy of Gadjah Mada University and from Prof. Masasi Kawaichi from the Laboratory of Gene Function in Animal, Graduate School of Biological Sciences, Nara Institute of Science and Technology. WiDr cells in a medium of Rosewell Park Memorial Institute (RPMI) were added with 10% of heat-activated fetal bovine serum (FBS) (PAA laboratories), 1% v/v penicillin-streptomycin (Nacalay Tesque), and 1.0 mM of L-glutamine (Nacalay Tesque). All cells were cultured in an incubator at 5% of CO2, in 37 °C.
Cytotoxic test materials: Dimethyl sulfoxide (DMSO) was used to dissolve C. gigantea extract. The concentration used in this study was maximum 1% in the culture medium, 0.025% trypsin in the culture medium was used to harvest the cells. Phosphate buffered saline (PBS) was used as wash buffer solution. 3-(4.5-dimetiltiazole-2-il)-2.5-difeniltetrazolium bromide (MTT) was used as a reagent that reacted with the succinate dehydrogenase enzyme in the cells.
Materials for cell cycle regulation testing using Flow Cytometry method: A solution of propidium iodine (PI) (Sigma) comprising of 50μg/ml PI, 20μg/ml, and 0.1% Triton-X (Pro GC-Merck) in PBS was used. Filters were used to filter out cells in a solution of PI to be separated into single cells.
Materials for apoptosis induction test using flow cytometry method: A solution of propidium iodine (PI) (Sigma) consisting of 50μg/ml PI, 20μg/ml, and 0.1% Triton-X (Pro GC-Merck) in PBS was used. AnnexinV assay (Annexin V-FITC Apoptosis Detection Kit Biovision) and binding buffer were used.
Equipment
The instruments for the processes of extraction, separation, isolation and purification of active compounds include glassware, rotary, analytical scale, vacuum columns, open columns, chambers, vials, spray stains, capillary tubes, fume hoods, TLC Visualizer (Camag), 254 and 365nm UV lights (Camag), high performance Liquid Chromatography (Shimadzu LC-06), RP shim-pack columns 4.6x250 mm, Prominanence DGU-20A5 degasser, Pump LC-6AD, shim-pack column 4,6x250 mm, Prominence DiodeArray Detector SPD-M20A.
The instruments for the cytotoxicity test and the operating mechanism: CO2 incubator (New Bronswick, Galaxy 170 R), centrifuge (Hermle Siemensstr-25 D-78 564), sonicator, Laminar Air Flow cabinet (Mascotte LH-S), a micropipette (soccorex), autoclave, hemocytometer, inverted microscope (Olympus CKX41-2), ELISA reader (Robonik), flow cytometer.
The instruments to identify the active ingredient and active compound: NMR JEOL Spectrometer with radio wave frequency of 400 Hz (JNM-ECS400), a set of UPLC- QToF-MS/MS instrumentation system (waters).
Methods
Extraction
A total of 5kg of C. gigantea root powder was macerated using 14.5 liters of 70% ethanol for 48 hours. Then, it was filtered and the filtrate was then separated. The residue was then dried and re-macerated with 14.5 liters of 70% ethanol for 48 hours. This process was repeated for four times. After that, the filtrate was collected and evaporated in a vacuum rotary evaporator to obtain a thick extract. The condensed extract was then dried in an oven at 40°C. Then, 100 ml of distilled water was added to the extract and then mixed to obtain liquid ethanol extract. The extract was then fractionated by using dichloromethane 100:500 (ethanol extract: dichloromethane) and two fractions were yielded; soluble and insoluble dichloromethane fractions. The soluble dichloromethane fraction was then fractionated with ethyl acetate with the ratio of 100:600 (insoluble dichloromethane fraction: ethyl acetate) thus, resulting ethyl acetate insoluble fraction and ethyl acetate soluble fraction. The ethyl acetate insoluble fraction was re-fractioned with butanol solvent with the ratio of 100:600 (dichloromethane insoluble fraction: ethyl acetate). Thus, the ethyl acetate soluble fraction and water fraction were obtained. The four fractions were evaporated with a rotary evaporator to get a concentrated extract. The fractions was then dried in an oven at a temperature of 40°C.
Isolation and Purification
The Separation of Ethyl Acetate Fraction by Using Column Vacuum Chromatography
The ethyl acetate fraction was separated using column vacuum chromatography with stationary phase silica gel G 60 where the mobile phase solvent gradient was based on the polarity degree (chloroform and methanol solvent gradients with 10% reduction, starting from 100% Chloroform up to 100% methanol). On each the resulted sub-fraction, thin layer chromatography using stationary phase silica gel F254 and mobile phase chloroform: methanol (9:1) were conducted. The TLC profiles were then observed with 254 nm and 366 nm UV lamps with 10% sulfuric acid used as stain surface. The sub-factions resulting the same profiles were then grouped together, evaporated and weighed. Cytotoxicity assay was performed on the resulted sub-fractions by using WiDr human colonic cells. The most active sub-fraction found would undergo further separation.
Active Sub-fraction Separation by Open Column Chromatography
The active sub-fractions were then separated with open column chromatography by using a stationary phase silica gel 60 and a mobile phase solvent gradient based on optimized polarity (n-hexane: ethyl acetate 4:1 and 2:8); ethyl acetate: chloroform (9:1) chloroform: methanol 1: 1, methanol). The Stationary phase was 25 cm tall with a diameter of 3.8 cm. On each vial bin resulted, thin layer chromatography was conducted using stationary phase silica gel F254 and mobile phase hexane: ethyl acetate (4:1) and chloroform: methanol (9:1). The TLC profile was observed with 254 nm and 366 nm UV lamps while the stain surface used was 10% sulfuric acid. The isolates producing single stains would be followed up by further chemical structure identification. Isolates that did not show any stain would be followed up by purification using preparative TLC.
Active Compound Identification with UPLCMS/MS
The identification of SF 3.4.2 isolate was conducted using UP-LCMS/MS device with the type of ACQUITY UPLC I-Class (Waters) with a diode-array detector (DAD) 2,996 (waters);column: Sunfire C18 sizing 50mm long, with 2 mm diameter and μm particle size (waters). Mobile phase: acetonitrile gradient; water; Formic acid, 1ml/min flow rate, injection volume 10 μl. MS system Xevo G2-S QTOF (waters), analyzer: TOF with positive and negative mode (ES +) electrosprayers; gas flow 794L/min.
Identification with NMR-1 H Spectroscopy
Approximately 3mg of isolates were dissolved in 0.5ml of CDCl3 and then put into a sample tube. Tetramethylsilane (TMS) was added to the solution as an internal reference. Then, the instrument was stationed between two magnetic poles where the spectrum was recorded.
Anticancer Activity Assay (MTT Method)
Cytotoxic Test with MTT method
As much as 100 mL of WiDr colon cancer cell suspension at a density of 3×104 cells/100 mL media was distributed into wells on 96-well plate and then incubated for 24 hours. After incubation, the wells were put into a 100 mL test solution at various concentration series. As a positive control, 100 mL of culture medium was added, followed by 100 mL of cisplatin on various concentration series in wells already containing 100mL cell suspension. As a control cell, 100 mL of culture medium was added in wells containing 100 mL of cell suspension while 100 mL of DMSO was added in wells containing 100 mL of culture medium and 100 mL of cell suspension as a control solvent with the delusions which matched the concentration delusions of the test solution. Then, these substances were incubated for 24 hours in an incubator with a stream of 5% CO2 and 95% O2. At the end of the incubation, culture medium was discarded and then 10mL solution of MTT (5 mg/mL PBS) was added, and the medium was replaced with 190mL of complete medium RPMI 1,640. Then, the cells were incubated for 3-4 hours. MTT reaction was stopped by adding SDS stopper reagent (100 mL). The microplates were then wrapped with tissue paper and incubated for 1 night at room temperature with dark lighting. The living cells reacted to MTT to form purple stain. The test result was then read by ELISA reader at a wavelength of 595nm (Nugroho, 2013) .
Cell cycle analysis by Flow Cytometry
In cell cycle analysis, propidium iodide (PI) staining was used. This dye can be used to analyze a set amount of DNA in each cell. AS many as 5x105 cells/wells were implanted in 6-well plates and then incubated until the cells went back to normal. The cells were treated with DMSO (0.25%) solvent and active isolates at IC50. The cells were then incubated for another 24 hours. At the end of the incubation period, the media were taken and then transferred to the centrifuge tubes (2,000 rpm for 3 minutes) where the supernatant was then discarded. In wells whose medium had been taken, PBS was added and then transferred to the same microtubes from the same treatment. Then, they were centrifuged where the supernatant was then discarded. This stage was repeated once again and then the cells were harvested with trypsin. The cells were then transferred into the same microtubes which then centrifuged (2,000 rpm for 3 minutes). The residue of cell harvest in the wells was then rinsed with PBS and underwent centrifuge again where then the PBS was discarded. The cell residue in the microtubes were then fixed with 70% ethanol, -20°C, and then incubated for 30 minutes at room temperature or overnight at 4°C. Then, the residue was centrifuged (2,000 rpm for 3 minutes). The precipitated cells were washed with PBS for 2 times. Then, PI reagent was carefully added, and immediately homogenized. Microtubes containing cell suspension was then wrapped in aluminum foil and incubated in 37°C waterbath for 20 minutes. The cell suspension was homogenized and transferred back into the flow cytometer tube using nylon filters which was then ready to be analyzed by using flow cytometer.
Analysis of cell Apoptosis induction by Flow Cytometry
As many as 5x105 cells/wells were grown in 6-well plates and then the cells were incubated until they got back to normal. The cells were treated with DMSO solvent (0.25%) and active isolates. The cells were then incubated for another 24 hours. At the end of the incubation period, the media was taken and transferred to centrifuge and decentrifuge (2,000 rpm, for 3 minutes) and then the supernatant was discarded. In wells where the medium had been taken, PBS was added, and it was then transferred to the same microtube from one treatment. Then it was centrifuged and the supernatant was discarded. This stage was repeated once more and then the cells were harvested with trypsin. Next, the cells were transferred into the same microtubes and then centrifuged (2,000 rpm for 3 minutes). The cell harvest residue left in the wells was washed with PBS and centrifuged again. Then the PBS was discarded. The residue was then carefully added with PI-Annexin V reagent, and immediately homogenized. Microtube containing cell suspension was wrapped in aluminum foil and incubated in 37°C waterbath for 5 minutes. The cell suspension was homogenized and then transferred back into the flow cytometer tube using nylon filters which then ready to be analyzed by using flow cytometer.
Analysis of caspase-8 by immunocytochemistry
24-well plates containing cell from the CO2 incubator were taken. Complete media from the well was removed with Pasteur pipette. PBS was added to the wells of 500 μL to wash the cells. PBS was removed with Pasteur pipette carefully. A sample of 1,000 μL was pipetted into the well. Complete media was added for cell control (2 cell controls). Cells were incubated in the CO2 incubator. Cell incubation was stopped. The media from the wells are disposed and PBS was loaded at 500 μl into each well to wash the cells. Cover slips was taken using tweezers placed in a used 6-well plate and labeled on each treatment. Cold methanol (300 μl) was added and incubated for 10 minutes in freezer. Methanol was removed slowly, and prevent cover slip from reversing. If the staining is on the next day, then the cover slip was stored in freezer. PBS 500 μl was added to the cover slip and left for 5 minutes. PBS was removed and disposed. Cells were washed with PBS to rinse up the trace of methanol. A 500 μl of distilled water was added and left for 5 minutes. The distilled water was discardedand washed with distilled water 2 times. Solution of hydrogen peroxide (blocking solution) was dripped and incubated for 10 minutes. Prediluted blocking serum was dripped and incubated for 10 minutes. The solutionwere discarded. This process was done to prevent non-specific staining. Primary monoclonal anticaspase 8 antibody (C4106 Sigma, monoclonal antibody caspase 8 031k4887, purified from hybridoma cell culture) was dripped and incubated for 5 minutes. PBS was discarded. Secondary antibodies were dropped and biotin labeled (biotinylated universal secondary antibody). Incubated for 10 minutes and 500 μl PBS was added, incubated for 5 minutes. PBS was discarded. Reagents containing peroxidase enzyme streptavidin complex was dripped and incubated for 10 min. PBS was added at 500 μl and incubated for 5 minutes. PBS was discarded. DAB chromogen substrate solution was dripped, and incubated for 10 minutes. Aquadest at 500 μl was added and then discarded. Haematoxylin solution was dripped and incubated for 3 minutes. Aquadest at 500 μl was added, and discarded. Cover slips were taken with tweezers carefully, and dipped in xylol. Cover slips dipped in alcohol. Cover slip was dipped. Cover slips were placed on top of glass object, dripped with mounting mediaand covered with square cover slip. Expression of caspase protein -8 was observed with a light microscope (CCRC, 2009).
Results
Isolation and Structural Analysis of Calotroposid A
Compound Isolation
Calotroposide A compound is a yellow amorphous crystalline isolate obtained from F2 separation of the roots of C.gigantea through three stages. In the first separation stage, column vacuum chromatography method was applied resulting seven sub-fractions i.e.: SF1-SF7. The cytotoxicity test results on SF1-SF7 showed that SF4 had high anticancer activity with IC50 14.3 μg/ml. In the second phase, the separation process was performed on the sub-fractions SF4 using open column with stationary phase silica gel 60 and mobile phase solvent gradients which were the mixtures of hexane and ethyl acetate 4:1. Then, a mixture of ethyl acetate and chloroform 9:1 was used, continued with a mixture of chloroform and methanol 1:1 and last methanol. From this separation process, five sub-fractions were produced i.e.: SF4.1, SF4.2, SF4.3, SF 4.4, and SF4.5. The results of cytotoxicity assay on SF4.1 to SF 4.5 showed that all sub-fractions had the potential to be anticancer agents with IC50 value below NCI (<20μg/ml) (Fadeyi et al, 2013). Next, the most active sub-fraction was selected, i.e.: sub-fraction 4.5 with the value of IC50 (3.49 μg/ml). The data on the cytotoxicity assay results on isolate SF4.1 to SF 4.5 can be seen in Table 1 as follows.
Table 1.
Values of IC50 of SF4.1-SF 4.5 Treatments on the Growth of WiDr Colon Cancer Cells
| Mean of Viability WiDr cells line (%)*± SD | ||||||||
|---|---|---|---|---|---|---|---|---|
| Isolate / Dosis | 0.78125 µg | 1.56 µg | 3.125 µg | 6.25 µg | 12.5 µg | 25 µg | 50 µg | IC50**± SD(µg/ml) |
| SF4.1 (isolate 1) | 80.55 ± 5.72 | 76.87 ± 5.15 | 65.66 ± 2.78 | 54.75 ± 2.03 | 48.3 ± 3.3 | 37.45 ± 1 | 24.1 ± 1.86 | 9.41 ± 1.69 |
| SF4.2 (isolate 2) | 83.55 ± 2.69 | 70.32 ± 0.73 | 61.6 ± 0.89 | 50.75 ± 2.36 | 40.3 ± 1.79 | 25.45 ± 3.23 | 14.1 ± 1.43 | 6.01 ± 0.74 |
| SF4.3 (isolate 3) | 86.22 ± 0.4 | 72.12 ± 0.99 | 60.2 ± 0.78 | 40.74 ± 0.53 | 37.32 ± 1.18 | 28.45 ± 2.3 | 13.3 ± 1.56 | 5.65 ± 0.22 |
| SF4.4 (isolate 4) | 87.43 ± 1.14 | 76.43 ± 0.16 | 56.83 ± 1.48 | 45.36 ± 1.06 | 37.65 ± 2.21 | 21.45 ± 3.28 | 1.3 ± 0.98 | 5.53 ± 0.53 |
| SF4.5 (isolate 5) | 76.52 ± 1.29 | 64.39 ± 2.06 | 51.23 ± 2.02 | 38.51 ± 0.03 | 28.35 ± 1.8 | 19.2 ± 0.76 | 9.32 ± 0.65 | 3.49 ± 0.22 |
, The average value of Viability (%) ± standard deviation with three replication;
, The average value of IC50 ± standard deviation with three replication
The data in the table above indicates that the IC50 values of sub-fraction SF4.1 to SF4.5 had IC50 values below the value specified by the NCI. Thus, it can be stated that sub-fraction SF 4.1- 4.5 are effective as anticancer. From this study, in addition, the most active sub-fraction was selected which was sub-fraction SF 4.5.
Based on the test result and SF.4 separation result, isolate SF4.5 had been proven to provide higher anticancer activity (3.49 μg/ml) compared to the other isolates. In addition, SF4.5 isolate weighed more than the other isolates, reaching 711.6 mg. Therefore, the next step is the isolate SF4.5 purification with preparatory chromatography method. From this isolation, isolate 4.5.1 was formed which was an isolate dominated with intense purplish-blue color stain on visible observation, and dark brown color on the observation under UV366 light. Isolate 4.5.1 was identified as Calotroposide A compound.
Figure 1.
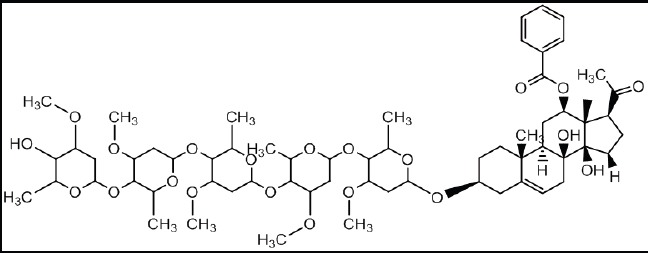
Chemical Structure of Isolate 4.5.1: Calotroposide A (Chemical Formula: C63H96O21)
Figure 2.
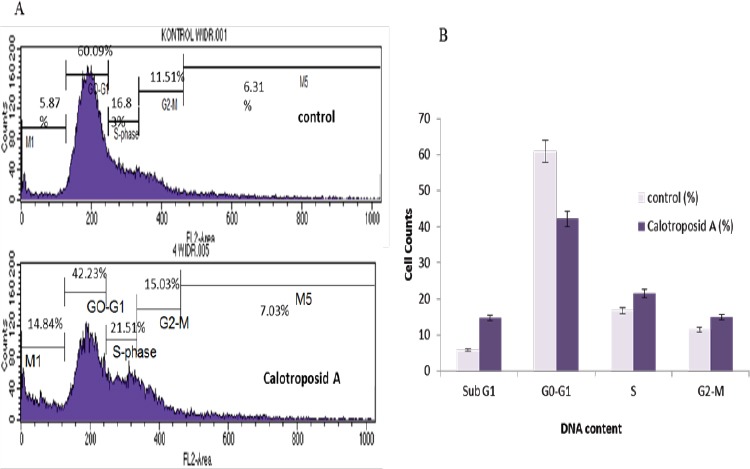
The Induction Effect of Cell Death and Distribution Changes on Cell Cycle Phases on WiDr Cells Due to Calotroposid A Treatment at IC50 17.23 µg/ml. As many as 5x105 cells/well of WiDr cells were incubated for 24 hours and then treated with Calotroposide A on IC50. The distribution profile of each phase (Sub G1, G0-G1, S, and G2/M) in the cell cycle was analyzed using flow cytometry with propidium iodide staining (PI).
Compound Identification
The identification result by using TLC and HPLC showed that this compound was a compound with polar characteristic. The TLC identification with stationary phase: RP-18; and mobile phase: ACN: water (7:3) had shown Rf: 0.75. The result of UPLCMS/MS chromatogram indicated that Calotroposide A compound was extracted at the retention time (0.5 min). The compound’s ESIMS spectrum indicated molecular ion of m/z 1189.6490 (calcd for C63H96O21, 1188.6444) that matched to the molecular formula of m/z C63H96O21. These molecular ions were fragmented into m/z 684.2009; m/z 436.3346; m/z 391.2794; m/z 182.9828; and m/z 141.9567.
The NMR-1H spectrum on the compound‘s aglycone showed three kinds of methyl on proton shear which were δ =1.86 (-CH3, 1H, Singlet); δ = 1.59 (-CH3, 1H, Singlet); δ = 1:04 (-CH3, 1H, Singlet). The occurring cyclohexane proton was δ=5:33 (-CH, 1H, Singlet). The characteristic of the benzene substituent occurrence on the compound was shown by proton signal at δ = 7.93/C2’6 ‘(= CH benzene, 1H, double doublet); δ = 7.56; 7.8/C3’C5 ‘(= CH benzene, 1H, triplet); δ = 7:46/C4 ‘(= CH benzene, 1H, triplet). A hydroxyl proton occurred at δ = 3.22 (OH, 1H, singlet).
Based on NMR-1H spectrum, this compound contained 5 types of oxypregnanane oligoglycoside sugars characterized with proton shear occurrence: 5 anomeric signals, 5 methyl doublet signals and 5 methoxy signals. The 5 anomeric spectrum signals were indicated by the proton shear at δ=4.75 (O-CH-O, 1H, dd); =4.65 (O-CH-O, 1H, dd); =4.63 (O-CH-O, 1H, dd); =4.57 (O-CH-O, 1H, dd); =4.77 (O-CH-O, 1H, dd). The 5 methyl doublet spectrum signals were indicated by the proton shear at δ=1:04 (CH3 -CO; H3, doublet) δ=1.13 (CH3 -CO; H3, doublet); δ=1.17 (CH3 -CO; H3, doublet); δ=1.19 (CH3 -CO; H3, doublet); δ=1.261 (CH3 -CO; H3, doublet). The 5 methoxy spectrum signals were indicated by a shift in the sugar protons at δ=3.40 (-OCH3, H3, Singlet); δ=3.32 (-OCH3, H3, Singlet), δ=3.40 (-OCH3, H3, Singlet); δ=3.40 (-OCH3, H3, Singlet); δ=3.40 (-OCH3, H3, Singlet).
The compound molecular ion was m/z 1189.6490 that matched the molecular formula of m/z C63H96O21 while the proton shear at NMR-1H spectrum was identical with the Calotroposide H compound had been isolated from the roots of Calotropis procera. In addition, it was also identical with Calotroposide A electrically insulated from Calotropis gigantea (Ibrahim et al., 2015; Kitagawa et al., 1992). The difference between Calotroposide H and A was that Calotroposide H was the epimer of Calotroposide A. Based on the species resemblance and also the similarity on molecular weight and proton shear, it can be concluded that this compound is a chemical compound of Calotroposide A. The chemical structure of Calotroposide A is presented in the figure below:
In vitro Assay of Calotroposid A Anticancer Compound
An in vitro anticancer activity test was conducted on Calotroposide A compound towards WiDr cancer cells by using MTT assay method. The activity test results showed that the increase on the concentration of the tested Calotroposid A compound might induce a decrease in WiDr cell viability. To identify the potential barriers of Calotroposide A compound growth, IC50 value was calculated by using probit analysis. The probit analysis result recapitulation can be seen in Table 2 below.
Table 2.
Average Percentage of WiDr Colon Cancer Cells Viability as a Result of Isolate Calotroposid A Treatment
| No | ISOLAT | Mean of Viability WiDr cells line (µg/mL) | IC50 (µg/ml)* | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 1.79 | 3.58 | 6.25 | 12.5 | 25 | 50 | 100 | |||
| 1 | Calotroposid A | 92.24 | 74.66 | 78.08 | 67.58 | 47.95 | 12.33 | 17.58 | 17.23±0.03 |
, The average value of IC50 ± standard deviation with three replication
Based on the data in the table above, it can be indicated that the IC50 value of isolate Calotroposide A was below the value specified by the NCI. Thus, it can be stated that isolate Calotroposide A may be effective as anticancer.
Apoptosis Induction Mechanism
The Regulation Test Results of WiDr Cancer Cell Cycle due to Calotroposide A Isolate Treatment
Cell Cycle analysis by using flow cytometri showed that treatment of 17.23 µg/ml Calotroposid A for 24 h caused increasing of sub-G1, S-phase and G2-M accumulation until 14.84%, 21.51% and 15.03% respectively compared to control cells (Figure 3A-B). The High cells accumulation on sub-G1 represents the death cells.
Figure 3.
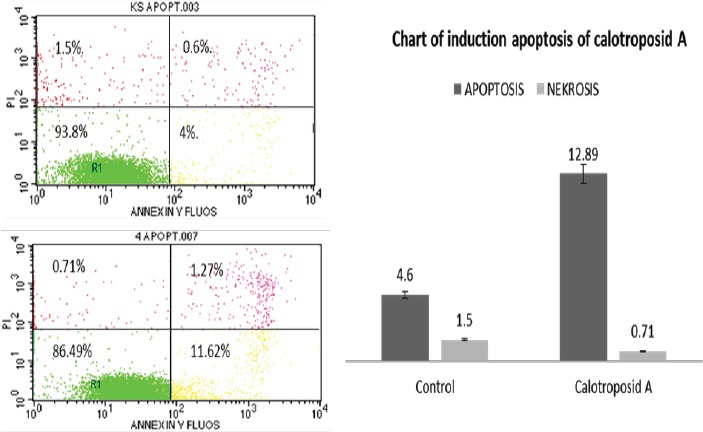
The Results of Cell Distribution and Induction Analysis of Cell Death/ Cell Quest by using Flow Cytometry (A) WiDr Control Cells (B) Calotroposid A Treatment on WiDr Cells; (R1) Living Cells, (R2) Early Apoptosis, (R3) Late Apoptosis and (R4) Necrosis.
The Induction Test Results of WiDr Cancer Cell Apoptosis due to Isolate Calotroposid A Treatment
On the cell apoptosis analysis results induced by Calotroposide A compound treatment on WiDr cancer cells, it can be seen that early apoptotic cells increased by 11.62%, with 1.27% of late apoptosis while necrosis declined by 0.71%. The increase in cell apoptosis was also correlated with increased cells accumulation in sub-G1 phase (Figure. 4A-B; Figure. 3A-B). Cytotoxic activity of isolate calotroposid A possibly through inducing of apoptosis and cells cycle accumulation in S-phase and G2/M-phase.
Figure 4.
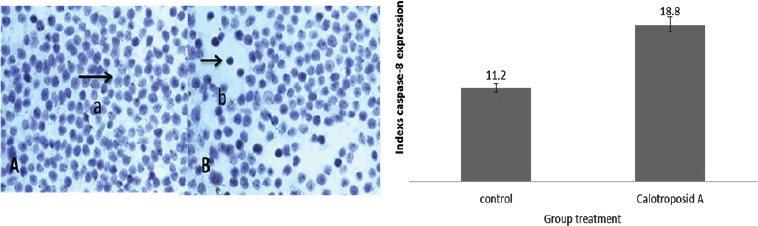
The Treatment Effects of Calotroposide A Compound on Caspase-8 Expression on WiDr Cells. WiDr Cells with (5x104) density were incubated on coverslips in 12 well plates for 24 hours to adapt. Then, Calotroposid A compound was treated on IC50 17.23 μg/ml, and then incubated for another 15 hours while caspase-8 staining was conducted using immunocytochemistry method in accordance with the research procedure. (A) cell control without caspase-8 antibody, (B) cells with Calotroposide A compound treatment with anti-caspase-8 antibody indicated an increase on caspase-8 protein expression as seen from the cytosol brown stain. The arrow (b) indicates caspase-8 expression. The microscope used was a light microscope with 400x magnification.
Figure 5.
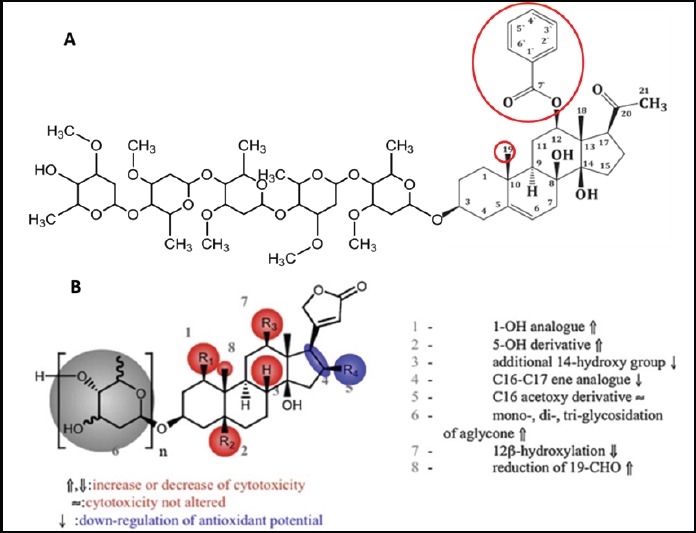
The Relationship between Calotroposide A and Cytotoxic Activity (modified from (Juncker et al., 2009))
The Analysis Results of Caspase-8 Protein Expression of WiDr Cells on Calotroposide A Compound Treatment
The immunocytochemistry test results indicated that the Calotroposide A treatment on IC50 17.23μg/ml on WiDr cancer cells had caused an increase on caspase-8 protein expression amounted to 18.8% compared to the control group. The increase on caspase-8 protein expression can be seen from the brown in the cytosol. The immunocytochemistry assay results on Calotroposide A treatment is presented in the figure below:
Discussion
Anticancer activity and the operating mechanism of Calotroposide A compound on WiDr cells is still unknown by previous research. Calotroposid A compound is a terpenoid glycoside compound consisting of aglycone part in form of terpenoid core structure and glycone part are composed of five sugar of cymarosa-cymarosa-oleandrosa-oleandrosa-cymarosa. Calotroposid A compound and its epimer i.e.: Calotroposide H has been found by previous researchers to be inactive in vitro on A549 NSCLC cells (IC50: 3.3μM), U 373 GBM cells (IC50:21.8 m) and prostate cancer cells PC-3 (IC50:17.6μM) (Ibrahim et al, 2015).
From the results of this study, it was indicated that Calotroposide A compound was proven to induce anticancer activity towards WiDr colon cancer cells with IC50:17.23 μg/ml. Based on the data of cell cycle regulation tests of cell apoptosis induction and caspase 8 expression, it could be explained that the Calotroposide A compound treatment towards the proliferation of WiDr colon cancer cells had led to an increase in S phase, G2/M phase 21.51% and 15.03%. As a result of these improvements, cancer cell apoptosis increased. In molecular level, the indicator of cell apoptosis increase was the occurrence of caspase 8 expression increased.
Considering the chemical structure, Calotroposide A is a triterpenoid glycoside compound. The amount of sugars in the aglycone part has affected the anticancer activity, where the more sugar chain built, the more the activities increase (Juncker et al., 2009). The pharmacophore group of this compound is suspected to be in atom C number 12, i.e.: benzoate and methyl groups on atom C number 19. The structure of active Calotroposide A compound and its pharmacophore group are presented in the figure below.
The glycone part on Calotroposide A compound serves to increase the solubility of aglycone part in body fluids, where body fluids are composed of 80% water which is a polar compound. Calotroposide A solubility increase in body fluids will increase the speed compound distribution to achieve network/target cell. The pharmacore group in the aglycone of Calotroposide A compound is capable of inducing apoptosis through the activation of death receptor (CD 95/Fas). The activation of Fas by Fas ligand (FasL) affects caspase-8 activation. Furthermore, caspase-8 breaks the inactive form of the Bid into two parts which are the part containing BH3 domain as an active form of Bid (tBid). The tBid Protein in translocation to the mitochondria stimulates the release of Cyt c which then forms a complex with Apaf-2. This complex further activates caspase-9 and caspase-7, and thus, the apoptosis induction takes place. The caspase-9 activation also leads to caspase-3 activation, followed by the caspase-2 activation. The increased activation of caspase-2 will trigger the G2/M checkpoint maintenance. The G2-phase checkpoint is a very important phase in ensuring the accuracy of DNA replication and division which prepares genomes into daughter cells. If inaccuracy occurs in DNA replication, there will be apoptosis induction. The increasing number of cells in G2/M phase of the normal DNA replication indicates the increase of inaccuracy, and thus, the number of cells undergoing apoptosis also increases (Cullen and Martin, 2009).
In conclusion, calotroposide A compound is capable of inhibiting the growth of WiDr colon cancer cells in IC50:17.23 μg/ml through apoptosis induction and increased expression of caspase-8.
References
- 1.Cullen SP, Martin SJ. Caspase activation pathways:some recent progress. Cell Death Differ. 2009;16:935–8. doi: 10.1038/cdd.2009.59. [DOI] [PubMed] [Google Scholar]
- 2.CCRC. Prosedur tetap pengamatan ekspresi protein dengan metode imunositokimia, cancer chemoprevention research center, facultyof pharmacy Gadjah Mada University. 2009:1–7. (Text I Indonesia) [Google Scholar]
- 3.Fadeyi SA, Fadeyi OB, Adejumo AA, et al. In vitro anticancer screening of 24 locally used Nigerian medicinal plants. BMC Complement Altern Med. 2013;13:79. doi: 10.1186/1472-6882-13-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Habib MR, Aziz MA, Karim MR. Inhibition of Ehrlich's ascites carcinoma by ethyl acetate extract from the flower of Calotropis gigantea L. in mice. J Appl Biomed. 2010;8:47–54. [Google Scholar]
- 5.Habib MR, Karim MR. Evaluation of antitumour activity of Calotropis gigantea L. root bark against Ehrlich ascites carcinoma in Swiss albino mice. Asian Pac J Trop Med. 2011;4:786–90. doi: 10.1016/S1995-7645(11)60194-6. [DOI] [PubMed] [Google Scholar]
- 6.Ibrahim SR, Mohamed GA, Shaala LA, et al. Calotroposides H–N, new cytotoxic oxypregnane oligoglycosides from the root bark of Calotropis procera. Steroids. 2015;96:63–72. doi: 10.1016/j.steroids.2015.01.012. [DOI] [PubMed] [Google Scholar]
- 7.Juncker T, Schumacher M, Dicato M, Diederich M. UNBS1450 from Calotropis procera as a regulator of signaling pathways involved in proliferation and cell death. Biochem Pharmacol. 2009;78:1–10. doi: 10.1016/j.bcp.2009.01.018. [DOI] [PubMed] [Google Scholar]
- 8.Kitagawa I, Zhang F, Park JD, et al. Indonesian medicinal plants I. chemical structures of calotroposides Aand B, two new oxypregnane-oligoglycosides from the root of Calotropis gigantea (Asclepiadaceae) Chem Pharm Bull. 1992;40:2007–13. doi: 10.1248/cpb.40.2007. [DOI] [PubMed] [Google Scholar]
- 9.Muti R, Griana TP, Ula QN, Andhyarto Y. The effect of calotropis gigantea leaves extract on fibrosarcoma growth and caspase 3 expression. Int J Pharmacol Clin Res. 2016;8:167–71. [Google Scholar]
- 10.Muti R, Sukardiman Widyawaruyanti A, Zulaikah S. Comparison of ethanol extract from roots leaves and flowers of. Alchemy. 2016;18:1–4. [Google Scholar]
- 11.Mutiah R, Sukardiman Widyawaruyanti A. Cytotoxic effect of crude extract and fraction from calotropis gigantea leaves on human colon cancer widr cell lines. Int J Pharm Pharm Sci. 2017;9:83–6. [Google Scholar]
- 12.Nugroho AE, Hermawan A, Putri DDP, Novika A. Combinational effects of hexane insoluble fraction of Ficus septica Burm and doxorubicin chemotherapy on T47D breast cancer cells Combinational. Asian Pac J Trop Biomed. 2003;3:297–302. doi: 10.1016/S2221-1691(13)60066-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Radjakmanugunsudarso M. Cabe puyang warisan nenek moyang. Jogjakarta. 1968:4–5. (Text in Indonesia) [Google Scholar]
- 14.Seeka C, Sutthivaiyakit S. Cytotoxic cardenolides from the leaves of Calotropis gigantea. Chem Pharm Bull. 2010;58:725–8. doi: 10.1248/cpb.58.725. [DOI] [PubMed] [Google Scholar]
- 15.Wang ZN, Wang MY, Mei WL, et al. A new cytotoxic pregnanone from Calotropis gigantea. Molecules. 2008;13:3033–3039. doi: 10.3390/molecules13123033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong SK, Lim YY, Abdullah NR, Nordin FJ. Assessment of antiproliferative and antiplasmodial activities of five selected Apocynaceae species. BMC Complement Altern Med. 2011;11:3–5. doi: 10.1186/1472-6882-11-3. [DOI] [PMC free article] [PubMed] [Google Scholar]