

ABSTRACT
The molecular machinery linking macroautophagy (autophagy hereafter) to apoptosis is still being elucidated. A recent study found that the transcription factor FOXO3/FOXO3A (forkhead box O3), which regulates autophagy, is itself regulated by basal autophagy to determine apoptosis sensitivity. Autophagy inhibition confers cell sensitivity to anti-cancer agents, and this effect is explained by the ability of FOXO3 to transactivate the pro-apoptotic gene BBC3/PUMA. Here, we discuss the possibility that FOXO3 acts as a cell surveillance mechanism to correct autophagy perturbations (i.e., autophagy inhibition), and confers apoptosis sensitization if this autophagy imbalance is not rectified.
KEYWORDS: Apoptosis, autophagy, CRISPR/Cas9, FOXO3, homeostasis, MDM2, PUMA, transcription
One adaptation mechanism that cells use to deal with stress is autophagy. Cells activate autophagy in response to a host of environmental cues, and these responses are thought to protect the cell. An important implication of this idea is that if the cell cannot handle the stress, then it should die instead. This suggests that cells must measure how well autophagy is working and tie that to the likelihood that it will die. This idea has been widely studied in the context of cancer where there are many ongoing clinical trials aiming to inhibit autophagy in combination with anti-cancer therapies. The rationale for these studies is that if we inhibit autophagy and thus remove the protection against stress that autophagy provides, the cancer cells will be more likely to undergo apoptosis in response to treatment with an anti-cancer agent. Hundreds of published studies show just this – if autophagy is inhibited in cancer cells by inhibition of specific ATG genes or with drugs such as chloroquine, those cells undergo apoptosis more easily when they are treated with an additional death stimulus. However, a major problem for the field is that there is little mechanistic understanding of why this strategy works.
Our latest study, which extends from a previous report, found a transcriptional mechanism linking autophagy to apoptosis [1]. This mechanism provides new insights into the connections between autophagy and apoptosis, and explains why autophagy inhibition can enhance tumor cell apoptosis by other anti-cancer drugs. Importantly, this mechanism also suggests that homeostatic control of autophagy is directly linked to the core apoptosis machinery. This finding means that when cells are defective in autophagy, they are also primed to undergo apoptosis. We found that upon genetic or pharmacological autophagy inhibition, the levels of the pro-apoptotic protein BBC3/PUMA increase, and this increase is essential for sensitizing cells to undergo apoptosis following a death stimulus. Surprisingly, the obvious mechanism for such an effect – i.e., BBC3 protein is degraded by selective autophagy – does not apply. Instead, BBC3 is actively transcribed upon autophagy inhibition by the forkhead box transcription factor FOXO3, which upregulates the BBC3 gene via a single forkhead response element (FHRE) in an intron. Importantly, CRISPR/Cas9-mediated deletion of this FHRE in the endogenous BBC3 locus is sufficient to block BBC3 upregulation upon autophagy inhibition. We then leveraged this mechanism to show that this specific mechanism whereby autophagy inhibition increases activity at a single transcription factor binding site in one gene in the human genome explains the ability of autophagy inhibitors to enhance DNA damaging agent-induced apoptosis. And, we were able to show that the same mechanism can allow an autophagy inhibitor to change the mode of action of an MDM2-targeted therapy from growth arrest to apoptosis.
FOXO3 poises cells for rapid induction of autophagy following cytokine deprivation. And, like some other transcription factors, it does so by transactivating autophagy-related genes. We wondered how the FOXO3 transcription factor is regulated so as to increase its activity upon autophagy inhibition. We found that FOXO3 is itself a substrate for basal autophagic degradation, and when this is blocked by either pharmacological autophagy inhibitors or by inactivation of autophagy regulators such as ATG5 or ATG7, then FOXO3 translocates to the nucleus where it can activate target genes. This finding adds another interesting feature to the mechanism we uncovered; FOXO3 – a transcription factor that regulates autophagy – is itself regulated by autophagic degradation. This suggests that FOXO3 is at the center of a homeostatic feedback loop to correct autophagy perturbations by transactivating autophagy targets when basal autophagy is inhibited. However, if autophagy inhibition persists, then the cell also activates pro-apoptotic genes such as BBC3 via the same transcription factor and thus becomes sensitized to undergo cell death (Figure 1). We therefore propose that basal autophagic turnover of FOXO3 serves as a surveillance mechanism to detect and correct autophagy flux disruptions and, at the same time, ensure that those cells where autophagy is deficient, are primed to undergo apoptosis.
Figure 1.
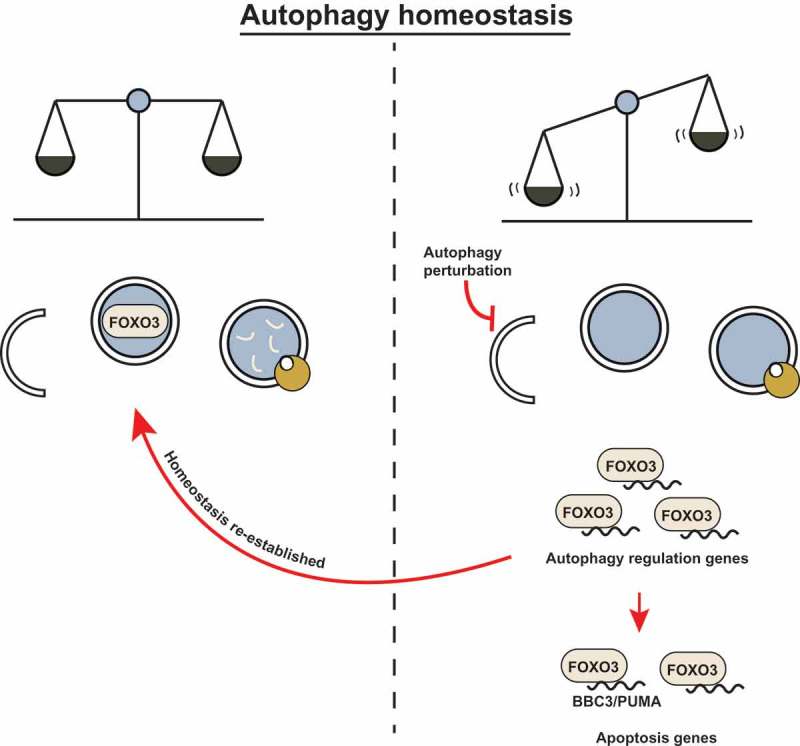
FOXO3 may act as a cell surveillance mechanism to maintain autophagy homeostasis. (left) Basal autophagy flux is upheld and homeostasis is maintained. FOXO3 is targeted for degradation by basal autophagy to confer low apoptosis sensitivity. (right) FOXO3 avoids autophagic turnover upon autophagy inhibition (genetic deletion of essential autophagy genes or pharmacological inhibition). FOXO3 transactivates autophagy-related genes and attempts to compensate for the perturbation in autophagy flux; if this correction is not achieved, FOXO3 leads to BBC3/PUMA transactivation and apoptosis sensitization.
This mechanism provides a specific link between autophagy and apoptosis that we can exploit to increase the ability of anti-cancer drugs to kill tumor cells, but several questions remain. Our studies showed that the transcriptional kinetics of various apoptotic and non-apoptotic FOXO3 targets vary following pharmacological and genetic autophagy perturbation; how is this regulated and what does it mean for cell fate? Also, we should understand how FOXO3 is selectively recruited to phagophores, and what signals allow it to avoid autophagic turnover and become activated upon autophagy inhibition.
Funding Statement
This work was supported by NIH grants RO1CA150925 and RO1CA190170;National Institutes of Health [RO1CA190170];National Institutes of Health [RO1CA150925].
Disclosure statement
No potential conflict of interest was reported by the authors.
Reference
- [1].Fitzwalter BE, Towers CG, Sullivan KD, et al. Autophagy inhibition mediates apoptosis sensitization in cancer therapy by relieving FOXO3a turnover. Dev Cell. 2018;44:555–65 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]