

ABSTRACT
tRNAHis guanylyltransferase (Thg1) has unique reverse (3’-5’) polymerase activity occurring in all three domains of life. Most eukaryotic Thg1 homologs are essential genes involved in tRNAHis maturation. These enzymes normally catalyze a single 5’ guanylation of tRNAHis lacking the essential G−1 identity element required for aminoacylation. Recent studies suggest that archaeal type Thg1, which includes most archaeal and bacterial Thg1 enzymes is phylogenetically distant from eukaryotic Thg1. Thg1 is evolutionarily related to canonical 5’-3’ forward polymerases but catalyzes reverse 3’-5’polymerization. Similar to its forward polymerase counterparts, Thg1 encodes the conserved catalytic palm domain and fingers domain. Here we investigate the minimal requirements for reverse polymerization. We show that the naturally occurring minimal Thg1 enzyme from Ignicoccus hospitalis (IhThg1), which lacks parts of the conserved fingers domain, is catalytically active. And adds all four natural nucleotides to RNA substrates, we further show that the entire fingers domain of Methanosarcina acetivorans Thg1 and Pyrobaculum aerophilum Thg1 (PaThg1) is dispensable for enzymatic activity. In addition, we identified residues in yeast Thg1 that play a part in preventing extended polymerization. Mutation of these residues with alanine resulted in extended reverse polymerization. PaThg1 was found to catalyze extended, template dependent tRNA repair, adding up to 13 nucleotides to a truncated tRNAHis substrate. Sequencing results suggest that PaThg1 fully restored the near correct sequence of the D- and acceptor stem, but also produced incompletely and incorrectly repaired tRNA products. This research forms the basis for future engineering efforts towards a high fidelity, template dependent reverse polymerase.
KEYWORDS: Reverse polymerization, tRNA editing, tRNA repair, nucleotidyltransferase, protein engineering
Introduction
DNA and RNA replication, transcription, and reverse transcription have long been understood as unidirectional nucleotide polymerization processes, exclusively proceeding with the addition of nucleotides in the 5’-3’ direction. Despite the advantages of 3’-5’ elongation in general cellular processes such as DNA replication and telomere formation, reverse (3’-5’) polymerization is almost non-existent in nature. The only known example is the tRNA editing enzyme tRNAHis guanylyltransferase (Thg1). Thg1 is evolutionarily related to canonical polymerases.1-4 All Thg1 homologs encode the conserved catalytic palm domain,4-7 which they use to catalyze a single nucleotide addition to the 5’ end of tRNAHis. Some Thg1 enzymes are capable of catalyzing extended and template dependent nucleotide addition and are, thus, bona fide reverse nucleotide polymerases.1,2,6,8
In eukaryotes, Thg1 is an essential enzyme in tRNA maturation.9 Thg1 adds a single guanine residue (G−1) to the 5'-end of premature tRNAHis 1,10. G−1 is the critical identity element that histidyl-tRNA synthetase (HisRS) uses to recognize its cognate tRNA, enabling HisRS to differentiate tRNAHis from the pool of other cellular tRNAs.11 Although bacteria and most archaea retain a genome encoded G−1 in the tRNAHis gene, thg1 is present in many of their genomes, and Thg1 enzymes from diverse species are indeed catalytically active.1,3,8 Bacterial and archaeal (archaeal-type) Thg1 are biochemically and phylogenetically distinct from their eukaryotic (eukaryotic-type) counterparts.1,3,12-17 Major differences between the two are found in RNA-template preference, ATP dependence and the capability to promote extended reverse nucleotide polymerization.1,3,7,12,14-16 While the eukaryote-type Thg1 exclusively adds G−1 to the 5'-end of tRNAHis in a non-templated fashion10,18,19, archaeal-type Thg1 exhibits extended reverse polymerase activity with template-dependent nucleotide addition. Eukaryotic Thg1 homologs generally require ATP to perform an initial activation step of the tRNA prior to GTP addition, whereas archaeal-type Thg1 enzymes can utilize ATP or GTP to activate tRNAHis lacking a G−1 residue (tRNAHisΔG−1) prior to the guanylation reaction.2,3,18,20
A current theory is that the extended reverse polymerization activity of archaeal-type Thg1 may be related to a role for Thg1 in tRNA repair and editing. Detailed biochemical experiments have established that archaeal-type Thg1 adds nucleotides to the 5’ end of immature or incorrectly processed tRNA.1,3,7,12-16 Taken together these studies show that Thg1 can uniquely perform 3’-5’ nucleotide addition and elongation, and that nucleotide polymerization in nature can proceed both in forward and reverse directions.
The mechanism of reverse polymerization was revealed in complex structures of Thg1 bound to tRNA.6,7 The co-crystal structures of Thg1 in complex with its substrate tRNA and ATP or GTP showed surprising structural similarity to forward DNA and RNA polymerases despite no obvious conservation of the amino acid sequence between Thg1 and other canonical polymerases.4-7 Like its forward polymerase relatives, each Thg1 subunit folds into a hand-shape containing a catalytic palm domain, and substrate binding finger/thumb domains (Fig. 1A). In both Thg1-tRNA and T7-DNA complexes, the direction of substrate approach to the catalytic core is related to the direction of polymerization.6,7 tRNA substrates enter Thg1's active site from the opposite side that DNA and RNA enter other canonical 5'-3' polymerases. Furthermore, the finger and thumb domains of Thg1 are on the opposite sides of the catalytic core compared to canonical polymerases.6 Structural, biochemical, and phylogenetic data indicate that reverse polymerization appeared early in evolution.1,6 While much progress has been made in the characterization of archaeal and eukaryotic-type Thg1 homologs, little is known about the sequence motifs or key residues required to facilitate reverse polymerization and substrate selection.
Figure 1.
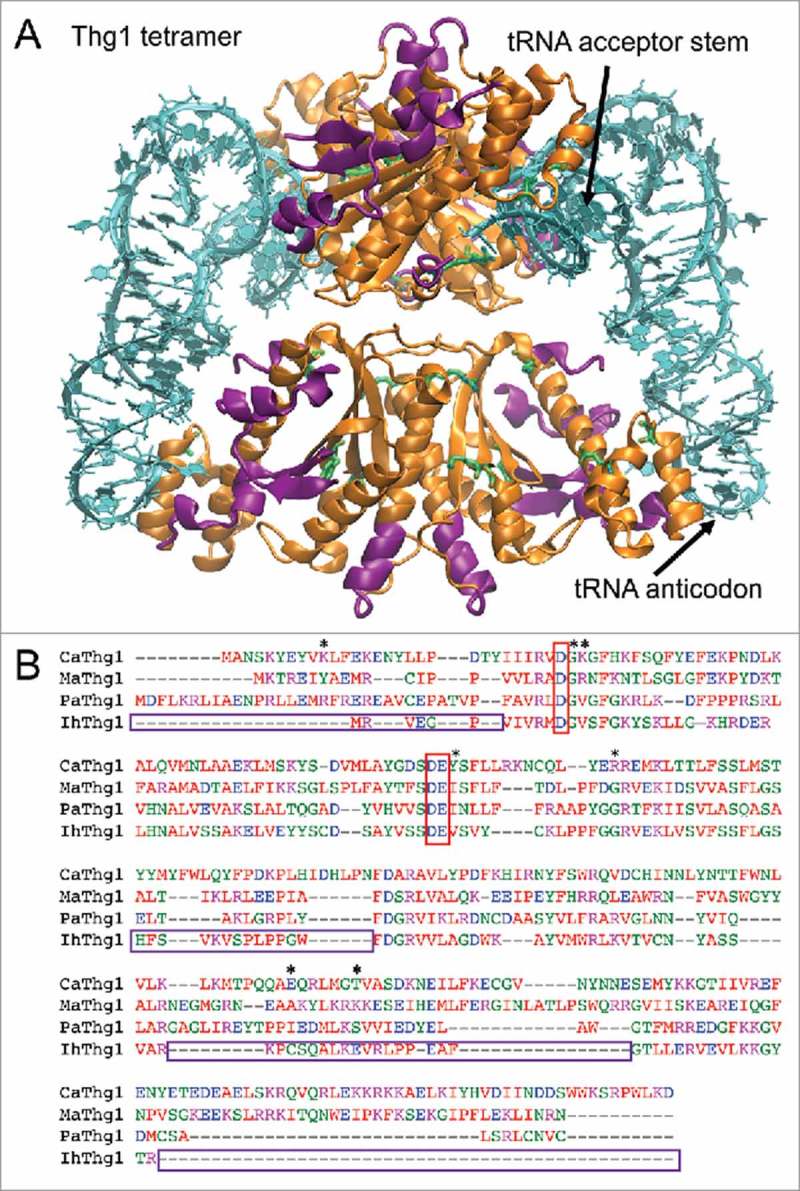
I. hospitalis Thg1 lacks multiple domains compared to eukaryotic Thg1. (A) Crystal structure of tetrameric CaThg1 with two tRNA substrates (pdb 3wc1)6. Amino acids absent or not aligned in IhThg1 are colored in purple. (B) Multiple Sequence alignment using CLUSTAL multiple sequence alignment by MUSCLE (3.8)30. Sections absent or not well aligned in IhThg1 are boxed. Amino acids subjected to mutational analysis are marked with a star, catalytic residues are boxed in red.
In this study, we identified a naturally occurring minimal Thg1 enzyme from Ignicoccus hospitalis. We also identified key Thg1 mutants capable of extended reverse polymerization. Finally, we found that Pyrobaculum aerophilum Thg1 (PaThg1) catalyzes high fidelity tRNA repair.
Results
Naturally occurring Thg1 homologs vary in amino acid sequence and length
Despite differences in amino acid composition and substrate specificity, most Thg1 homologs are of similar length (Fig. 1B) and encode a conserved palm and finger domain.1 An interesting exception is the naturally occurring Thg1 enzyme from Ignicoccus hospitalis (IhThg1), which lacks several sections of the protein, and aligns poorly with other Thg1 sequences (Figs. 1B, 2A), but was identified in a large Thg1 phylogenetic analysis.1
Figure 2.
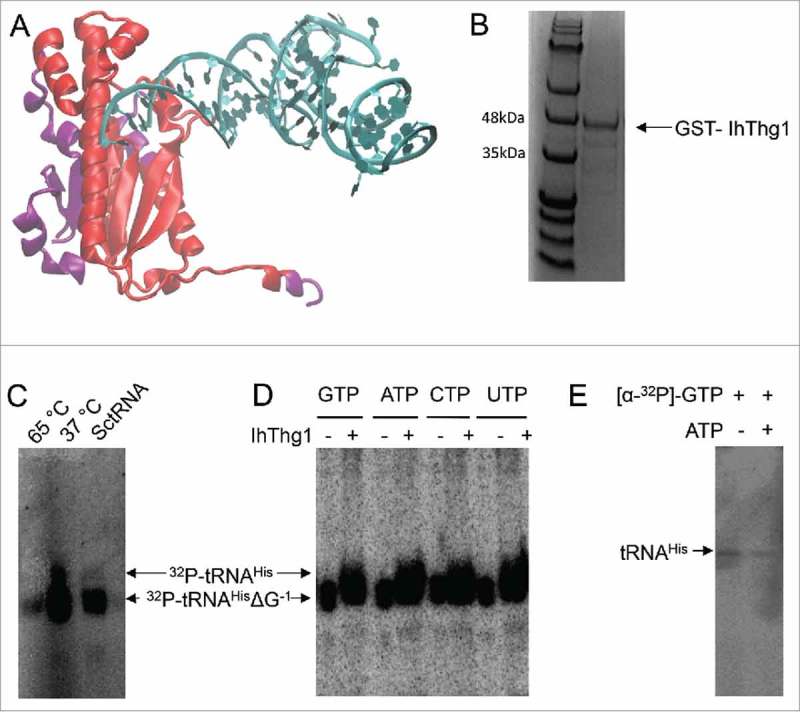
The naturally shortened I. hospitalis enzyme is enzymatically active. (A) Projection of the IhThg1 domains onto CaThg1 (pdb 3wc1). Sections missing in IhThg1 are depicted in purple. (B) IhThg1 was purified as a GST-fusion protein to apparent homogeneity. (C, D, and E) Autoradiography of IhThg1 reaction products separated on a 12% denaturing polyacrylamide gel with 8M Urea. For enzyme activity assays, proteins were incubated with 3’-monophosphorylated tRNAHisΔG−1 transcripts and the indicated nucleotides. Successful nucleotide addition results in a shift of the radiolabelled transcript or the appearance of a radiolabelled transcript. (C) IhThg1 incubated with GTP, ATP, and labelled 3’-32P -tRNAHisΔG−1 displays enzyme activity at 37 °C, but not at 65 °C, and exhibited reduced activity with S. cerevisiae tRNAHisΔG−1. (D) IhThg1 was incubated with labelled E. coli 3’-32P-tRNAHisΔG−1 and individual nucleotides GTP, ATP, CTP, and UTP and displayed nucleotidyltransferase activity with all four nucleotides. (E) IhThg1 was incubated with tRNAHisΔG−1 and [α-32P]-GTP in the absence and presence of ATP. IhThg1 enzyme activity is independent of ATP.
We produced and purified IhThg1 to investigate whether this minimal Thg1 enzyme is catalytically active despite lacking large sections of the Thg1 protein (Fig. 2B). In an in vitro enzyme activity assay with radiolabelled Escherichia coli tRNAHisΔG−1, IhThg1 was active at 37 °C but not at 65 °C (Fig. 2C, D). IhThg1 displayed reduced enzyme activity with yeast tRNAHisΔG−1, which encodes an A73 discriminator base compared to E. coli tRNAHisΔG−1 with a C73 discriminator base (Fig. 2C). In order to examine the nucleotide specificity of IhThg1, we incubated the recombinant enzyme with radiolabelled E. coli tRNAHisΔG−1 and the individual nucleotides GTP, ATP, CTP and UTP (Fig. 2D). IhThg1 added all four nucleotides to E. coli tRNAHisΔG−1 in vitro at comparable efficiency. Previous studies have shown that eukaryotic Thg1 enzymes require ATP for substrate activation, whereas archaeal-type Thg1 enzymes are ATP independent.1,3,13,20 To test for ATP dependence, we incubated IhThg1 with tRNAHisΔG−1 and [α-32P]-GTP in the presence and absence of 100 µM ATP. Radioactive product formation was observed in the presence or absence of ATP (Fig. 2E), suggesting that IhThg1 does not require ATP-dependent activation, but can utilize GTP, as observed for other archaeal-type Thg1 enzymes.3,15
Minimal protein requirements for reverse polymerization
The co-crystal structure of Thg1 with its substrate tRNA revealed that the catalytic palm domain can promote forward or reverse extension of a polynucleotide chain and that substrate orientation correlates with the direction of polymerization.6 Though the finger domain aids in substrate coordination and orientation, it is unclear whether it is required for Thg1's catalytic activity. To determine whether the finger domain is dispensable for catalytic activity, we created constructs including only the palm domains from Methanosarcina acetivorans Thg1 (MaPalm, amino acids 1–141) or PaThg1 (PaPalm, amino acids 1–150). The domain organization is depicted in Fig. 3A. The purified enzymes were incubated with radiolabelled E. coli tRNAHisΔG−1 and GTP. Both truncated enzymes were catalytically active, as evidenced by the appearance of a radiolabelled product band in the polyacrylamide gel. Palm domains alone were active at a similar level compared to the full length MaThg1, adding a single nucleotide to tRNAHisΔG−1 in the absence of the fingers domain in vitro (Fig. 3B).
Figure 3.
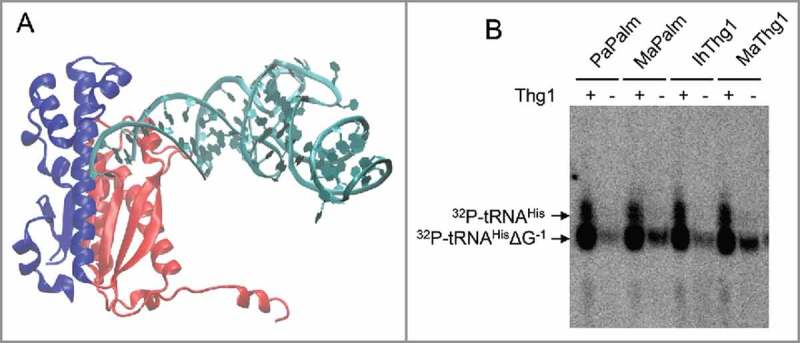
The palm domain of Thg1 is sufficient for nucleotidyltransferase activity. (A) The palm domain (red) and finger domains (blue) of CaThg1. The palm domain contains the catalytic nucleotidyltransferase domain, whereas the finger domain is mainly required for substrate positioning. (B) Autoradiography of Thg1 reaction products separated on a 12% denaturing polyacrylamide gel with 8M Urea. For enzyme activity assays, proteins at a concentration of 0.5 mg/mL were incubated with p-tRNAHisΔG−1 3’-32P labelled transcripts and GTP for 30 minutes. Successful nucleotide addition results in a shift of the radiolabelled product. Radiolabelled E. coli tRNAHisΔG−1 tRNA was incubated with GTP and Thg1 palm domains from P. aerophilum (PaPalm), M. acetivorans (MaPalm), or full-length IhThg1 or M. acetivorans Thg1 (MaThg1). The palm domain is sufficient for nucleotide addition.
P. aerophilum Thg1 repairs truncated tRNA substrates
A major limitation in engineering Thg1 towards a high fidelity reverse polymerase acting on substrates outside of tRNA is the substrate recognition element, namely the anticodon of tRNAHis. 1-3,9,10,20 To circumvent the requirement of a tRNA-like structure with a GUG-anticodon, we sought to employ a guide/target system, which has been successfully employed with enzymes that act on tRNAs, particularly RNases. In these cases, the enzymes display enzymatic activity when provided with a guide/target RNA hybrid that mimics the general tRNA structure (Fig. 4A).21,22 The guide RNA provides sequence elements forming the tRNA anticodon, T-loop and half of the D-loop and acceptor stem, with the latter two being complementary to the 5’end of the target RNA (Fig. 4A). Since Thg1 almost exclusively relies on the anticodon for tRNAHis recognition, sequences in the guide acceptor stem and D-stem can vary to accommodate complementary sequences to various target RNAs.1-3,9,10,20
Figure 4.
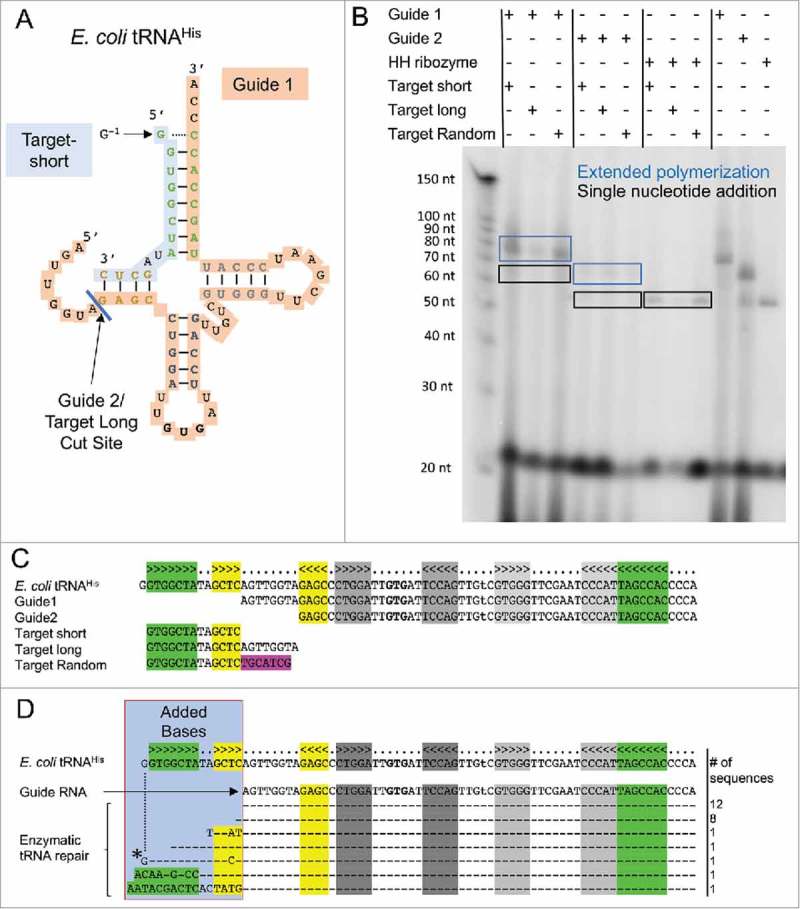
P. aerophilum Thg1 (PaThg1) exhibits tRNA repair activity. (A) In a split tRNA assay, E. coli tRNAHisΔG−1 is divided into a guide RNA, spanning from the D-loop to the CCA-end and a target RNA consisting of the second half or the D-stem and the acceptor stem. Split sites for guide 1 (target-short) and guide 2 (target-long) RNAs are depicted. (B) For enzyme activity assays, PaThg1 was incubated as indicated with guide, target or non-substrate (Hammerhead (HH) ribozyme) RNA and [α-32P]-GTP. Successful nucleotide addition results in a radiolabelled product as visualized by denaturing gel electrophoresis and subsequent phosphorimaging. No radiolabelled products for target RNAs were observed. Guide RNA without target RNAs are potent substrates for PaThg1, and yield two products for guide RNAs (boxed). (C) Alignment of full-length E. coli tRNAHisΔG−1, guide 1 and 2 RNAs, target-long, target-short and target-random. (D) Alignment of full-length E. coli tRNAHisΔG−1, guide 1 and sequences of repaired guide 1 RNA. The reaction product of guide-1 RNA, GTP, ATP, CTP, UTP and [α-32P]-GTP with PaThg1 was extracted from a denaturing polyacrylamide gel, circularized, amplified with primers binding to the anticodon loop, and ligated into Topo-TA for sequencing. Added bases are boxed and aligned to E. coli tRNAHisΔG−1. Dashes indicated bases matching E. coli tRNAHis.
We designed two different guide RNAs based on E. coli tRNAHisΔG−1, which encode sequences starting from the 3’ half of the D-stem to the 3’CCA-end of the tRNA (guide 1) and from the D-loop to the 3’CCA-end (guide 2) (Fig. 4A). The target RNAs encode sequence elements of the 5’ acceptor stem towards the 5’ D-stem (target-short), and additionally the D- loop sequence (target-long), or a scrambled sequence in place of the D-loop (target-random). The guide/target system is visualized in Fig. 4A, with sequence details given in Fig. 4C. To test whether Thg1 can be directed to larger range of RNA substrates and circumvent the requirement of a tRNA like structure with the guide/target system, we incubated PaThg1 with various guide/target combinations.
In these assays, radiolabelled GTP and unlabeled GTP, ATP, UTP and CTP were added to a final concentration of 0.1 mM. As a control, we assayed Hammerhead ribozyme RNA, which does form a tRNA like-structure, but instead features a stem-loop structure. PaThg1 adds a single nucleotide to the guide RNAs and, interestingly, also to the hammerhead ribozyme RNAs in the presence or absence of target RNAs. We found no evidence for nucleotide addition to the target RNAs in any of our experiments (Fig. 4B), suggesting that the guide/target system is not applicable for redirecting Thg1 activity. It is interesting to note that the hammerhead ribozyme stem-loop structure is a sufficient substrate for guanylation, while the relatively short target sequences are insufficient to be recognized as a substrate on their own (Fig. 4).
In addition to a reaction product stemming from likely a single guanylation addition, a second extended product formation was observed for all guide RNAs, but not the hammerhead ribozyme RNA in the presence or absence of the target RNAs. To analyze the nature of the extended reaction product, we carried out a large-scale reaction with guide 1 substrate, all four nucleotides and [α-32P]-GTP. We excised the extended reaction product from the gel, circularized and reverse transcribed the RNA products, which were subsequently sequenced. Representative sequencing results were aligned to the wild type E. coli tRNAHis sequence (Fig. 4D). In this Fig., dashed lines represent nucleotides that are identical to the sequence of E. coli tRNAHisΔG−1 and alterations are denoted according to the sequencing results. Of the 25 sequenced reaction products, 12 of the retrieved sequences aligned to the guide RNA with no additional nucleotides. Eight sequences encoded an additional C, which represents a single nucleotide repair. The remaining 5 sequences are extended reaction products, with various lengths and accuracy of near perfect repair. At least one sequence (denoted with a star, Fig. 4D) would be a suitable substrate for HisRS, indicating a repair efficiency of at least 4%, even though the low sample number and assay conditions may not be representative of events occurring in the cell.
Thg1 sequence determinants for extended reverse polymerization
While most Thg1 homologs function in vivo to specifically add one guanylate nucleotide to pre-tRNAHis, some enzymes display additional template dependent reverse polymerization activity and add several nucleotides to a truncated tRNA 5’-end.16 These homologs all belong to the archaeal-type Thg1 subgroup, which differs in substrate and cofactor requirements from their eukaryotic counterparts.1,20 Not all archaeal-type Thg1 enzymes display this ability.1,3,15,17 A recent study suggested that the final step of guanylation, the 5’ pyrophosphate removal of the added nucleotide, prevents extended polymerization in yeast Thg1.18 Based on this data from the yeast Thg1, we used the related Candida albicans Thg1 (CaThg1) to identify residues involved in extended reverse polymerization. We thus identified and mutated amino acid residues in the vicinity of the active site that we hypothesized may be involved in catalyzing pyrophosphate removal (Fig. 1A) and that were not mutated in previous studies.6,18,23
We produced and purified recombinant CaThg1 mutants K10A, K31A, G32A, Y78A, R92A, E178A and T184A (Fig. 5A). All of the mutants were enzymatically active and added nucleotides to a yeast tRNA substrate. Mutants K31A, R92A, G32A, and E178A produced a product between 70–80 nts, indicating single nucleotide addition (Fig. 5B). The R92A mutant was notably more active than any other mutant or the wildtype enzyme. Mutants Y78A, T184A, E178A and K10A produced a product stemming from an extended polymerization reaction that does not appear as a reaction product of wildtype CaThg1, or mutants K31A, R92A or G32A. These amino acid residues are not conserved between species, and likely play a general role in nucleotide and RNA positioning. Pyrophosphate removal has been noted as an inefficient process,18 and alterations in nucleotide and substrate positioning may already be sufficient to further lower the elimination process and allow for an extended polymerization.
Figure 5.
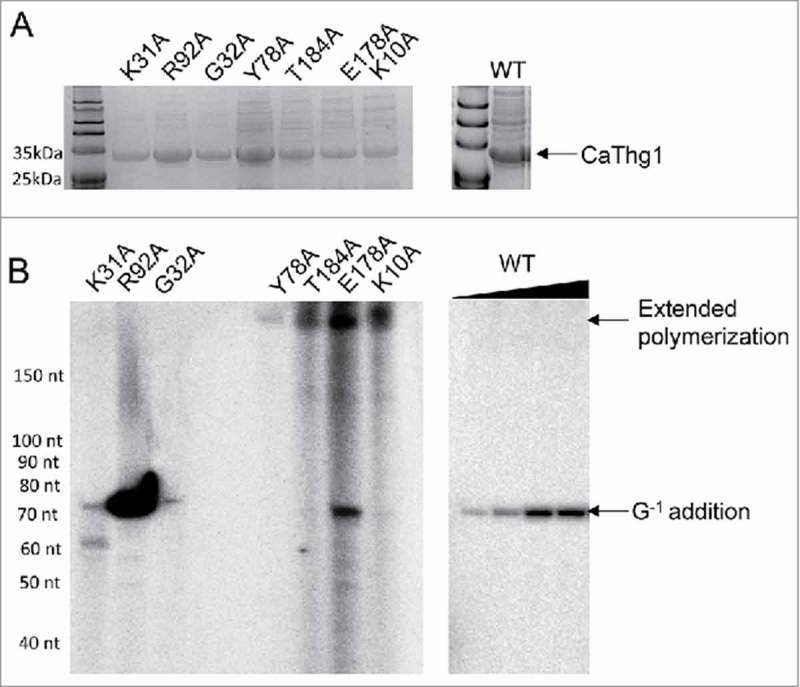
C. albicans Thg1 (CaThg1) mutants are capable of extended reverse polymerization. (A) SDS Page of CaThg1 mutants purified via His-tag chromatography. (B) Enzyme activity assay of CaThg1 mutants. Wildtype CaThg1 and mutants were incubated as indicated with S. cerevisiae tRNAHisΔG−1 and [α-32P]-GTP. Successful nucleotide addition results in a radiolabelled product as visualized by denaturing gel electrophoresis and subsequent phosphorimaging. Mutants Y78A, T184A, E178A and K10A exhibited extended reverse polymerization activity.
Discussion
I. hospitalis Thg1 is active despite lacking conserved RNA recognition motifs
In most archaea and bacteria, RNase P displays an altered cleavage pattern specific to tRNAHis and leaves the genome encoded G−1 residue intact.1,3,20 The hyperthermophilic archaeon I. hospitalis encodes the G−1 residue of tRNAHis in its genome,24 so it is possible that RNase P retains the genome encoded HisRS identity element upon 5’-end tRNA processing. I. hospitalis encodes a minimized Thg1 homolog, yet potentially does not require Thg1 activity to maintain tRNAHis identity.24 The full length IhThg1 (162 aa, 18 kDA) is roughly 40% shorter than CaThg1 (268 aa, 32.5 kDa, 30.6% identity in 85 residues overlap) and 25% shorter than PaThg1 (212 aa, 24 kDa, 35% identity in 169 residues overlap). Though small in size, most of the palm domain, including the catalytic carboxylates (Fig. 1B, red boxes), is conserved in the minimal IhThg1 homolog.
Despite its small size, we here show that IhThg1 is enzymatically active, and efficiently guanylates a tRNAHisΔG−1 substrate. Although I. hospitalis is a hyperthermophile, IhThg1 was not active at 65°C. It is possible that specific chaperones found only in I. hospitalis are required for proper folding of IhThg1.25 Similar to its archaeal homologs MaThg1 and PaThg1, IhThg1 is ATP independent and prefers an E. coli tRNA substrate with a C73 discriminator base over S. cerevisiae tRNAHis with a A73 discriminator base (Fig. 2).1,3,7,15,20 Interestingly, IhThg1 added all four nucleotides to E. coli tRNAHisΔG−1, indicating that the nucleotide addition does not depend on Watson-Crick base-pairing with the discriminator base (C73) (Fig. 2D). The relaxed nucleotide specificity suggests that IhThg1 assumes a broader function than tRNAHis guanylation in vivo. Even though it remains to be determined which nucleotide is the preferred substrate in the cell, an elegant series of studies of several Dictyostelium discoideum Thg1 homologs has shown that Thg1-like enzymes can assume roles in tRNA repair and polymerization activities beyond tRNAHisΔG−1 guanylation,12,14,16,26 and this may be the case for IhThg1 as well.
It is evident from the CaThg1 crystal structure in complex with tRNA that amino acids K10, N202, and K209 read the base G36 of the tRNA, which is the third anticodon base.6 IhThg1 lacks several motifs required for anticodon recognition (Fig. 6A), including the amino acids required for G36 coordination. IhThg1 is thus likely unable to discriminate the GUG anticodon from GUN, which would allow guanylation of additional tRNAs, including tRNAHis GUG, tRNATyr GUA, tRNAAsn GUU, and tRNAAsp GUC. Because the addition of G−1 is sufficient to convert any tRNA into a substrate for HisRS,27 this Thg1 activity may lead to mistranslation. A tRNATyr containing the G−1 residue will be converted to His-tRNATyr, and can lead to mistranslation by incorporating His at designated Tyr codons throughout the proteome. Indeed, not only is there precedent for this type of mistranslation, but it can even have a selective advantage. A recent study showed that the closely related hyperthermophile archaeon Aeropyrum pernix adapts to low temperatures by inducing mistranslation by temperature-dependent mis-charging of tRNALeu with methionine.28
Figure 6.
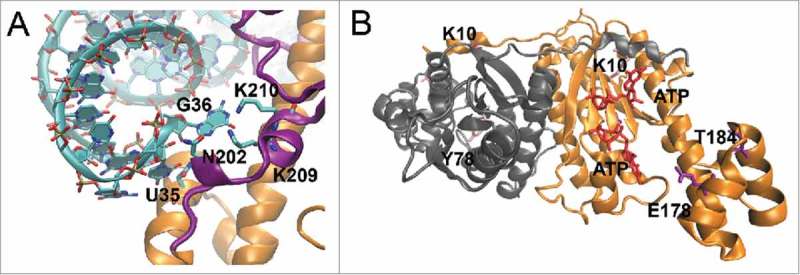
Mutational analysis of Thg1 enzymes. (A) I. hospitalis Thg1 lacks homologs to C. albicans (CaThg1) amino acid residues N202, K209 and K210 that read base G36 of the tRNAHis substrate6. (B) P. aerophilum Thg1 mutants K10 and E178 are involved in coordination of the incoming GTP nucleotide6.
Taking into consideration that Thg1 is likely not required to maintain tRNAHis identity in I. hospitalis, and displays relaxed nucleotide specificity, IhThg1 may assume alternate functions in I. hospitalis related to tRNA repair and potentially adaptation to changes in the environment.
The palm domain of Thg1 is sufficient for catalytic activity
Both DNA and RNA polymerases, as well as nucleotidyltransferases share the catalytic palm domain.1,3 Considering that IhThg1 lacks major parts of the protein and remains catalytically active, we investigated whether the fingers domain is required for catalytic activity. We purified and assayed the palm domains of P. aerophilum and M. acetivorans Thg1 in the absence of the fingers domain, and found that the truncated proteins are catalytically active in vitro. MaThg1 was initially characterized as a split protein that consisted of two protein halves separated by a UAG stop codon followed by a short, theoretically noncoding, DNA sequence.2 The predicted split occurs between the palm and finger domains, but in vivo MaThg1 is expressed as a full-length protein with UAG decoded as pyrrolysine.2 We previously showed that the split protein halves could be reassembled to form a functional and enzymatically active Thg1, yet the palm domain of MaThg1 showed no significant activity on its own.2 Due to instability of the protein construct, we were previously unsuccessful in obtaining active MaThg1 palm domain. However, we can now report significant nucleotidyltransferase activity by the MaThg1 palm domain alone (Fig. 3). This activity was only observable upon assaying the protein immediately after purification. Following prolonged storage, the MaThg1 palm domain degrades and rapidly loses activity. The data clearly show that the Thg1 palm domain is sufficient for guanylation activity. The full length Thg1, including the fingers domain, is likely required for stability and substrate recognition in vivo.
P. aerophilum Thg1 repairs truncated tRNA substrates
Thg1 homologs, given their unique catalytic activity, are ideal targets for engineering novel methods to label RNA molecules. A major limitation in the application of Thg1 to 5’ labeling of RNA molecules is its substrate specificity to tRNAHis. Thg1 has been shown to specifically recognize the tRNAHis anticodon.10 Developing a method that can extend Thg1s substrate range to non-tRNA molecules would make the enzyme useful in RNA labeling applications. While the 5’end of mRNAs is defined by the transcription start site, the 3’ end is less unique, and often indistinguishable between mRNAs due to the presence of the poly(A) tail. Thg1 could be employed to universally label RNA molecules at the 5’ end at a specific pulse. The 5’-end of RNAs is targetable for sequence specific labeling, as it is unique to the RNA species, compared to the universal poly(A) tail which does not allow for sequence specific labeling. Reverse polymerases could specifically and covalently label a specific RNA, which would be highly useful in diagnostics, but requires a protein that can be directed towards a variety of sequences, rather than being restricted to tRNAHis. An engineered Thg1 system that can be directed towards a variety of substrates would thus be highly useful and add to the current toolbox of RNA and DNA manipulating enzymes.29 We are currently engineering Thg1 enzymes to use a guide RNA-based system to target specific cellular RNAs for 5’-end labeling. The guide based technology has previously found application in gene silencing therapies, utilizing a tRNA-like structure to direct RNAses towards a non-tRNA substrate.21,22
The experiments presented here represent our initial efforts to adapt Thg1 to function with a guide RNA target system. Strikingly, while we found no evidence that Thg1 utilized the provided guide sequence to guanylate a target RNA, we instead discovered an unexpected template dependent tRNA repair activity in the wild type Thg1 (Fig. 4). We expected to observe RNA labeling of the shorter target RNAs (13-22 nucleotides), and no end labeling of the guide RNAs (55 and 63 nucleotides, respectively). Much to our surprise, we found no evidence that the target RNAs were labelled, but instead observed a reaction product corresponding to a singly labelled guide RNA. Additionally, we observed an extended reaction product indicating multiple nucleotide additions to the guide RNA. These reaction products were found in the presence and absence of the target RNAs. The sequencing results of the extended reaction product confirmed that Thg1 repairs the guide RNA, adding the missing nucleotides at the 5’ end of the truncated tRNA substrate. With only a tRNA fragment as substrate, Thg1 produces the full length tRNA in a template dependent fashion. In addition, Thg1 produced nearly correct as well as incomplete and non-templated products. In many cases, only few nucleotides were added in a template dependent manner, but we found evidence that templated RNA polymerization was extended up to the acceptor stem, adding up to 13 nucleotides in an almost error free manner. All four nucleotides were provided in the reaction and were catalytically added to the truncated tRNA substrate by PaThg1.
Out of 25 sampled sequences, one repaired tRNA is almost error free and suitable as a HisRS substrate, indicating that PaThg1 is capable of a catalyzing a template dependent tRNA repair. A similar tRNA repair and editing mechanism has been found in Thg1 homologs from Dictyostelium discoideum12,14,16,26, Acanthamoeba castellanii,14 M. acetivorans and Bacillus thuringiensis.15 In these cases tRNA repair was restricted to a few nucleotide additions, while we report a novel extended repair function, in which Thg1 is able to reverse polymerize a 13 nucleotide segment spanning parts of the D-loop and acceptor stem. These data point to an even more elaborate repair activity of Thg1-like proteins than previously assumed. It is interesting to note that we also observed radiolabelled product in reactions containing the hammerhead ribozyme RNA as a substrate, in what appears to be a single nucleotide addition reaction. In contrast to eukaryotic and many bacterial and archaeal Thg1 enzymes,1 PaThg1 appears to be capable of adding nucleotides to substrates other than tRNAHis and can be denoted as a low fidelity, template dependent reverse polymerase.
Engineering extended reverse polymerization
Even though extended reverse polymerization has been shown for a few Thg1 homologs, the eukaryotic Thg1 enzymes are restricted to the addition of a single guanylate residue on tRNAHis. 1 A previous study showed that removal of the 5’pyrophosphate after tRNAHis guanylation is an essential step in limiting reverse polymerization to a single nucleotide.18 We identified new residues involved in this final step of eukaryotic Thg1 activity.
Residues from CaThg1 were chosen based on their vicinity to the active site and tRNA binding regions. Mutation of CaThg1 residues K10, Y78, E178 and T184 lead to an extended polymerization reaction (Fig. 5B), with E178A exhibiting residual single guanylation activity. Residue T184 is involved in the coordination of the G+1 residue of the tRNA substrate.6 Residues K10 and E178 are in close proximity to the GTP binding site. Y78 is likely involved in coordinating the activating ATP as it is located adjacent to the catalytic carboxylates D76 and E77 (Fig. 6B). A previous study showed that pyrophosphate removal in yeast Thg1 is slow, and only effective when the guanylate is added in a Watson-Crick independent manner.18 In a reaction allowing for Watson-Crick base-pairing pyrophosphate removal is too inefficient to prevent extended polymerization. In this study, we employed S. cerevisiae tRNAHisΔG−1 to determine enzymatic activity of the CaThg1 wild type and mutant proteins, which encodes an A73 discriminator base. Consequently, the first guanylation reaction is a non-Watson-Crick base pairing reaction, whereas further polymerization can occur in a templated reaction opposite to the CC nucleotides of the 3’CCA-end of the tRNA. CaThg1 mutants produced an extended reaction product of ∼50-100 nucleotides in our reactions, which appeared to be an extended, untemplated polymerization reaction.
Taking into account that pyrophosphate removal is slow,18 a disturbance of tRNA or nucleotide substrate coordination may further reduce efficiency of the elimination reaction and allow for an efficient RNA polymerization. Interestingly, two of the mutants displaying extended polymerization are mutations of amino acids in the palm domain (K10, Y78), and two are located in the fingers domain (E178, T184). None of these residues are well conserved between Thg1 enzymes (Fig. 1B), indicating that they likely play a general role in RNA and nucleotide positioning.
Based on its proximity to the active site, K10 may play a role in catalysis. Pyrophosphate removal is thought to occur by nucleophilic water attacking the phosphodiester bond, releasing pyrophosphate and yielding a mono-phosphorylated 5’-tRNA end. K10 can potentially provide a base, which could deprotonate a nucleophilic water, which in turn attacks the phosphodiester bond and result in removal of the pyrophosphate. While the exact mechanism remains to be elucidated, the mutant Thg1 extended polymerization activity may be the result of an inefficient or ablated pyrophosphate removal activity.18 These mutants also represent prototypes in our on-going efforts to engineering Thg1 into a processive and perhaps high fidelity reverse polymerase.
Conclusion
We found that the conserved polymerase palm domain in Thg1 is sufficient for nucleotidyltransferase activity, and further that single point mutations of amino acid residues in either palm or fingers domain are sufficient to promote extended polymerization activity. We identified PaThg1 as an enzyme capable of a template dependent extended tRNA repair activity. Taken together, these findings clearly show that Thg1 activity can be evolved towards enhanced processivity and a broader substrate range. Our mutagenesis efforts provide a framework for future engineering of a versatile reverse polymerase. A reverse polymerase has clear biotechnological and biological applications. These functions include 3’-5’ sequencing, promoter identification, and sequence specific and sequence unspecific 5’ RNA labeling. Additional engineering efforts will include extending the Thg1 substrate range to include DNA and modified nucleotides, which will be highly valuable in research and diagnostic applications.
Materials and methods
Plasmids and protein purification
P. aerophilum (PaThg1), M. acetivorans Thg1 (MaThg1) truncated MaThg1 (MaPalm and MaFingers) and C. albicans Thg1 (CaThg1) plasmid constructs and purification were described previously.2,3,6 I. hospitalis Thg1 (IhThg1) was ordered as a codon optimized gene (Genewiz) for expression in Escherichia coli and cloned into pGEX-6P-2, which was modified to include a –terminal His-tag using XhoI and EcoRI restriction sites. A His-tag was added by PCR between GST and IhThg1, and the construct was transformed into E. coli BL21 Codon Plus cells. Protein production was induced at OD600 = 0.6 and cells were grown at 16 °C for 18 hours. Cells were harvested, and broken by sonication. Since most of the produced protein was found to be insoluble, protein was extracted from the insoluble fraction. The cell pellet from 2 L cells was suspended in 10 mL Thg1 buffer2,3 containing 7M hydroxyurea. The solution was incubated for 1 hour, centrifuged, and the protein extract loaded onto a Ni-NTA column. The column was washed with 5 column volumes of Thg1 buffer with 7M urea. Proteins were eluted with Thg1 buffer containing 7M hydroxyurea and 250 mM imidazole. Elution fractions were pooled and dialyzed twice against 100x volume Thg1 buffer without urea to allow protein refolding. Mutants of CaThg1 were generated using Quickchange Site directed mutagenesis kit (Stratagene). Truncated PaThg1 PaPalm (amino acids 1 to 150) was cloned into pet20b using XhoI and EcoRI restriction sites. Purification of truncated constructs was carried out as described for other Thg1 enzymes2,3,6 and proteins were assayed the same day to prevent degradation.
tRNA in vitro transcription
tRNA constructs and in vitro transcription of P. aerophilum, S. cerevisiae and E. coli tRNAHisΔG−1 were described previously.2,3,6 Sequences for guide RNA expression were cloned into puc19 as a T7 runoff construct. E. coli tRNAHis sequence was used as a base to design guide and target sequences. Guide RNAs sequences are EcHisTguide1 (63 nt) 5’-AGTTGGTAGAGCCCTGGATTGTGATTCCAGTTGtCGTGGGTTCGAATCCCATTAGCCACCCCA-3’ and EcHisTguide2 (55 nt) 5’-GAGCCCTGGATTGTGATTCCAGTTGtCGTGGGTTCGAATCCCATTAGCCACCCCA-3’. Target sequences were ordered as 5’-monophosphorylated RNAs from Sigma Aldrich. Target-long, 5’-GTGGCTATAGCTCAGTTGGTA-3’; Target-short, 5’-GTGGCTATAGCTC-3’; Target-random, 5’-GTGGCTATAGCTCTGCATCG-3’. In vitro transcribed RNAs were gel purified and refolded by heating and adding MgCl2 to a final concentration of 5 mM.
Enzyme activity assays
Thg1 activity assays were carried out as described previously.2,3,6 Briefly, Thg1 enzymes were incubated with tRNAHis substrates lacking the G−1 residue, and [α-32P]-GTP. In some cases, the tRNA was 3’-end labelled using CCA-adding enzyme and [γ-32P]-ATP, as described previously.
Circularization/guide Target system
PaThg1 was incubated with the RNA substrates Guide 1, GTP, ATP, CTP, UTP, and [α-32P]-GTP for 1 hour. Reactions were separated by polyacrylaminde gel electrophoresis and the extended product was eluted from the RNA gel as carried out for in vitro transcribed RNAs.2,3,6 RNAs were circularized with T4 RNA Ligase (New England Biolabs, Cat No M0204S), and the 5’-3’ ligation was reverse transcribed and amplified with primers binding to the tRNA anticodon 5’-TGTGATTCCAGTTGTCGTGGGTTCGAATCCC-3’ and 5’-TCACAATCCAGGGCTCTACCAACTGAGC-3’. The resulting product was ligated into pCR2.1®-TOPO® TA (ThermoFisher Cat No 451641) for sequencing.
Disclosure statement
The authors report no conflict of interest.
Acknowledgments
We thank Lauren Seidl for technical help, and Brian Shilton, Murray Junop and Patrick O'Donoghue for helpful discussions and encouragement. This work was supported by the Natural Sciences and Engineering Research Council of Canada under grant RGPIN 04776-2014 and the J.P. Bickell Foundation.
Funding details
This work was supported by the Natural Sciences and Engineering Research Council of Canada under grant RGPIN 04776-2014 and the J.P. Bickell Foundation.
References
- 1.Heinemann IU, Nakamura A, O'Donoghue P, Eiler D, Söll D. tRNAHis-guanylyltransferase establishes tRNAHis identity. Nucleic Acids Res. 2012;40:333–44. doi: 10.1093/nar/gkr696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heinemann IU, O'Donoghue P, Madinger C, Benner J, Randau L, Noren CJ, Söll D. The appearance of pyrrolysine in tRNAHis guanylyltransferase by neutral evolution. Proc Natl Acad Sci U S A. 2009;106:21103–8. doi: 10.1073/pnas.0912072106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heinemann IU, Randau L, Tomko RJ Jr., Söll D. 3'-5' tRNAHis guanylyltransferase in bacteria. FEBS Lett. 2010;584:3567–72. doi: 10.1016/j.febslet.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hyde SJ, Eckenroth BE, Smith BA, Eberley WA, Heintz NH, Jackman JE, Doublie S. tRNAHis guanylyltransferase (THG1), a unique 3'-5' nucleotidyl transferase, shares unexpected structural homology with canonical 5'-3' DNA polymerases. Proc Natl Acad Sci U S A. 2010;107:20305–10. doi: 10.1073/pnas.1010436107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee K, Lee EH, Son J, Hwang KY. Crystal structure of tRNAHis guanylyltransferase from Saccharomyces cerevisiae. Biochem Biophys Res Commun. 2017. doi: 10.1016/j.bbrc.2017.06.054. [DOI] [PubMed] [Google Scholar]
- 6.Nakamura A, Nemoto T, Heinemann IU, Yamashita K, Sonoda T, Komoda K, Tanaka I, Söll D, Yao M. Structural basis of reverse nucleotide polymerization. Proc Natl Acad Sci USA. 2013;110:20970–5. doi: 10.1073/pnas.1321312111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hyde SJ, Rao BS, Eckenroth BE, Jackman JE, Doublie S. Structural studies of a bacterial tRNAHIS guanylyltransferase (Thg1)-like protein, with nucleotide in the activation and nucleotidyl transfer sites. PLoS One. 2013;8:e67465. doi: 10.1371/journal.pone.0067465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heinemann IU, Söll D, Randau L. Transfer RNA processing in archaea: unusual pathways and enzymes. FEBS Lett. 2010;584:303–9. doi: 10.1016/j.febslet.2009.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gu W, Jackman JE, Lohan AJ, Gray MW, Phizicky EM. tRNAHis maturation: an essential yeast protein catalyzes addition of a guanine nucleotide to the 5' end of tRNAHis. Genes Dev. 2003;17:2889–901. doi: 10.1101/gad.1148603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackman JE, Phizicky EM. tRNAHis guanylyltransferase adds G-1 to the 5' end of tRNAHis by recognition of the anticodon, one of several features unexpectedly shared with tRNA synthetases. RNA. 2006;12:1007–14. doi: 10.1261/rna.54706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Francklyn C, Schimmel P. Enzymatic aminoacylation of an eight-base-pair microhelix with histidine. Proc Natl Acad Sci U S A 1990;87:8655–9. doi: 10.1073/pnas.87.21.8655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Long Y, Abad MG, Olson ED, Carrillo EY, Jackman JE. Identification of distinct biological functions for four 3'-5' RNA polymerases. Nucleic Acids Res. 2016;44:8395–406. doi: 10.1093/nar/gkw681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kimura S, Suzuki T, Chen M, Kato K, Yu J, Nakamura A, Tanaka I, Yao M. Template-dependent nucleotide addition in the reverse (3'-5') direction by Thg1-like protein. Sci Adv. 2016;2:e1501397. doi: 10.1126/sciadv.1501397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Long Y, Jackman JE. In vitro substrate specificities of 3'-5' polymerases correlate with biological outcomes of tRNA 5'-editing reactions. FEBS Lett. 2015;589:2124–30. doi: 10.1016/j.febslet.2015.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rao BS, Maris EL, Jackman JE. tRNA 5'-end repair activities of tRNAHis guanylyltransferase (Thg1)-like proteins from Bacteria and Archaea. Nucleic Acids Res. 2011;39:1833–42. doi: 10.1093/nar/gkq976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abad MG, Long Y, Willcox A, Gott JM, Gray MW, Jackman JE. A role for tRNAHis guanylyltransferase (Thg1)-like proteins from Dictyostelium discoideum in mitochondrial 5'-tRNA editing. RNA. 2011;17:613–23. doi: 10.1261/rna.2517111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abad MG, Rao BS, Jackman JE. Template-dependent 3'-5' nucleotide addition is a shared feature of tRNAHis guanylyltransferase enzymes from multiple domains of life. Proc Natl Acad Sci U S A. 2010;107:674–9. doi: 10.1073/pnas.0910961107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith BA, Jackman JE. Saccharomyces cerevisiae Thg1 Uses 5'-Pyrophosphate Removal To Control Addition of Nucleotides to tRNAHis. Biochemistry. 2014;53:1380–91. doi: 10.1021/bi4014648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith BA, Jackman JE. Kinetic analysis of 3'-5' nucleotide addition catalyzed by eukaryotic tRNAHis guanylyltransferase. Biochemistry. 2012;51:453–65. doi: 10.1021/bi201397f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jackman JE, Gott JM, Gray MW. Doing it in reverse: 3'-to-5' polymerization by the Thg1 superfamily. RNA. 2012;18:886–99. doi: 10.1261/rna.032300.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dreyfus DH, Tompkins SM, Fuleihan R, Ghoda LY. Gene silencing in the therapy of influenza and other respiratory diseases: Targeting to RNase P by use of External Guide Sequences (EGS). Biologics: targets & therapy. 2007;1:425–32. [PMC free article] [PubMed] [Google Scholar]
- 22.Nakashima A, Takaku H, Shibata HS, Negishi Y, Takagi M, Tamura M, Nashimoto M. Gene silencing by the tRNA maturase tRNase ZL under the direction of small-guide RNA. Gene therapy. 2007;14:78–85. doi: 10.1038/sj.gt.3302841. [DOI] [PubMed] [Google Scholar]
- 23.Jackman JE, Phizicky EM. Identification of critical residues for G-1 addition and substrate recognition by tRNAHis guanylyltransferase. Biochemistry. 2008;47:4817–25. doi: 10.1021/bi702517q. [DOI] [PubMed] [Google Scholar]
- 24.Lowe TM, Chan PP. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016;44:W54–7. doi: 10.1093/nar/gkw413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vieille C, Zeikus GJ. Hyperthermophilic enzymes: sources, uses, and molecular mechanisms for thermostability. Microbiol Mol Biol Rev. 2001;65:1–43. doi: 10.1128/MMBR.65.1.1-43.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abad MG, Long Y, Kinchen RD, Schindel ET, Gray MW, Jackman JE. Mitochondrial tRNA 5'-editing in Dictyostelium discoideum and Polysphondylium pallidum. J Biol Chem. 2014;289:15155–65. doi: 10.1074/jbc.M114.561514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Connolly SA, Rosen AE, Musier-Forsyth K, Francklyn CS. G-1:C73 recognition by an arginine cluster in the active site of Escherichia coli histidyl-tRNA synthetase. Biochemistry. 2004;43:962–9. doi: 10.1021/bi035708f. [DOI] [PubMed] [Google Scholar]
- 28.Schwartz MH, Pan T. Temperature dependent mistranslation in a hyperthermophile adapts proteins to lower temperatures. Nucleic Acids Res. 2016;44:294–303. doi: 10.1093/nar/gkv1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pauff S, Withers JM, McKean IJ, Mackay SP, Burley GA. Synthetic biological approaches for RNA labelling and imaging: design principles and future opportunities. Curr Opin Biotechnol. 2017;48:153–8. doi: 10.1016/j.copbio.2017.04.003. [DOI] [PubMed] [Google Scholar]
- 30.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]