

ABSTRACT
The macroautophagic/autophagic machinery cannot only target cell-endogenous components but also intracellular pathogenic bacteria such as Listeria monocytogenes. Listeria are targeted both by canonical autophagy and by a noncanonical form of autophagy referred to as LC3-associated phagocytosis (LAP). The molecular mechanisms involved and whether these processes contribute to anti-listerial immunity or rather provide Listeria with a replicative niche for persistent infection, however, remained unknown. Recently, using an in vivo mouse infection model, we have been able to demonstrate that Listeria in tissue macrophages are targeted exclusively by LAP. Furthermore, our data show that LAP is required for killing of Listeria by macrophages and thereby contributes to anti-listerial immunity of mice, whereas canonical autophagy is completely dispensable. Moreover, we have elucidated the molecular mechanisms that trigger LAP of Listeria and identified the integrin ITGAM-ITGB2/Mac-1/CR3/integrin αMß2 as the receptor that initiates LAP in response to Listeria infection.
KEYWORDS: ß2 integrin Mac-1/CR3, acid sphingomyelinase, LC3-associated phagocytosis, Listeria monocytogenes, lysosome, macrophages, NAPDH oxidase Nox2, noncanonical autophagy, reactive oxygen species
Macrophages are white blood cells that are crucial for immunity as they patrol the tissues to detect, phagocytose and kill invading pathogens. Some pathogens such as Listeria, however, specialize in escaping killing by macrophages. To this end, Listeria use their pore-forming toxin, listeriolysin O (LLO), and the phospholipases PlcA and PlcB to destroy the membrane of the phagosome and then colonize the cytosol (Figure 1). Cytosolic Listeria can be targeted by canonical autophagy resulting in killing in autolysosomes. However, virulent Listeria largely avoid this fate by surrounding themselves, in a process mediated by the virulence factor ActA, with a halo of host actin and stalling phagophore assembly via PlcA and PlcB.
Figure 1.
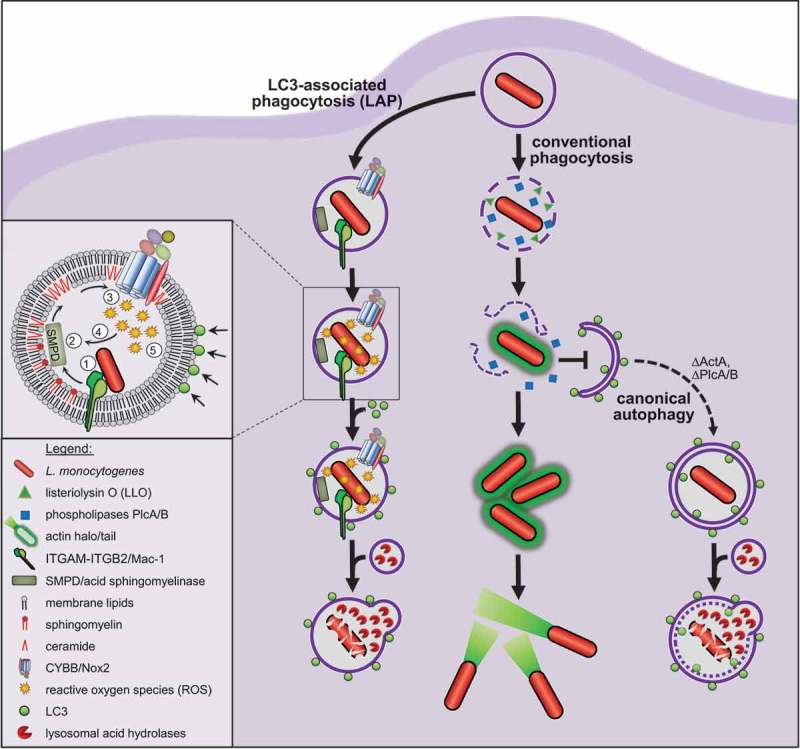
Model for LAP of Listeria. Listeria use their virulence factors LLO and PlcA/B to destroy the membrane of conventional phagosomes before these can fuse with lysosomes, and then colonize the cytosol. Cytosolic Listeria avoid targeting by canonical autophagy by surrounding themselves, in a process mediated by the virulence factor ActA, with a halo of host actin and by stalling phagophore assembly via PlcA/B. Consequently, canonical autophagy does not contribute to anti-listerial activity of macrophages. By contrast, recruitment of LC3 to Listeria-containing phagosomes by LAP markedly enhances anti-listerial activity of macrophages and immunity of mice. LAP of Listeria (1) is initiated by ITGAM-ITGB2 that induces (2) breakdown of the membrane lipid sphingomyelin into ceramide by SMPD. (3) The resulting ceramide-enriched membrane platforms facilitate CYBB assembly and activation. (4) CYBB-derived ROS amplify SMPD activity in a positive feedback loop and (5) induce recruitment of LC3 by LAP. LAP then promotes fusion of the phagosome with lysosomes, which increases exposure of Listeria to bactericidal lysosomal acid hydrolases and thus anti-listerial activity of macrophages.
Indeed, we were recently able to show that canonical autophagy is completely dispensable for killing of Listeria by tissue macrophages in vivo and that Listeria, in fact, are not targeted by canonical autophagy at all [1]. Nonetheless, Listeria are targeted by the autophagic machinery as they colocalize with LC3. Strikingly, this targeting is completely independent of the capability of Listeria to damage the phagosome, as avirulent or even dead Listeria also colocalize with LC3. Therefore, we investigated the molecular mechanisms underlying LC3 recruitment to Listeria in vivo in more detail. LC3 is recruited i) exclusively to Listeria in single-membrane phagosomes, ii) independently of the ULK complex components ULK1/2 and RB1CC1/FIP200 and iii) only upon generation of reactive oxygen species (ROS) by the phagocyte NADPH oxidase CYBB/Nox2. These all are hallmarks of LAP, a noncanonical form of autophagy that results in attachment of LC3 to the single membrane of phagosomes, which requires some components of the autophagic machinery (e.g., ATG5, ATG7 and BECN1/Beclin-1) but not others (e.g., ULK1/2, RB1CC1 and ATG14) and depends on ROS production by CYBB. Thus, our data demonstrate that Listeria in tissue macrophages in vivo are targeted exclusively by LAP, but not canonical autophagy.
We next addressed the question of whether LAP contributes to anti-listerial immunity or, as has been suggested, rather provides Listeria with a replicative niche for persistent infection. Our data show that deficiency for LAP markedly reduces anti-listerial activity of tissue macrophages. Furthermore, deficiency for LAP in macrophages, but not that for canonical autophagy, increases susceptibility of mice to infection with Listeria. Thus, our data show that LAP in vivo substantially contributes to anti-listerial activity of macrophages and immunity of mice.
We also elucidated the molecular mechanisms underlying the anti-listerial function of LAP. LC3-positive Listeria-containing phagosomes fuse more often with lysosomes than conventional LC3-negative phagosomes, and deficiency for LAP markedly impairs fusion of Listeria-containing phagosomes with lysosomes. Consequently, Listeria in LC3-positive phagosomes are exposed to higher levels of lysosomal acid hydrolases than Listeria in conventional phagosomes or in LAP-deficient macrophages. Because lysosomal acid hydrolases are of particular importance for killing of Listeria by macrophages, our data indicate that LAP enhances the anti-listerial activity of macrophages by promoting fusion of Listeria-containing phagosomes with lysosomes.
Concerning the pathway triggering LAP in response to Listeria infection, we were able to show that previously reported LAP-initiating receptors such as Toll-like receptors recognizing pathogen-associated molecular patterns or FCGR/Fcγ receptor recognizing antibody-decorated pathogens are completely dispensable for LAP of Listeria. Furthermore, they also are completely dispensable for activation of the CYBB-mediated ROS production that is a prerequisite for LAP of Listeria. Instead, our data demonstrate that the pathway that initiates LAP of Listeria emanates from ITGAM-ITGB2, a receptor that can recognize a number of different ligands including diverse microbial molecules. Genetic deficiency for ITGAM or antibody-mediated blockade markedly impair infection-induced ROS production by CYBB and LAP. As a connecting link between ITGAM and CYBB activation and subsequent induction of LAP, we identified SMPD/acid sphingomyelinase, a lipid-converting enzyme hydrolyzing the major membrane lipid sphingomyelin into ceramide and phosphorylcholine. In response to Listeria infection, ITGAM induces SMPD activation resulting in an altered phagosomal membrane lipid composition that facilitates assembly and activation of CYBB. CYBB-derived ROS then induce LC3 recruitment to Listeria-containing phagosomes by LAP.
Interestingly, deficiency for ITGAM or SMPD does not completely impair LAP of Listeria indicating that LAP is not exclusively initiated via this pathway and that a, yet to be identified, ITGAM-SMPD-independent pathway of LAP initiation exists.
Taken together, we were able to demonstrate that LAP is an important anti-listerial mechanism of macrophages that contributes to immunity against Listeria infection. On the molecular level, LAP in response to Listeria infection is initiated by ITGAM. ITGAM induces SMPD-mediated membrane rearrangements that allow CYBB activation. CYBB-derived ROS then induce recruitment of LC3 to Listeria-containing phagosomes, which promotes phagosome-lysosome fusion. The resulting increased exposure of Listeria to lysosomal acid hydrolases underlies the anti-listerial function of LAP.
Our work highlights LAP as a particularly bactericidal form of phagocytosis. Nonetheless, only a subpopulation of approximately 20–25% of phagocytosed Listeria is targeted by LAP. Why not all phagocytosed Listeria are targeted by LAP and how macrophages decide which Listeria are to be targeted by LAP and which not, remain open questions. Perspectively, our data indicate that increasing the relative contribution of LAP to phagocytosis may be a promising strategy to enhance immunity to bacterial infection.
Funding Statement
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) [SFP 670].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Gluschko A, Herb M, Wiegmann K, et al. The β 2 Integrin Mac-1 Induces Protective LC3-Associated Phagocytosis of Listeria monocytogenes. Cell Host Microbe. 2018;23:324–337.e5. [DOI] [PubMed] [Google Scholar]