

ABSTRACT
Mitochondrial dynamics is required to adapt the manifold functions of mitochondria to cell needs and regulate their turnover by mitophagy. Actually, only if fragmented, mitochondria are engulfed by phagophores, the precursors to autophagosomes, and subsequently degraded. This process is essential to maintain a correct and healthy number of mitochondria that, otherwise, might be harmful. They, indeed, represent the main source of reactive oxygen species that – according to the mitochondrial free radical theory of aging – can cause aging when chronically overproduced. In a recent study, we demonstrated that S-nitrosylation, the reversible modification of cysteine residues by nitric oxide (NO), hyperactivates mitochondrial fragmentation by targeting DNM1L/Drp1 (dynamin 1-like) at Cys644, but inhibits mitophagy, the concomitant occurrence of these conditions driving cell senescence. We demonstrated that cell senescence, as well as mouse and human aging are characterized by an epigenetically-driven decrease in ADH5/GSNOR (alcohol dehydrogenase 5 [class III], chi polypeptide), suggesting that ADH5 may act as new longevity gene.
KEYWORDS: ADH5, aging, cell senescence, Drp1, GSNOR, mitochondrial dynamics, mitophagy, nitric oxide, S-nitrosylation, Tet1
Aging is a complex and multifactorial process that ineluctably drives the decline of biological systems. Among several hypotheses formulated to provide a molecular rationale explaining why a cell – and the whole organism – gets aged, the mitochondrial free radical theory of aging represents one of the most prevalent and commonly accepted. It states that reactive oxygen species (ROS), which are physiologically, and inevitably, generated as byproducts of cellular respiration, target mitochondria and negatively affect their function. As a result, dysfunctional mitochondria produce ROS at a higher amount; this condition brings about the accumulation of damaged biomolecules and is detrimental to cellular constituents. Although there is a large body of evidence in support to this theory, the real impact of ROS and oxidative stress in cell senescence and aging remains controversial. Moreover, it is still largely debated which are the molecular players modulating mitochondrial (dys)function and, in turn, affecting cell senescence. In this context, autophagy, and in particular mitophagy, has been proposed as being beneficial and acting as an anti-aging process by removing damaged mitochondria and limiting cellular worsening. However, no mechanisms aimed at sustaining the physiological turnover of mitochondria by mitophagy have been so far identified to be linked to longevity.
In our work, we found out that S-nitrosylation increases the rate of mitochondrial fragmentation and inhibits mitophagy, this being tightly associated with an aging phenotype [1]. We performed the experiments in a transgenic mouse model, in which the expression of the main denitrosylase, ADH5, is suppressed, and the resulting basal level of S-nitrosylation coherently increases. To date, this represents the most physiologically relevant model of S-nitrosylation. The choice to use ADH5-deficient (adh5−/-) mice had been also made on the basis of a constellation of previous evidence arguing for a number of changes that typified aging in these mice, such as: i) leucopenia and reduced immune response; ii) increased morbidity upon endotoxic shock; iii) DNA damage repair defects; iv) predilection to cancer; v) insulin resistance; vi) skeletal muscle wasting. In our study, we also observed that young adh5−/- mice exhibit extensive accumulation of SNCA/α-synuclein- and ubiquitin-positive protein aggregates in the brain cortex, which are associated with locomotor deficits, both phenomena being correlated to premature neurodegeneration.
We then moved to cells and evaluated whether and how ADH5-deficiency could be causative of senescence. We focused on primary cortical neurons (PCNs), which are post-mitotic cells used as model of aging in vitro, and mouse embryonic fibroblasts (MEFs), commonly accepted as model of replicative senescence. In both cases, we observed that adh5−/- cells show signs of senescence, (e.g., positivity to GLB1/β-gal staining and nuclear size increase) earlier than the wild-type counterparts. Concomitantly, they exhibit depolarized and dysfunctional mitochondria, which is consistent with what has already been reported about the detrimental effects of aberrant S-nitrosylation on mitochondria. As a matter of fact, inhibition of NOS (nitric oxide synthase) activity with L-NG-nitroarginine methyl ester (L-NAME) – carried out to prevent S-nitrosylation – totally restores mitochondrial and senescent phenotypes, unambiguously indicating that S-nitrosylation-denitrosylation dynamics link mitochondrial function to aging.
Moreover, detailed analyses of mitochondrial morphology, performed by confocal microscopy coupled to 3D rendering, indicate that ADH5-deficiency deeply affects organelle size and shape. In particular, adh5−/- MEFs and PCNs show a highly fragmented mitochondrial network associated with accumulation of the S-nitrosylated form of both DNM1L and PARK2/parkin. The former is a small GTPase involved in mitochondrial fragmentation reported as being activated by S-nitrosylation at Cys644. The latter is an E3-ubiquitin ligase that participates in mitochondrial targeting for removal by autophagy. At variance with DNM1L, PARK2 activity is repressed upon excessive S-nitrosylation, e.g., upon high (pathological) fluxes of NO. By combining these effects, we observed that mitochondria of adh5−/- cells are highly fragmented but not properly recognized by the autophagic machinery. Electron microscopy images indicated that the majority of adh5−/- cells display mitochondria with severely compromised cristae, which are rarely localized inside autophagosomes. Mitophagy induction by uncoupling compounds produces swelling and disruption of mitochondrial cristae in adh5−/- cells but, again, mitochondria are essentially excluded from autophagic vesicles. Live-imaging fluorescence microscopy analyses confirmed that mitochondria are highly fragmented but not recognized by autophagy. Ectopic expression of the pro-fusion protein OPA1 (OPA1, mitochondrial dynamin like GTPase), or the non-nitrosylable C644A mutant of DNM1L, as well as DNM1L pharmacological inhibition with Mdivi1, restore an elongated mitochondrial phenotype. Similarly, treatments with L-NAME, or the thiol reductant dithiothreitol (that should complement ADH5 denitrosylating activity) reestablish a correct mitophagic flux, thereby reinforcing the hypothesis that adh5−/- cells are deficient in targeting damaged mitochondria for removal. It has been recently reported that PINK1 undergoes inhibitory S-nitrosylation at Cys658, this also resulting in mitophagy impairment. Based on this evidence, we cannot exclude the possibility that additional mechanisms and players, other than PARK2, might be regulated by S-nitrosylation and involved in mitophagy.
The final step of our study was to understand the biological relevance of our results. To this end, we moved to a more physiological context, i.e., wild-type models, in which we could evaluate whether any changes in ADH5 were physiologically associated with aging. In our study, we observed an age-dependent decrease of ADH5 expression in primary cells undergoing senescence, as well as in mice and humans during their life span, which is accompanied by a rise in S-nitrosylated proteins. Going into further detail in the regulatory mechanisms responsible for the age-related modulation of ADH5 expression, we found out that it is epigenetically controlled by cytosine methylation of CpG islands located in the promoter region. The demethylating activity of TET1 (tet methylcytosine dioxygenase 1), a member of 2-oxoglutarate-dependent dioxygenases (already suggested to play a role in aging), is required to keep low the methylation levels, and, in turn, sustain Adh5 transcription. We demonstrated that during cell senescence, or over the life span of mice and humans, both TET1 and ADH5 expression are reduced, whereas, strikingly, they are unaffected in exceptionally long-lived individuals (centenarians).
Altogether, our results indicate that ADH5-dependent denitrosylation significantly contributes to the healthy state of mitochondria and cell longevity (Figure 1). They also point to the role of S-nitrosylation as a novel regulatory posttranslational modification selectively modulating mitophagy (Figure 1), and is thus a potential target for age-related pathophysiological states.
Figure 1.
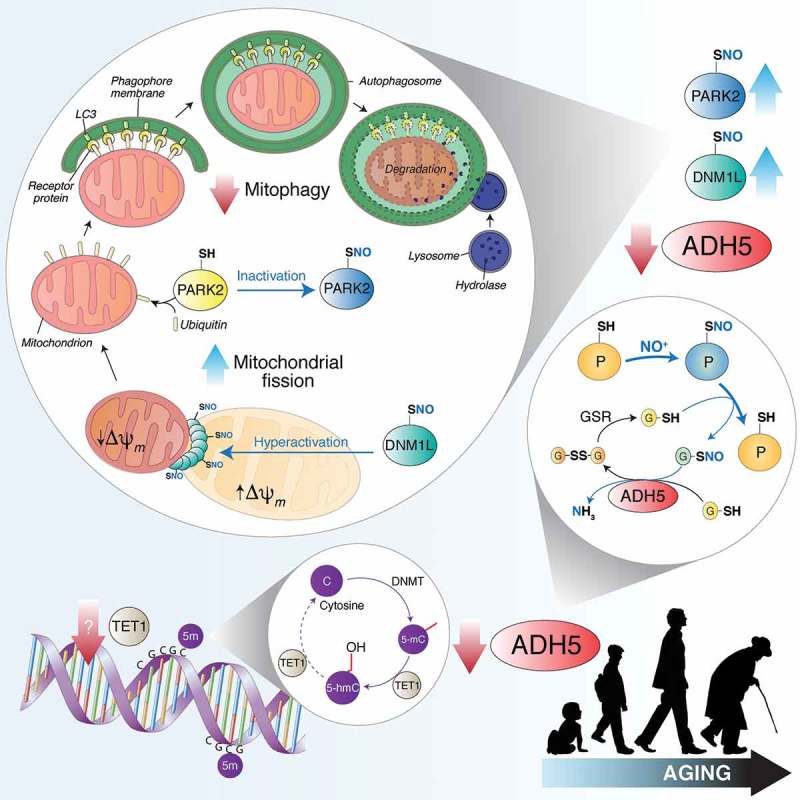
TET1 catalyzes the conversion of 5-methylcytosine (5-mC) into 5-hydroxymethylcytosine (5-hmC), thus counteracting gene silencing due to cytosine (C) methylation by DNMTs (DNA methyltransferases). Age-dependent TET1 decline results in ADH5 downregulation. By reducing S-nitrosoglutathione (GSNO) to glutathione disulfide (GSSG) and ammonia (NH3), ADH5 indirectly controls the amount of S-nitrosylated proteins (PSNOs), generated upon reaction with an NO moiety (in the figure shown as NO+). ADH5 decrease causes the accumulation of PSNOs, i.e., DNM1L and PARK2. DNM1L S-nitrosylation (SNO) stimulates its GTPase activity, resulting in a massive mitochondrial fragmentation. Fragmented mitochondria, characterized by low transmembrane potential (Δψm), are modified by ubiquitination on specific protein receptors by PARK2. S-nitrosylation of PARK2 inhibits its E3 ubiquitin-ligase activity, blocking, in such a way, mitochondria recognition by the autophagic machinery. Other mitophagy-related proteins (i.e., PINK1, not shown in this scheme) are sensitive to S-nitrosylation and could play a role in ADH5-dependent mitophagy control during aging.
Funding Statement
This work has been supported by Danish Cancer Society Grant Kræftens Bekæmpelses Videnskabelige Udvalg (KBVU) Grant [R146-A9414], and Associazione Italiana per la Ricerca sul Cancro (AIRC) Grant [IG20719].
Disclosure statement
No potential conflict of interest was reported by the authors.
Reference
- [1].Rizza S, Cardaci S, Montagna C, et al. S-nitrosylation drives cell senescence and aging in mammals by controlling mitochondrial dynamics and mitophagy. Proc Natl Acad Sci. 2018;115:E3388–E3397. [DOI] [PMC free article] [PubMed] [Google Scholar]