

ABSTRACT
Retrograde transport of tRNAs from the cytoplasm to the nucleus was first described in Saccharomyces cerevisiae and most recently in mammalian systems. Although the function of retrograde transport is not completely clear, it plays a role in the cellular response to changes in nutrient availability. Under low nutrient conditions tRNAs are sent from the cytoplasm to nucleus and presumably remain in storage there until nutrient levels improve. However, in S. cerevisiae tRNA retrograde transport is constitutive and occurs even when nutrient levels are adequate. Constitutive transport is important, at least, for the proper maturation of tRNAPhe, which undergoes cytoplasmic splicing, but requires the action of a nuclear modification enzyme that only acts on a spliced tRNA. A lingering question in retrograde tRNA transport is whether it is relegated to S. cerevisiae and multicellular eukaryotes or alternatively, is a pathway with deeper evolutionary roots. In the early branching eukaryote Trypanosoma brucei, tRNA splicing, like in yeast, occurs in the cytoplasm. In the present report, we have used a combination of cell fractionation and molecular approaches that show the presence of significant amounts of spliced tRNATyr in the nucleus of T. brucei. Notably, the modification enzyme tRNA-guanine transglycosylase (TGT) localizes to the nucleus and, as shown here, is not able to add queuosine (Q) to an intron-containing tRNA. We suggest that retrograde transport is partly the result of the differential intracellular localization of the splicing machinery (cytoplasmic) and a modification enzyme, TGT (nuclear). These findings expand the evolutionary distribution of retrograde transport mechanisms to include early diverging eukaryotes, while highlighting its importance for queuosine biosynthesis.
KEYWORDS: Intron, splicing, tRNA, transport, retrograde, queuosine
Introduction
Following transcription, tRNAs undergo several maturation steps before partaking in protein synthesis. These include trimming of the 5′ and 3′ leader and trailer sequences,1,2 3′-end CCA addition,3 and the acquisition of a number of post-transcriptional nucleotide modifications.4 Some tRNAs also contain introns, which are removed by a specialized tRNA splicing machinery.5 In Eukarya, many tRNA processing events are localized to the nucleus, for example end trimming and some, but not all, modifications; nuclear export may thus serve as an important checkpoint that ensures that only properly processed tRNAs make it to the cytoplasm and engage in translation.
Paradoxically, the intracellular localization of tRNA splicing varies among different organisms, and although in many cases it takes place in the nucleus, in S. cerevisiae all the factors involved in tRNA splicing reside in the cytoplasm.6-8 Furthermore, the localization of maturation components is intricately connected to dedicated mechanisms that export tRNAs from the nucleus.9-11 In S. cerevisiae tRNAs are also imported back to the nucleus from the cytoplasm12-15 by retrograde nuclear transport; a pathway that is generally constitutive. Retrograde nuclear transport is necessary for 1-methylguanosine (m1G) formation at position 37 of tRNAPhe; a required first step in the synthesis of the hypermodified nucleotide wybutosine (yW) in S. cerevisiae.16 Beyond its role in tRNA modification, retrograde transport has also been implicated in cellular responses to nutrient deprivation. Low levels of certain nutrients (e.g., glucose or amino acids) lead to nuclear tRNA accumulation, potentially serving as a protective mechanism against tRNA degradation.12,13,17,18 Upon removal of stress, cells can then reverse tRNA nuclear retention, releasing the withheld tRNA to the cytoplasm presumably contributing to a rapid translational response. Although this process has been observed in S. cerevisiae, rat hepatoma, Chinese hamster ovary cells, and humans, little is known about its occurrence in early divergent eukaryotes and/or its evolutionary conservation.19-21
Reminiscent of S. cerevisiae, cytoplasmic localization of tRNA splicing has been recently described in the single-cell protist Trypanosoma brucei,22 an organism that encodes a single intron-containing tRNA (tRNA tyrosine, tRNATyr GUA).23 While studying modifications that target this tRNA, we observed that the native intron-containing tRNA is devoid of modifications, yet the mature tRNA contains the modified nucleotide queuosine (Q) at the first position of the anticodon (Q34).24 This suggested that this modification only occurs after tRNA splicing. Q modification in Eukarya is catalyzed by the enzyme tRNA-guanine transglycosylase (Fig. 1. eTGT),25,26 which in Xenopus has a strict preference for spliced over intron-containing tRNA.27 In this manuscript, we focus on TbTGT, a paralog of the canonical eukaryotic TGT and show that, unlike other eukaryotic counterparts, it localizes to the nucleus of T. brucei. In line with this observation, we also show substantial amounts of spliced Q-containing tRNA in nuclear fractions, but importantly, like in other eukaryotes, intron-containing tRNATyr is not a substrate for TGT. Taken together these findings lead to a model for a dynamic interplay between localization, splicing and tRNA modification and provides the first example of tRNA nuclear retrograde transport in an early branching eukaryote; a process that may be more evolutionarily widespread than currently thought.
Figure 1.
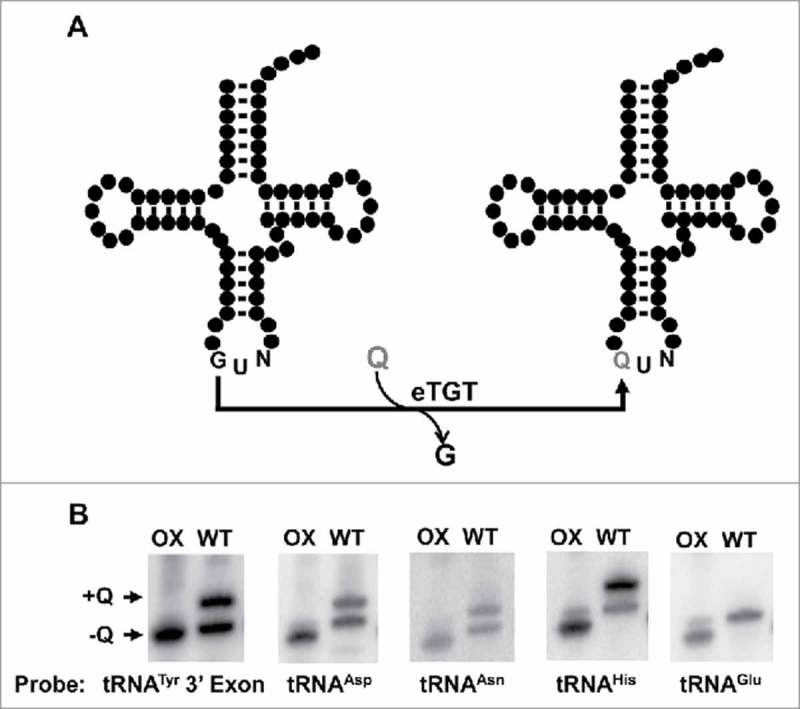
Queuosine-containing tRNAs in T. brucei. A. Model for the proposed queuosine (Q) insertion pathway in eukaryotes. Queuine is taken from an extracellular source (i.e., food or gut microbiome) and inserted into tRNA by the eukaryotic tRNA-guanine transglycosylase (eTGT). B. Total RNA extracted from T. brucei was analyzed for the presence of Q by APB-gel electrophoresis followed by Northern hybridization. Probes corresponding to the four known Q-containing tRNAs in other eukaryotes (tRNAAsp, -Asn,-His and-Tyr) were used to determine the presence (+Q) or absence (-Q) of queuosine in T. brucei. A portion of RNA was oxidized by sodium periodate treatment serving as a negative control (OX); WT refers to the wild type non-oxidized sample. The non-Q-containing tRNAGlu was used as a loading control.
Results
T.brucei encodes a TGT homolog
Queuosine formation in Eukarya is catalyzed by tRNA-guanine transglycosylase (TGT), which exchanges guanine for queuine at position 34 of a sub-set of tRNAs encoded with a G at position 34 (tRNAHis, tRNAAsp, tRNAAsn and tRNATyr) (Fig. 1),28,29 while related modifications (pre-Q0 and pre-Q1) are catalyze by similar enzymes in Archaea and Bacteria respectively.30,31 In general, the sequence U33G34U35 is essential for TGT recognition,32 where the almost universal U33, which forms the U-turn in the anticodon loop in most tRNAs, is part of the recognition motif. It is thought that for activity, TGT requires an unpaired G34 target.26,27 However, eukaryal tRNATyr universally contains an intron, which forms a base pair critical for splicing with G34 in the exon, suggesting that Q formation occurs after splicing. Prior to this work we showed that in T. brucei the tRNA splicing endonuclease and ligase localize to the cytoplasm;22 expectedly if Q formation follows splicing, TGT may also be a cytoplasmic enzyme.
To assess the intracellular distribution of Q, we first determined its presence in total T. brucei RNA. In Eukarya and bacteria Q is found at the first position of the anticodon.28,29 We took advantage of an affinity chromatography system, which exploits the ability of amino-phenyl boronic acid (APB) to bind cis-diols, such as those present in queuosine.33 In these gels, an electrophoretic-mobility shift is observed when RNAs contain Q. This shift is in addition to that caused by the cis-diols naturally occurring at the ends of RNAs (i.e., due to the 3′ and 2′ terminal hydroxyls). We separated total RNA from T. brucei by APB-gel electrophoresis, followed by Northern analysis with radioactive oligonucleotide probes specific for tRNATyr, tRNAAsp, tRNAAsn and tRNAHis. As expected, tRNATyr, −Asp, −Asn and -His displayed a slower migrating band during electrophoresis, indicative of Q (Fig. 1B). As a negative control, separate samples were treated with the oxidizing agent sodium m-periodate prior to electrophoresis. This treatment converts cis-diols into cis-dialdehydes; the latter have no affinity for boronate.33 As an additional control, we also probed for tRNAGlu, which lacks G34 and is not a known substrate for TGT (Fig. 1B). In this case a small shift was observed that is ascribed to that expected from the terminal hydroxyls naturally occurring at the 3′ end of most RNAs, also noticeable when comparing the oxidized control to queuosine-containing tRNAs.
We then used the sequences of the various eukaryotic TGT subunits to search the TriTryp database, revealing the presence of a potential TGT homolog in the T. brucei genome, TbTGT (Tb927.5.3520). To establish the role of TbTGT on Q biosynthesis, a portion of the coding sequence of this protein was cloned into the RNAi vector p2T7-177, transfected into T. brucei 29-13 cells and clonal lines established as described.34 In these cells, RNAi can be induced by addition of tetracycline to the growth media. A growth curve comparing uninduced and RNAi-induced cells did not show a major alteration to growth when TbTGT was knocked down (Fig. 2A). Five days post induction, total RNA was collected from both RNAi-induced and uninduced cells and analyzed by APB-gel electrophoresis followed by Northern hybridization utilizing the same oligonucleotide probes as in Fig. 1B (Fig. 2B). A substantial reduction in Q levels was observed when comparing the RNAi-induced samples to either the uninduced or the wild type (Fig. 2B).
Figure 2.
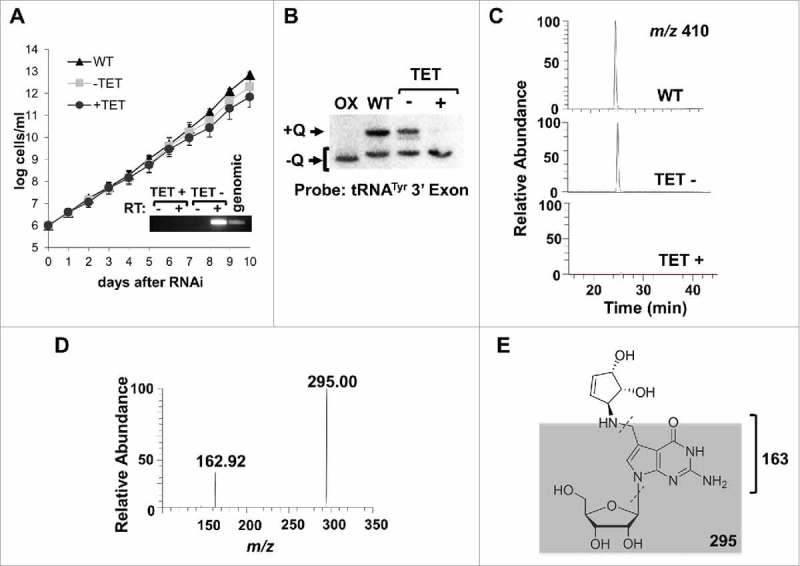
TbTGT is necessary for queuosine formation. A. A growth curve of T. brucei cells where TbTGT expression has been knockdown by RNAi (TET+) compared to wild-type cells or an uninduced control (TET-). The inset shows the reduction in TbTGT levels as determined by reverse transcription PCR (RT-PCR), where RT- refers to a control for DNA contamination where reverse transcriptase was left out of the reaction and RT+ refers to reaction to which reverse transcriptase was added prior to PCR. “Genomic” refers to a DNA positive control for PCR. B. APB-gel Northern hybridization showing the effect of RNAi knockdown of TbTGT on the queuosine content of tRNATyr. Oxidixed RNA was used as a negative control (OX). C. Extracted ion chromatogram for Q 410 m/z comparing samples from TbTGT RNAi induced (TET+) and uninduced (TET-) conditions to total wild type RNA (WT). A peak corresponding to m/z of 410 is not detectable in the RNAi-induced sample. D. Collision induced dissociation of the ion in C (410 m/z) reveals a characteristic fragmentation pattern of Queuosine. E. The neutral loss of C-N bond gives an m/z of 295; glycosidic bond breakage then yields the ion 163.
To validate the APB gel and Northern results, total RNA isolated from wild type, uninduced and TbTGT RNAi-induced cells was also analyzed by LC-MS/MS.35 A peak eluting at 25 min with a 410 m/z was observed, consistent with the presence of Q in tRNA from wild type or uninduced cells.36 This peak was further analyzed by collision-induced dissociation yielding a fragmentation pattern characteristic of Q, including the neutral loss of the C-N bond yielding a m/z of 295 and the glycosidic bond breakage yields the ion 163, confirming the presence of queuosine in total tRNA from T. brucei (Fig. 2C-E). A similar peak was absent in RNA isolated from the TbTGT RNAi cells indicating that this enzyme is essential for Q formation in T. brucei (Fig. 2C).
TbTGT localizes to the nucleus
Given our previous report that tRNA splicing occurs in the cytoplasm22 and that intron-containing tRNA, prior to splicing, is devoid of modifications,24 we determined the intracellular localization of TbTGT. This was done with the goal of establishing the order of events that lead to tRNATyr maturation. In eukaryotes, some TGTs localize to the cytoplasm while others are found in the outer mitochondrial membrane.26 TbTGT was tagged with a V5 epitope, expressed in T. brucei and analyzed by immunofluorescence with anti-V5 antibodies.37 These cells were also stained with MitoTracker Red and DAPI, to mark the position of the mitochondrial and nuclear compartments. We found that TbTGT was strictly found in the nucleus (Fig. 3) as judged by its co-localization with the nuclear DAPI signal. Importantly, no significant levels of TbTGT were observed in either the cytoplasm or the mitochondrial membrane leading to the conclusion that TbTGT is a nuclear enzyme (Fig. 3).
Figure 3.
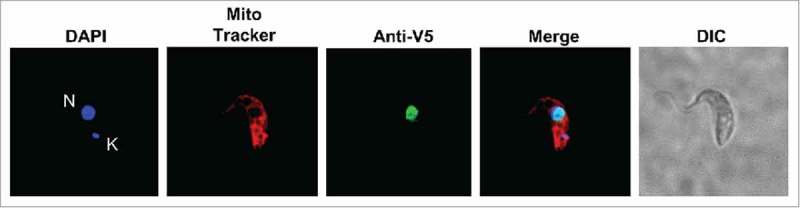
TbTGT is a nuclear enzyme. Immunofluorescence localization performed with cells expressing a V5-epitope tagged TbTGT. Anti-V5 antibodies were used to detect TbTGT. Mitotracker was used to stain the mitochondria (red) while DAPI stained the nuclear (N) and mitochondrial DNA (K) (blue). DIC refers to a phase-contrast image. The figure is representative of at least five different experiments.
Intron-containing tRNATyr does not contain queuosine
The intracellular distribution of TbTGT and the tRNA splicing machinery to two separate compartments then raises the possibility of a dynamic intracellular distribution of tRNATyr in the process of maturation. Either Q formation takes places in the nucleus prior to cytoplasmic export or it occurs after cytoplasmic splicing. The former implies that intron-containing tRNA is a substrate for Q formation, contrary to the known specificity of TGT for an unpaired G34; the latter suggests that once splicing takes place in the cytoplasm, the tRNA undergoes retrograde transport to the nucleus to be modified.
To test these possibilities, we exploited the earlier observation that RNAi of TbTrl1 leads to accumulation of intron-containing tRNA in T. brucei. Total RNA from the TbTrl1 RNAi-induced cells was separated by APB-gel electrophoresis and analyzed by Northern hybridization with a probe specific for the 3′ exon of tRNATyr and compared to RNA collected from an RNAi uninduced control. This probe does not discriminate between intron-containing and spliced tRNATyr, but can differentiate between the two based on their size; as such it can detect both species simultaneously. These experiments revealed the presence of two bands in the uninduced sample; a slow migrating band corresponding to Q34-containing spliced tRNATyr and a faster migrating band corresponding to G34-containing spliced tRNATyr (Fig. 4A). In the TbTrl1 RNAi-induced sample, a faint band corresponding to spliced Q-containing tRNA is present but in addition a strong band corresponding to intron-containing tRNA is also observed (Fig. 4A). Importantly, the migration of the intron-containing tRNA band does not shift to a position indicative of the presence of Q when compared to the oxidized control when the same membrane is probed with an intron-specific probe (Fig. 4B). This observation supports the view that intron-containing tRNA is not a substrate for TGT and Q formation occurs following splicing. This conclusion is also in line with our previous observation that native intron-containing tRNATyr was devoid of detectable modifications.24
Figure 4.
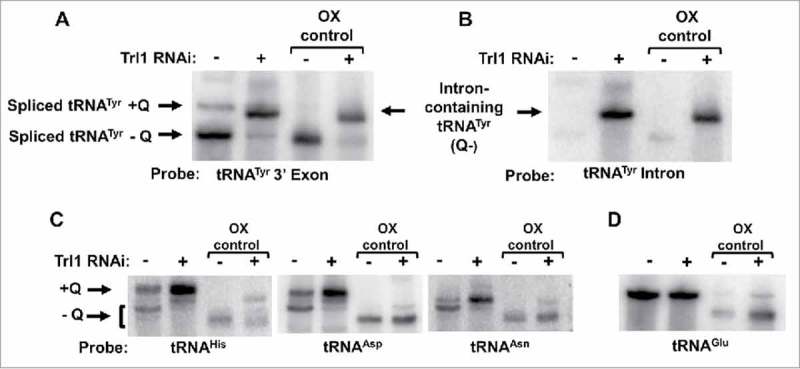
Intron-containing tRNA lacks Queuosine. A. APB-gel/Northern hybridization was performed on total RNA collected from TbTrl1 RNAi inlduced (+) and uninduced (-) cells. The arrows indicate spliced tRNA and intron-containing tRNA generated by the RNAi knockdown of TbTrl1. Shifted Q-containing (+Q) bands and non-Q-containing-bands (-Q) are as indicated. Samples were treated with sodium periodate to serve as a negative Q control (OX). The experiment was performed with a probe specific for the 3′ exon of tRNATyr. B. The same membrane as in (A) was probed with an intron-specific probe to assess whether intron-containing tRNA contained Q. The higher band located above the intron-containing tRNATyr band is not likely related to Q, as it is also found in the oxidized control lanes. C. Detection of Q in other potential Q-containing tRNAs (tRNAAsp,-Asn and-His) using the samples as in (A). As before, the Q-containing band (+Q) appears as a shifted band as indicated. D. The membrane was further hybridized with a probe specific for tRNAGlu serving as a loading and non-Q containing tRNA control.
To rule out the possibility that the lack of Q in intron-containing tRNATyr is due to some general or secondary effect from the TbTrl1 RNAi knockdown, we tested the levels of Q in the non-intron containing tRNAs that are also targets of TGT. Interestingly, the Q content of these tRNAs appeared to increase after down regulation of TbTrl1 following RNAi (Fig. 4C), while the levels of the non-Q containing tRNAGlu remained unaffected (Fig. 4D). Although we do not fully understand these results, the observed Q increase in other TbTGT tRNA substrates after TbTrl1 knockdown may be due to one less tRNA competing for TbTGT or alternatively it may be due to the fact that Q is in limiting concentrations in the cells and the absence of one substrate inherently leads to increased Q availability for others. However, at the moment we cannot discriminate between either possibility. Taken together, these observations support the view that intron-containing tRNATyr is not a substrate for TbTGT and the lack of Q is not due to a more general defect in Q formation related to the down regulation of an important protein.
Retrograde nuclear import is necessary for queuosine formation in tRNATyr
We then explored the second possibility that because tRNA splicing occurs in the cytoplasm and TbTGT is nuclear, then tRNATyr may require retrograde transport to the nucleus to get modified. We purified total RNA from wild type T. brucei subcellular fractions as previously described.22 The resulting RNA was again analyzed by APB-gel electrophoresis followed by Northern hybridization using the same 3′ exon-specific probe, as before. Surprisingly, we found significant amounts of mature tRNATyr in the nuclear fraction (Fig. 5A). Given the reported cytoplasmic localization of splicing, this observation is consistent with export of the intron-containing tRNA to the cytoplasm after transcription, followed by cytoplasmic splicing and the subsequent re-import of the spliced tRNA to the nucleus to get modified. Thus, we also analyzed the different fractions for the presence of Q. We found that both non-Q and Q-containing tRNA were present in the nuclear fraction (Fig. 5A). Similar results were obtained when the same membrane was probed for tRNAGlu, which is not a TGT substrate, indicating that differences in tRNA levels cannot be ascribed to a general instability of tRNAs during fractionation (Fig. 5B). To ensure the purity of the fractions and rule out the possibility of compartment cross-contamination, the fractions were also probed for compartment-specific markers. Northern hybridization with a probe specific for SnoRNA, a nucleolar marker, revealed negligible levels of this RNA in the cytoplasmic fraction with the majority found in the nuclear fraction (Fig. 5C). Similarly, Western blot experiments with protein samples from the same fractionation, using antibodies for the compartment-specific protein markers: anti-Nog1 (nuclear/nucleolar marker) and enolase (cytoplasmic marker), confirmed the purity of the fractions and ruled out the possibility that the observed presence of spliced Q-containing tRNATyr in the nuclear fractions was due to fraction cross-contamination during cell breakage and purification (Fig. 5D).
Figure 5.
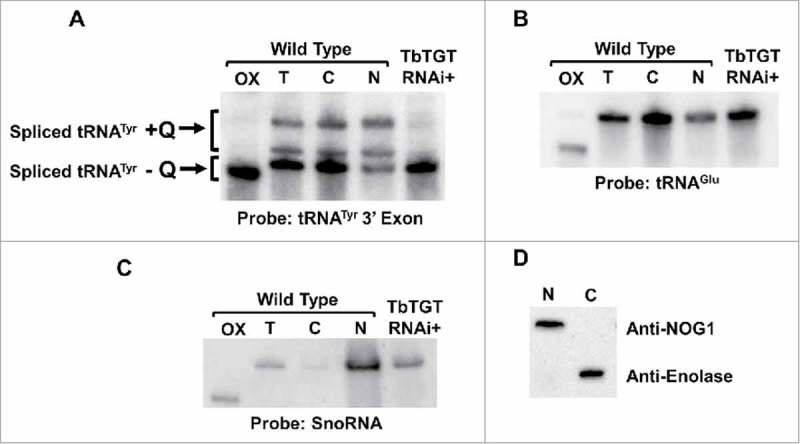
The nucleus contains significant amounts of spliced tRNATyr. A. Total RNA from unfractionated “T,” nuclear “N,” and cytoplasmic “C” subcellular fractions analyzed by APB-gel followed by Northern hybridization. Arrows indicate the spliced Q-containing (+Q) and non-Q-containing (-Q) tRNA bands. A portion of the RNA was treated with sodium periodate and used as a negative control for Q (OX). TbTGT RNAi+ refers to total RNA collected from TbTGT RNAi induced cells as shown. B. The same membrane was probed for tRNAGlu, a non-Q-containing tRNA, which is used as a loading control. C. The membrane was probed for SnoRNA (nuclear/nucleolus marker) to assess fraction purity. D. Western blot with antibodies against enolase (cytoplasmic marker) and Nog1 (nuclear/nucleolar marker) and the same subcellular fractions as above used as an additional control for fraction purity.
We also examined the role Q might play in nuclear retrograde transport. Specifically, we determined if lack of Q was a signal for nuclear import of spliced tRNATyr to obtain Q. We purified RNA from subcellular fractions of the TbTGT RNAi knockdown; if lack of Q were a signal for retrograde transport, we would expect spliced tRNATyr to accumulate in the nucleus with reduced levels in the cytoplasm. Northern hybridization analysis probing for tRNATyr exon revealed no major differences in the band intensities between TbTGT RNAi induced and uninduced cells (Fig. 6A) suggesting that lack of Q was not a signal to send tRNA to the nucleus. If Q was not directly affecting retrograde transport, it may indirectly contribute to it by altering the aminoacylation state of the tRNA. To test this, we purified total RNA from wild type, TbTGT RNAi induced, and uninduced cells under acidic conditions and performed acid-gel electrophoresis followed by Northern hybridization to determine if the lack of Q alters the aminoacylation state of the tRNA. By this method, tRNA that retain their amino acid appear as a shifted band compared to deacylated tRNA allowing for visualization of aminoacylation levels. The acid gel Northern hybridization probed for tRNATyr did not reveal major differences in aminoacylation among these samples (Fig. 6B). These results, taken together, suggest that the absence of Q is not a signal for retrograde import itself, nor is Q a requirement for aminoacylation.
Figure 7.
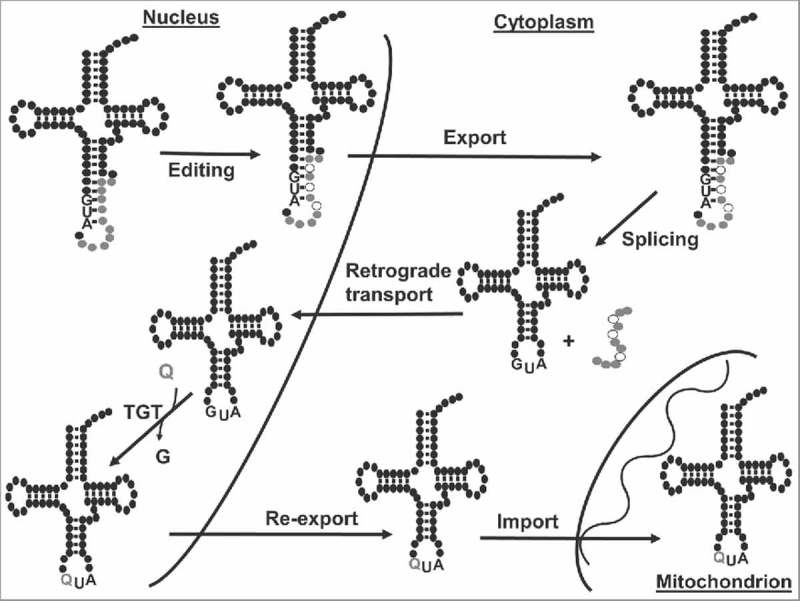
Nuclear retrograde transport is necessary for queuosine formation in tRNATyr. The figure shows a model for the maturation of tRNATyr in T. brucei. The tRNA is transcribed in the nucleus containing an 11-nucleotide long intron. In the nucleus, two or three nucleotides within the intron undergo non-canonical editing before export to the cytoplasm; editing is required for intron cleavage. Once in the cytoplasm, the edited intron-containing tRNATyr is spliced and then re-imported back into the nucleus where the nucleus-localized TGT replaces G34 for Q34. Finally, Q-containing tRNATyr is re-exported to the cytoplasm.
Figure 6.
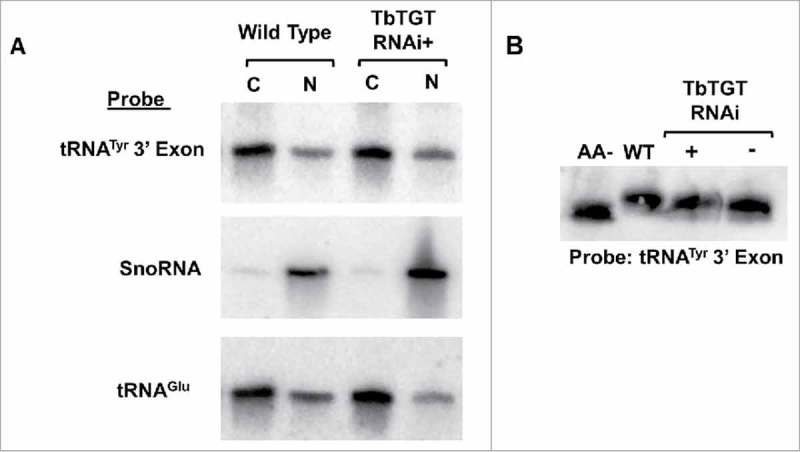
Neither lack of queuosine nor aminoacylation are signals for retrograde transport. A. Nuclear (N) and cytoplasmic (C) subcellular fractions of total RNA isolated from TbTGT RNAi induced cells (RNAi+) were compared to similar samples isolated from wild type cells and analyzed by Northern hybridization with probes specific for either the tRNATyr 3′ exon, SnoRNA or tRNAGlu as indicated. B. The aminoacylation levels of tRNATyr after TbTGT RNAi (RNAi+), an uninduced control (RNAi-) or wild-type cells were determined by acid-gel electrophoresis and Northern hybridization using a 3′exon-specific tRNATyr probe. A portion of RNA was deacylated by in a basic pH buffer and served as a negative control. Shifted bands correspond to the aminoacylated tRNA.
Discussion
One of the defining features of eukaryotic cells is their extensive intracellular compartmentalization, whereby membrane boundaries provide a higher order of organization, but make it a requisite to also have specialized transport systems. The latter ensure the purposeful movement of all sorts of molecules across selectively permeable membranes, driven by the need to provide a given function in a specific compartment. Intracellular transport dynamics may also dictate where and when many macromolecules become fully mature.38 In the case of tRNA, maturation involves the addition of, sometimes, numerous posttranscriptional modifications. These may take place at any step of the maturation pathway: In the nucleus during or immediately following transcription, in the cytoplasm following nuclear export, or even in the genome-containing organelles (plastids or mitochondria). Although for many years tRNA modification was thought to temporally occur in a linear fashion pinpointed by transcription and subsequent nuclear export to the cytoplasm, the discovery that tRNAs could be retrogradely transported back to the nucleus raised the possibility that addition of modifications could be more dynamic. For example, in S. cerevisiae methylation at position 37 of tRNAPhe to form 1-methylguanosine (m1G) occurs in the nucleus, the place where the Trm5 enzyme resides. However, since Trm5 cannot act on an intron-containing tRNA and splicing is cytoplasmic, once the intron is removed the tRNA travels back to the nucleus to be modified.16
Our findings on Q formation in T. brucei are in line with published work showing that TGT is not able to modify an intron-containing tRNA.27 Confocal localization studies conducted in monkey kidney cells (Cos7) showed that endogenous TGT is associated with the outer mitochondrial membrane.26 These studies were supported by the observed efficiency in the rates of Q incorporation when tRNAs were microinjected into the cytoplasm of Xenopus laevis.29,39 We now show that in T. brucei a nucleus-localized TGT paralog is involved in Q formation in the nucleus and at least in the case of tRNATyr, Q formation requires the tRNA to travel from and to the nucleus to get spliced and modified. Interestingly, three additional tRNAs are also TGT substrates but do not contain an intron; these may not require retrograde nuclear transport to be modified. It is then feasible that the differential localization of TGT and the splicing machinery, coupled to the necessity to form Q in tRNATyr, has partly provided the selective pressure to maintain a retrograde transport mechanism in this organism. The pathway described here is analogous to that seen in yeast for the biosynthesis of wybutosine providing another example of a modification requiring retrograde transport.16
Retrograde transport is thought to play a particularly important function during times of nutritional stress. In S. cerevisiae, a decrease in the levels of amino acids, inorganic phosphate, and/or glucose causes tRNA accumulation in the nucleus.12,13,18 Because retrograde transport is constitutive, such nuclear accumulation has been attributed to a reduction in the rate of re-export when nutrients are low.15 Presumably, by withholding tRNAs in the nucleus, cells can avoid tRNA degradation and re-synthesis.40 Upon removal of stress, however, these tRNAs are rapidly released back to the cytoplasm without the need for re-synthesis.12,13,17,18 Our findings presented here only show the connection between retrograde transport and formation of queuosine in one tRNA. We cannot rule out the possibility that in T. brucei retrograde transport also serves other purposes, for example to better deal with nutritional imbalances. These organisms have two different well-marked developmental stages: one in the insect vector, the other in the mammal host. Each stage demands specific metabolic adaptations from the parasites.41 For instance, in the insect T. brucei respires and primary gets all its ATP by oxidative phosphorylation, while in the host, glycolysis and substrate-level phosphorylation take over and mitochondrial functions are down regulated.42,43 It is provocative to think that retrograde transport of tRNAs may play a role in such environmental adaptations.
Materials and methods
Cell culture and growth media
Wild type, procyclic T. brucei 29-13 cells were grown in SDM-79 supplemented with 10% fetal bovine-serum. Cell growth was monitored by counting with a hemocytometer every 24 hours. RNAi constructs were generated by cloning of 851bp region of the of the coding sequence of TbTGT into the plasmid vector p2T7-177 using the oligonucleotides: TbTGT-F: CCCAAGCTTTACCGTACCTAACACCCGAAC and TbTGT-R: CGCGGATCCCAAAATACAACTGTTCATCTCCTGG. Cloning sites HindIII and BamHI are underlined in the sequence. RNAi was induced by the addition of 1 μg/ml of tetracycline to the media, generating dsRNA from head-to-head orientated T7 promoters.34,44 The Trl1 RNAi cell line was generated as previously described.22
Nuclei fractionation
Cells were collected after reaching mid to late log (6 × 106 to 1 × 107 cells/mL), pelleted by centrifugation and washed twice in phosphate buffer saline (PBS). Pellets were resuspended in a solution containing 1mM PIPES pH 7.4, 1 mM CaCl2, 500 mM Hexylene glycol, 125 mM sucrose. The suspension was passed through a Standsted homogenizer machine at 20 PSI. The resulting lysate was pelleted by centrifugation at 2000 xg for 20 minutes and suspended in 5 mL of supernatant. This was layered on top of a 35% Percoll gradient containing 1mM PIPES pH 7.4, 5 mM CaCl2, 500 mM Hexylene glycol and 700 mM sucrose, followed by centrifugation at 60,000 × g for 35 minutes. The nuclei band (the bottom most band) was obtained by side puncture. This was in turn washed in gradient buffer and suspended in either TE for RNA analysis, or SDS-loading buffer for protein analysis.
Northern hybridization and APB-gel analysis
Total RNA was isolated as previously described,45 separated on a denaturing 8M urea 8% polyacrylamide gel, and electroblotted to Zeta-probe membranes. The membrane was then UV cross-linked for 1 min followed by oven baking 80°C for 30min. For APB-gel analysis, samples were deacylated prior to electrophoresis by incubation in 100mM Tris pH 9.0 for 30 min. Oxidation control RNA was deacylated and added to a solution containing 50mM Tris pH 5.0 and 2 mM NaIO4 for 2 hours at 37°C in the dark. The oxidation reaction was quenched with 2.5 mM glucose before use. 50 mg of 3-aminophenylboronic acid (Sigma) was added to 10 mL of 8M-urea polyacrylamide mix before gel pouring. Electrophoresis on APB Gels was carried out for approximately 5 hours at 75 V at 4°C. Northern hybridization was performed according to the manufacturer (Bio-Rad) using with 32P-labeled oligonucleotides. Following hybridization, membranes were exposed overnight on a phosphoimager screen. Blots were analyzed using a Typhoon FLA 9000 scanner and the ImageQuant TL software (GE Healthcare). Probes used for Northern hybridization were as follows: tRNATyr-3′ exon GTGGTCCTTCCGGCCGGAATCGAA; tRNAHis GGGAAGACCGGGAATCGAAC; tRNAAsp CGGGTCACCCGCGTGACAGG; tRNAAsn AACCAACGACCTGTAGGTTAACAGC; tRNAGlu TTCCGGTACCGGGAATCGAAC;
SnoRNA CAACGTCCATCTGCGACGGCTTTA.
Western blot analysis
Protein fractions were separated on a 10% SDS-polyacrylamide gel. Polyclonal rabbit antibodies to Enolase and Nog1 were diluted 1:5,000 and 1:10,000 respectively. Secondary anti-rabbit IgG antibodies were used at a dilution of 1:5,000 (GE Healthcare) and visualized with ECL (Pierce).
Immunofluorescence
TbTGT was cloned into pT7-V5c vector using the oligos F: GCTAAGCTTATGCACGGAGTGTATCCTATT and R: GCTGGATCCCAACTGTGCAGCACCCA. Expressing cells were stained with 500 nM MitoTracker Red dye (Life Technologies) for 30 min with shaking at 27°C. Cells were then harvested at 2-5 × 106 cells/mL and washed twice in PBS. This was followed by permeabilization by addition of 0.1% Triton X-100 and three more PBS washes. Blocking by 5.5% FBS and 0.05% Tween 20 in PBS was done for 1 hr in a humid chamber. A 1:4000 dilution was used for primary mouse anti-V5 antibody and for secondary goat anti-mouse antibody AlexaFluor488. Cells were then air dried briefly and mounted with Vectashield containing DAPI. Images were taken with a Nikon Ti microscope and analyzed via ImageJ (NIH) and Nikon Elements AR.
Acid gel for monitoring tRNA aminoacylation
Cells were harvested at mid-log phase (6 × 106 cells/mL) and pelleted. Pellets were washed twice with PBS before finally being suspended in 0.3 mL of 0.1 M sodium acetate pH 4.5 containing 0.1 M EDTA, followed by extraction twice with phenol equilibrated in the same buffer. RNA was then ethanol precipitated and resuspended in 0.1 M sodium acetate pH 4.5, 8 M urea loading dye. A portion of the RNA was further deacylated prior to loading by incubation for 30 min at 37°C in 0.1 M Tris-HCl pH 9.0, this sample served as a negative control for aminoacylation. RNA samples were separated by electrophoresis for 48 hours at 50V on an 8 M urea, 12% polyacrylamide gel containing 0.1 M sodium acetate.
LC-MS/MS analysis
One liter of cells at 1 × 107 cells/mL was harvested and total RNA isolated as described previously.37 Purified total tRNA was degraded to nucleosides by digestion by digestion with nuclease P1, followed by treatment with CIAP. The total nucleoside digest was analyzed in an ESI-LC/MS system Thermo-LTQ XL ion-trap mass spectrometer, interphased with a Hitachi D-7000 HPLC equipped with a diode array detector set at 260 nm. Chromatographic separation was performed on Hypersil Gold C18 (Thermo Fischer), 1.9 μm, 150 × 2.1 mm at 300 μL/min flow rate. The mobile phase used were as follows: (mobile phase A) 5.3 mM ammonium acetate (pH 5.3) and (mobile phase B) 40% acetonitrile. The column eluent was split post column, 1/3 to the electrospray ion source and 2/3 to the DAD detector. Each replicate of the injection contained 10 μg of the digest, resuspended in 10 μL of mobile phase A. Mass spectra was recorded in positive mode, capillary temperature of 275°C, spray voltage 3.7-4.0 kV, sheath gas, auxiliary gas and sweep gas of 45, 25 and 10 arbitrary units respectively. Data dependent MS/MS of the two most abundant ions were recorded throughout the run.
Acknowledgments
We would like to thank Anita Hopper for insightful discussions and guidance concerning tRNA retrograde transport and to all members of the Alfonzo and Paris laboratories for their comments and useful suggestions. We would also like to thank P. Michels for the anti-enolase and M. Parsons for the anti-Nog1 antibodies. This work was partially supported by NIH grant GM084065 grant to J.D.A, a Czech Science Foundation (15-21450Y) grant to Z.P, a GAJU 036/2017/P grant from the University of South Bohemia to S.S.K. and an NSF Award (CHE 1507357) to P.A.L.
References
- 1.Maraia RJ, Lamichhane TN. 3′ processing of eukaryotic precursor tRNAs. Wiley Interdiscip Rev RNA. 2012;2:362–75. doi: 10.1002/wrna.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klemm BP, Wu N, Chen Y, Liu X, Kaitany KJ, Howard MJ, Fierke CA. The diversity of ribonuclease P: Protein and RNA catalysts with analogous biological functions. Biomolecules. 2016;6. doi: 10.3390/biom6020027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Betat H, Mörl M. The CCA-adding enzyme: A central scrutinizer in tRNA quality control. BioEssays. 2015;37:975–82. doi: 10.1002/bies.201500043. [DOI] [PubMed] [Google Scholar]
- 4.McKenney K, Alfonzo J. From Prebiotics to Probiotics: The Evolution and Functions of tRNA Modifications. Life. 2016;6:13. doi: 10.3390/life6010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lopes RRS, Kessler AC, Polycarpo C, Alfonzo JD. Cutting, dicing, healing and sealing: The molecular surgery of tRNA. Wiley Interdiscip Rev RNA. 2015;6:337–49. doi: 10.1002/wrna.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tohru Yoshihisa, Kaori Yunoki-Esaki, Chie Ohshima, Nobuyuki Tanka and TE. Possibility of Cytoplasmic pre-tRNA Splicing: the Yeast tRNA Splicing Endonuclease Mainly Localizes on the Mitochondria. Mol Biol Cell. 2003;14:3266–79. doi: 10.1091/mbc.E02-11-0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huh W-K, Falvo J V., Gerke LC, Carroll AS, Howson RW, Weissman JS, O'Shea EK. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–91. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- 8.Mori T, Ogasawara C, Inada T, Englert M, Beier H, Takezawa M, Endo T, Yoshihisa T. Dual functions of yeast tRNA ligase in the unfolded protein response: Unconventional cytoplasmic splicing of HAC1 pre-mRNA is not sufficient to release translational attenuation. Mol Biol Cell. 2010;21:3722–34. doi: 10.1091/mbc.E10-08-0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kutay Ulrike; Lipowsky Gerd; Izaurralde Elisa; F. Bischoff Ralf; Schwarzmaier Petra; Hartmann Enno; Görlich D. Identification of a tRNA-Specific Nuclear Export Receptor. Mol Cell. 1998;1:359–69. doi: 10.1016/S1097-2765(00)80036-2. [DOI] [PubMed] [Google Scholar]
- 10.Calado A, Treichel N, Müller EC, Otto A, Kutay U. Exportin-5-mediated nuclear export of eukaryotic elongation factor 1A and tRNA. EMBO J. 2002;21:6216–24. doi: 10.1093/emboj/cdf620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lipowsky G, Bischoff FR, Izaurralde E, Kutay U, Schäfer S, Gross HJ, Beier H, Görlich D. Coordination of tRNA nuclear export with processing of tRNA. RNA. 1999;5:539–49. doi: 10.1017/S1355838299982134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaheen HH, Hopper AK. Retrograde movement of tRNAs from the cytoplasm to the nucleus in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2005;102:11290–5. doi: 10.1073/pnas.0503836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Michael L. Whitney, Rebecca L. Hurto, Hussam H. Shaheen and AKH. Rapid and Reversible Nuclear Accumulation of Cytoplasmic tRNA in Response to Nutrient Availability. Mol Biol Cell. 2007;18:2678–86. doi: 10.1091/mbc.E07-01-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eswara MBK, McGuire AT, Pierce JB, Mangroo D. Utp9p facilitates Msn5p-mediated nuclear reexport of retrograded tRNAs in Saccharomyces cerevisiae. Mol Biol Cell. 2009;20:5007–25. doi: 10.1091/mbc.E09-06-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takano A, Endo T, Yoshihisa T. tRNA Actively Shuttles Between the Nucleus and Cytosol in Yeast. Science. 2005;309:140–2. doi: 10.1126/science.1113346. [DOI] [PubMed] [Google Scholar]
- 16.Ohira T, Suzuki T. Retrograde nuclear import of tRNA precursors is required for modified base biogenesis in yeast. Proc Natl Acad Sci U S A. 2011;108:10502–7. doi: 10.1073/pnas.1105645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hurto RL, Tong AHY, Boone C, Hopper AK. Inorganic phosphate deprivation causes tRNA nuclear accumulation via retrograde transport in Saccharomyces cerevisiae. Genetics. 2007;176:841–52. doi: 10.1534/genetics.106.069732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murthi A, Shaheen HH, Huang HY, Preston MA, Lai TP, Phizicky EM, Hopper AK. Regulation of tRNA Bidirectional Nuclear-Cytoplasmic Trafficking in Saccharomyces cerevisiae. Mol Biol Cell. 2010;21:639–49. doi: 10.1091/mbc.E09-07-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghavidel A, Kislinger T, Pogoutse O, Sopko R, Jurisica I, Emili A, Hopper AK, Phizicky EM, Sarkar S, Azad a K, et al.. Retrograde nuclear accumulation of cytoplasmic tRNA in rat hepatoma cells in response to amino acid deprivation. Proc Natl Acad Sci U S A. 2007;96:162–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barhoom S, Kaur J, Cooperman BS, Smorodinsky NI, Smilansky Z, Ehrlich M, Elroy-Stein O. Quantitative single cell monitoring of protein synthesis at subcellular resolution using fluorescently labeled tRNA. Nucleic Acids Res. 2011;39:e129. doi: 10.1093/nar/gkr601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyagawa R, Mizuno R, Watanabe K, Ijiri K. Formation of tRNA granules in the nucleus of heat-induced human cells. Biochem Biophys Res Commun. 2012;418:149–55. doi: 10.1016/j.bbrc.2011.12.150. [DOI] [PubMed] [Google Scholar]
- 22.Lopes RRS, Silveira G, De O, Eitler R, Vidal RS, Kessler A, Hinger S, Paris Z, Alfonzo JD, Polycarpo C. The essential function of the Trypanosoma brucei Trl1 homolog in procyclic cells is maturation of the intron-containing tRNATyr. RNA. 2016;22:1190–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schneider A, McNally KP, Agabian N. Splicing and 3′-processing of the tyrosine tRNA of Trypanosoma brucei. J Biol Chem. 1993;268:21868–74. [PubMed] [Google Scholar]
- 24.Rubio MAT, Paris Z, Gaston KW, Fleming IMC, Sample P, Trotta CR, Alfonzo JD. Unusual noncanonical intron editing is important for tRNA splicing in trypanosoma brucei. Mol Cell. 2013;52:184–92. doi: 10.1016/j.molcel.2013.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Howes NK, Farkas WR. Studies with a homogeneous enzyme from rabbit erythrocytes catalyzing the insertion of guanine into tRNA. J Biol Chem. 1978;253:9082–7 [PubMed] [Google Scholar]
- 26.Boland C, Hayes P, Santa-Maria I, Nishimura S, Kelly VP. Queuosine formation in eukaryotic tRNA occurs via a mitochondria-localized heteromeric transglycosylase. J Biol Chem. 2009;284:18218–27. doi: 10.1074/jbc.M109.002477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nishikura K, De Robertis EM. RNA processing in microinjected Xenopus oocytes. Sequential addition of base modifications in a spliced transfer RNA. J Mol Biol. 1981; 145:405–20. doi: 10.1016/0022-2836(81)90212-6. [DOI] [PubMed] [Google Scholar]
- 28.Noguchi S, Hirotat Y, Nishimura S. Isolation and Characterization of an Escherichia coli Mutant Lacking tRNA-Guanine Transglycosylase. Function and biosynthesis of queuosine in tRNA. J Biol Chem. 1982;257:6544–50. [PubMed] [Google Scholar]
- 29.Haumont E, Droogmans L, Grosjean H. Enzymatic formation of queuosine and of glycosyl queuosine in yeast tRNAs microinjected into Xenopus laevis oocytes: The effect of the anticodon loop sequence. Eur J Biochem. 1987; 168:219–25. doi: 10.1111/j.1432-1033.1987.tb13408.x. [DOI] [PubMed] [Google Scholar]
- 30.Watanabe M, Matsuo M, Tanaka S, Akimoto H, Asahi S, Nishimura S, Katze JR, Hashizume T, Crain PF, Mccloskey JA, et al.. Biosynthesis of Archaeosine, a Novel Derivative of 7-Deazaguanosine Specific to Archaeal tRNA, Proceeds via a Pathway Involving Base Replacement on the tRNA Polynucleotide Chain *. 1997;272:20146–51. [DOI] [PubMed] [Google Scholar]
- 31.Okada N, Noguchi S, Kassai H, Shindo-Okada N, Ohgi T, Goto T, Nishimura S. Novel mechanism of post-transcriptional modification of tRNA. 1979;254:3067–73. [PubMed] [Google Scholar]
- 32.Nakanishi S, Ueda T, Hori H, Yamazaki N, Watanabe N, Okada K. A UGU sequence in the anticodon loop is a minimum requirement for recognition by Escherichia coli tRNA-guanine transglycosylase. J Biol Chem. 1994;269:32221–5. [PubMed] [Google Scholar]
- 33.Kӧssel GLI and H Affinity electrophoresis for monitoring terminal phosphorylation and the presence of queuosine in RNA. Application of polyacrylamide containing a covalently bound boronic acid. Nucleic Acids Res. 1985;13:6881–98. doi: 10.1093/nar/13.19.6881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wickstead B, Ersfeld K, Gull K. Targeting of a tetracycline-inducible expression system to the transcriptionally silent minichromosomes of Trypanosoma brucei. Mol Biochem Parasitol. 2002;125:211–6. doi: 10.1016/S0166-6851(02)00238-4. [DOI] [PubMed] [Google Scholar]
- 35.Ross R, Cao X, Yu N, Limbach PA. Sequence mapping of transfer RNA chemical modifications by liquid chromatography tandem mass spectrometry. Methods. 2016;107:73–8. doi: 10.1016/j.ymeth.2016.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Phillipson DW, Edmonds CG, Crain PF, Smith DL, Davis DR, McCloskey JA. Isolation and structure elucidation of an epoxide derivative of the hypermodified nucleoside queuosine from Escherichia coli transfer RNA. J Biol Chem. 1987;262:3462–71. [PubMed] [Google Scholar]
- 37.Sample PJ, Kořený L, Paris Z, Gaston KW, Rubio MA, Fleming IM, Hinger S, Horáková E, Limbach PA, Lukeš J, Alfonzo JD. A common tRNA modification at an unusual location: the discovery of wyosine biosynthesis in mitochondria. Nucleic Acids Res. 2015;43(8):4262–73. doi: 10.1093/nar/gkv286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rubio MAT, Hopper AK. tRNA travels from the cytoplasm to organelles. 2012;2:802–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carbon P, Haumont E, Henau S De, Keith G, Grosjean H. Enzymatic replacement in vitro of the first anticodon base of yeast tRNAAsp: application to the study of tRNA maturation in vivo, after microinjection into frog oocytes. 1982;10:3715–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chu H-Y, Hopper AK. Genome-wide investigation of the role of the tRNA nuclear-cytoplasmic trafficking pathway in regulation of the yeast Saccharomyces cerevisiae transcriptome and proteome. Mol Cell Biol. 2013;33:4241–54. doi: 10.1128/MCB.00785-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith TK, Bringaud F, Nolan DP, Figueiredo LM. Metabolic reprogramming during the Trypanosoma brucei life cycle [version 2; referees: 4 approved] Referee Status. 2017;6:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bochud-Allemann N, Schneider A. Mitochondrial substrate level phosphorylation is essential for growth of procyclic Trypanosoma brucei. J Biol Chem. 2002;277:32849–54. doi: 10.1074/jbc.M205776200. [DOI] [PubMed] [Google Scholar]
- 43.Clayton CE, Michels P. Metabolic compartmentation in African trypanosomes. Parasitol Today. 1996;12:465–71. doi: 10.1016/S0169-4758(96)10073-9. [DOI] [PubMed] [Google Scholar]
- 44.Shi H, Djikeng A, Mark T, Wirtz E, Tschudi C, Ullu E. Genetic interference in Trypanosoma brucei by heritable and inducible double-stranded RNA. RNA. 2000;6:1069–76. doi: 10.1017/S1355838200000297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chomczynski P, Sacchi N. Single-Step Method of RNA Isolation by Acid Guanidinium Extraction. Anal Biochem. 1987;159:156–9. doi: 10.1016/0003-2697(87)90021-2. [DOI] [PubMed] [Google Scholar]