

ABSTRACT
Ischemic Heart Disease (IHD) remains the developed world’s number one killer. The improved survival from Acute Myocardial Infarction (AMI) and the progressive aging of western population brought to an increased incidence of chronic Heart Failure (HF), which assumed epidemic proportions nowadays. Except for heart transplantation, all treatments for HF should be considered palliative because none of the current therapies can reverse myocardial degeneration responsible for HF syndrome. To stop the HF epidemic will ultimately require protocols to reduce the progressive cardiomyocyte (CM) loss and to foster their regeneration. It is now generally accepted that mammalian CMs renew throughout life. However, this endogenous regenerative reservoir is insufficient to repair the extensive damage produced by AMI/IHD while the source and degree of CM turnover remains strongly disputed. Independent groups have convincingly shown that the adult myocardium harbors bona-fide tissue specific cardiac stem cells (CSCs). Unfortunately, recent reports have challenged the identity and the endogenous myogenic capacity of the c-kit expressing CSCs. This has hampered progress and unless this conflict is settled, clinical tests of repair/regenerative protocols are unlikely to provide convincing answers about their clinical potential. Here we review recent data that have eventually clarified the specific phenotypic identity of true multipotent CSCs. These cells when coaxed by embryonic cardiac morphogens undergo a precisely orchestrated myogenic commitment process robustly generating bona-fide functional cardiomyocytes. These data should set the path for the revival of further investigation untangling the regenerative biology of adult CSCs to harness their potential for HF prevention and treatment.
KEYWORDS: Cardiac stem cells, myogenic differentiation, cardiac regeneration
Introduction
Cardiovascular diseases (CVD) are the number one cause of mortality worldwide with an estimated 17.3 million deceases per year representing 31.5% of all global deaths [1]. The most important CVD, in terms of mortality, morbidity and average life expectancy, is myocardial infarction (MI). In response to cardiomyocyte (CM) loss by MI, the heart tries to compensate the lost muscle mass with pathological cardiac remodelling [2]. The latter entails reactive CM hypertrophy, ongoing cell death and replacement fibrosis, which produce a diseased myocardial milieu that impairs regeneration events [3,4], leading over time to heart failure (HF), a chronic and progressive condition in which the heart fails to sustain appropriate organs’ blood supply. Despite all novel and current pharmacological treatments [5], there is no curative treatment for chronic HF that has now reached epidemic proportions and for which a global call to action has been launched to find an effective, affordable and widely available therapy.
During physiological mammalian growth, approximately 40% of all adult CMs are generated in neonatal life; after this period and during early adulthood, cardiac growth is characterized by a transition from a hyperplastic to a hypertrophic phase, with formation of bi-nucleated CMs that permanently withdraw from the cell cycle, becoming terminally differentiated cells [6,7]. However, the adult heart maintains a constant CM turnover rate, the amount of which has been extrapolated to be less than 1% per year whereby this extrapolation is based on mathematical formulas and not on actual direct counting. Independently from the real CM turnover rate, it remains indisputable that CM loss by myocardial ischemia and infarction are not adequately refreshed by new endogenous CM formation, that is insufficient to prevent heart failure [8–10]. Considering the decrease in the number of viable CMs after injury, associated with the (mala)adaptive hypertrophy of the remaining CMs that inefficiently perform their specialized function [11], heart transplantation is the only therapeutic option to revert HF, despite all biological limitation, post-surgery problems and complications of cardiac transplant together with the severe shortage of donors compared to the increasing number of HF patients in need of organ transplantation.
In the last two decades, it became a priority the search for novel effective methods able to regenerate new myocardium with the aim to improve cardiac function, thus prolonging lifespan. Since the advent of stem cell biology, the heart has been intensively investigated as potential target of stem and progenitor cell-based therapies [12–16]. Cell replacement therapy currently represents the holy grail for myocardial degenerative diseases, with its stated goal to generate clinically relevant and functional contractile cells. Several cell types, including embryonic stem cells, foetal myoblasts, endothelial progenitor cells, bone marrow-derived cells, amniotic fluid-derived stem cells, have been evaluated by transplantation into the post-infarcted myocardium to test their capacity to replace lost CMs and recover the myocardial tissue [17–21]. Despite the encouraging results obtained from experiments carried out in small animals, the outcomes from the majority, if not all, clinical trials have been modest.
The discovery that the adult heart possesses a pool of resident cardiac stem cells (CSCs) participating actively in cardiac homeostasis and repair, overturned the traditional view of the heart as a non-regenerative organ and opened the era of CSC-based therapy [12,22–25]. Endogenous CSCs were first identified by the expression of a typical stemness marker, the stem cell factor receptor c-kit [12]. However, the heart contains a heterogeneous population of c-kit positive (c-kitpos) cells, mainly composed of endothelial progenitor cells. This heterogeneity of c-kitpos cardiac cells has contributed significantly to the raise of a hot controversy on the actual identity and myogenic plasticity of resident CSCs. Despite the persistence of somatic cardiac stem cells and cardiac progenitor cells (CPCs) in the adult heart [26–28], the quantitative contribution of these cells to structural repair in adult mammalian hearts, and in humans in particular, is highly debated [29–31]. The overall negative message from recent genetic fate mapping studies to putatively track CSCs or CPCs have significantly contributed to a muddled state whereby CSC existence is doubted and their contribution to myocardial regeneration is at most considered negligible [32–37].
In this review, we describe the phenotypic characterization of the true multipotent resident CSCs isolated by the presence/absence of several surface markers. In this phenotypic definition, c-kit expression is necessary but not sufficient to identify true adult CSCs. The qualifier “c-kit” can be indeed misleading and we propose that these cells, when meeting the listed phenotypic characteristics, should simply be called adult “cardiac stem cells” or CSCs. This phenotype needs to be taken into account in the design of any CSC cell-fate tracking approach. We describe the most important and evolutionary conserved factors and molecules with their related signalling cascades that are indispensable for activation and differentiation of CSCs to replenish CMs in the adult myocardium. These factors and their molecular pathways may in the short future represent therapeutic agents/targets for cardiac regenerative medicine in order to obtain functional myocardial regeneration in the everyday clinical scenario.
Phenotypic identity of endogenous cardiac stem cells
The adult mammalian heart contains a pool of so-called “Cardiac Stem/Progenitor Cells” consisting of myogenic as well as vascular progenitors derived from the activation of a more-primitive stem cell [12,38]. The first report of mammalian CSCs in rodents [12], was rapidly followed by other studies which described several surface markers for the identification and the isolation of cells with cardiac tissue-specific progenitor properties in the adult mammalian heart. Each reporting group focused the interest on specific markers (or cell properties) which allegedly made their cell “unique” and different from those previously described (Table 1 and Figure 1) [12,23,26,27,31,39–55]. Unfortunately, the use of these markers, each supposedly identifying a specific stem cell, has created confusion in the scientific community regarding which cell is the real cardiac stem cell and what is its origin. The most common misunderstanding originates from the uncertain difference between CSCs and progenitor cells. CSCs are clonogenic, self-renewing and multipotent, giving rise to a minimum of three different cardiogenic cell lineages (myocytes, smooth muscle and endothelial cells) both in vitro and in vivo and exhibit significant cardiac tissue regenerative capacity. On the other hand, a cardiac progenitor cell is an immature but already committed myocardial cell that can proliferate and mature into its respective precursor which, in turn, develops into one of the main cardiac cell lineages. More importantly, the identification of different cardiac stem/progenitor cells, by the expression of different surface markers, suggest that these phenotypically different progenitor cells are likely to be the phenotypic and developmental variation of a unique cardiac stem cell type.
Table 1.
Summary of CSC populations.
| Phenotype | Markers | Source | Derivation |
|---|---|---|---|
| c-Kitpos CSCs* | CD31–, CD34–, CD45–, Sca-1+, Abcg2+, CD105+, CD166+, GATA4+, NKX2-5+/ – or low, MEF2C+ [12,23,39–42,49] | Mouse, rat, dog, pig, sheep and human | Embryonic, Neonatal, Adult |
| Sca-1pos CSCs* | CD31–, CD34–, CD45–, FLK1–, c-Kit+/ – or low, GATA4+, NKX2-5+/ – or low, MEF2C+ [43–45] | Mouse and human | Embryonic, Adult |
| Side population cells | CD31–, CD34+, CD45+, Abcg2+, Sca-1+, c-Kit+, NKX2-5–, GATA4– [26,46,47] | Mouse, human | Embryonic, Adult |
| CardioSphere-derived cells (CDCs) | CD105+, CD34+, CD45+, Abcg2+, Sca1+, c-Kitlow [27,48] | Mouse, rat, dog, pig and human | Neonatal, Adult |
| Colony-forming unit fibroblasts (CFUFs) | Sca-1+, PDGFR-α+, CD31–, c-Kitlow, CD45–, FLK1–, CD44+, CD90+, CD29+ and CD105 + [50] | Mouse | Embryonic, Adult |
| Cardiac mesangioblasts | CD31+, CD34+, CD44+, CD45–, Sca-1+, c-Kit+ [51] | Mouse and human | Embryonic, Neonatal, Juvenile |
| Isl1pos CPCs** | CD31–, Sca-1–, c-Kit–, GATA4+, NKX2-5+ [52,53] | Mouse, rat and human | Embryonic, Neonatal |
| Epicardium-derived cells (EPDCs) | c-Kit±, CD34+, CD44+, CD90+,CD105+,CD46+ [54,55] | Mouse and human | Embryonic, Adult |
Figure 1.
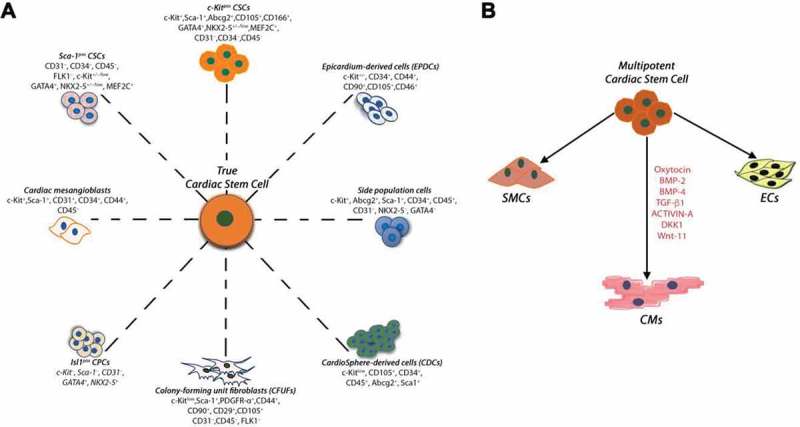
(A) Summary of the endogenous CSCs/CPCs populations identified in the developing/adult mammalian heart, including the human heart. The different cardiac stem/progenitor cells show a mixed yet mostly overlapping expression of several membrane markers and transcription factors (which were differently used for their initial identification). (B) Lin−c-kitpos CSCs are multipotent, giving rise to a minimum of three different cardiogenic cell lineages: cardiomyocytes (CMs), smooth muscle and endothelial cells (SMCs and ECs, respectively). In myogenic differentiation medium, administration of BMP-2, BMP-4, TGF-β1, Wnt-11, Activin-A and DKK-1 induce functional myogenic commitment of multipotent CSCs.
The first report on the endogenous cardiac stem cells (CSCs) [12] described acardiac cell population isolated and identified through the expression of the commons stem cell markers c-kit [56], Sca-1 [57] and MDR-1 [58]. Importantly, the isolation of these cells was initially based exclusively by the c-kit expression in a mixed cells population sorted using immunomagnetic microbeads or FACS sorter from old mice [12]. Stem cell properties were tested in the isolated c-kit cardiac cells after their harvest in culture followed by their amplification for several passages. Clearly, the age of the animal donors and the in vitro expanding protocols may have selected for a more homogenous and activated cell population when compared to the real freshly isolated cells. Yet, despite the latter crucial technical aspect and despite the fact that c-kit is a marker for a heterogeneous cell population within the heart [59,60], often it has been assumed that the lone identification of c-kit expression in a cardiac cell equates to the identification of a CSC [32,35–37,61]. This broad extrapolation has produced considerable controversy about the nature, physiological role and regenerative capacity of what has come to be called the “c-kitpos cardiac cells” [32,35–37,61,62].
The identification of a cell population expressing c-kit is necessary but insufficient to define a specific c-kit positive stem cell [25,49], because the isolation of the total c-kitpos cardiac cells in the heart harvest a mixed cell population containing at least two or more precursors, each differentiating into a different cell lineage. On the other hand, the combined use of positive/negative cell surface markers, together with c-kit, identifies the only population of cells with the properties of adult multipotent CSCs within the adult heart [25]. In rodent (as well as human) cardiomyocyte-depleted freshly isolated cell preparations, only a small fraction (~10%) of the total c-kitpos cells in the heart possess sizable tissue-specific stem/progenitor characteristics and properties. On the other hand, ~90% of c-kitpos cells co-express blood markers of the myeloid, lymphoid and erythroid lineages and endothelial progenitor lineage markers such as CD45 and CD31 (Linpos) [25]. Thus, a negative sorting (Lin−) for CD45 and CD31 is necessary to eliminate from the c-kit cardiac cells the vast majority of lineage-committed cells, whose presence dilutes the identification of CSCs. Half of the Lin− c-kitpos cardiac cells (that are enriched for CSCs) express Sca-1 or PDGF-Rα and roughly 20% express both Sca-1 and PDGF-Rα (Figure 2). Incidentally, PDGF-Rα has been used as marker to mark the clonogenic and cardiogenic potential of cardiac stem/progenitor cells expressing Sca-1 [63]. Additonally, PDGF-Rα provides an efficient means to purify epicardium-derived adult multipotent clonogenic cardiac progenitor cells [50]. Using a simple technical strategy by a single negative selection with CD45 antibody it is possible to harvest cells in culture that are practically negative for CD31 and CD34 [25,49]. This is a widely-applicable and inexpensive experimental approach to obtain a less heterogeneous c-kit positive cell population that express in different percentages Sca-1, Abcg2, PDGFR-α, Flk-1, MDR-1 and CD166. Of these CD45negCD31negc-kitpos cardiac cells, 10% are clonogenic and serially expandable at single cell level. These cells are enriched for cells capable to differentiate, in vitro and in vivo, into mature and functional CMs as well as vascular cells [25,43,49,55,64]. Within the freshly isolated CD45negCD31negc-kitpos cardiac cells there are harboured cells that express pluripotency genes such as Oct-4, Nanog, Klf-4 and Sox-2, as well as stemness regulatory genes involved in cell proliferation and self-renewal and the typical transcription factors of the early stages of cardiac myogenic differentiation (Tert, Bmi-1, Gata-4, Mef2c, Nkx2.5) [52,65–68]. On the contrary, the lineage positive (CD45 and CD31 positive) Linposc-kitpos cardiac cells are unable to form cardiospheres in vitro and do not possess clonal expansion or myogenic differentiation potential whereby are only able to commit to endothelial cells.
Figure 2.

(A) Flow cytometry dot plots (representative of n = 3) show expression of CD45, CD31, c-kit, Sca-1 and PDGF-Rα in the myocyte-depleted total cardiac cells obtained through enzymatic digestion of a mouse heart by retrograde perfusion. (B) After CD45 and CD31 negative sorting, the flow cytometry analysis shows the efficiency of CD45 and CD31 removal from the cell preparation. The CD45/CD31 lineage negative cardiac cells still express c-kit, Sca-1 and PDGF-Rα. Importantly half of the CD45neg/CD31neg c-kitpos cardiac cells (that are enriched for CSCs) express Sca-1 or PDGF-Rα. More importantly, roughly 20% of the CD45neg/CD31neg c-kitpos cardiac cells express both Sca-1 and PDGF-Ra. This negative/positive multiple-marker expression of freshly isolated cells is similarly shown by multipotent single cell-derived CSC clones propagated in vitro, and it represents the minimal or “essential phenotype for the identification and isolation of mammalian adult endogenous CSCs”(adapted from Vicinanza et al. 2017 [25]).
Using a clonal analysis, the only reliable method to identify a stem cell [69], is possible to distinguish between the alternatives in c-kitpos cardiac cells population of the heart. Practically all single cells within Lin−c-kitpos cloned CSCs express c-kit, PDGF-Rα, CD166, SSEA-1, Nestin, Bmi-1, Tert, Gata-4 and Nkx2.5. All cloned CSCs also express the pluripotency genes Oct3/4, Nanog, Klf-4 and Sox-2 [12,25,49,70,71] and are negative for CD45, CD31 and CD34. These cloned CSCs generate cardiospheres at high frequency, which give rise to secondary and tertiary cardiospheres. In the relative dedicated differentiation medium, cloned CSCs reproducibly and consistently turn on the expression proteins specific of the CMs, endothelial and smooth muscle cell lineages.
Therefore, the cells with a demonstrable multipotent CSC phenotype represent only ~1% of the total myocardial c-kitpos population. In order to evaluate the participation of CSCs in heart homeostasis/repair, it is essential to study or to produce cells with clinical relevance and applications in heart regeneration. In our study we propose the above described as the definitive phenotype for the identification and isolation of adult endogenous CSCs (Figure 2) [25], which needs to be taken into account in the design of any CSC fate mapping study or other approach.
Quantifying CSC contribution to adult heart cell homeostasis and regeneration: methods matter
In the last decade, the existence and the potential of tissue-specific endogenous CSCs as regenerative agents has been well documented and by now widely accepted [23,70–72]. Despite some controversy on the actual myogenic potential of CSCs [35–37,62,73], even the most strenuous scepticism has been finally removed with the acceptance that the adult heart harbours clonogenic CSCs able to specify into functional cardiomyocytes in vitro and in vivo [73]. Thus, the discussion moved from the identity to the nature and extent of CSC role in myocardial homeostasis and repair.
“Genetic fate mapping” technology uses a class of molecules known as “site-specific recombinases” that, through the capacity to produce precise DNA excisions, are capable of transforming into a specific cell/tissue a silenced reporter transgene into a constitutively expressed one. Fate map strategy systems are therefore an extremely powerful tool for biologists, establishing the correspondence between individual cells at one stage of development or adult life, and their progeny at later stages of development or life. Thus, genetic fate map systems, mainly Cre/Lox technology, have been used to analyze the contribution of c-kit expressing CSCs in the heart homeostasis/repair.
Using a lentiviral construct carrying the Cre recombinase driven by the c-kit promoter [23] to prospectively and specifically tag c-kit expressing cells in the adult myocardium of mice with a floxed reporter gene [23], we rigorously documented that true c-kitpos CSCs efficiently differentiate into bona-fide cardiomyocytes in vitro and in vivo [23] These and additional results led to our conclusion that endogenous CSCs are necessary and sufficient for cardiomyocyte regeneration/replenishment after injury [23].
Despite the reproducibility of the published data showing the proper identity and myogenic potential of CSCs, considerable confusion has arisen recently about the physiological role and regenerative capacity of what has come to be called the “c-kitpos cardiac cells” [32,33,35–37,62,73–75]. This controversy was initiated by reports that c-kit expressing cardiac cells possess a robust cardiomyogenic potential in the neonatal period but it becomes significantly reduced in the adult [61,75], a change which coincides with an increase in myocardial c-kitpos mast cells lowering the relative abundance of true CSCs among the “c-kitpos cardiac cells”. More problematically, three recent publications which did not attempt to replicate our published work, have challenged the conclusion that the CSCs are responsible for replacement of CMs lost by wear and tear and after injury [35–37]. van Berlo et al. [35], Sultana et al. [36] and Liu et al. [37] showed that the tagged c-kitpos cells generated on the short and long term a small/minimal number of CMs. Therefore, they concluded that there is adult cardiomyogenesis which undoubtedly is generated by the (some?) so-called “c-kitpos CSCs”. However, this differentiation is negligible at best. Whether their results were due to a low myogenic potential of all the “c-kitpos CSCs” or to a few tagged “c-kitpos cells” with a high myogenic potential was nevertheless not addressed. Despite the significant negative impact that these papers have had on the field of myocardial biology and repair/regeneration, they have some critical shortcomings which should have been addressed, but were not, before their publication [60,76]. Indeed, using a fate mapping strategy, it is important to establish the extent to which Cre expression matches that of the endogenous gene promoter. If Cre levels trigger recombination in only a subset of those cells in which the specific promoter is normally active, the resultant fate map will underestimate the descendant population. Thus, it is critical to determine whether all or only part of an initial cell cohort, identified by expression of the cell type-specific gene used to drive Cre, is being fate mapped. This is a significant issue if the Cre-driving gene exhibits a heterogeneous level of expression in different cell types of the cohort. It is possible, indeed likely, that the cells with lowest expression of Cre in this heterogeneous population might fail to have their fate tracked because their level of Cre is below the threshold needed to trigger site-specific recombination and ensuing reporter expression [76] It is then fundamental to check for all these potential pitfalls to avoid false negative results [76].
Importantly, van Berlo et al. [35], Sultana et al. [36] and Liu et al. [37] have knocked-in (KI) Cre into exon 1 of the c-kit mouse locus (c-kitCre allele). Using these mice, the three publications have concluded that endogenous c-kitpos cells mainly differentiate into endothelial cells and minimally, if at all, form new cardiomyocytes [35–37]. Some even state that the “cardiac c-kitpos cells” are not CSCs at all but endothelial cells and their precursors [36]. Instead of cardiomyogenic potential, these papers report that “the cardiac c-kitpos cells” have a largely vasculogenic and adventitial cell lineage predisposition. This result was not unexpected considering that >90% of c-kitpos cells in the adult heart are CD45 positive and CD31 positive [25]. Moreover, the fraction of c-kitpos cells which become genetically tagged in these mouse strains, resemble the bone marrow derived c-kitpos/Sca-1pos/Flk-1pos cells identified by Fazel and colleagues [77]. These cells, in response to injury, home to the heart and contribute to the revascularization of the damaged area.
In order for a cell to be defined a stem cell, it must exhibit “stem cell” properties: clonogenicity, self-renewal and multipotency in vitro and in vivo. Identifying, tracing and characterizing stem/progenitor cells according to expression of a single surface receptor such as c-kit [35–37,61,75], is not sufficient to identify the CSCs. As shown above, the majority of the total CSCs (~90%) are mast cells and endothelial (progenitor) cells while only ~1% are demonstrably multipotent clonogenic CSCs but the mentioned publications assumed that all, or most, c-kitpos cells in the heart are CSCs. Thus, relying on genetic tagging to determine the prospective fate or regenerative potential of c-kitpos cells within any tissue, including the heart, and for any quantification is a major biological and practical pitfall. At a minimum, the authors should have determined first, by single cell testing, what fraction of the “c-kitpos CSCs” are true CSCs and what fraction of these recombine the marker gene after induction of Cre. The publications have challenged the role and/or existence of the CSCs, all using a cell-specific genetic cell-fate mapping strategy whereby Cre (constitutive or TAM-inducible) was knocked-in the Exon1 of the c-kit locus [35–37]. Their faulty rational was that Cre/lox KIs are fool proof to track the fate of the myocardial “c-kitpos cells” and to identify and quantify their myogenic contribution [33]. However, all the Cre KIs in the c-kit locus reported so far, including those under discussion [35–37], have rendered the targeted allele a null mutant, resulting in hemizygous expression and a c-kit protein deficiency [60,78–80]. These c-kit hypomorphs exhibit growth and differentiation defects in many stem and somatic cell types [60,78–80]. More critically, the Cre-dependent recombination efficiency is directly proportional to the level of Cre expression from the mutated c-kit allele. [60,76,81]. Therefore, because of the low level of c-kit expression in most stem cell types [82,83], and particularly in the c-kitpos CSCs [83], and the low abundance of CSCs among the “c-kitpos cells”, it was highly questionable whether the hemizygous c-kitCre-KI strategy could recombine a meaningful fraction of the c-kitpos CSCs to track their fate [60] among the noise generated by the easier to recombine mast and endothelial progenitor cells (EPCs).
Our recent data shows that c-kit is expressed in CSCs at a significantly lower level than in the mast cells and EPCs [83]. Inexplicably, the efficiency of Cre-recombination of the CSCs, a critical assumption of their experiment, was not determined. We have therefore addressed this issue and characterized the effect of the c-kitCre-KI-insertion on CSC biology and cardiomyogenic potential [83]. Our data shows that c-kit expression level in the CSCs is too low to produce enough Cre to effectively recombine the floxed marker and tag the CSCs and their progeny [83]. c-kitCre-KI model [35,36] only minimally if not negligibly tag and fate map resident CSCs. Furthermore, Cre-KI into the first intron of the c-kit locus in all cases has produced a null c-kit allele which is responsible for the W phenotype of these mice; the c-kitCre null-allele fatally impairs in vitro and in vivo CSC growth, self-renewal, myogenicity and regenerative potential, properties which are rescued by BAC-mediated single-copy c-kit transgenesis [83]. These fate map strategies investigate neither the identity nor the fate of CSCs because the protocol used fails to tag the vast majority, if not all, of them. The low number of c-kitpos progenitor-generated cardiomyocytes detected in the c-kitCre-KI mice, simply reflects the absence of efficient recombination in the CSCs to track their progeny in foetal and adult life together with the defective myogenesis consequence of the mutated c-kit allele in the CSC [83]. Furthermore, proper c-kit expression is necessary for cardiomyogenesis, a conclusion in agreement with our recent finding that a gain-of-function mutation in the c-kit kinase domain increases CSC’s myogenic and angiogenic potential in vitro and in vivo [25,84].
Taken together the results shown here reinforce our previous conclusion [23] that the CSCs are necessary and sufficient for robust cardiomyogenesis and to support myocardial regeneration/repair in response to diverse types of damage. Confirmation of these conclusions should clear the way for the potential development of CSC-based myocardial regenerative protocols.
Cardiac morphogens drive adult CSC commitment in bona-fide cardiomyocytes
Cardiac development is a highly interconnected process regulated by several morphogens families. Well-orchestrated signalling pathways regulate various interaction between transcriptional and growth factors, underlying the future cardiac identity of progenitor cells by promoting their proliferation or differentiation. The embryonic cardiac progenitor cells originate from mesoderm and are detected first in the cardiac crescent, where myocardial cell differentiation takes place, through the stage-specific progressive expression of cardiomyogenic transcription factors such as Nkx2.5 and Gata-4 [85,86]. Gata-4 acts with Nkx2.5 to initiate the cardiac specification program; Tbx-5, as well as Mef2c factor, are required for full myogenic differentiation [65,87,88]. Cardiac progenitor cell proliferation in mesoderm is regulated by canonical Wnt pathway, FGF and Hedgehog pathways. On the contrary, Notch and non-canonical Wnt signalling regulate the differentiation events during heart formation [89–92]. Integrated with the above cardiopoietic growth factors and molecular targets’ signaling, non-coding RNAs and epigenetics play a crucial role in cardiac development and myogenic specification of embryonic cardiac progenitors [93–96]. However, this aspect is beyond the scope of this review and the readers are therefore referred to relevant and elegant reviews on this topics [93–97].
The key molecular steps involved in cardiac development have been employed in vitro [44,45] to test whether the known cardiac myogenic morphogens were able to specify mammalian adult cardiac progenitors, including human, into CMs through the inhibition/stimulation of the relative specific pathways in defined conditions [44,45]. The possibility to translate the in vitro mechanistic evidence of CSC differentiation in future applications to be used in in vivo experiments, will be indeed an important strategy for CSC activation in situ. Cloned Lin−c-kitpos CSCs express and respond in vitro to the Wnt/β-catenin and TGF-β/SMADs signalling pathways, as it would be expected from true cardiac-specific stem/progenitor cells [25]. Modulating the molecular signalling of these known cardiac morphogens through gain and loss of function experiments we investigated their involvement in the self-renewal potential and cardiomyogenic specification of the CSCs to generate fully differentiated contracting CMs in vitro [25,98–101]. Cultured CSCs express the cell-surface receptor for TGF-β/SMAD signalling, TGF-β-R1. The TGFβ family, comprising also BMPs and Activin A, plays critical and specific roles in cardiac development and CM commitment [102]. TGFβ1 is expressed at high levels in the heart during embryonic development and in the adult life. TGFβ2 null mice exhibit multiple cardiac developmental abnormalities [103]. During heart development, the signalling cascade induced by BMPs antagonizes the pro-proliferative role of FGF signalling and promotes progressive myocardial differentiation and maturation [104]. Accordingly, in vitro administration of TGFβ1 or Activin-A specifically induces SMAD2 phosphorylation in CSCs activating the myogenic commitment of these cells. We showed that by transfecting c-kitposCSCs with a lentiviral vector carrying a short hairpin RNA specific for SMAD2 (Smad2shRNA), determining the destruction of TGF-β signalling dependent on SMAD, CSC cardiomyogenic commitment is prevented [25]. Indeed, Smad2shRNA reduces CSC myocyte specification and completely blocks TGF-β1 and Activin-A-dependent positive myogenic effect. Interestingly, either increasing or blocking TGF-β/SMAD-2 axis has no effect on CSC self-renewal and amplification.
CSCs also express the cell-surface receptor of Wnt/β-catenin canonical pathway, Frizzled (FZD), as well as its co-receptor LRP-6. Wnt proteins are a large family of glycoproteins related to the Drosophila wingless gene [105]. Canonical Wnt pathway begins when Wnt binds the Frizzled receptors (Fzd), triggering a variety of intracellular responses that end into the stabilization of the levels of β-catenin protein, a transcriptional coactivator that entering the nucleus interacts with T cell transcription factor (TCF/LEF), activating the Wnt target genes. Positive and negative modulators of Wnt/β-catenin axis act through respectively stabilizing or degrading β-catenin and its LEF/TCF-dependent transcriptional activity. Soluble Wnt-3a as well as Wnt-3a-conditioned medium improves CSCs expansion and clonogenicity while canonical Wnt pathway inhibition by Dickkopf 1 (Dkk1) or by constitutively active GSK-3β signalling induces myogenic commitment of CSCs by promoting differentiation with the inhibition of proliferation and clonogenicity potential [25,106,107]. On the other hand, conditional expression of β-catenin, results in expansion of CSCs preventing their myogenic commitment.
These tests provided the basis for selecting a set of myogenic inducers which were used together in a stepwise and stage-specific myogenic differentiation assay on cardiospheres [49]. The latter assay was employed as an in vitro developmental model system to follow cardiomyocyte specification of the c-kitposCSCs, in a similar manner with which embryo bodies are used to assess myogenic specification of pluripotent stem cells [108]. Thus, using this cardiosphere myogenic assay we characterized the CSCs’ cardiomyogenic induction program over 14 through 21 days [25].
In myogenic differentiation medium, administration of BMP-2, BMP-4 and Activin-A, significantly increase the expression of myogenic lineage markers and the number of troponin positive cells (Figure 1 and Figure 3). Modulation of each of them is indispensable to generate fully differentiated contracting cardiomyocytes. We have shown that using different concentrations of BMP-4 and Activin-A for 4 days, the fraction of troponin positive cells increase to ~40%. Adding Dkk-1 from day 5 increase to ~60% the troponin positive cells. Removing BMP-4 and Activin-A at day 5 further increased the number of troponin positive cells to ~70%. Through immunofluorescence staining for the cardiac-myofilament proteins, CSC-derived single CMs display a well-organized sarcomeric structure with clear cross-striations and connexin-43 gap junctions by day 21 in culture (Figure 3) [25].
Figure 3.
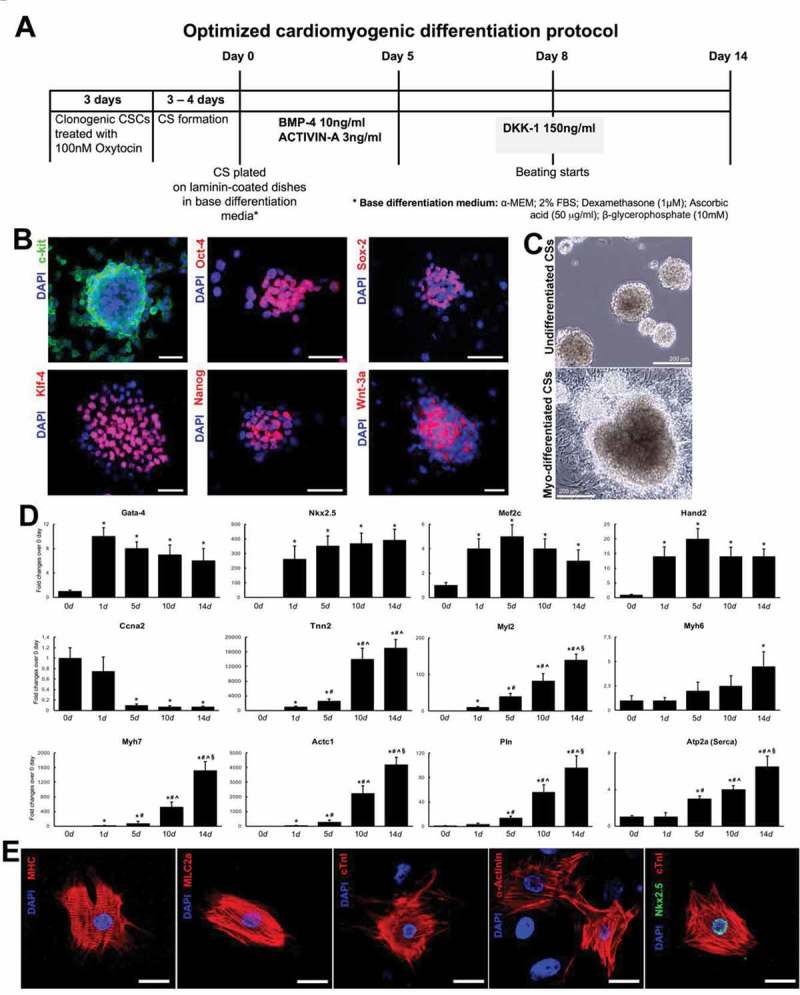
(A) Schematics of optimized cardimyogenic differentiation protocol to obtain in vitro CSC-derived cardiomyocytes. (B) c-kitpos (green) CSC cardiospheres express multipotent stemness markers and Wnt3a (red). Bar = 50µm. (C) Representative light microscopy images showing (top) clonal CSC-derived cardiospheres grown in suspension and (bottom) differentiated cardiospheres 14 days after the treatment with the optimized stage-specific TGF-β-Family/Wnt Inhibitor cocktail. (D) Cumulative RT-PCR data for the expression of myogenic genes in CSC-derived cardiospheres from day 0 to day 14 in the optimized stage-specific TGF-β-Family/Wnt Inhibitor cocktail. Ccna2, Cyclin A2; Tnnt2, cardiac troponin t2; Myl2, myosin light chain 2; Myh, myosin heavy chain; Actc1, cardiac Actin; Pln, phospholamban; Atp2a, ATPase Sarcoplasmic/Endoplasmic Reticulum Ca2+ Transporting 1 (also known as SERCA Ca(2+)-ATPases). *p < 0.05 vs. 0d; #p < 0.05 vs. 1d; ^p < 0.05 vs. 5d; §p < 0.05 vs. 10d. (E) Dissociated cells from CSC-derived beating cardiospheres stain positive for the cardiomyocyte lineage, exhibiting sarcomeric structures. MHC, myosin heavy chain; MLC2A, myosin light chain 2A; cTnI, cardiac troponin I. DAPI (blue staining) depicts cell nuclei. Bar = 20 µm..(adapted from Vicinanza et al. 2017 [25]).
We have then specifically assessed whether cardiomyogenic differentiation of CSCs in vitro follows a coherent and coordinated stage-specific sequence to determine whether the induction program in CSC myogenic commitment mimics the one seen in embryonic or pluripotent stem cell cultures. Starting at day 0, CSC-derived cardiospheres in cardiomyogenic induction media were harvested at day 0, 1, 5, 10 and 14. RT-PCR analysis of main cardiac transcription factors and myocyte contractile genes show that Gata-4, Nkx2.5, Mef2C and Hand2 are mainly induced in the early time points whereby contractile genes, cTnnt2, Myl2, Myh7 and Actc1, and reached their peak of expression at the late stages (Figure 3). Intriguingly, RNA Seq analysis of whole transcriptome show that CSC-derived CMs are still immature when compared to adult cardiomyocytes yet they are practically indistinguishable from neonatal cardiomyocytes [25].
The differentiation program activated in CSCs in culture by the stage-specific cocktail of cardiopoietic morphogen inducers produced rhythmic and spontaneous beating cell clusters [25]. The shape and time course of the action potential of CSC-derive CMs was similar to that of neonatal rat ventricular myocyte (NRVM) action potentials measured using optical mapping techniques [25]. Spontaneously contractile CMs derived from cloned CSCs exhibited robust calcium transients [25]. Calcium transient decay is brought about by re-sequestering Ca2+ into the sarcoplasmic reticulum (SR) by SERCA and extrusion from the cell by the sodium calcium exchanger (NCX), with minor contributions from additional slow mechanisms. Mechanisms of calcium transient decay show SERCA and NCX to contribute approximately equally for CSC-derived CMs. Application of specific relative activators/inhibitors revealed the presence of robust physiologic stimulatory β-adrenergic and inhibitory muscarinic acetylcholinergic signalling pathways.
Our data, as summarized above, show that the contemporary inhibition of the Wnt canonical pathway and activation of TGF-β family cascade promotes functional cardiomyogenic commitment in adult CSCs. However, other morphogens such as Hippo, Notch and Hedgehog signalling have become targets to promote CSC myogenic differentiation in vitro and in vivo. A link between Wnt/β-catenin pathway and Notch signalling has been indeed well demonstrated [109]. Notch normally attenuates the proliferative effect of canonical Wnt signalling [110]. Pharmacological inhibition of Notch in vivo interferes with endogenous progenitor commitment and repair after myocardial infarction [111]. Wnt signalling is also modulated by Hippo pathway, an important mediator of cardiac growth and embryonic heart size through the regulation of cell proliferation and apoptosis [112]. The principal Hippo effector Yap (Yes-associated protein), when is dephosphorylated, interacts with β-catenin negatively regulating Wnt/β-catenin signalling [113,114]. Considered a key component in maintaining pluripotency, the Hippo pathway can regulate proliferation and self-renewal of stem cells, while targeting Yap effector using small molecules or by induction its degradation through phosphorylation, may restore CSC differentiation. The Hippo and Wnt/β-catenin pathways may be modulated using several growth factors in order to promote CSCs re-entry in cell cycle and proliferation or their exit from cell cycle inducing differentiation. Activation of Hippo effectors YAP/TAZ may to be a potential strategy to promote tissue regeneration [115,116]. Hedgehog signalling is necessary for cardiomyocyte differentiation, is upregulated during ischemic heart injury such as MI and coordinates in vivo morphogenesis of the heart [117,118] influencing cell fate but also cell growth and survival [117,118].
Overall, these results provide indisputable evidence that Lin−c-kitpos CSCs are cardiomyogenic and respond to known cardiac morphogens involved in myogenic commitment during heart development to generate contracting cardiac muscle cells as would be expected from true cardiac-specific stem/progenitor cells (Figure 1). This conclusion is further strengthened by the normal structural organization and functional activity of the cardiomyocytes generated in vivo in response to CSC transplantation (Figure 4) [12,23]. Importantly, the cardiomyocyte differentiation program operative in c-kitpos CSCs follows a step by step finely-regulated molecular cascade that is closely reminiscent of the known molecular program regulating cardiac development from primary heart tube to the fetal/neonatal heart. Accordingly, the in vitro myogenic specification of clonogenic adult CSCs produces bona fide cardiomyocytes whose structural, molecular and functional maturity is practically indistinguishable from neonatal mammalian cardiomyocytes. The latter shows that the phenotypic maturation of in vitro CSC-derived CMs is at least similar (if not superior) to the one reached by in vitro CMs derived either from myogenic commitment of pluripotent stem cell cultures (murine and human) as well as from the different protocols using either transcription factors or small molecules to induce the direct reprogramming of somatic cells [119,120].
Figure 4.
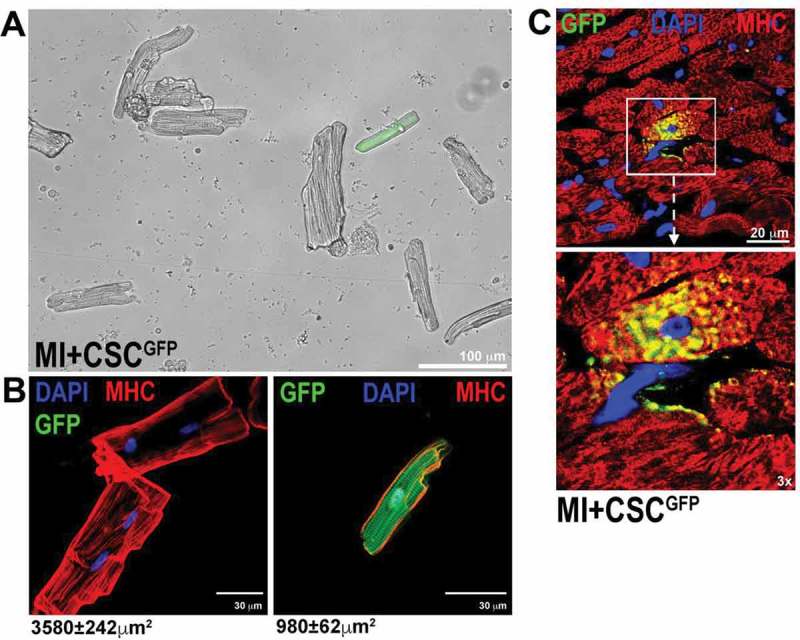
(A) Light microscopy image of freshly isolated adult cardiomyocytes from a dissociated heart 28 days after myocardial infarction (MI) and CSCGFP injection (MI+CSCGFP) show a CSC-derived GFP-positive cardiomyocyte. (B) Confocal microscopy images show GFPpos cardiomyocytes isolated from MI+CSCGFP rat hearts at 28 days after MI. Note that GFPpos cardiomyocytes are of smaller size and mono-nucleated when compared to surviving bi-nucleated GFPneg cardiomyocytes of the host. (C) Representative confocal images show at high magnification a CSC-derived newly formed GFPpos cardiomyocyte in the infarct-border zone 28 days after MI treated with CSCGFP.(adapted from Vicinanza et al. 2017 [25]).
C-kit function in endogenous cardiac stem cell and cardiac regeneration
c-kit has been mainly used in CSC biology as marker or a extracellular cluster of differentiation (CD) molecule to detect and isolate these cells [12,23,49,60,121,122]. However, c-kit is a gene necessary for life whose role in CSC function for cardiac repair and regeneration has been recently started to be elucidated [23,25]. The proto-oncogene c-kit produces a protein belonging to the class of the type III tyrosine kinase receptors, which are mainly composed of an extracellular ligand binding domain, a transmembrane region and an intracellular kinase signalling domain [74]. Alternate splicing of murine c-kit mRNA results in two isoforms of c-kit that are expressed in different ratios in various cell types and differ in their signalling capabilities [79,123]. The binding of c-kit receptor with its natural ligand, the stem cell factor (SCF), which also exhibits two distinct isoforms with different biological functions, induces receptor homodimerization and autophosphorylation of the intracellular tyrosine kinase domains leading to the modulation of different signalling pathways, which are essentials in the regulation of cell survival, function, migration and proliferation and in stem cell maintenance, hematopoiesis, gametogenesis, mast cell development and melanogenesis [79,80,124]. Mice lacking c-kit gene present germ cell and melanocyte defects and die in the first days of postnatal life because of impaired hematopoiesis [79,80,124] and defective c-kit signalling leads to compromized cardiac function [125,126]. Genetically mutant mice deficient in c-kit signalling show a worsened cardiac remodelling after MI [77]. On the other hand, transgenic mice with over-activation of c-kit signalling exhibit an improved myocardial repair after MI [127,128].
To investigate the role and the function of c-kit signalling in CSC biology and heart repair, we have generated a transgenic mice carrying a mutated version of c-kit gene that leads to the constitutive activation of the c-kit receptor in cardiac tissue and isolated cardiac cells derived from c-kit-activated transgenic mice [84]. c-kit receptor exerts a beneficial protective/regenerative role in myocardial tissue after injury, improving cardiac remodelling and repair and fostering activation and differentiation of CSCs mainly through MAPK and AKT signalling activation. Indeed, ERK1/2 and AKT phosphorylation, which are the main downstream molecular effectors of c-kit receptor signalling, are increased in heart tissue and isolated/cultured CSCs derived from the c-kit-activated transgenic mice [84], and these signalling pathways are essential in the modulation of the activation and endothelial/myogenic differentiation of CSCs. The signalling generated from the constitutive c-kit receptor activation, increases cardiac repair in vivo as well as angiogenic and myogenic response to injury with a significant improvement of survival, confirming the bi-potent fate of c-kitpos cardiac cells to support both endothelial and cardiomyocyte differentiation [84].
In many cell types, the expression of c-kit is lost upon cell differentiation, indeed c-kit is widely used as a “cell surface marker” to identify stem cells such as hematopoietic stem cells, myeloid progenitor cells, dendritic cells, mast cell and pro-B/T cells. Accordingly, c-kit protein expression is lost in mature cardiomyocytes derived from CSC myogenic commitment [23]. However, c-kit is transiently induced during the cardiac morphogens-regulated stages of progressive specification of uncommitted CSCs into cardiomyocytes [25,49]. The latter is also in line with the independent in vivo evidence that c-kit appears to be involved with terminal differentiation of neonatal cardiomyocytes becoming mature adult cardiomyocytes [129,130]. This phenomenon also brought to the evidence that c-kit mRNA is induced in a small number of adult cardiomyocytes mainly in response to injury [130,131], somehow appearing to participate in the molecular network characterized by the reactivation of several foetal genes in pathological overloaded cardiomyocytes [130,131].
The gain of function data above described clearly show that the positive modulation of c-kit function exerts beneficial effects on CSC regenerative potential. On the other hand, the c-kit null allele produced by Cre insertion in the c-kit locus to develop c-kitCre-KI mice offers a typical loss of function assay to establish whether normal c-kit content is necessary for adult CSC biology. To this aim, we further tested the efficiency of Lin−c-kitpos CSCs obtained from c-kitCre-KI mice compared with wild type Lin−c-kitpos CSCs (wtCSCs) before in vitro and then transplanting them in vivo in a murine myocardial infarction model [83]. In both the in vitro and in vivo tests, we show that Cre knock-in induced a typical White (W) mutation in c-kitCre-KI mice that significantly reduced CSC amplification while nearly abolished cardiomyogenic potential of these cells. Remarkably, restoring the normal level of c-kit expression in CSCs obtained from c-kitCre-KI mice through transfection of a BAC construct spanning the entire c-kit gene locus, the regenerative effects of these cells in vitro and in vivo are reestablished [83].
Overall, these data show that adult CSC biology and regenerative potential in vitro and in vivo requires and is dependent upon a diploid level of c-kit expression (Figure 5). However, the main findings emanating from the use of c-kitCre mice raise an intriguing and un-expected aspect of c-kit in cardiomyogenesis. Indeed, the available evidence seems to portrait a dichotomy on c-kit role during heart embryonic development as opposed to adult cardiac regeneration. c-kit deletion as it occurs in homozygous W-mutated mice [74] is incompatible with life and mice die prematurely in the very last foetal days or very early in the neonatal life. However, those mice appear to have a heart anatomically and macroscopically normally developed [35]. On the other hand, c-kit-defective adult hearts and adult CSCs (from heterozygous c-kitCre-KI mice) have a significant defect in their regeneration potential in vivo. The latter is unpredicted when considering all the attempts currently undergoing to decipher the pathways of developmental cardiac generation and neonatal heart regeneration to instruct effective protocols of adult cardiac regeneration.
Figure 5.
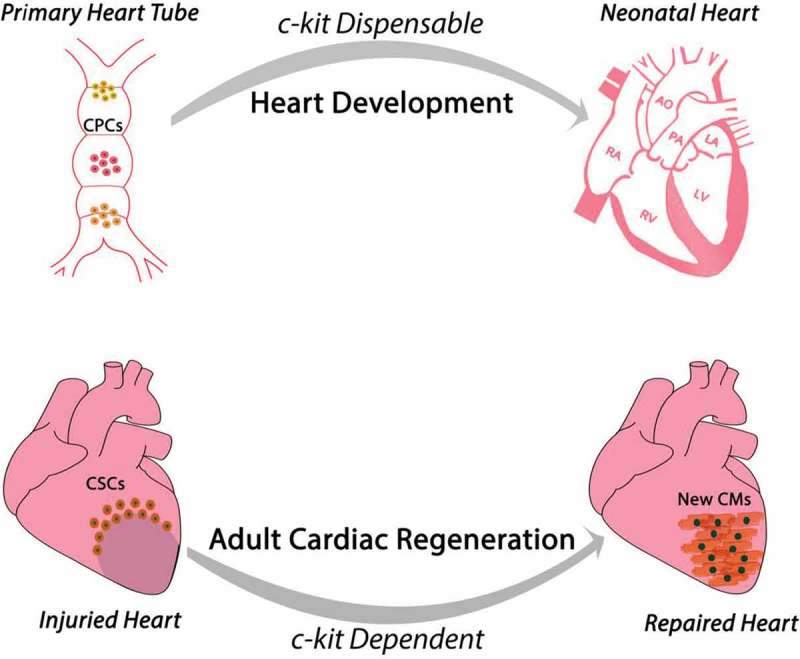
Proposed function and role of c-kit during heart embryonic development and adult cardiac regeneration. The available evidence portraits a dichotomy of c-kit role in these two processes despite the current hypothesis that cardiac regeneration resembles cardiac development. During development c-kit expression appears dispensable for complete heart formation despite c-kit deletion is incompatible with life. On the contrary, adult CSCs biology and myocardial regenerative potential requires and is dependent upon a diploid level of c-kit expression.
Quiescence vs aging of the CSCs: exiting & entering or permanently staying out of the cell cycle
All the above data show how CSCs can be coaxed to become cardiomyocytes using cardiac morphogens and their related molecular cascades. However, these cardiac developmental cues are active during a process of physiologic organ generation and maturation. No data exists to show that these mechanisms are similarly active in the adult myocardial tissue and especially in response to cardiac injury. In other words, it is still unknown whether adult endogenous cardiac regeneration recapitulate cardiac development/generation. In multicellular organisms, post-natal tissue homeostasis and regeneration following injury are mainly mediated by adult (or tissue specific) stem cells, which are maintained within uninjured tissues in a quiescent and undifferentiated state [132–136]. Quiescent adult stem cells however are rapidly activated upon tissue injury, leading to the production of transit amplifying progenitors that in turn differentiate into functional mature cells capable of tissue regeneration [132,133,137]. A small population of transit amplifying adult stem cells exits the cell cycle and re-enters quiescence to maintain a reserve of quiescent tissue-specific stem cells that can respond to future demands [132,133,137]. From an evolutionary perspective, this “quiescence cycle” helps to ensure a steady state number of tissue-specific stem cells available for tissue regeneration, and act as a protective mechanism against genotoxic stresses and progressive replicative senescence [132,133,137,138].
The efficacy of the endogenous mechanisms of heart repair in general, and the ones mediated by CSCs in particular, is limited by the hostile cardiac microenvironment generated in pathological conditions, characterized by ischemia, inflammation, fibrosis and inadequate angiogenesis [139]. This microenvironment prevents CSC activation from their quiescence impairing also their mobilization and homing or producing eventually a senescence state during aging. To understand the mechanisms and the biology of CSC quiescence and aging is therefore pivotal in order to develop approaches for their manipulation and/or to therapeutically target them in vivo to enhance and improve their myogenic differentiation potential for cardiac tissue maintenance or repair. In the healthy adult myocardium, ~90% of the c‐kitpos CSCs are quiescent [23,25]. In response to the diffuse injury by catecholamine overdrive, most c-kitpos endogenous CSCs enter the cell cycle, multiply and progress to the commitment toward the myogenic lineages, and finally differentiate into new cardiomyocytes [23] among other cell types. Interestingly, once the Isoproterenol-induced myocyte loss and cardiac function failure have been corrected by the regenerated cells, the number of activated CSCs diminishes progressively with the concomitant increase of quiescent endogenous CSCs [23]. Therefore, the return to myocardial homeostasis is accompanied by the return of activated CSCs to their quiescent state. This loop of the endogenous CSCs from dormancy to activated and back to quiescence appears therefore similar to what it has been described for other stem cell populations from other self-renewing organs [140,141]. As the prevalent paradigm still views development and differentiation as a unidirectional process going from broader to restricted potential, this “backward trip” of the CSCs, from activated to quiescent, awaits a molecular explanation. Thus, the reversibility between the activated and dormant states of the CSCs, might just highlight the important role that these cells play in the homeostasis of a tissue which is essential for organism survival. Gaining a better understanding of the mechanisms by which activated endogenous CSCs return to the quiescent state is an intriguing biological issue which will open new avenues to exploit the regenerative potential of these cells.
As it is well demonstrated for the majority of adult stem cells, also CSCs appear to be located in a hypoxic niches organized in cluster, higher in the atrial and apical myocardium [132,134,136,142]. Within the niches, chemical and physical signals modulate the behaviour and fate of stem cells promoting self-renewal or their migration for differentiation. The niches harbours cycling cells and cells that are rarely or slowly cycling that remain dormant, in a G0 state, until they have to become functional, for example in response to stress or injury [12,42,136,143]. A small population of these activated stem cells exits the cell cycle and re-entry in a quiescent state to maintain a reserve of stem cells, indispensable for tissue homeostasis and regeneration. The existence of a quiescent state was first postulated by Howard and Pelc using a radioactive labelling techniques to study DNA replication during cell division [144]. Quiescence, described as “sleeping beauty state”, is an active and reversible state relevant for stem cells function and integrity, characterized by the absence of cell proliferation. Several molecules, transcription factors and growth factors play a key role in stem cells fate decisions. Any environmental change leads to mobilization of resident cells within the niche and integrins protein and adherens junctions play critical roles in maintaining the location, adhesiveness and proliferative potential of stem cells within tissues. A precise network of signals evolutionarily conserved into the niches are involved in maintaining stem cell quiescence or re-entry in cell cycle [145,146], including the above cited morphogens. Key signals networks including Wnt, Notch and FGF pathways are important in stem cell renewal/quiescence and differentiation. As above reported, Wnt signalling modulation is key in regulating CSC activation and differentiation [25,147]. Notch signalling is involved in tissue maintenance and contributes to cell fate decisions during tissue regeneration, amplifying progenitors in many tissue compartments in a context- time-dependent manner and balancing the maintenance of stemness or differentiation via Nkx2.5 gene activation [148–151]. The Hippo signalling has been shown to have a relevant role in mediating stem cell function within their niche [113,152,153]. The retinoblastoma (RB) family members, on the other hand, have the ability to maintain adult stem cells in a quiescent state and their niches in a repopulation mode in order to protect stem cells from premature differentiation [137,154]. The Rb is the major target of two proteins involved in cell cycle regulation: cyclin-dependent kinase (CDK) 2 and 4. The proteins p21, p27 and p57, members of Cip/Kip family of cyclin-dependent kinase (CDK) inhibitors, also regulate stem cells quiescence [137]. In particular p57 is the most abundant family member in quiescence. Downstream target of the CDK inhibitors are CDK2/4, and therefore they maintain Rb in a un-phosphorylate state to keep cell cycle quiescence [137,154]. The understanding of how the different cardiac morphogens regulate cell cycle quiescence and activation will be pivotal to foster endogenous regeneration from CSCs after injury.
On the other hand, if cardiac cell homeostasis is dependent on myocyte and vascular cell regeneration from the activation of resident endogenous CSCs, it can be predicted that loss of CSC function, either as a consequence of their death or because they become non-productive, should result in progressive myocyte loss and impaired ventricular function [155]. Proliferation of mammalian cells, including human, is dependent on having functional telomeres [156–159]. Adult somatic stem cells, like ES cells, are self-renewing. Most express telomerase, which restores telomere length after each cell replication and prevents “replicative senescence”. However, hematopoietic stem cells (HSC) have a finite lifespan and develop a senescent phenotype [160]. Telomere shortening in the absence of telomerase activity is one of the major causes of loss of proliferation in mammalian cells [161,162]. Telomere attrition occurs with aging and as a consequence of oxidative stress and it has been proposed to be a cause of replicative cell aging. Progressive loss of telomere sequences, like in telomerase knockout mice (Terc-/-), eventually leads to loss of organism viability after 3–6 generations [158]. Telomere shortening in the endogenous CSCs from the second and fifth generation of Terc-/- mice is coupled with a profound attenuation in new myocyte formation, increased apoptosis and hypertrophy of the remaining myocytes [158]. These cellular changes are concomitant with wall thinning, ventricular dilatation and dysfunction, which resulted in decompensated eccentric hypertrophy and heart failure [158]. It has also been shown that aged cells, in addition of having lost their self-renewal capacity, they might also have a damaging influence over their neighbouring cells by producing deleterious secreted products. Indeed, Baker and colleagues [163] designed a novel transgene (INK-ATTAC) for the inducible elimination of p16Ink4a-positive senescent cells upon administration of a drug. In the BubR1 progeroid mouse background (mouse strain characterized by premature tissue senescence and disease), the drug-induced clearance of p16Ink4a-positive senescent cells delayed the ageing-associated disorders [163].
We and others have found that in 22-month old mice and old humans (>70 years), a majority of c-kitpos CSCs are “aged”, characterized by impaired proliferation, p16INK4a expression, telomere shortening, “permanent” withdrawal from the cell cycle (putatively senescent), impaired differentiation capacity and increased apoptosis [155,164]. To assess whether the age-related aging of the endogenous CSCs was ruled by an internal cell clock or was in response to the environmental milieu, we studied the natural aging on c-kitpos CSC and its effect on growth and differentiation in wild type (WT) mice as compared to transgenic mice overexpressing Insulin-like Growth Factor-1 (IGF-1) (TG) [155]. At 22 months, 70% of CSCs in the WTs, but only 16% in the TGs, were p16INK4a positive, indicating increased CSC senescence and a block in the cell cycle with age. At 22 months, CSC apoptosis was 5-fold higher in the WTs than the TGs; this resulted in a 33% decrease in myocyte number and cardiac failure in the WTs, but normal ventricular performance and myocyte number in the TGs. Therefore, senescence of endogenous CSCs with age is not regulated by an internal cell or organismal clock but can be modulated by the environment with important physiological consequences. In the WT animals it results in the development of cardiac dysfunction and failure, while this progression is altered favourably in IGF-1-TG mice [155]. Ongoing work also highlight the role that Bmi-1 and Wnt family plays in CSC aging, as it has been established for other tissue specific adult stem cell populations. [165,166].
Therefore, the CSC senescence concomitant with increased organismal age results in decreased CSC functionality, but can be restored by the specific growth factors. In addition to organismal age (physiological), CSC senescence is dramatically accelerated by a variety of pathological stresses such as work overload, and acute as well as chronic ischemia [167]. As mentioned above, there appears to be a feed-back mechanism in which the appearance of aged cells begets more aged cells because of their deleterious effects on their neighbours. These evidences suggest that endogenous CSC aging is a response to the environment and that its phenotype is not irreversible because the “aged CSCs” can be rejuvenated and returned to functionality in response to a limited set of growth factors. Therefore, in order to take full advantage of this biology it will be necessary not only to understand the homeostasis and biology of the aged CSCs but also for their effect on the bystander “healthy” cells.
The results summarized above do not address whether CSC ageing is the result of a stochastic cell autonomous process which affects only the “aged” cells. If this were the case, the aged cell in a given tissue would be impaired and its number and probability to develop the “aged” phenotype would increase with age. However, the “normal/non-aged” cells from the same tissue should be indistinguishable from the “normal-cycling competent” cells of a young donor. On the other hand, if the “aged” phenotype were the result of an age and/or cell-cycle dependent process, which affects the whole endogenous CSC cohort, the aged myocardium will be constituted by a majority of aged CSCs, which are already at or close to express the senescent phenotype. The distinction between these two models of CSC aging is not only academic but has important clinical consequences. Indeed, the future of cardiovascular regenerative therapeutic approaches based on the stimulation of the endogenous regenerative capacity of the myocardium is likely to depend on the answer to this question. If CSC “aging” is a stochastic cell autonomous process it should be possible to rejuvenate the endogenous CSC population by stimulating the growth of the “non-aged” cells or reverting the phenotype of those already aged into self-renewing. However, if CSC “aging” is an age or cell cycle dependent process which affects all or most of the endogenous CSC population, most or all regenerative therapies based on the stimulation of the non-aged cells (calling them out from quiescence) are likely to result in further exhaustion of the self-renewal capability of these cells and accelerated loss of regenerative capacity.
Overall, it will be a better understanding of the biology of these processes, i.e. quiescence and aging, which will pen-ultimately lead to developing realistic and clinically applicable strategies, utilizing the proper stimulation of CSCs and leading to meaningful and functional myocardial regeneration.
Conclusions
Taken together, the data summarized in this review support several conclusions: a) the adult myocardium harbours a tissue specific c-kitposCSC population with robust myogenic potential; b) the CSCs, when properly stimulated, have a robust myogenic differentiation capacity generating fully functional bona fide cardiomyocytes; c) the c-kitCre-KI cell-fate mapping strategy used by van Berlo et al., and their followers [35–37] cannot be used to qualitatively or quantitatively determine the fate and progeny of the CSCs in vivo or in vitro; d) a diploid level of c-kit protein expression and its ensuing physiological target activation is necessary both in vitro and in vivo for the growth, self-renewal, differentiation and regenerative properties of adult the CSCs; e) the very low number of endogenous c-kitpos CSC-generated cardiomyocytes detected in the c-kitCre-KI mice simply reflects the failure to recombine the CSCs to track their progeny and the severe defect in CSCs myogenesis produced by the c-kitCre-allele.
Despite these proof-of-principle results, autologous CSC transplantation has to be considered a personalized therapy because the very high incidence of CHF in the developed world makes it general use not affordable economically and in manpower [168]. Furthermore, considering the time needed for harvesting and expanding the autologous CSCs, this therapy cannot and will not be available in the early post-MI when it is likely to be most effective in protecting cardiomyocytes at risks and preventing pathological remodelling, the primary cause of chronic HF. For these reasons, it is imperative to develop a therapeutic protocol, which can result in a functional and histological autologous repair triggered by a therapeutic agent that can be mass-produced and is stable, off-the-shelf, available at all times, affordable, effective and safe. However, it is mandatory to gain more reproducible insights in the regulation of CSC biology in the adult mammalian in order to eventually arrive to a therapy with the characteristics delineated above that acts through the patient’s own endogenous CSCs triggering a physiologically meaningful histological and functional autologous myocardial repair.
Funding Statement
This work was supported by grants PRIN 2015 (2015ZTT5KB_004), FIRB Futuro-in-Ricerca 2012 (RBFR12I3KA), PON03PE_00009_2—iCARE from the Ministero dell’Istruzione, dell’Università e della Ricerca (M.I.U.R., IT), and Finalized Research 2010 (GR-2010-2318945) from the Ministero della Salute (IT).
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
- 1.Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, De Ferranti SD, Floyd J, Fornage M, Gillespie C, et al. Heart disease and stroke statistics-2017 update: a report from the American heart association. Circulation. [Internet] 2017;135:e146–603. [cited 2018 January 17]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28122885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Katz AM. The cardiomyopathy of overload: an unnatural growth response in the hypertrophied heart. Ann Intern Med. [Internet] 1994;121:363–371. [cited 2018 January 17]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8042826 [DOI] [PubMed] [Google Scholar]
- 3.Grossman W, Jones D, Mclaurin LP. Wall stress and patterns of hypertrophy in the human left ventricle. 1975. [cited 2018. January 17]. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC436555/pdf/jcinvest00142-0071.pdf [DOI] [PMC free article] [PubMed]
- 4.Hutchins GM, Bulkley BH. Infarct expansion versus extension: two different complications of acute myocardial infarction. Am J Cardiol. [Internet] 1978;41:1127–1132. [cited 2018 January 17]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/665522 [DOI] [PubMed] [Google Scholar]
- 5.Tang WW, Francis GS. Novel pharmacological treatments for heart failure. Expert Opin Investig Drugs. [Internet] 2003;12:1791–1801. [cited 2018 January 29]. Available from: http://www.tandfonline.com/doi/full/10.1517/13543784.12.11.1791 [DOI] [PubMed] [Google Scholar]
- 6.Soonpaa MH, Field LJ. Assessment of cardiomyocyte DNA synthesis in normal and injured adult mouse hearts. Am J Physiol Circ Physiol. [Internet] 1997;272:H220–6. [cited 2018 January 17]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9038941 [DOI] [PubMed] [Google Scholar]
- 7.Nadal-Ginard B. Commitment, fusion and biochemical differentiation of a myogenic cell line in the absence of DNA synthesis. Cell. [Internet] 1978;15:855–864. [cited 2018 January 24]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/728992 [DOI] [PubMed] [Google Scholar]
- 8.Ali SR, Hippenmeyer S, Saadat LV, Luo L, Weissman IL, Ardehali R. Existing cardiomyocytes generate cardiomyocytes at a low rate after birth in mice. Proc Natl Acad Sci U S A. [Internet] 2014;111:8850–8855. [cited 2018 January 17]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24876275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabé-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, et al. Evidence for cardiomyocyte renewal in humans. Science (80-). [Internet] 2009;324:98–102. [cited 2018 January 17]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19342590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kajstura J, Gurusamy N, Ogorek B, Goichberg P, Clavo-Rondon C, Hosoda T, D’Amario D, Bardelli S, Beltrami AP, Cesselli D, et al. Myocyte turnover in the aging human heart. Circ Res. [Internet] 2010;107:1374–1386. [cited 2018 January 17]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21088285 [DOI] [PubMed] [Google Scholar]
- 11.Gomer RH. Cell division: not being the wrong size. Nat Rev Mol Cell Biol. [Internet] 2001;2:48–55. [cited 2018 January 18]. Available from: http://www.nature.com/doifinder/10.1038/35048058 [DOI] [PubMed] [Google Scholar]
- 12.Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. [Internet] 2003;114:763–776. [cited 2018 January 18]. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0092867403006871 [DOI] [PubMed] [Google Scholar]
- 13.Urbanek K. Cardiac stem cells possess growth factor-receptor systems that after activation regenerate the infarcted myocardium, improving ventricular function and long-term survival. Circ Res. [Internet] 2005;97:663–673. [cited 2018 January 18]. Available from: http://circres.ahajournals.org/cgi/doi/10.1161/01.RES.0000183733.53101.11 [DOI] [PubMed] [Google Scholar]
- 14.Srivastava D, Ivey KN. Potential of stem-cell-based therapies for heart disease. Nat. 2006;4417097:2006. [DOI] [PubMed] [Google Scholar]
- 15.Menasche P. Cell-based therapy for heart disease: a clinically oriented perspective. Mol Ther. [Internet] 2009;17:758–766. [cited 2018 January 23]. Available from: http://linkinghub.elsevier.com/retrieve/pii/S1525001616317737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sohn RL, Jain M, Liao R. Adult stem cells and heart regeneration. Expert Rev Cardiovasc Ther. [Internet] 2007;5:507–517. [cited 2018 January 29]. Available from: http://www.tandfonline.com/doi/full/10.1586/14779072.5.3.507 [DOI] [PubMed] [Google Scholar]
- 17.Nir S, David R, Zaruba M, Franz W-M, Itskovitz-Eldor J. Human embryonic stem cells for cardiovascular repair. Cardiovasc Res. [Internet] 2003;58:313–323. [cited 2018 January 23]. Available from: https://academic.oup.com/cardiovascres/article-lookup/doi/10.1016/S0008-6363(03)00264-5 [DOI] [PubMed] [Google Scholar]
- 18.Menasché P, Hagège AA, Scorsin M, Pouzet B, Desnos M, Duboc D, Schwartz K, Vilquin JT, Marolleau JP. Myoblast transplantation for heart failure. Lancet. [Internet] 2001;357:279–280. [cited 2018 January 23]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11214133 [DOI] [PubMed] [Google Scholar]
- 19.Zhang S, Ge J, Sun A, Xu D, Qian J, Lin J, Zhao Y, Hu H, Li Y, Wang K, et al. Comparison of various kinds of bone marrow stem cells for the repair of infarcted myocardium: single clonally purified non-hematopoietic mesenchymal stem cells serve as a superior source. J Cell Biochem. [Internet] 2006;99:1132–1147. [cited 2018 January 23]. Available from:: http://www.ncbi.nlm.nih.gov/pubmed/16795039 [DOI] [PubMed] [Google Scholar]
- 20.Nadal-Ginard B, Torella D, Ellison G. Cardiovascular regenerative medicine at the crossroads. Clinical trials of cellular therapy must now be based on reliable experimental data from animals with characteristics similar to human’s. Rev Española Cardiol (English Ed). [Internet] 2006;59:1175–1189. [cited 2018 January 18]. Available from: http://linkinghub.elsevier.com/retrieve/pii/S1885585707600668 [PubMed] [Google Scholar]
- 21.Petsche Connell J, Camci-Unal G, Khademhosseini A, Jacot JG. Amniotic fluid-derived stem cells for cardiovascular tissue engineering applications. Tissue Eng Part B Rev. [Internet] 2013;19:368–379. [cited 2018 January 24]. Available from:: http://www.ncbi.nlm.nih.gov/pubmed/23350771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Urbanek K, Torella D, Sheikh F, De Angelis A, Nurzynska D, Silvestri F, Beltrami CA, Bussani R, Beltrami AP, Quaini F, et al. Myocardial regeneration by activation of multipotent cardiac stem cells in ischemic heart failure. Proc Natl Acad Sci U S A. [Internet] 2005;102:8692–8697. [cited 2018 January 19]. Available from:: http://www.ncbi.nlm.nih.gov/pubmed/15932947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ellison GM, Vicinanza C, Smith AJ, Aquila I, Leone A, Waring CD, Henning BJ, Stirparo GG, Papait R, Scarfò M, et al. Adult c-kitpos cardiac stem cells are necessary and sufficient for functional cardiac regeneration and repair. Cell. [Internet] 2013;154:827–842. [cited 2018 January 18]. Available from:: http://www.ncbi.nlm.nih.gov/pubmed/23953114 [DOI] [PubMed] [Google Scholar]
- 24.Nadal-Ginard B, Ellison GM, Torella D. The cardiac stem cell compartment is indispensable for myocardial cell homeostasis, repair and regeneration in the adult. Stem Cell Res. [Internet] 2014;13:615–630. [cited 2018 January 18]. Available from: https://www.sciencedirect.com/science/article/pii/S1873506114000440 [DOI] [PubMed] [Google Scholar]
- 25.Vicinanza C, Aquila I, Scalise M, Cristiano F, Marino F, Cianflone E, Mancuso T, Marotta P, Sacco W, Lewis FC, et al. Adult cardiac stem cells are multipotent and robustly myogenic: c-kit expression is necessary but not sufficient for their identification. Cell Death Differ. [Internet] 2017;24:2101–2116. [cited 2018 January 18]. Available from:: http://www.ncbi.nlm.nih.gov/pubmed/28800128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin CM, Meeson AP, Robertson SM, Hawke TJ, Richardson JA, Bates S, Goetsch SC, Gallardo TD, Garry DJ. Persistent expression of the ATP-binding cassette transporter, Abcg2, identifies cardiac SP cells in the developing and adult heart. DevR Biol. [Internet] 2004;265:262–275. [cited 2018 January 18]. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0012160603005815 [DOI] [PubMed] [Google Scholar]
- 27.Messina E, De Angelis L, Frati G, Morrone S, Chimenti S, Fiordaliso F, Salio M, Battaglia M, Latronico MVG, Coletta M, et al. Isolation and expansion of adult cardiac stem cells from human and murine heart. Circ Res. [Internet] 2004;95:911–921. [cited 2018 January 24]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15472116 [DOI] [PubMed] [Google Scholar]
- 28.Torella D, Ellison GM, Nadal-Ginard B, Indolfi C. Cardiac stem and progenitor cell biology for regenerative medicine. trends Cardiovasc Med. [Internet] 2005;15:229–236. [cited 2018 January 18]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16182134 [DOI] [PubMed] [Google Scholar]
- 29.Noseda M, Abreu-Paiva M, Schneider MD. The quest for the adult cardiac stem cell. Circ J. [Internet] 2015;79:1422–1430. [cited 2018 January 18]. Available from: https://www.jstage.jst.go.jp/article/circj/79/7/79_CJ-15-0557/_article [DOI] [PubMed] [Google Scholar]
- 30.Garbern JC, Lee RT. Cardiac stem cell therapy and the promise of heart regeneration. Cell Stem Cell. Internet. 2013;12:689–698. [cited 2018 January 18]. Available from: https://www.sciencedirect.com/science/article/pii/S1934590913002014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Le T, Chong J. Cardiac progenitor cells for heart repair. Cell Death Discov. [Internet] 2016;2:16052 Available from http://www.nature.com/articles/cddiscovery201652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Berlo JH, Molkentin JD. An emerging consensus on cardiac regeneration. Nat Med. Internet. 2014;20:1386–1393. [cited 2018 January 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25473919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Berlo JH, Molkentin JD. Most of the dust has settled. Circ Res. [Internet] 2016;118:17–19. [cited 2018 January 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26837741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cai C-L, Molkentin JD. The elusive progenitor cell in cardiac regeneration: slip slidin’ away. Circ Res. [Internet] 2017;120:400–406. [cited 2018 January 18]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28104772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Berlo JH, Kanisicak O, Maillet M, Vagnozzi RJ, Karch J, Lin SCJ, Middleton RC, Marbán E, Molkentin JD. C-kit+ cells minimally contribute cardiomyocytes to the heart. Nature. 2014;509:337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sultana N, Zhang L, Yan J, Chen J, Cai W, Razzaque S, Jeong D, Sheng W, Bu L, Xu M, et al. Resident c-kit+ cells in the heart are not cardiac stem cells. Nat Commun. 2015. [Internet] [cited 2018 January 18];6:8701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Q, Yang R, Huang X, Zhang H, He L, Zhang L, Tian X, Nie Y, Hu S, Yan Y, et al. Genetic lineage tracing identifies in situ Kit-expressing cardiomyocytes. Cell Res. 2016. [Internet] [cited 2018 January 19];26:119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hierlihy AM, Seale P, Lobe CG, Rudnicki MA, Megeney LA. The post-natal heart contains a myocardial stem cell population. FEBS Lett. 2002. October 23;530(1–3):239–243. [DOI] [PubMed] [Google Scholar]
- 39.Linke A, Müller P, Nurzynska D, Casarsa C, Torella D, Nascimbene A, Castaldo C, Cascapera S, Böhm M, Quaini F, et al. Stem cells in the dog heart are self-renewing, clonogenic, and multipotent and regenerate infarcted myocardium, improving cardiac function. Proc Natl Acad Sci U S A. 2005. June 21;102(25):8966–8971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ellison GM, Torella D, Dellegrottaglie S, Perez-Martinez C, Perez De Prado A, Vicinanza C, Purushothaman S, Galuppo V, Iaconetti C, Cd W, et al. Endogenous cardiac stem cell activation by insulin-like growth factor-1/hepatocyte growth factor intracoronary injection fosters survival and regeneration of the infarcted pig heart. J Am Coll Cardiol. 2011. August 23;58(9):977–986. . Epub 2011 Jun 30. [DOI] [PubMed] [Google Scholar]
- 41.Hou X, Appleby N, Fuentes T, Longo LD, Bailey LL, Hasaniya N. Kearns-Jonker1M. Isolation, characterization, and spatial distribution of cardiac progenitor cells in the sheep heart. J Clin Exp Cardiolog. 2012. October;11(Suppl 6):004. [PMC free article] [PubMed] [Google Scholar]
- 42.Bearzi C, Rota M, Hosoda T, Tillmanns J, Nascimbene A, De Angelis A, Yasuzawa-Amano S, Trofimova I, Siggins RW, Lecapitaine N, et al. Human cardiac stem cells. Proc Natl Acad Sci U S A. [Internet] 2007;104:14068–14073. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17709737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oh H, Bradfute SB, Gallardo TD, Nakamura T, Gaussin V, Mishina Y, Pocius J, Michael LH, Behringer RR, Garry DJ, et al. Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proc Natl Acad Sci. [Internet] 2003;100:12313–12318. [cited 2018 January 18]. Available from: http://www.pnas.org/cgi/doi/10.1073/pnas.2132126100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Vliet P, Roccio M, Smits AM, Van Oorschot AAM, Metz CHG, Van Veen TAB, Sluijter JPG, Doevendans PA, Goumans MJ. Progenitor cells isolated from the human heart: a potential cell source for regenerative therapy. Neth Heart J. 2008. May;16(5):163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smits AM, Van Vliet P, Metz CH, Korfage T, Sluijter JP, Doevendans PA, Goumans MJ. Human cardiomyocyte progenitor cells differentiate into functional mature cardiomyocytes: an in vitro model for studying human cardiac physiology and pathophysiology. Nat Protoc. 2009;4(2):232–243. [DOI] [PubMed] [Google Scholar]
- 46.Pfister O, Mouquet F, Jain M, Summer R, Helmes M, Fine A, Colucci WS, Liao R. CD31- but Not CD31+ cardiac side population cells exhibit functional cardiomyogenic differentiation. Circ Res. [Internet] 2005;97:52–61. [cited 2018 January 24]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15947249 [DOI] [PubMed] [Google Scholar]
- 47.Sandstedt J, Jonsson M, Kajic K, Sandstedt M, Lindahl A, Dellgren G, Jeppsson A, Asp J. Left atrium of the human adult heart contains a population of side population cells. Basic Res Cardiol. [Internet] 2012;107:255 [cited 2018 January 24]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22361742 [DOI] [PubMed] [Google Scholar]
- 48.Chimenti I, Gaetani R, Barile L, Forte E, Ionta V, Angelini F, Frati G, Messina E, Giacomello A. Isolation and expansion of adult cardiac stem/progenitor cells in the form of cardiospheres from human cardiac biopsies and murine hearts. Somatic Stem Cells. [Internet] Totowa, NJ: Humana Press; 2012. [cited 2018 January 24]. p. 327–338. Available from http://link.springer.com/10.1007/978-1-61779-815-3_19 [DOI] [PubMed] [Google Scholar]
- 49.Smith AJ, Lewis FC, Aquila I, Waring CD, Nocera A, Agosti V, Nadal-Ginard B, Torella D, Ellison GM Isolation and characterization of resident endogenous c-Kit+ cardiac stem cells from the adult mouse and rat heart. Nat Protoc. 2014;9:1662–1681. [DOI] [PubMed] [Google Scholar]
- 50.Chong JJ, Chandrakanthan V, Xaymardan M, Asli NS, Li J, Ahmed I, Heffernan C, Menon MK, Scarlett CJ, Rashidianfar A, et al. Adult cardiac-resident MSC-like stem cells with a proepicardial origin. Cell Stem Cell. 2011. December 2;9(6):527–540. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sampaolesi M, Biressi S, Tonlorenzi R, Innocenzi A, Draghici E, Cusella De Angelis MG, Cossu G. Cell therapy of primary myopathies. Arch Ital Biol. 2005. September;143(3–4):235–242. [PubMed] [Google Scholar]
- 52.Laugwitz K-L, Moretti A, Lam J, Gruber P, Chen Y, Woodard S, Lin L-Z, Cai C-L, Lu MM, Reth M, et al. Postnatal isl1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature. [Internet] 2005;433:647–653. [cited 2018 January 18]. Available from: http://www.nature.com/doifinder/10.1038/nature03215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bu L, Jiang X, Martin-Puig S, Caron L, Zhu S, Shao Y, Roberts DJ, Huang PL, Domian IJ, Chien KR. Human ISL1 heart progenitors generate diverse multipotent cardiovascular cell lineages. Nature. 2009. July 2;460(7251):113–117. . [DOI] [PubMed] [Google Scholar]
- 54.Bollini S, Vieira JM, Howard S, Dubè KN, Balmer GM, Smart N, Riley PR. Re-activated adult epicardial progenitor cells are a heterogeneous population molecularly distinct from their embryonic counterparts. Stem Cells Dev. 2014. August 1;23(15):1719–1730. . Epub 2014 May 8. [DOI] [PubMed] [Google Scholar]
- 55.Smart N, Bollini S, Dubé KN, Vieira JM, Zhou B, Davidson S, Yellon D, Riegler J, Price AN, Lythgoe MF, et al. De novo cardiomyocytes from within the activated adult heart after injury. Nature. 2011. [Internet] [cited 2018 January 24];474:640–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kondo M, Wagers AJ, Manz MG, Prohaska SS, Scherer DC, Beilhack GF, Shizuru JA, Weissman IL. BIology of hematopoietic stem cells and progenitors : implications for clinical application. Annu Rev Immunol. [Internet] 2003;21:759–806. [cited 2018 January 24]. Available from: http://www.annualreviews.org/doi/10.1146/annurev.immunol.21.120601.141007 [DOI] [PubMed] [Google Scholar]
- 57.Morrison SJ, Wandycz AM, Hemmati HD, Wright DE, Weissman IL. Identification of a lineage of multipotent hematopoietic progenitors. Development. [Internet] 1997;124:1929–1939. [cited 2018 January 24]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9169840 [DOI] [PubMed] [Google Scholar]
- 58.Sellers SE, Tisdale JF, Agricola BA, Metzger ME, Donahue RE, Dunbar CE, Sorrentino BP. The effect of multidrug-resistance 1 gene versus neo transduction on ex vivo and in vivo expansion of rhesus macaque hematopoietic repopulating cells. Blood. [Internet] 2001;97:1888–1891. [cited 2018 January 24]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11238136 [DOI] [PubMed] [Google Scholar]
- 59.Sandstedt J, Jonsson M, Lindahl A, Jeppsson A, Asp J. C-kit+ CD45− cells found in the adult human heart represent a population of endothelial progenitor cells. Basic Res Cardiol. [Internet] 2010;105:545–556. [cited 2018 January 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20119835 [DOI] [PubMed] [Google Scholar]
- 60.Nadal-Ginard B, Ellison GM, Torella D. Absence of evidence is not evidence of absence: pitfalls of Cre knock-ins in the c-Kit locus. Circ Res. [Internet] 2014;115:415–418. [cited 2018 January 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24965482 [DOI] [PubMed] [Google Scholar]
- 61.Jesty SA, Steffey MA, Lee FK, Breitbach M, Hesse M, Reining S, Lee JC, Doran RM, Nikitin AY, Fleischmann BK, et al. c-kit+ precursors support postinfarction myogenesis in the neonatal, but not adult, heart. Proc Natl Acad Sci. [Internet] 2012;109:13380–13385. [cited 2018 January 18]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22847442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Keith MCL, Bolli R. “String Theory” of c-kit pos Cardiac Cells. Circ Res. [Internet] 2015;116:1216–1230. [cited 2018 January 24]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25814683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Noseda M, Harada M, McSweeney S, Leja T, Belian E, Stuckey DJ, Abreu Paiva MS, Habib J, Macaulay I, De Smith AJ, et al. PDGFRα demarcates the cardiogenic clonogenic Sca1+ stem/progenitor cell in adult murine myocardium. Nat Commun. 2015. May 18;6: 6930. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Torella D, Ellison GM, Karakikes I, Nadal-Ginard B. Cardiovascular development: towards biomedical applicability - Resident cardiac stem cells. Cell Mol Life Sci. 2007;64:661–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Buckingham M, Meilhac S, Zaffran S. Building the mammalian heart from two sources of myocardial cells. Nat Rev Genet. 2005;6:826–835. [DOI] [PubMed] [Google Scholar]
- 66.Moretti A, Caron L, Nakano A, Lam JT, Bernshausen A, Chen Y, Qyang Y, Bu L, Sasaki M, Martin-Puig S, et al. Multipotent embryonic Isl1+ progenitor cells lead to cardiac, smooth muscle, and endothelial cell diversification. Cell. [Internet] 2006;127:1151–1165. [cited 2018 January 24]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17123592 [DOI] [PubMed] [Google Scholar]
- 67.Zhou B, Ma Q, Rajagopal S, Wu SM, Domian I, Rivera-Feliciano J, Jiang D, Von Gise A, Ikeda S, Chien KR, et al. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature. [Internet] 2008;454:109–113. [cited 2018 January 24]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18568026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhou B, Von Gise A, Ma Q, Rivera-Feliciano J, Pu WT. Nkx2-5- and Isl1-expressing cardiac progenitors contribute to proepicardium. Biochem Biophys Res Commun. [Internet] 2008;375:450–453. [cited 2018 January 24]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18722343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bianco P, Cao X, Frenette PS, Mao JJ, Robey PG, Simmons PJ, Wang C-Y. The meaning, the sense and the significance: translating the science of mesenchymal stem cells into medicine. Nat Med. [Internet] 2013;19:35–42. [cited 2018 January 18]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23296015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Matsuura K, Nagai T, Nishigaki N, Oyama T, Nishi J, Wada H, Sano M, Toko H, Akazawa H, Sato T, et al. Adult Cardiac Sca-1-positive cells differentiate into beating cardiomyocytes. J Biol Chem. [Internet] 2004;279:11384–11391. [cited 2018 January 18]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/14702342 [DOI] [PubMed] [Google Scholar]
- 71.Oyama T, Nagai T, Wada H, Naito AT, Matsuura K, Iwanaga K, Takahashi T, Goto M, Mikami Y, Yasuda N, et al. Cardiac side population cells have a potential to migrate and differentiate into cardiomyocytes in vitro and in vivo. J Cell Biol. [Internet] 2007;176:329–341. [cited 2018 January 18]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17261849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smith RR, Barile L, Cho HC, Leppo MK, Hare JM, Messina E, Giacomello A, Abraham MR, Marban E. Regenerative potential of cardiosphere-derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation. [Internet] 2007;115:896–908. [cited 2018 January 24]. Available from: http://circ.ahajournals.org/cgi/doi/10.1161/CIRCULATIONAHA.106.655209 [DOI] [PubMed] [Google Scholar]
- 73.Kanisicak O, Vagnozzi RJ, Molkentin JD. Identity crisis for regenerative cardiac cKit+ c. Circ Res. [Internet] 2017;121:1130–1132. [cited 2018 January 18]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29074532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lennartsson J, Rönnstrand L. Stem cell factor receptor/c-Kit: from basic science to clinical implications. Physiol Rev. [Internet] 2012;92:1619–1649. [cited 2018 January 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23073628 [DOI] [PubMed] [Google Scholar]
- 75.Zaruba MM, Soonpaa M, Reuter S, Field LJ. Cardiomyogenic potential of C-Kit+-expressing cells derived from neonatal and adult mouse hearts. Circulation. [Internet] 2010;121:1992–2000. [cited 2018 January 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20421520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Aquila I, Marino F, Cianflone E, Marotta P, Torella M, Mollace V, Indolfi C, Nadal-Ginard B, Torella D. The use and abuse of Cre/Lox recombination to identify adult cardiomyocyte renewal rate and origin. Pharmacol Res. [Internet] 2018;127:116–128. [cited 2018 January 24]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28655642 [DOI] [PubMed] [Google Scholar]
- 77.Fazel S, Cimini M, Chen L, Li S, Angoulvant D, Fedak P, Verma S, Weisel RD, Keating A, Li R-K. Cardioprotective c-kit+ cells are from the bone marrow and regulate the myocardial balance of angiogenic cytokines. J Clin Invest. [Internet] 2006;116:1865–1877. [cited 2018 January 24]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16823487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bernex F, De Sepulveda P, Kress C, Elbaz C, Delouis C, Panthier JJ. Spatial and temporal patterns of c-kit-expressing cells in WlacZ/+ and WlacZ/WlacZ mouse embryos. Development. [Internet] 1996;122:3023–3033. [cited 2018 January 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8898216 [DOI] [PubMed] [Google Scholar]
- 79.Reith AD, Rottapel R, Giddens E, Brady C, Forrester L, Bernstein A. W mutant mice with mild or severe developmental defects contain distinct point mutations in the kinase domain of the c-kit receptor. Genes Dev. [Internet] 1990;4:390–400. [cited 2018 January 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/1692559 [DOI] [PubMed] [Google Scholar]
- 80.Bernstein A, Chabot B, Dubreuil P, Reith A, Nocka K, Majumder S, Ray P, Besmer P. The mouse W/c-kit locus. Ciba Found Symp. [Internet] 1990;148:158-66-72 [cited 2018 January 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/1690623 [PubMed] [Google Scholar]
- 81.Schmidt-Supprian M, Rajewsky K. Vagaries of conditional gene targeting. Nat Immunol. [Internet] 2007;8:665–668. [cited 2018 January 19]. Available from: http://www.nature.com/articles/ni0707-665 [DOI] [PubMed] [Google Scholar]
- 82.Shin JY, Hu W, Naramura M, Park CY. High c-Kit expression identifies hematopoietic stem cells with impaired self-renewal and megakaryocytic bias. J Exp Med. [Internet] 2014;211:217–231. [cited 2018 January 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24446491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vicinanza C, Aquila I, Cianflone E, Scalise M, Marino F, Fumagalli F, Giovannone ED, Cristiano F, Iaccino E, Torella A, et al. c-kit Cre knock-ins fail to fate-map cardiac stem cells. Nature. 2018;555:E1–E5. [DOI] [PubMed] [Google Scholar]
- 84.Di Siena S, Gimmelli R, Nori SL, Barbagallo F, Campolo F, Dolci S, Rossi P, Venneri MA, Giannetta E, Gianfrilli D, et al. Activated c-Kit receptor in the heart promotes cardiac repair and regeneration after injury. Cell Death Dis [Internet] 2016;7:1–15. Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Harvey RP. Organogenesis: patterning the vertebrate heart. Nat Rev Genet. [Internet] 2002;3:544–556. [cited 2018 January 24]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12094232 [DOI] [PubMed] [Google Scholar]
- 86.Lopez-Sanchez C, Garcia-Martinez V. Molecular determinants of cardiac specification. Cardiovasc Res. [Internet] 2011;91:185–195. [cited 2018 January 24]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21525125 [DOI] [PubMed] [Google Scholar]
- 87.Brown CB, Wenning JM, Lu MM, Epstein DJ, Meyers EN, Epstein JA. Cre-mediated excision of Fgf8 in the Tbx1 expression domain reveals a critical role for Fgf8 in cardiovascular development in the mouse. Dev Biol. [Internet] 2004;267:190–202. [cited 2018 January 24]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/14975726 [DOI] [PubMed] [Google Scholar]
- 88.Verzi MP, McCulley DJ, De Val S, Dodou E, Black BL. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev Biol. [Internet] 2005;287:134–145. [cited 2018 January 24]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16188249 [DOI] [PubMed] [Google Scholar]
- 89.Rochais F, Dandonneau M, Mesbah K, Jarry T, Mattei M-G, Kelly RG. Hes1 is expressed in the second heart field and is required for outflow tract development. PLoS One. [Internet] 2009;4:e6267 [cited 2018 January 24]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19609448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.High FA, Jain R, Stoller JZ, Antonucci NB, Lu MM, Loomes KM, Kaestner KH, Pear WS, Epstein JA. Murine Jagged1/Notch signaling in the second heart field orchestrates Fgf8 expression and tissue-tissue interactions during outflow tract development. J Clin Invest. [Internet] 2009;119:1986–1996. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19509466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Koyanagi M, Bushoven P, Iwasaki M, Urbich C, Zeiher AM, Dimmeler S. Notch signaling contributes to the expression of cardiac markers in human circulating progenitor cells. Circ Res. [Internet] 2007;101:1139–1145. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17967789 [DOI] [PubMed] [Google Scholar]
- 92.David R, Brenner C, Stieber J, Schwarz F, Brunner S, Vollmer M, Mentele E, Müller-Höcker J, Kitajima S, Lickert H, et al. MesP1 drives vertebrate cardiovascular differentiation through Dkk-1-mediated blockade of Wnt-signalling. Nat Cell Biol. [Internet] 2008;10:338–345. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18297060 [DOI] [PubMed] [Google Scholar]
- 93.Hausenloy DJ, Yellon DM. Cardioprotective growth factors. Cardiovasc Res. [Internet] 2009;83:179–194. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19218286 [DOI] [PubMed] [Google Scholar]
- 94.Aguirre A, Montserrat N, Zacchigna S, Nivet E, Hishida T, Krause MN, Kurian L, Ocampo A, Vazquez-Ferrer E, Rodriguez-Esteban C, et al. In vivo activation of a conserved MicroRNA program induces mammalian heart regeneration. Cell Stem Cell. [Internet] 2014;15:589–604. [cited 2018 January 25]. Available from: https://www.sciencedirect.com/science/article/pii/S1934590914004548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Klattenhoff CA, Scheuermann JC, Surface LE, Bradley RK, Fields PA, Steinhauser ML, Ding H, Butty VL, Torrey L, Haas S, et al. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell. [Internet] 2013;152:570–583. [cited 2018 January 25]. Available from: https://www.sciencedirect.com/science/article/pii/S0092867413000044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cao DJ. Epigenetic regulation and heart failure. Expert Rev Cardiovasc Ther. [Internet] 2014;12:1087–1098. [cited 2018 January 29]. Available from: http://www.tandfonline.com/doi/full/10.1586/14779072.2014.942285 [DOI] [PubMed] [Google Scholar]
- 97.Zhang Y, Zhong JF, Qiu H, Robb MacLellan W, Marbán E, Wang C. Epigenomic reprogramming of adult cardiomyocyte-derived cardiac progenitor cells. Sci Rep. [Internet] 2016;5:17686 [cited 2018 January 25]. Available from: http://www.nature.com/articles/srep17686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Qyang Y, Martin-Puig S, Chiravuri M, Chen S, Xu H, Bu L, Jiang X, Lin L, Granger A, Moretti A, et al. The renewal and differentiation of Isl1+ cardiovascular progenitors are controlled by a Wnt/β-catenin pathway. Cell Stem Cell. [Internet] 2007;1:165–179. [cited 2018 January 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18371348 [DOI] [PubMed] [Google Scholar]
- 99.Klaus A, Muller M, Schulz H, Saga Y, Martin JF, Birchmeier W. Wnt/-catenin and Bmp signals control distinct sets of transcription factors in cardiac progenitor cells. Proc Natl Acad Sci. [Internet] 2012;109:10921–10926. Available from http://www.pnas.org/cgi/doi/10.1073/pnas.1121236109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kattman SJ, Witty AD, Gagliardi M, Dubois NC, Niapour M, Hotta A, Ellis J, Keller G. Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell. [Internet] 2011;8:228–240. [cited 2018 January 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21295278 [DOI] [PubMed] [Google Scholar]
- 101.Shenghui H, Nakada D, Morrison SJ. Mechanisms of stem cell self-renewal. Annu Rev Cell Dev Biol. [Internet] 2009;25:377–406. [cited 2018 January 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19575646 [DOI] [PubMed] [Google Scholar]
- 102.Mohsin S, Siddiqi S, Collins B, Sussman MA. Empowering adult stem cells for myocardial regeneration. Circ Res. [Internet] 2011;109:1415–1428. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22158649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Beis D, Kalogirou S, Tsigkas N. Insights into heart development and regeneration In: Introduction to translational cardiovascular research [Internet] Cham: Springer International Publishing; 2015. [cited 2018 January 25]. p. 17–30. Available from http://link.springer.com/10.1007/978-3-319-08798-6_2 [Google Scholar]
- 104.Tirosh-Finkel L, Zeisel A, Brodt-Ivenshitz M, Shamai A, Yao Z, Seger R, Domany E, Tzahor E. BMP-mediated inhibition of FGF signaling promotes cardiomyocyte differentiation of anterior heart field progenitors. Development. [Internet] 2010;137:2989–3000. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20702560 [DOI] [PubMed] [Google Scholar]
- 105.Sethi JK, Vidal-Puig A. Wnt signalling and the control of cellular metabolism. Biochem J. [Internet] 2010;427:1–17. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20226003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Torella D, Ellison GM, Karakikes I, Nadal-Ginard B. Growth-factor-mediated cardiac stem cell activation in myocardial regeneration. Nat Clin Pract Cardiovasc Med. [Internet] 2007;4:S46–51. [cited 2018 January 25]. Available from: http://www.nature.com/articles/ncpcardio0772 [DOI] [PubMed] [Google Scholar]
- 107.Mii Y, Taira M. Secreted Frizzled-related proteins enhance the diffusion of Wnt ligands and expand their signalling range. Development. [Internet] 2009;136:4083–4088. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19906850 [DOI] [PubMed] [Google Scholar]
- 108.Yang L, Soonpaa MH, Adler ED, Roepke TK, Kattman SJ, Kennedy M, Henckaerts E, Bonham K, Abbott GW, Linden RM, et al. Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature. [Internet] 2008;453:524–528. [cited 2018 January 25]. Available from: http://www.nature.com/doifinder/10.1038/nature06894 [DOI] [PubMed] [Google Scholar]
- 109.Kwon C, Qian L, Cheng P, Nigam V, Arnold J, Srivastava D. A regulatory pathway involving Notch1/β-catenin/Isl1 determines cardiac progenitor cell fate. Nat Cell Biol. [Internet] 2009;11:951–957. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19620969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yuasa S, Itabashi Y, Koshimizu U, Tanaka T, Sugimura K, Kinoshita M, Hattori F, Fukami S, Shimazaki T, Okano H, et al. Transient inhibition of BMP signaling by Noggin induces cardiomyocyte differentiation of mouse embryonic stem cells. Nat Biotechnol. [Internet] 2005;23:607–611. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15867910 [DOI] [PubMed] [Google Scholar]
- 111.Urbanek K, Cabral-da-Silva MC, Ide-Iwata N, Maestroni S, Delucchi F, Zheng H, Ferreira-Martins J, Ogorek B, D’Amario D, Bauer M, et al. Inhibition of Notch1-dependent cardiomyogenesis leads to a dilated myopathy in the neonatal heart. Circ Res. [Internet] 2010;107:429–441. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20558824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Von Gise A, Lin Z, Schlegelmilch K, Honor LB, Pan GM, Buck JN, Ma Q, Ishiwata T, Zhou B, Camargo FD, et al. YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc Natl Acad Sci U S A. [Internet] 2012;109:2394–2399. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22308401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Imajo M, Miyatake K, Iimura A, Miyamoto A, Nishida E. A molecular mechanism that links Hippo signalling to the inhibition of Wnt/β-catenin signalling. EMBO J. [Internet] 2012;31:1109–1122. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22234184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, Martin JF. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science. [Internet] 2011;332:458–461. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21512031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang Y, Yu A, Yu FX. The Hippo pathway in tissue homeostasis and regeneration. Protein Cell. 2017;8:349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, Porrello ER, Mahmoud AI, Tan W, Shelton JM, et al. Hippo pathway effector Yap promotes cardiac regeneration. Proc Natl Acad Sci [Internet] 2013;110:13839–13844. Available from: http://www.pnas.org/lookup/doi/10.1073/pnas.1313192110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kusano KF, Pola R, Murayama T, Curry C, Kawamoto A, Iwakura A, Shintani S, Ii M, Asai J, Tkebuchava T, et al. Sonic hedgehog myocardial gene therapy: tissue repair through transient reconstitution of embryonic signaling. Nat Med. [Internet] 2005;11:1197–1204. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16244652 [DOI] [PubMed] [Google Scholar]
- 118.Johnson NR, Wang Y. Controlled delivery of sonic hedgehog with a heparin-based coacervate [Internet] New York, NY: Humana Press; 2015. [cited 2018 January 25]. p. 1–7. Available from http://link.springer.com/10.1007/978-1-4939-2772-2_1 [DOI] [PubMed] [Google Scholar]
- 119.Zhang Y, Cao N, Huang Y, Spencer CI, Fu JD, Yu C, Liu K, Nie B, Xu T, Li K, et al. Expandable cardiovascular progenitor cells reprogrammed from fibroblasts. Cell Stem Cell. 2016;18:368–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cao N, Huang Y, Zheng J, Spencer CI, Zhang Y, Fu J-D, Nie B, Xie M, Zhang M, Wang H, et al. Supplementary material for conversion of human fibroblasts into functional cardiomyocytes by small molecules. Science. 2016;1502:1–10. [DOI] [PubMed] [Google Scholar]
- 121.Nadal-Ginard B, Ellison GM, Torella D. The cardiac stem cell compartment is indispensable for myocardial cell homeostasis, repair and regeneration in the adult. Stem Cell Res [Internet] 2014;13:615–630. Available from. [DOI] [PubMed] [Google Scholar]
- 122.Dey D, Han L, Bauer M, Sanada F, Oikonomopoulos A, Hosoda T, Unno K, De Almeida P, Leri A, Wu JC. Dissecting the molecular relationship among various cardiogenic progenitor cells. Circ Res. [Internet] 2013;112:1253–1262. [cited 2018 January 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23463815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Reith AD, Ellis C, Lyman SD, Anderson DM, Williams DE, Bernstein A, Pawson T. Signal transduction by normal isoforms and W mutant variants of the Kit receptor tyrosine kinase. EMBO J. [Internet] 1991;10:2451–2459. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/1714377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dolci S, Pellegrini M, Di Agostino S, Geremia R, Rossi P. Signaling through extracellular signal-regulated kinase is required for spermatogonial proliferative response to stem cell factor. J Biol Chem. [Internet] 2001;276:40225–40233. [cited 2018 January 25]. Available from: http://www.jbc.org/lookup/doi/10.1074/jbc.M105143200 [DOI] [PubMed] [Google Scholar]
- 125.Ye L, Zhang EY, Xiong Q, Astle CM, Zhang P, Li Q, From AHL, Harrison DE, Zhang JJ. Aging kit mutant mice develop cardiomyopathy. PLoS One. [Internet] 2012;7:e33407 [cited 2018 January 25]. Available from: http://dx.plos.org/10.1371/journal.pone.0033407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cimini M, Fazel S, Zhuo S, Xaymardan M, Fujii H, Weisel RD, Li R-K. c-Kit dysfunction impairs myocardial healing after infarction. Circulation. [Internet] 2007;116:I-77-I-82 [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17846329 [DOI] [PubMed] [Google Scholar]
- 127.Xiang F-L, Lu X, Hammoud L, Zhu P, Chidiac P, Robbins J, Feng Q. Cardiomyocyte-specific overexpression of human stem cell factor improves cardiac function and survival after myocardial infarction in mice. Circulation. [Internet] 2009;120:1065–1074. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19738140 [DOI] [PubMed] [Google Scholar]
- 128.Yaniz-Galende E, Chen J, Chemaly E, Liang L, Hulot J-S, McCollum L, Arias T, Fuster V, Zsebo KM, Hajjar RJ. Stem cell factor gene transfer promotes cardiac repair after myocardial infarction via in situ recruitment and expansion of c-kit+ cells. Circ Res. [Internet] 2012;111:1434–1445. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22931954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Naqvi N, Li M, Yahiro E, Graham RM, Husain A. Insights into the characteristics of mammalian cardiomyocyte terminal differentiation shown through the study of mice with a dysfunctional c-kit. Pediatr Cardiol. [Internet] 2009;30:651–658. [cited 2018 January 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19165540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Li M, Naqvi N, Yahiro E, Liu K, Powell PC, Bradley WE, Martin DIK, Graham RM, Dell’Italia LJ, Husain A. c-kit is required for cardiomyocyte terminal differentiation. Circ Res. [Internet] 2008;102:677–685. [cited 2018 January 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18258857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tallini YN, Greene KS, Craven M, Spealman A, Breitbach M, Smith J, Fisher PJ, Steffey M, Hesse M, Doran RM, et al. c-kit expression identifies cardiovascular precursors in the neonatal heart. Proc Natl Acad Sci U S A. [Internet] 2009;106:1808–1813. [cited 2018 January 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19193854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fuchs E, Tumbar T, Gausch G. Howard hughes medical institute. socializing with the neighbors: stemcells and their niche. Cell. [Internet] 2004;116:769–778. Available from http://ac.els-cdn.com/S0092867404002557/1-s2.0-S0092867404002557-main.pdf?_tid=65f65714-5d26-11e5-a5bb-00000aab0f26&acdnat=1442485700_e60f19ef722d6bb1a010a891e40f0793 [DOI] [PubMed] [Google Scholar]
- 133.Fuchs E, Chen T. A matter of life and death: self-renewal in stem cells. EMBO Rep. [Internet] 2012;14:39–48. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23229591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Moore KA, Lemischka IR. Stem cells and their niches. Science (80-). 2006;311:1880–1885. [DOI] [PubMed] [Google Scholar]
- 135.Leri A, Rota M, Hosoda T, Goichberg P, Anversa P. Cardiac stem cell niches. Stem Cell Res. 2014;13:631–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Urbanek K, Cesselli D, Rota M, Nascimbene A, De Angelis A, Hosoda T, Bearzi C, Boni A, Bolli R, Kajstura J, et al. Stem cell niches in the adult mouse heart. Proc Natl Acad Sci [Internet] 2006;103:9226–9231. Available from: http://www.pnas.org/cgi/doi/10.1073/pnas.0600635103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cheung T, Rando T. Molecular regulation of stem cell quiescence. Nat Rev Mol Cell Biol. [Internet] 2013;14:1–26. Available from http://www.nature.com/nrm/journal/v14/n6/abs/nrm3591.html [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Tan S, Barker N. Engineering the niche for stem cells. Growth Factors. [Internet] 2013;31:175–184. [cited 2018 January 25]. Available from: http://www.tandfonline.com/doi/full/10.3109/08977194.2013.859683 [DOI] [PubMed] [Google Scholar]
- 139.Kanda P, Davis DR. Cellular mechanisms underlying cardiac engraftment of stem cells. Expert Opin Biol Ther. [Internet] 2017;17:1127–1143. [cited 2018 January 29]. Available from: https://www.tandfonline.com/doi/full/10.1080/14712598.2017.1346080 [DOI] [PubMed] [Google Scholar]
- 140.Barker N, Bartfeld S, Clevers H. Tissue-resident adult stem cell populations of rapidly self-renewing organs. Cell Stem Cell. [Internet] 2010;7:656–670. [cited 2018 January 25]. Available from: https://www.sciencedirect.com/science/article/pii/S1934590910006387 [DOI] [PubMed] [Google Scholar]
- 141.Wilson A, Laurenti E, Oser G, Van Der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. [Internet] 2008;135:1118–1129. [cited 2018 January 25]. Available from: https://www.sciencedirect.com/science/article/pii/S009286740801386X [DOI] [PubMed] [Google Scholar]
- 142.Sanada F, Kim J, Czarna A, Chan NY-K, Signore S, Ogórek B, Isobe K, Wybieralska E, Borghetti G, Pesapane A, et al. c-Kit-positive cardiac stem cells nested in hypoxic niches are activated by stem cell factor reversing the aging myopathy. Circ Res. [Internet] 2014;114:41–55. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24170267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bearzi C, Leri A, Lo Monaco F, Rota M, Gonzalez A, Hosoda T, Pepe M, Qanud K, Ojaimi C, Bardelli S, et al. Identification of a coronary vascular progenitor cell in the human heart. Proc Natl Acad Sci U S A. [Internet] 2009;106:15885–15890. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19717420 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 144.Howard A, Pelc SR. Synthesis of desoxyribonucleic acid in normal and irradiated cells and its relation to chromosome breakage. Int J Radiat Biol Relat Stud Physics, Chem Med. [Internet] 1986;49:207–218. [cited 2018 January 25]. Available from: http://www.tandfonline.com/doi/full/10.1080/09553008514552501 [Google Scholar]
- 145.Watt FM, Hogan BL. Out of Eden: stem cells and their niches. Science. [Internet] 2000;287:1427–1430. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10688781 [DOI] [PubMed] [Google Scholar]
- 146.Park I, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, Morrison SJ, Clarke MF. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. [Internet] 2003;423:302–305. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12714971 [DOI] [PubMed] [Google Scholar]
- 147.Ozhan G, Weidinger G. Wnt/β-catenin signaling in heart regeneration. Cell Regen. [Internet] 2015;4:3 [cited 2018 January 25]. Available from: http://linkinghub.elsevier.com/retrieve/pii/S2045976917300068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Boni A, Urbanek K, Nascimbene A, Hosoda T, Zheng H, Delucchi F, Amano K, Gonzalez A, Vitale S, Ojaimi C, et al. Notch1 regulates the fate of cardiac progenitor cells. Proc Natl Acad Sci [Internet] 2008;105:15529–15534. Available from: http://www.pnas.org/cgi/doi/10.1073/pnas.0808357105 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 149.De La Pompa JL, Epstein JA. Coordinating tissue interactions: notch signaling in cardiac development and disease. Dev Cell. [Internet] 2012;22:244–254. [cited 2018 January 25]. Available from: http://linkinghub.elsevier.com/retrieve/pii/S1534580712000482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.High FA, Epstein JA. The multifaceted role of Notch in cardiac development and disease. Nat Rev Genet. [Internet] 2008;9:49–61. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18071321 [DOI] [PubMed] [Google Scholar]
- 151.Campa VM, Gutiérrez-Lanza R, Cerignoli F, Díaz-Trelles R, Nelson B, Tsuji T, Barcova M, Jiang W, Mercola M. Notch activates cell cycle reentry and progression in quiescent cardiomyocytes. J Cell Biol. [Internet] 2008;183:129–141. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18838555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhao B, Tumaneng K, Guan K-L. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat Cell Biol. [Internet] 2011;13:877–883. [cited 2018 January 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21808241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Affolter M, Basler K. The Decapentaplegic morphogen gradient: from pattern formation to growth regulation. Nat Rev Genet. [Internet] 2007;8:663–674. [cited 2018 January 25]. Available from: http://www.nature.com/articles/nrg2166 [DOI] [PubMed] [Google Scholar]
- 154.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. [Internet] 1995;81:323–330. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/7736585 [DOI] [PubMed] [Google Scholar]
- 155.Torella D, Rota M, Nurzynska D, Musso E, Monsen A, Shiraishi I, Zias E, Walsh K, Rosenzweig A, Sussman MA, et al. Cardiac stem cell and myocyte aging, heart failure, and insulin-like growth factor-1 overexpression. Circ Res. [Internet] 2004;94:514–524. [cited 2018 January 19 Available from: http://circres.ahajournals.org/cgi/doi/10.1161/01.RES.0000117306.10142.50 [DOI] [PubMed] [Google Scholar]
- 156.Kajstura J, Pertoldi B, Leri A, Beltrami C-A, Deptala A, Darzynkiewicz Z, Anversa P. Telomere shortening is an in vivo marker of myocyte replication and aging. Am J Pathol. [Internet] 2000;156:813–819. [cited 2018 January 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10702397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Leri A, Barlucchi L, Limana F, Deptala A, Darzynkiewicz Z, Hintze TH, Kajstura J, Nadal-Ginard B, Anversa P. Telomerase expression and activity are coupled with myocyte proliferation and preservation of telomeric length in the failing heart. Proc Natl Acad Sci U S A. [Internet] 2001;98:8626–8631. [cited 2018 January 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11447262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Leri A, Franco S, Zacheo A, Barlucchi L, Chimenti S, Limana F, Nadal-Ginard B, Kajstura J, Anversa P, Blasco MA. Ablation of telomerase and telomere loss leads to cardiac dilatation and heart failure associated with p53 upregulation. EMBO J. [Internet] 2003;22:131–139. [cited 2018 January 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12505991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Van Der Harst P, De Boer RA, Samani NJ, Wong LSM, Huzen J, Codd V, Hillege HL, Voors AA, Van Gilst WH, Jaarsma T, et al. Telomere length and outcome in heart failure. Ann Med. [Internet] 2010;42:36–44. [cited 2018 January 29]. Available from: http://www.tandfonline.com/doi/full/10.3109/07853890903233887 [DOI] [PubMed] [Google Scholar]
- 160.Morrison SJ, Prowse KR, Ho P, Weissman IL. Telomerase activity in hematopoietic cells is associated with self-renewal potential. Immunity. [Internet] 1996;5:207–216. [cited 2018 January 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/8808676 [DOI] [PubMed] [Google Scholar]
- 161.Hornsby PJ. Telomerase and the aging process. Exp Gerontol. [Internet] 2007;42:575–581. [cited 2018 January 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17482404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hiyama E, Hiyama K. Telomere and telomerase in stem cells. Br J Cancer. [Internet] 2007;96:1020–1024. [cited 2018 January 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17353922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, Van De Sluis B, Kirkland JL, Van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. [Internet] 2011;479:232–236. [cited 2018 January 25]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22048312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Cesselli D, Beltrami AP, D’Aurizio F, Marcon P, Bergamin N, Toffoletto B, Pandolfi M, Puppato E, Marino L, Signore S, et al. Effects of age and heart failure on human cardiac stem cell function. Am J Pathol. 2011. July;179(1):349–366. . Epub 2011 May 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Park I-K, Morrison SJ, Clarke MF. Bmi1, stem cells, and senescence regulation. J Clin Invest. [Internet] 2004;113:175–179. [cited 2018 January 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/14722607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zhang D, Wang H, Tan Y. Wnt/β-catenin signaling induces the aging of mesenchymal stem cells through the DNA damage response and the p53/p21 pathway. PLoS One. [Internet] 2011;6:e21397 [cited 2018 January 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21712954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Piao L, Zhao G, Zhu E, Inoue A, Shibata R, Lei Y, Hu L, Yu C, Yang G, Wu H, et al. Chronic psychological stress accelerates vascular senescence and impairs ischemia-induced neovascularization: the role of dipeptidyl peptidase-4/glucagon-like peptide-1-adiponectin axis. J Am Heart Assoc [Internet] 2017;6 [cited 2018 January 19]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28963101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Marotta P, Cianflone E, Aquila I, Vicinanza C, Scalise M, Marino F, Mancuso T, Torella M, Indolfi C, Torella D. Combining cell and gene therapy to advance cardiac regeneration. Expert Opin Biol Ther. [Internet] 2018. [cited 2018 January 26]:1–15. Available from http://www.ncbi.nlm.nih.gov/pubmed/29347847 [DOI] [PubMed] [Google Scholar]