

ABSTRACT
The Wnt signaling pathway controls stem cell identity in the intestinal epithelium and cancer stem cells (CSCs). The transcription factor Ascl2 (Wnt target gene) is fate decider of intestinal cryptic stem cells and colon cancer stem cells. It is unclear how Wnt signaling is translated into Ascl2 expression and keeping the self-renewal of CRC progenitor cells. We showed that the exogenous Ascl2 in colorectal cancer (CRC) cells activated the endogenous Ascl2 expression via a direct autoactivatory loop, including Ascl2 binding to its own promoter and further transcriptional activation. Higher Ascl2 expression in human CRC cancerous tissues led to greater enrichment in Ascl2 immunoprecipitated DNA within the Ascl2 promoter in the CRC cancerous sample than the peri-cancerous mucosa. Ascl2 binding to its own promoter and inducing further transcriptional activation of the Ascl2 gene was predominant in the CD133+CD44+ CRC population. R-spondin1/Wnt activated Ascl2 expression dose-dependently in the CD133+CD44+ CRC population, but not in the CD133−CD44− CRC population, which was caused by differences in Ascl2 autoregulation under R-spondin1/Wnt activation. R-spondin1/Wnt treatment in the CD133+CD44+ or CRC CD133−CD44− populations exerted a different pattern of stemness maintenance, which was defined by alterations of the mRNA levels of stemness-associated genes, the protein expression levels (Bmi1, C-myc, Oct-4 and Nanog) and tumorsphere formation. The results indicated that Ascl2 autoregulation formed a transcriptional switch that was enhanced by Wnt signaling in the CD133+CD44+ CRC population, thus conferring their self-renewal.
Keywords: Wnt signaling, Achaete scute-like 2, Autoregulation, Colorectal cancer, Self-renewal, Progenitor cell
Introduction
Colorectal cancer (CRC) is the third leading cause of death from cancer worldwide and a leading cause of morbidity and mortality in developed countries [1]. The cancer stem cell (CSC) hypothesis, in which a subpopulation of cancer cells that exhibit stem-like features sustain tumor formation, metastasis, and resistance to therapy [2–5], has been proposed to explain the functional heterogeneity and carcinogenesis of cancer. Several studies have investigated the protein-coding genes and their products that participate in the “stemness” maintenance and tumorigenicity of CRC progenitor cells [6–8]. Thus, it is important to identify the regulatory mechanisms and signaling pathways involved in CRC progenitor cells to develop the novel reagents to target the refractory CRC progenitor cell population [9].
Achaete scute-like 2 (Ascl2), a basic helix-loop-helix transcription factor and downstream target of Wnt signaling, plays a critical role in intestinal stem cells and CRC progenitor cells [10,11]. Ascl2 controls the fate of intestinal cryptic Lgr5+ stem cells from such gain- and loss-of-function experiments [10]. Ascl2 is also overexpressed in colorectal cancer [12–14]. In addition, Ascl2 overexpression has the potential to shift the hierarchy of stem and progenitor cells within liver metastases, resulting in self-renewal rather than differentiation, potentially affecting the clinical behavior of these tumors [14]. It has also been found to initiate T-helper-cell development, differentiation from mouse trophoblast stem cells to trophoblast progenitors, and intestinal neoplastic epithelial cell differentiation to a goblet cell phenotype [15–17]. Mash2 (homologue of Ascl2) can be regulated by the hpo/Mst signaling pathway as well as Tssc3, HIF 1α and 2α, L1 family of cell adhesion receptors and OVO-like 1 (OVOL1) in trophoblast stem (TS) cells [16,18–23].
Our previous work indicated that Ascl2 controlled the fate of CRC progenitor cells via a miR-302-dependent mechanism, Ascl2 was predominantly expressed in CD133+ CRC progenitor cells, and Ascl2 knockdown in CRC cells led to the inhibition of their “stemness” [11]. This has led to the conclusion that Ascl2 functions as a master regulator of the “stemness” of CRC progenitor cells. Ascl2 in CRC cells is significantly induced by the induction of chemokine receptor CXCR4 or KIAA1199 [24,25]. However, the molecular mechanism of Ascl2 over-expression in CRC progenitor cells is still unclear.
Schuijers et al showed that Ascl2 is able to transactivate its own promoter in intestinal cryptic Lgr5+ stem cells and that together with β-catenin/Tcf, it activates genes that are fundamental to the stem cell state, suggesting a positive and R-spondin1/Wnt activated autoregulatory loop in intestinal cryptic Lgr5+ stem cells that further maintains their “stemness” [26]. This result leads us to infer that Ascl2 autoregulation is also possibly present in CRC progenitor cells.
Here, we identified that the Ascl2 gene was transactivated by a positive autoactivatory loop, which was confirmed by the fact that endogenous Ascl2 expression was activated by exogenous Ascl2 in CRC cells, including Ascl2 binding with its own promoter and transcriptional activation of the Ascl2 gene. This positive autoactivatory loop for activating the Ascl2 gene is predominant in the CD133+CD44+ CRC population. R-spondin1/Wnt activated Ascl2 expression dose-dependently only in the CD133+CD44+ CRC population and maintained its “stemness”. Ascl2 autoregulation amplified Wnt signaling in CRC progenitor cells and controlled the stability of this cell state, thus conferring the ability for self-renewal.
Results
Identification the autoregulation of ascl2 expression
To identify the autoregulation of Ascl2, we designed the exogenous Ascl2 protein to be expressed, it include the three tandem repeats of the FLAG tag (DYKDDDDK) at the N-terminus of its sequence (Figure 1(a)). Thus, the exogenous Ascl2 protein was able to be recognized by both anti-Ascl2 and anti-FLAG antibodies, whereas the endogenous Ascl2 protein could only be recognized by the anti-Ascl2 antibody and not the anti-FLAG antibody (Figure 1(a)). The exogenous Ascl2 protein (approximately 23 kDa) produced in Ad-Ascl2/Caco-2 can be recognized by both anti-Ascl2 and anti-FLAG antibodies, but the endogenous Ascl2 protein (approximately 20 kDa) can only be recognized by anti-Ascl2 antibody and not by the anti-FLAG antibody (Figure 1(a)). Thus the significant increase of endogenous Ascl2 protein gave definite evidence of the presence of the positive autoregulation of Ascl2 in Ad-Ascl2/Caco-2 cells compared with control cells transfected by an empty vector (Figure 1(b)). The endogenous and vector mRNA gene products are distinguished using specific primer pairs, and the corresponding proteins are discriminated by molecular weight, which differs due to the three FLAG tags encoded by the expression vector. Endogenous Ascl2 mRNA expression in HT-29, LS174T or Caco-2 cells upon transfection with either an empty vector or an Ad-vector expressing FLAGs-Ascl2 was significantly increased in Ad-Ascl2/HT-29, Ad-Ascl2/LS174T and Ad-Ascl2/Caco-2 cells when compared with control cells (Figure 1(c)). Western blots showed increased endogenous Ascl2 protein expression in HT-29, LS174T or Caco-2 cells upon transfection with an Ad-vector expressing Ascl2 (Figure 1(d)). The results confirmed that positive Ascl2 autoregulation was present in CRC cell lines, which had low Ascl2 expression levels.
Figure 1.

Effect of Ascl2 exogenous expression on the endogenous Ascl2 level in CRC and non-gastrointestinal cell lines and identification of Ascl2 binding to its own promoter. (a) Schematic representation of the exogenous Ascl2 construct. cDNA encoding human Ascl2 preceded by seventy-two nucleotides encoding three FLAG tags (DYKDDDDK) at the N-terminus was cloned into the expression Ad-vector. (b) Identification of exogenous and endogenous Ascl2 expression in Ad-Ascl2/Caco-2 and control cells. The exogenous Ascl2 protein can be recognized by both anti-Ascl2 and anti-FLAG antibodies, but the endogenous Ascl2 protein can only be recognized by the anti-Ascl2 antibody, M: marker. (c) Endogenous Ascl2 mRNA expression in HT-29, LS174T and Caco-2 cells upon transfection with either an empty vector or an Ad-vector expressing Ascl2. GAPDH was used as the mRNA level control. (d) Western blots showing endogenous levels of Ascl2 in HT-29, LS174T and Caco-2 cells upon transfection with either an empty vector or an Ad-vector expressing Ascl2. GAPDH was used as the loading control. (e) A schematic representation of the Ascl2 proximal promoter (−2588~+1) in which the clustered E-box(s) were present and marked as black dots; these clusters were the relevant regions that were selected for the ChIP assay. Chromatin immunoprecipitation was carried out with an isotype control immunoglobulin G (IgG) or an anti-Ascl2 antibody in DNA from HT-29 cells (f) and LS174T cells (g). Purified DNA was analyzed by PCR using specific primers covering eight regions of the proximal human Ascl2 promoter.
Ascl2 binding to its own promoter and further transcription
Ascl2 can transcriptionally regulate miR-200 family members and CDX2 gene expression by binding to the E-box(s) residing on their promoters [17,27], but it remains unclear whether Ascl2 autoregulation occurs via a transcriptional mechanism, including Ascl2 binding to its own promoter and exerting transcriptional regulation. Thus, we analyzed a cohort of transcription factor response elements located within the 2588-bp region upstream of the transcription start site of the human Ascl2 gene using promoter analysis. We found sixteen clustered E-boxes within the proximal 2588-bp region upstream of the transcription start site of the human Ascl2 gene (Figure 1(e)). We performed a ChIP assay to determine whether Ascl2 binds directly to its own promoter. ChIP experiments 1–8 provided direct evidence of Ascl2 binding to its proximal promoter (Figure 1(f,g)). The binding of Ascl2 to its own promoter in Ascl2-interfered HT-29 (shRNA-Ascl2/HT-29) and LS174T (shRNA-Ascl2/LS174T) cells was significantly reduced when compared with their control cells (p < 0.01) (Figure 2(a,b)). However, the binding of Ascl2 to its own promoter in Ascl2-overexpressed SW480 (lv-Ascl2/SW480) and Lovo (lv-Ascl2/Lovo) cells was significantly increased when compared with their control cells (no difference in ChIP 8 using lv-Ascl2/SW480 cells and ChIP 6 using lv-Ascl2/Lovo cells) (*: p < 0.05; **: p < 0.01) (Figure 2(c,d)). To locate the specific auto-regulators of Ascl2 expression, the upstream amplifier region of Ascl2 (−2588/+1) was inserted into a luciferase reporter pGL3 vector using relevant primer pairs (Figure 2(e)). The full-length human Ascl2 promoter (−2588/+1) generated significantly lower luciferase activity levels in both shRNA-Ascl2/HT-29 and shRNA-Ascl2/LS174T cells than in control cells (**: p < 0.01) (Figure 2(f,g)). In contrast, the full-length human Ascl2 promoter (−2588/+1) generated significantly higher luciferase activity levels in both lv-Ascl2/SW480 and lv-Ascl2/Lovo cells than in control cells (**: p < 0.01) (Figure 2(h, i)). These ChIP and luciferase assays provided direct evidence of the presence of Ascl2 auto-transcriptional activation in CRC cells.
Figure 2.

Altered Ascl2 binding to its own promoter and/or autoregulated transcriptional activation of the Ascl2 promoter in cell lines and CRC tissues. ChIP assays were performed using Ascl2-interfered HT-29 (shRNA-Ascl2/HT-29) or LS174T (shRNA-Ascl2/LS174T) cells (a and b) and using Ascl2-enforced lv-Ascl2/SW480 and lv-Ascl2/Lovo cells (c and d) to analyze relative Ascl2 binding to its own promoter compared with control cells. Purified DNA was analyzed by qPCR using specific primers covering eight regions of the proximal human Ascl2 promoter. Fold enrichments were expressed as the ratio of the IP:Ascl2 signal to that of the IP:IgG signal and calculated by extrapolation from a standard curve of the input DNA dilutions. (e) A schematic representation of the human Ascl2 promoter construct that was used in this study. (f and g) shRNA-Ascl2/HT-29 and shRNA-Ascl2/LS174T cells as well as control cells were transfected with the Ascl2 promoter-Luc construct. Reduced Ascl2 promoter activities were evident from luciferase reporter assays (**: p < 0.01). (h and i) lv-Ascl2/SW480 and lv-Ascl2/Lovo cells as well as control cells were transfected with the Ascl2 promoter-Luc construct, and increased Ascl2 promoter activities were evident from luciferase reporter assays (**: p < 0.01). (j and l) The representative normal intestinal mucosa and CRC tissues were immunohistochemically stained using the anti-Ascl2 antibody (magnification: 200x). (K and M) ChIP assays using the cancerous samples from two patients to analyze the relative Ascl2 binding to its own promoter compared with their normal intestinal mucosa. Purified DNA was analyzed by qPCR using specific primers covering eight regions of the human Ascl2 promoter. Fold enrichments were expressed as described before (**: p < 0.01). Error bars represent the SD.
Ascl2 binding to its own promoter in human CRC tissues
To confirm the relevance of the autoregulatory mechanism in vivo, ChIP was performed against Ascl2 in two human CRC samples. These two CRC samples were selected from 21 cancerous samples, in which Ascl2 expression was immunohistochemically detected [17], and they had relatively higher Ascl2 expression in their cancerous tissues (Figure 2(j, l)). Since Ascl2 is exclusively present in the Lgr5+ cryptic stem cell of colon mucosa, ChIP was also performed on the whole mucosa fragment containing mucosa, and submucosa tissues and served as a control. As shown in Figure 2(k) for case 1, ChIPs 1–8 gave evidence of Ascl2 binding to its own promoter and showed greater enrichment in immunoprecipitated DNA in the cancerous sample than peri-cancerous mucosa (**: p < 0.01). Additionally, in Figure 2(m) for case 2, ChIPs 1–6 and 8 confirmed Ascl2 binding to its own promoter and showed a greater enrichment in immunoprecipitated DNA in the cancerous sample than peri-cancerous mucosa (ChIP 7 gave a negative result) (**: p < 0.01).
Ascl2 autoregulation preferred CD44+CD133+ CRC progenitor cells
Fluorescence-activated cell sorting and flow cytometry of HT-29 and Caco-2 cells were used to separate the cells into the CD133−CD44− and CD133+CD44+ HT-29 cell populations as well as the CD133−CD44− and CD133+CD44+ Caco-2 cells (Figure 3(a,b)). CD133+CD44+ HT-29 cells and CD133+CD44+ Caco-2 cells were considered as CRC progenitor cells. Ascl2 mRNA expression levels and protein expression in CD133+CD44+ CRC cells were significantly higher than for CD133−CD44− CRC cells (**: p < 0.01) (Figure 3(c,d)). CD133−CD44− or CD133+CD44+ CRC cells were transfected with the Ascl2 promoter-Luc construct, and increased Ascl2 promoter activity was evident from luciferase reporter assays in the CD133+CD44+ CRC cell population compared with the CD133−CD44− CRC cell population (**: p < 0.01) (Figure 3(e)). To identify whether the activated Ascl2 transcription was partially caused by the increased Ascl2 protein in the CD133+CD44+ CRC cell population, ChIP assays using the CD133+CD44+ CRC cell population to analyze the relative Ascl2 binding to its own promoter compared with the CD133−CD44− CRC cell population indicated there were significant increases of Ascl2 binding using ChIPs 1–7 in the CD133+CD44+ CRC cell population (**: p < 0.01) (Figure 3(f,g)). These data confirmed that the increased Ascl2 in the CD133+CD44+ CRC cell population was, to some degree, caused by Ascl2 autoregulation activation.
Figure 3.

Ascl2 expression, transcriptional activation of the Ascl2 promoter and Ascl2 binding to its own promoter in CD133−CD44− and CD133+CD44+ CRC cell populations. (a and b) Fluorescence-activated cell sorting and flow cytometry of human CRC cells (HT-29 and Caco-2) were used to separate cells into CD133−CD44− and CD133+CD44+ HT-29 cells as well as CD133−CD44− and CD133+CD44+ Caco-2 cells. (c) Relative Ascl2 mRNA expression in CD133−CD44− and CD133+CD44+ CRC cells. (d) Ascl2 protein expression in CD133−CD44− and CD133+CD44+ CRC cells. (e) CD133−CD44− and CD133+CD44+ CRC cells were transfected with the Ascl2 promoter-Luc construct, and increased Ascl2 promoter activity was evident from luciferase reporter assays in the CD133+CD44+ CRC cell population compared with the CD133−CD44− CRC cell population (**: p < 0.01). (f and g) ChIP assays using the CD133+CD44+ CRC cell population to analyze the relative Ascl2 binding to its own promoter in comparison with the CD133−CD44− CRC cell population indicated that there was a significant increase of Ascl2 binding using ChIPs 1–7 in the CD133+CD44+ CRC cell population. Purified DNA was analyzed by qPCR using specific primers covering eight regions of the human AsCL2 promoter (**: p < 0.01).
R-spondin1/wnt dose-dependently enhanced ascl2 expression and its auto-activation in HT-29 and LS174T cells
It is clear that Ascl2 is a target gene of Wnt signaling and R-spondin1 (Rspo1), an extracellular protein that enhances β-catenin-dependent Wnt signaling. To identify how R-spondin1/Wnt activates Ascl2 expression and whether R-spondin1/Wnt activated Ascl2 expression is related to the Ascl2 autoregulation mechanism, we detected relative Ascl2 mRNA expression levels and its protein in HT-29 and LS174T cells treated with recombinant human R-spondin1/Wnt protein (hRspo1) at different concentrations (0 ~ 1000 ng/ml) for 16 hours, and the levels were quantitated by q-PCR and western blots. As shown in Figure 4(a–f), Ascl2 mRNA and protein expression in HT-29 and LS174T cells persistently increased in a dose-dependent manner. The hRspo1-treated HT-29 and LS174T cells were transfected with the Ascl2 promoter-Luc construct, and Ascl2 promoter activities were increased due to hRspo1 treatment in a dose-dependent manner (Figure 4(g,h)). Thus, Ascl2 autoregulation further amplified R-spondin1/Wnt-activated Ascl2 expression in a dose-dependent manner.
Figure 4.
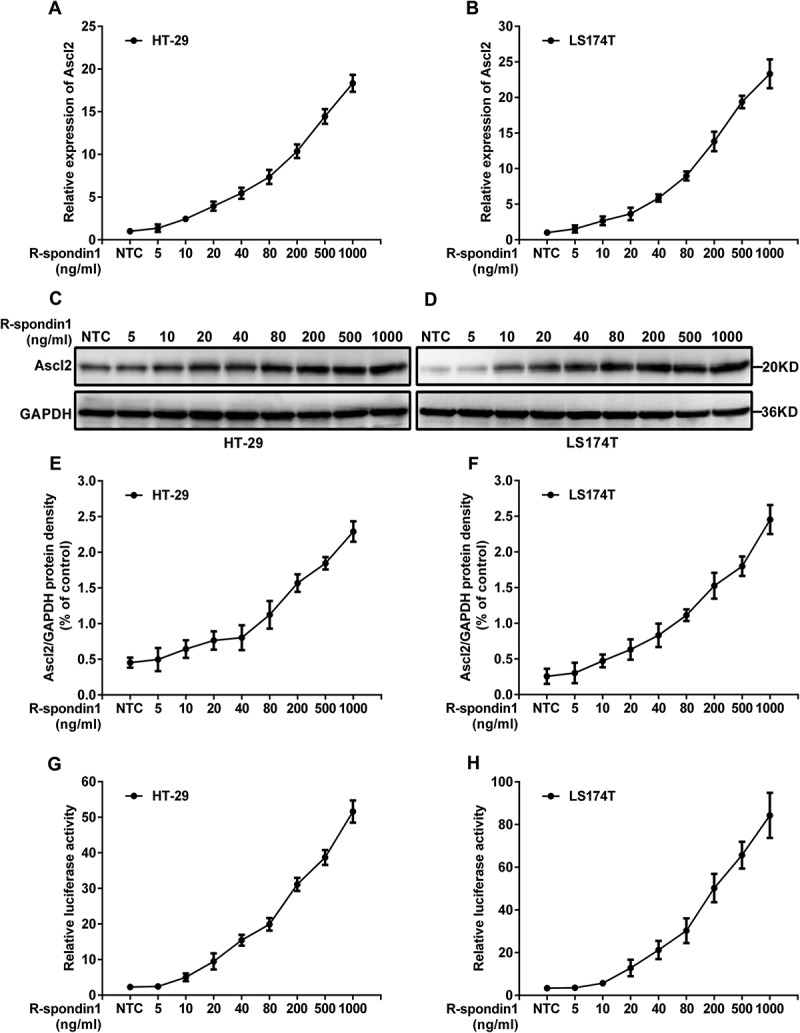
R-spondin1/Wnt dose-dependently enhanced Ascl2 expression and activated its own promoter in HT-29 and LS174T cells. (a and b) Relative Ascl2 mRNA expression levels in HT-29 (a) and LS174T (b) cells, which were treated with recombinant human R-spondin1/Wnt protein (hRspo1) at the indicated concentrations for 16 hours, were quantitated by q-PCR. (c and d) Ascl2 protein in HT-29 (c) and LS174T (d) cells treated with hRspo1 at the indicated concentrations for 3 hours were detected by western blots. (e and f) Quantitation of the western blots of the Ascl2 protein from hRspo1-treated HT-29 (e) and LS174T (f) cells. (G and H) Regulation by Ascl2 of its own promoter in HT-29 (g) and LS174T (h) cells treated with hRspo1 at the indicated concentrations for 16 hours. hRspo1-treated HT-29 and LS174T cells were transfected with the Ascl2 promoter-Luc construct, and the Ascl2 promoter activities were increased due to more hRspo1 treatment. NTC, not treated control.
R-spondin1/wnt-induced ascl2 was uniquely present in the CD133+CD44+ CRC cell population
Relative Ascl2 mRNA expression levels in CD133+CD44+ and CD133−CD44− HT-29 cells as well as CD133+CD44+ and CD133−CD44− Caco-2 cells treated with hRspo1 at the indicated concentrations for 16 hours were quantitated by q-PCR. Ascl2 mRNA expression levels in both CD133−CD44− HT-29 and CD133−CD44− Caco-2 cells dose-dependently increased at low concentrations (0-~20 or 40 ng/ml) of hRspo1, and then, they reached a plateau and became unchangeable despite increases of the hRspo1 concentration (20 or 40 ~ 1000 ng/ml) (Figure 5(a,c)). Whereas, Ascl2 mRNA expression levels in both CD133+CD44+ HT-29 and CD133+CD44+ Caco-2 cells dose-dependently and persistently increased with increasing concentrations of hRspo1 (0 ~ 1000 ng/ml) (Figure 5(b,d)). Ascl2 protein expression in these cancer cell populations exhibited a similar phenomenon after treatment with hRspo1 at different concentrations (0 ~ 1000 ng/ml). Ascl2 protein expression in both CD133−CD44− HT-29 and CD133−CD44− Caco-2 cells was dose-dependently increased at low concentrations (0 ~ 40 or 80 ng/ml) of hRspo1 and then became unchangeable despite more hRspo1. Whereas, Ascl2 protein expression in both CD133+CD44+ HT-29 and CD133+CD44+ Caco-2 cells dose-dependently and persistently increased with increasing concentrations of hRspo1 (0 ~ 1000 ng/ml) (Figure 5(e–l)). These results indicated that Ascl2 was capable of self-activation and functioned as a positive feedback loop, which would be activated when a threshold level of Wnt signaling was reached and allowed two different states that corresponded to CRC progenitor cells (Ascl2-ON) and non-CRC progenitor cells (Ascl2-OFF).
Figure 5.

Ascl2 expression and its own promoter activities in CD133+CD44+ and CD133−CD44− CRC cell populations treated with hRspo1. (A-D) Relative Ascl2 mRNA expression levels in CD133+CD44+ HT-29 (B) and CD133−CD44− HT-29 (A) cells as well as CD133+CD44+ Caco-2 (D) and CD133−CD44− Caco-2 (C) cells treated with recombinant hRspo1 at the indicated concentrations for 16 hours were quantitated by q-PCR. (E-H) Ascl2 protein in CD133+CD44+ HT-29 (F) and CD133−CD44− HT-29 (E) cells as well as CD133+CD44+ Caco-2 (H) and CD133−CD44− Caco-2 (G) cells treated with different concentrations of recombinant hRspo1 were detected by western blots. (I-L) Quantitation of the western blots of Ascl2 protein from hRspo1-treated CD133+CD44+ HT-29 (J), CD133−CD44− HT-29 (I), CD133+CD44+ Caco-2 (H) and CD133−CD44− Caco-2 (G) cells. The hRspo1-treated CD133+CD44+ (N and P) and CD133−CD44− (M and O) CRC cell populations from HT-29 (M and N) and Caco-2 (O and P) were transfected with the Ascl2 promoter-Luc construct, and the Ascl2 promoter activities in the CD133−CD44− CRC cell populations (M and O) were increased at dosages of 0–20 ng/ml hRspo1. However, the promotor activities were nearly unchanged at dosages of 20–1000 ng/ml hRspo1. The Ascl2 promoter activities in the CD133+CD44+ CRC cell populations (N and P) were persistently increased at the dosages of 0–1000 ng/ml hRspo1. NTC, not treated control.
R-spondin1/wnt-induced ascl2 activation in the CD133+CD44+ CRC cell population was caused by uncontrolled autoregulation
To identify how R-spondin1/Wnt induced uncontrolled Ascl2 activation in the CD133+CD44+ CRC cell population, we performed a luciferase assay in hRspo1-treated CD133+CD44+ and CD133−CD44− CRC cell populations from HT-29 and Caco-2 cells transfected with the Ascl2 promoter-Luc construct, and Ascl2 promoter activities in the CD133−CD44− CRC cell populations from HT-29 and Caco-2 were increased at dosages of 0–20 or 40 ng/ml hRspo1, but they were almost unchangeable at dosages of 20–1000 ng/ml hRspo1 (Figure 5(m,o)). Ascl2 promoter activities in the CD133+CD44+ CRC cell populations from HT-29 and Caco-2 persistently increased at dosages of 0–1000 ng/ml hRspo1 (Figure 5(n,p)). The results gave an explanation that R-spondin1/Wnt-induced Ascl2 activation in the CD133+CD44+ CRC cell population was caused by uncontrolled Ascl2 auto-activation.
Self-renewal of CRC progenitor cells was determined by r-spondin1/wnt-induced ascl2 activation
To determine the biological significance of R-spondin1/Wnt-induced Ascl2 auto-activation, we compared the expression levels of “stemness”-associated genes in CD133+CD44+ and CD133−CD44− CRC cell populations treated with hRspo1 (0, 20, 80, or 500 ng/ml). The mRNA levels of “stemness”-associated genes (Bmi1, C-myc, Oct-4 and Nanog) in the CD133−CD44− CRC cell populations from HT-29 and LS174T cells treated with 20, 80, or 500 ng/ml hRspo1 were nearly similar (Figure 6(a,c)), but in CD133+CD44+ and CD133−CD44− CRC cell populations treated with hRspo1 (0, 20, 80, or 500 ng/ml), the mRNA levels of “stemness”-associated genes were increased with the addition of increasing amounts of hRspo1 in the culture media (**: p < 0.01) (Figure 6(b,d)). The protein expression of “stemness”-associated genes (Bmi1, C-myc, Oct-4 and Nanog) in hRspo1-treated CD133+CD44+ and CD133−CD44− CRC cell populations from HT-29 and Caco-2 cells gave the coincidental evidence of their transcriptional levels (Figure 6(e–l)).
Figure 6.
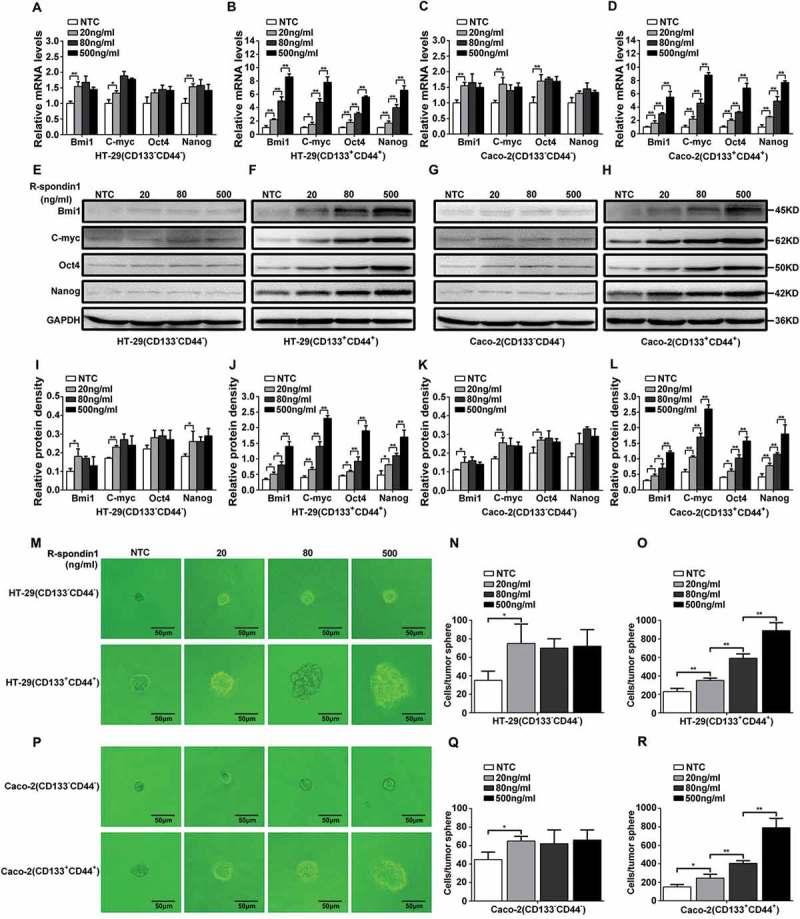
Expression of “stemness”-associated genes and tumorsphere formation in CD133+CD44+ and CD133−CD44− CRC cell populations treated with hRspo1. (A-D) Expression of “stemness”-associated genes (mRNA) in CD133+CD44+ (B and D) and CD133−CD44− (A and C) CRC cell populations from HT-29 and LS174T cells treated with 20, 80 or 500 ng/ml hRspo1. (E-H) Expression of “stemness”-associated genes (protein) in CD133+CD44+ (F and H) and CD133−CD44− (E and G) CRC populations treated with hRspo1. (I-L) The lower panel shows the quantitation of western blots (*: p < 0.05; **: p < 0.01). (M-R) CD133+CD44+ and CD133−CD44− CRC cell populations were treated with 20, 80 or 500 ng/ml hRspo1 and cultured in a special ultra-low attachment culture plate with conditional medium for tumorsphere formation. Tumorspheres formed in CD133+CD44+ HT-29 or Caco-2 cells were larger than those formed in CD133−CD44− HT-29 or Caco-2 cells (M and P). The cell numbers per tumorsphere formed in 20 ng/ml hRspo1-treated CD133−CD44− HT-29 or Caco-2 cells were more than for the non-treated control (NTC), but there were no significant differences among 20, 80 or 500 ng/ml hRspo1-treated CD133−CD44− HT-29 or Caco-2 cells (N and Q). The cell numbers per tumorsphere formed in 20, 80 or 500 ng/ml hRspo1-treated CD133+CD44+ CRC cell populations were more than in the non-treated control (NTC) and increased significantly in a dose-dependent manner (*: p < 0.05; **: p < 0.01) (O and R).
Tumorsphere formation assays in CD133+CD44+ and CD133−CD44− CRC cell populations from HT-29 and Caco-2 cells treated with hRspo1 (0, 20, 80, or 500 ng/ml) were used to compare their self-renewal abilities. Tumorspheres formed in CD133−CD44− HT-29 or Caco-2 cells had similar sizes despite different concentrations of hRspo1 (0, 20, 80, or 500 ng/ml) (Figure 6(m,p)), but the size of tumorspheres formed in CD133+CD44+ HT-29 or Caco-2 cells increased with increasing hRspo1 treatment (Figure 6(m,p)). The cell numbers per tumorsphere formed in 20 ng/ml hRspo1-treated CD133−CD44− HT-29 or Caco-2 cells were greater than for the untreated control, but there were no significant differences among 20, 80 or 500 ng/ml hRspol-treated CD133−CD44− HT-29 or Caco-2 cells. The cell numbers per tumorsphere formed in 0, 20, 80 or 500 ng/ml hRspo1-treated CD133+CD44+ CRC cell populations were more than for the untreated control and increased significantly in a dose-dependent manner (*: p < 0.05, **: p < 0.01) (Figure 6(n,o,q,r)). The result confirmed that Ascl2 autoactivation would confer stability to this cell state and confer the ability for self-renewal.
Discussion
The biological significance of Ascl2 has been recognized within the last few years. It has been identified, or suggested, to regulate genes expressed in the Lgr5+ intestinal stem cell gene expression program and genes expressed in the CRC cell expression program, such as miR-200 family members, CDX2 and miR-302 [11,17,27]. We and others have previously demonstrated that Ascl2 is a transcriptional regulator of binding to the E-box residing at the promoter of relevant genes [11,17,26].
In the present study, we showed that Ascl2 is autoregulated and behaves as its own transcriptional target in CRC cell lines and CRC tissues. Our results confirmed and extended the previously shown Ascl2 transactivation of its own promoter in Lgr5+ intestinal cryptic stem cells [26]. In this study, Ascl2 was shown to transactivate a 2588-bp Ascl2 promoter, which better mimics the endogenous promoter by covering a much more extensive regulatory region in a panel of CRC cell lines. Most interestingly, Ascl2 autoregulation is preferred in the CD133+CD44+ CRC cell population, which is widely recognized to be CRC progenitor cells [28–31]. Ascl2 binding with its own promoter and its further transactivation efficiency was more predominant in the CD133+CD44+ CRC cell population than in the CD133−CD44− CRC cell population. R-spondin1/Wnt activated Ascl2 expression dose-dependently in the CD133+CD44+ CRC population, but not in the CD133−CD44− CRC population, resulting in the maintenance of the “stemness” of the CD133+CD44+ CRC population, we provided a hypothesis detailing the role of R-spondin1/Wnt-enhanced Ascl2 autoregulation in controlling of the self-renewal of colorectal cancer progenitor cells (Figure 7). Ascl2 autoregulation in CRC cells and CRC progenitor cells as well as its biological significance provided the other evidence of Ascl2 auto-loop regulation besides its presence in Lgr5+ intestinal cryptic stem cells [26].
Figure 7.

Our proposed hypothesis detailing the role of R-spondin1/Wnt-enhanced Ascl2 autoregulation controls the self-renewal of colorectal cancer progenitor cells.
Confirmation of Ascl2 autoregulation in CRC cells was based on assessing Ascl2 activity on its endogenous level. The results obtained by further Ascl2 binding to its own promoter and Ascl2 transactivation reinforce this observation. As we know, Ascl2 is a Wnt signaling target and Ascl2 expression is controlled by the activation of Wnt signaling, which is among the most prominent driving forces of intestinal cryptic Lgr5+ adult stem cells [10]. The finding that Ascl2 binds to its own promoter in CRC cells (including the CD133+CD44+ CRC cell population) and human CRC tissues as well as leads to Ascl2 transactivation of its own promoter suggested that Ascl2 transactivation occurs in vivo and may have a role in amplifying Wnt signaling to achieve a sufficient Ascl2 expression level in CRC cells, especially in CRC progenitor cells (Ascl2-ON), but Ascl2 transactivation was absent in the CD133−CD44− CRC cell population (Ascl2-OFF). Positive autoregulation of transcription factors is an important physiological mechanism for maintaining a committed differentiation state during distinct stages of cell lineage and tissue development by reinforcing the formation of high concentrations of several master regulators of cell identity [32–36]. Ascl2 acts as a master regulator of intestinal crypt stem cells and CRC progenitor cells. In cancer cells, aberrant autoregulation loops initiated by tumor-associated expression of specific transcription factors and transcription cofactors, or the loss of repressor proteins that normally prevent binding of the factors to their respective core promoter elements, may promote malignant transformation [37,38]. The identified cooperation of the autoregulation loop of Ascl2 binding its own promoter with Wnt signaling is used to govern CRC progenitor cell identity, thus retaining their self-renewal.
It should be noted that although cell type-specific autoregulation of Ascl2 has been previously studied [26], an auto-regulatory mechanism of Ascl2 and cooperative mechanism with Wnt signaling predicts that self-renewal maintenance occurs to progress along a lineage of the CD133+CD44+ CRC cell population, as shown by the induced changes in Ascl2 overexpression. The involvement of Wnt-induced CSCs in colorectal cancer metastasis is widely accepted and implies a role for Wnt-induced CSCs in the propagation of metastasis [39]. Ascl2 autoregulation fuels Wnt signaling in the CD133+CD44+ CRC cell population and leads to the growth of CRC tumors by inducing the gene expression program of CRC progenitor cells. However, it is still unknown what causes the different Ascl2 autoregulation responses to the R-spondin1/Wnt stimulus in CD133+CD44+ and CD133−CD44− CRC cell populations.
Schuijers et al. [26] identified the Ascl2 autoregulation in Lgr5+ intestinal cryptic stem cells, our present study provided an evidence of the Ascl2 autoregulation in CD133+CD44+ CRC progenitor cell. There were some difference between our study and Schuijers’ report [26]. The target cells in Ascl2 autoregulation and its biological significance were different, Ascl2 autoregulation in Lgr5+ intestinal cryptic stem cell provides its mechanism for their renewal and keeps the homeostasis of intestinal mucosa, Ascl2 autoregulation in CD133+CD44+ CRC progenitor cells led to the renewal of CRC progenitor cells and conferred to their ability of migration, invasion, metastasis and tolerance to chemotherapy and radiotherapy. The methods to confirm Ascl2 autoregulation in Schuijers’ report [26] was based on ChIP-seq and single molecular FISH for indentifying transcript densities, our study to confirm Ascl2 autoregulation was based on luciferase assay, ChIP experiment and endogenous Ascl2 activation by exogenous Ascl2 expression. Ascl2 binding with its own promoter was not only present in CRC cells and CD133+CD44+ CRC progenitor cells, but we provided an evidence of Ascl2 binding with its own promoter in human CRC tissues, Ascl2 binding with its own promoter in Schuijers’ report is present in Lgr5+ intestinal cryptic stem cell and confirmed by ChIP-seq. R-spondin1/Wnt-enhanced Ascl2 autoregulation was only present in CRC progenitor cells and the mechanism was necessary for these cells to maintain their self-renewal.
Taken together, the results presented in this study clearly show that Ascl2 regulates its own expression in the gastrointestinal context and is bound to its own promoter in CRC cell lines and human CRC tissues. This mechanism is predominant in the CRC progenitor cells and is related to the maintenance of the “stemness” of CRC progenitor cells. The disturbance of Ascl2 autoregulation might give insight into CRC treatment for targeting CRC progenitor cells.
Material and methods
Cell culture
Human colon adenocarcinoma cell lines HT-29, LS174T and Caco-2 were obtained from American Type Culture Collection (ATCC) and grown by ATCC recommendations. The shRNA-Ctr/HT-29, shRNA-Ascl2/HT-29, shRNA-Ctr/LS174T, shRNA-Ascl2/LS174T, lv-Ascl2/Lovo, lv-Ctr/Lovo, lv-Ctr/SW480 and lv-Ascl2/SW480 cells were described previously and maintained in our lab [11,17].
Flow cytometry cell sorting and flow cytometry analysis
For the isolation of CD133+CD44+ and CD133−CD44− cell populations from HT-29 or Caco-2 cells, the single cell suspension was detached using 0.02% EDTA in phosphate-buffered saline (PBS), counted and washed in PBS. At least 106 cells were incubated with APC-conjugated anti-human CD133 antibody (mouse IgG2b, 130–098-129, Miltenyi Biotec, Auburn, CA, USA) and FITC-conjugated anti-human CD44 antibody (mouse IgG1, 130–098-210, Miltenyi Biotec, Auburn, CA, USA) at 4°C for 10 min in the dark. Isotype-matched mouse immunoglobulin IgG2b (130–098-890, Miltenyi Biotec, Auburn, CA, USA) and isotype-matched mouse immunoglobulin IgG1 (130–098-847, Miltenyi Biotec, Auburn, CA, USA) served as negative controls. After the washing steps, the labeled cells were analyzed and sorted with a fluorescence-activated cell sorter (FACS) (BD Biosciences, San Jose, CA, USA).
R-spondin1/wnt treatment in different cells
HT-29, LS174T, CD133−CD44− HT-29, CD133+CD44+ HT-29, CD133−CD44− Caco-2, and CD133+CD44+ Caco-2 cells were maintained at 37°C in a humidified atmosphere of 5% CO2 and subcultured at a dilution of 1:10. The cells were seeded into 6-well tissue-culture plates (for HT-29 and LS174T cells) or 6-well ultra-low attachment plates (for CD133−CD44− HT-29, CD133+CD44+ HT-29, CD133−CD44− Caco-2 and CD133+CD44+ Caco-2 cells) at a density of approximately 1 × 106 cells/dish. The cells were treated in their different culture media containing 5, 10, 20, 40, 80, 200, 500 or 1000 ng/ml R-spondin1/Wnt for 16 hours and used for further experiments.
Tumorsphere formation assay
The single cell suspension was suspended in DMEM/F12 (Hyclone, USA) supplemented with B27 (1×, Gibco), 20 ng/mL epidermal growth factor (EGF, Peprotech, USA), and 20 ng/ml basic fibroblast growth factor (bFGF, Peprotech, USA). The cells were treated with 20, 80 or 500 ng/ml recombinant human R-spondin1 protein for 16 hours and then plated in 24-well ultra-low attachment plates (Corning, USA) at a concentration of 1000 cells per well. Plates were analyzed 7–10 days later for tumorsphere formation and were quantified using an inverted microscope (Olympus) at 100× magnification. For subsequent quantification of the cell numbers per tumorsphere, tumorspheres were collected with a 40-µm sieve (BD Biosciences, San Jose, CA, USA) and disassociated with 0.25% trypsin/0.02% EDTA to make a single cell suspension. The viable cells were then counted using trypan blue exclusion.
Construction of adenoviral vectors and their transient transfection
Adenovirus particles expressing Ascl2 were produced by the GenePharma Co. Ltd (Shanghai, China). HT-29, LS174T and Caco-2 cells were transfected with adenovirus particles using the ADV4 (CMV/IRES/GFP) vector with Ascl2 plus three FLAGs inserts according to the manufacturer’s protocol. The cells were harvested 48 h after transfection to examine Ascl2 expression by real-time PCR and western blotting analysis.
Real-time PCR analysis
Total RNA was isolated from the cells using TRIzol (TaKaRa Biotechnology Co. Ltd, Dalian, China). cDNA was synthesized by using the Pri-meScript RT reagent kit (TaKaRa Biotechnology Co. Ltd, Dalian, China), PCR amplification was performed using the primer sets shown in Table 1 and the products were separated in 1% agarose gels. GAPDH was used as an internal control. qPCR was performed with the SYBR premix Ex TaqTM Green II (TaKaRa Biotechnology Co. Ltd, Dalian, China). Primers were designed with National Center for Biotechnology Information (NCBI) primer design software. Relative mRNA expression levels were calculated by the formula 2−ΔΔCt using SDS software (Applied Biosystems). The primers used for qPCR are shown in Table 1.
Table 1.
Sequences of the oligonucleotides used for RT-PCR, qPCR and chromatin immunoprecipitation (ChIP).
| RT-PCR | 5ʹ→ 3’ |
|---|---|
| Endogenous Ascl2 | F: 5ʹ-CACAGTTTTCCCCGTCG-3ʹ R: 5ʹ-GAAGCCCAAGTTCACCAG-3’ |
| Exogenous Ascl2 | F: 5ʹ-ACAAGGATGACGATGACAA-3ʹ R: 5ʹ-GCCTGGAAGCCCAAGTT-3’ |
| GAPDH | F: 5ʹ-CATCAAGAAGGTGGTGAAGCAG-3ʹ R: 5ʹ-AAAGGTGGAGGAGTGGGTGTC-3’ |
| qPCR | 5ʹ→ 3’ |
| Ascl2 | F: 5ʹ-CGTGAAGCTGGTGAACTTGG-3ʹ R: 5ʹ-GGATGTACTCCACGGCTGAG-3’ |
| Bmi1 | F: 5ʹ-CGTGTATTGTTCGTTACCTGGA-3ʹ R: 5ʹ-TTCAGTAGTGGTCTGGTCTTGT-3’ |
| C-myc | F: 5ʹ-GGCTCCTGGCAAAAGGTCA-3ʹ R: 5ʹ-CTGCGTAGTTGTGCTGATGT-3’ |
| Oct4 | F: 5ʹ-CTGGAGAAGGAGAAGCTGGA-3ʹ R: 5ʹ-CAAATTGCTCGAGTTCTTTCTG-3’ |
| Nanog | F: 5ʹ-CTCTCCTCTTCCTTCCTCCAT-3ʹ R: 5ʹ-TTGCGACACTCTTCTCTGC-3’ |
| ChIP | 5ʹ→ 3’ |
| ChIP1 | F: 5ʹ-GACAGGAAGTTCTGGGGTGA-3ʹ R: 5ʹ-CCTCTGCTGCTCTGGGTG-3’ |
| ChIP2 | F: 5ʹ-GCCCACCCATCAAGTCAC-3ʹ R: 5ʹ-AGCCCCTCGGTCATCTGT-3’ |
| ChIP3 | F: 5ʹ-GAGGCAGCAGAAGGAGGG-3ʹ R: 5ʹ-GCCCTGAGTCTGAATCCG-3’ |
| ChIP4 | F: 5ʹ-GTCGGCTTCCTCATCTGC-3ʹ R: 5ʹ-CCACCTGCTCCCATTACC-3’ |
| ChIP5 | F: 5ʹ-CCCCGAGGCTGGCAGTAAA-3ʹ R: 5ʹ-TCCCGAAGGTGACCAGATGCT-3’ |
| ChIP6 | F: 5ʹ-CAGCCTGGGTCACAAAAGA-3ʹ R: 5ʹ-GTGGCTCCCTCCATCTGC-3’ |
| ChIP7 | F: 5ʹ-AGATTAACGCACAGGTGGGG-3ʹ R: 5ʹ-GGGGAGCATCGCTAACAAAG-3’ |
| ChIP8 | F: 5ʹ-GGAAGGCTCAACCCAGGACC-3ʹ R: 5ʹ-CCCAACTGGCGAAATCTGC-3’ |
| GAPDH (ChIP for positive control) |
F: 5ʹ-TACTAGCGGTTTTACGGGCG-3ʹ R: 5ʹ-TCGAACAGGAGGAGCAGAGAGCGA-3’ |
Western blotting assay
The cell pellet was lysed in SDS sample buffer. Proteins were separated by SDS–PAGE and transferred to a PVDF membrane (Millipore, Bedford, MA). GAPDH was used as a control. The membrane was probed overnight at 4°C with a primary antibody. Protein bands were visualized using the chemiluminescent reagent of the Pierce ECL kit (Thermo Scientific), and quantification of the western blots was performed using ChemiDoxTM XRS with Image LabTM software. The primary antibodies used in this study were: mouse anti-Ascl2 (1:300, Millipore, MAB4418), mouse anti-FLAG (1:5000, Proteintech, 66,008–2-Ig), rabbit anti-Bmi1 (1:500, Proteintech, 10,832–1-AP), rabbit anti-C-myc (1:500, Proteintech, 10,828–1-AP), rabbit anti-Oct4 (1:500, Proteintech, 11,263–1-AP), rabbit anti-Nanog (1:500, Abcam, ab21624), and rabbit anti-GAPDH (1:3000, Proteintech, 10,494–1-AP). Detailed western blotting procedures have been described previously [24].
Transfection and luciferase assay
For studying whether Ascl2 promotes its own gene transcription, the Ascl2 5ʹ-flanking sequence (−2588/+1 bp region) was amplified using PCR and cloned into the luciferase reporter vector pGL3-Basic (Promega, Madison, WI). Briefly, primers containing KpnI and Nhe I adapters were used to amplify the Ascl2 promoter sequence from intestinal tissue DNA. The primer pair used to produce the promoter fragment (−2588/+1) was as follows: forward: 5ʹ-GGGGTACCTCCGGAGATCTTACCA-3ʹ; and reverse: 5ʹ-CGGCTAGCCGCGCCTGCATCCAC-3ʹ. The products were digested with KpnI and Nhe I, and then, they were ligated into the pGL3-Basic vector. Transfection of luciferase reporter containing the Ascl2 promoter and the luciferase assay were performed as described previously [24,25].
Chromatin immunoprecipitation (chip) assay
The ChIP assay was performed using a ChIP assay kit (Millipore) according to the manufacturer’s instructions. Soluble chromatin was prepared from shRNA-Ctr/HT-29, shRNA-Ascl2/HT-29, shRNA-Ctr/LS174T, shRNA-Ascl2/LS174T, lv-Ctr/SW480, lv-Ascl2/SW480, lv-Ctr/Lovo, lv-Ascl2/Lovo, CD133−CD44− HT-29, CD133+CD44+ HT-29, CD133−CD44− Caco-2, and CD133+CD44+ Caco-2 cells as well as CRC tissues. Chromatin was immunoprecipitated with anti-Ascl2 (Millipore, MAB4418). To study if Ascl2 binds to its own promoter, the final DNA extracts were amplified by PCR using primer pairs that included ChIP 1, ChIP 2, ChIP 3, ChIP 4, ChIP 5, ChIP 6, ChIP 7 or ChIP 8 consensus sequences in the human Ascl2 promoter. The primer sequences of the amplified PCR products are shown in Table 1. These assays were performed as described previously [24,25].
Immunohistochemistry
Immunohistochemical detection for Ascl2 was performed in human CRC tissues and their peri-cancerous mucosa (n = 21). The primary antibody against Ascl2 (Millipore) was diluted 1:50 in 5% BSA. The detailed immunohistochemical procedure was described previously [17]. This study was approved by the local clinical research ethics committee. All of the subjects provided informed consent before their colonoscopy or resection surgery.
Statistical analysis
GraphPad Prism 6.0 software was used for all statistical analyses. All differences were deemed significant when p < 0.05 and very significant when p < 0.01.
Funding Statement
This work was supported by the following awards: 81572903 and 81372557 (to R.W.) from the National Natural Science Foundation of China.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. [DOI] [PubMed] [Google Scholar]
- 2.Huang EH, Wicha MS.. Colon cancer stem cells: implications for prevention and therapy. Trends Mol Med. 2008;14:503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tan BT, Park CY, Ailles LE, et al. The cancer stem cell hypothesis: a work in progress. Lab Invest. 2006;86:1203–1207. [DOI] [PubMed] [Google Scholar]
- 4.O’Brien CA, Pollett A, Gallinger S, et al. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. [DOI] [PubMed] [Google Scholar]
- 5.Ricci-Vitiani L, Lombardi DG, Pilozzi E, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. [DOI] [PubMed] [Google Scholar]
- 6.Gangemi RM, Griffero F, et al. SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells. 2009;27:40–48. [DOI] [PubMed] [Google Scholar]
- 7.Todaro M, Alea MP, Di Stefano AB, et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell. 2007;1:389–402. [DOI] [PubMed] [Google Scholar]
- 8.Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011;17:313–319. [DOI] [PubMed] [Google Scholar]
- 9.de Sousa EM, Vermeulen L, Richel D, et al. Targeting Wnt signaling in colon cancer stem cells. Clin Cancer Res. 2011;17:647–653. [DOI] [PubMed] [Google Scholar]
- 10.van der Flier LG, van Gijn ME, Hatzis P, et al. Transcription factor achaete scute-like 2 controls intestinal stem cell fate. Cell. 2009;136:903–912. [DOI] [PubMed] [Google Scholar]
- 11.Zhu R, Yang Y, Tian Y, et al. Ascl2 knockdown results in tumor growth arrest by miRNA-302b-related inhibition of colon cancer progenitor cells. PloS One. 2012;7:e32170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jubb AM, Chalasani S, Frantz GD, et al. Achaete-scute like 2 (ascl2) is a target of Wnt signalling and is upregulated in intestinal neoplasia. Oncogene. 2006;25:3445–3457. [DOI] [PubMed] [Google Scholar]
- 13.Jubb AM, Hoeflich KP, Haverty PM, et al. Ascl2 and 11p15.5 amplification in colorectal cancer. Gut. 2011;60:1606–1607; author reply 1607. [DOI] [PubMed] [Google Scholar]
- 14.Stange DE, Engel F, Longerich T, et al. Expression of an ASCL2 related stem cell signature and IGF2 in colorectal cancer liver metastases with 11p15.5. Gain. Gut. 2010;59:1236–1244. [DOI] [PubMed] [Google Scholar]
- 15.Liu X, Chen X, Zhong B, et al. Transcription factor achaete-scute homologue 2 initiates follicular T-helper-cell development. Nature. 2014;507:513–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takao T, Asanoma K, Tsunematsu R, et al. The maternally expressed gene Tssc3 regulates the expression of MASH2 transcription factor in mouse trophoblast stem cells through the AKT-Sp1 signaling pathway. J Biol Chem. 2012;287:42685–42694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shang Y, Pan Q, Chen L, et al. Achaete scute-like 2 suppresses CDX2 expression and inhibits intestinal neoplastic epithelial cell differentiation. Oncotarget. 2015;6:30993–31006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Du X, Dong Y, Shi H, et al. Mst1 and mst2 are essential regulators of trophoblast differentiation and placenta morphogenesis. PloS One. 2014;9:e90701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cowden Dahl KD, Fryer BH, Mack FA, et al. Hypoxia-inducible factors 1alpha and 2alpha regulate trophoblast differentiation. Mol Cell Biol. 2005;25:10479–10491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Renaud SJ, Chakraborty D, Mason CW, et al. OVO-like 1 regulates progenitor cell fate in human trophoblast development. Proc Natl Acad Sci USA. 2015;112:E6175–E6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wei X, Ye J, Shang Y, et al. Ascl2 activation by YAP1/KLF5 ensures the self-renewability of colon cancer progenitor cells. Oncotarget. 2017;8(65):109301–109318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shang Y, Chen H, Ye J, et al. HIF-1α/Ascl2/miR-200b regulatory feedback circuit modulated the epithelial-mesenchymal transition (EMT) in colorectal cancer cells. Exp Cell Res. 2017;360(2):243–256. [DOI] [PubMed] [Google Scholar]
- 23.Basu S, Gavert N, Brabletz T, et al. The intestinal stem cell regulating gene ASCL2 is required for L1-mediated colon cancer progression. Cancer Lett. 2018;424:9–18. [DOI] [PubMed] [Google Scholar]
- 24.Dessein AF, Stechly L, Jonckheere N, et al. Autocrine induction of invasive and metastatic phenotypes by the MIF-CXCR4 axis in drug-resistant human colon cancer cells. Cancer Res. 2010;70:4644–4654. [DOI] [PubMed] [Google Scholar]
- 25.Birkenkamp-Demtroder K, Maghnouj A, Mansilla F, et al. Repression of KIAA1199 attenuates Wnt-signalling and decreases the proliferation of colon cancer cells. Br J Cancer. 2011;105:552–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schuijers J, Junker JP, Mokry M, et al. Ascl2 acts as an R-spondin/Wnt-responsive switch to control stemness in intestinal crypts. Cell Stem Cell. 2015;16:158–170. [DOI] [PubMed] [Google Scholar]
- 27.Tian Y, Pan Q, Shang Y, et al. MicroRNA-200 (miR-200) cluster regulation by achaete scute-like 2 (Ascl2): impact on the epithelial-mesenchymal transition in colon cancer cells. J Biol Chem. 2014;289:36101–36115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Basu S, Haase G, Ben-Ze’ev A. Wnt signaling in cancer stem cells and colon cancer metastasis. F1000Research. 2016;5 DOI: 10.12688/f1000research.7579.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pan Q, Meng L, Ye J, et al. Transcriptional repression of miR-200 family members by Nanog in colon cancer cells induces epithelial-mesenchymal transition (EMT). Cancer Lett. 2017;392:26–38. [DOI] [PubMed] [Google Scholar]
- 30.Song IS, Jeong YJ, Jeong SH, et al. FOXM1-induced PRX3 regulates stemness and survival of colon cancer cells via maintenance of mitochondrial function. Gastroenterology. 2015;149:1006–1016.e1009. [DOI] [PubMed] [Google Scholar]
- 31.Moon CM, Kwon JH, Kim JS, et al. Nonsteroidal anti-inflammatory drugs suppress cancer stem cells via inhibiting PTGS2 (cyclooxygenase 2) and NOTCH/HES1 and activating PPARG in colorectal cancer. Int J Cancer. 2014;134:519–529. [DOI] [PubMed] [Google Scholar]
- 32.Bellizzi A, Sebastian S, Ceglia P, et al. Co-expression of CD133(+)/CD44(+) in human colon cancer and liver metastasis. J Cell Physiol. 2013;228:408–415. [DOI] [PubMed] [Google Scholar]
- 33.Thayer MJ, Tapscott SJ, Davis RL, et al. Positive autoregulation of the myogenic determination gene MyoD1. Cell. 1989;58:241–248. [DOI] [PubMed] [Google Scholar]
- 34.Chen H, Ray-Gallet D, Zhang P, et al. PU.1 (Spi-1) autoregulates its expression in myeloid cells. Oncogene. 1995;11:1549–1560. [PubMed] [Google Scholar]
- 35.McCormick A, Brady H, Theill LE, et al. Regulation of the pituitary-specific homeobox gene GHF1 by cell-autonomous and environmental cues. Nature. 1990;345:829–832. [DOI] [PubMed] [Google Scholar]
- 36.Serfling E, Chuvpilo S, Liu J, et al. NFATc1 autoregulation: a crucial step for cell-fate determination. Trends Immunol. 2006;27:461–469. [DOI] [PubMed] [Google Scholar]
- 37.Barros R, da Costa LT, Pinto-de-Sousa J, et al. CDX2 autoregulation in human intestinal metaplasia of the stomach: impact on the stability of the phenotype. Gut. 2011;60:290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Angel P, Hattori K, Smeal T, et al. The jun proto-oncogene is positively autoregulated by its product, Jun/AP-1. Cell. 1988;55:875–885. [DOI] [PubMed] [Google Scholar]
- 39.Nicolaides NC, Gualdi R, Casadevall C, et al. Positive autoregulation of c-myb expression via Myb binding sites in the 5ʹ flanking region of the human c-myb gene. Mol Cell Biol. 1991;11:6166–6176. [DOI] [PMC free article] [PubMed] [Google Scholar]