

ABSTRACT
Chaperone-mediated autophagy (CMA) is a selective degradative process for cytosolic proteins that contributes to the maintenance of proteostasis. The signaling mechanisms that control CMA are not fully understood but might involve response to stress conditions including oxidative stress. Considering the role of CMA in redoxtasis and proteostasis, we sought to determine if the transcription factor NFE2L2/NRF2 (nuclear factor, erythroid derived 2, like 2) has an impact on CMA modulation. In this work, we identified and validated 2 NFE2L2 binding sequences in the LAMP2 gene and demonstrated in several human and mouse cell types that NFE2L2 deficiency and overexpression was linked to reduced and increased LAMP2A levels, respectively. Accordingly, lysosomal LAMP2A levels were drastically reduced in nfe2l2-knockout hepatocytes, which also displayed a marked decrease in CMA activity. Oxidant challenge with paraquat or hydrogen peroxide, or pharmacological activation of NFE2L2 with sulforaphane or dimethyl fumarate also increased LAMP2A levels and CMA activity. Overall, our study identifies for the first time basal and inducible regulation of LAMP2A, and consequently CMA activity, by NFE2L2.
Abbreviations: ACTB: actin, beta, ARE: antioxidant response element; ATG5: autophagy related 5; BACH1: BTB domain and CNC homolog 1; ChIP: chromatin immunoprecipitation; CMA: chaperone-mediated autophagy; DHE: dihydroethidium; DMF: dimethyl fumarate; ENCODE: Encyclopedia of DNA elements at the University of California, Santa Cruz; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; GBA: glucosylceramidase beta; GFP: green fluorescent protein; HMOX1: heme oxygenase 1; H2O2: hydrogen peroxide; HSPA8/HSC70: heat shock protein family A (Hsp70) member 8; KEAP1: kelch like ECH associated protein 1; LAMP2A: lysosomal associated membrane protein 2A; LAMP2B: lysosomal associated membrane protein 2B; LAMP2C: lysosomal associated membrane protein 2C; LAMP1: lysosomal associated membrane protein 1; MAFF: MAF bZIP transcription factor F; MAFK: MAF bZIP transcription factor K; NFE2L2/NRF2: nuclear factor, erythroid derived 2, like 2; NQO1: NAD(P)H quinone dehydrogenase 1; PQ: paraquat; PI: protease inhibitors; qRT-PCR: quantitative real-time polymerase chain reaction; RNASE: ribonuclease A family member; SFN: sulforaphane; SQSTM1/p62: sequestosome 1; TBP: TATA-box binding protein
KEYWORDS: LAMP2A, NRF2, proteostasis
Introduction
Intracellular oxidized proteins can be eliminated by a selective type of autophagy termed chaperone-mediated autophagy (CMA). The main characteristic of this mode of autophagy is the presence of a lysosomal receptor called LAMP2A (lysosomal associated membrane protein 2A). Soluble cytosolic proteins bearing a KFERQ-like motif are recognized by HSPA8/HSC70 (heat shock protein family A [Hsp70] member 8) [1–3]. HSPA8 delivers cargoes to the lysosome surface, where they interact with LAMP2A [4]. Then, LAMP2A multimerizes in the lysosomal membrane to form a translocation complex [5], so that unfolded substrates can enter the lysosome, assisted by lumenal HSPA8, and eventually be degraded by lysosomal proteases [6,7]. The limiting step in the whole process appears to be the availability of LAMP2A receptor at the lysosomal membrane, as lysosomal LAMP2A levels have been reported to correlate with CMA activity, and knockdown or overexpression of LAMP2A results in a decrease or increase in CMA proportional to the levels of expression [4,8]. LAMP2A is one of the 3 proteins that originate from alternative splicing of the LAMP2 gene. All LAMP2A, LAMP2B and LAMP2C isoforms share an identical lumenal region but differ in their cytosolic and transmembrane tail [9]. These isoforms result from cell-specific yet unknown regulation of differential splicing [10,11] but experimental evidence supports the conclusion that LAMP2A is the only LAMP2 variant required for CMA [4,8].
Although basal CMA activity can be detected in most cell types [12], this process is maximally activated in response to stress. Activation of CMA is associated with increased levels of LAMP2A and its multimerization to form membrane translocation complexes, lysosomal enrichment in HSPA8 and relocation of lysosomes to the perinuclear region [13–15]. It has been reported that a mild oxidant environment activates the expression of Lamp2a [14] and that this process is under the transcriptional regulation of NFAT in T cells [16]. However, a generic mechanism that might regulate the expression of LAMP2A under these circumstances remains unknown.
NFE2L2/NRF2 (nuclear factor, erythroid derived 2, like 2), participates in the control of metabolic redox processes including degradation of oxidized proteins. NFE2L2 controls the basal and stress-inducible expression of over 250 genes that share a common a cis-acting enhancer termed the antioxidant response element (ARE) [17]. These genes participate in several cytoprotective functions such as phase I, II and III detoxification reactions, glutathione and PRDX/peroxiredoxin-TXN/thioredoxin metabolism, NADPH production through the pentose phosphate pathway and ME (malic enzyme), fatty acid oxidation or iron metabolism [18]. Most of these reactions provide an armamentarium to control redox homeostasis. Importantly for this study, we and others previously reported that NFE2L2 can modulate the expression of several genes involved in macroautophagy [19–22]. Thus, we identified many NFE2L2-regulated genes involved in the different phases of the macroautophagy process, from cargo recognition to autolysosome clearance [22]. Considering this evidence and the role of CMA in the cellular response to oxidative stress [14], it might be feasible that NFE2L2 also participates in the regulation of CMA, but this possibility has not yet been investigated.
In this study, we show for the first time that NFE2L2 is required for CMA activation through the transcriptional control of LAMP2A expression, revealing a molecular pathway that connects oxidative stress with CMA induction. Bearing in mind that oxidized proteins accumulate in aging and in several age-related pathological conditions [23], when CMA activity has been shown to decline [24,25], this study provides a new strategy to activate CMA through NFE2L2 inducers to facilitate elimination of such damaged and otherwise toxic proteins.
Results
NFE2L2 binds to functional antioxidant response elements (ares) in the LAMP2 gene
We searched the Encyclopedia of DNA Elements at UCSC (ENCODE) [26] of the human genome for putative AREs in this gene (Figure 1a). The ENCODE database gathers experimental data from chromatin immunoprecipitation (ChIP) analysis of ARE binding transcription factors MAFF, MAFK and BACH1 although NFE2L2 is not analyzed. As shown in Figure 1a, some of these binding sites were located at histone acetylated and DNAse-sensitive regions in the LAMP2 gene, both factors being typical of active enhancers. We then used a Python-based bioinformatics analysis to scan these binding regions for the consensus ARE as established in the JASPAR database [27]. We detected 8 putative AREs in the LAMP2 gene (Table S1) and 3 of them showed a relative score higher than 85%, a commonly used threshold for transcription factor binding-site analysis [28,29]. Moreover, the analysis also retrieved already identified AREs in the bona fide target genes of NFE2L2, HMOX1 and SQSTM1/p62 (Table 1).
Figure 1.
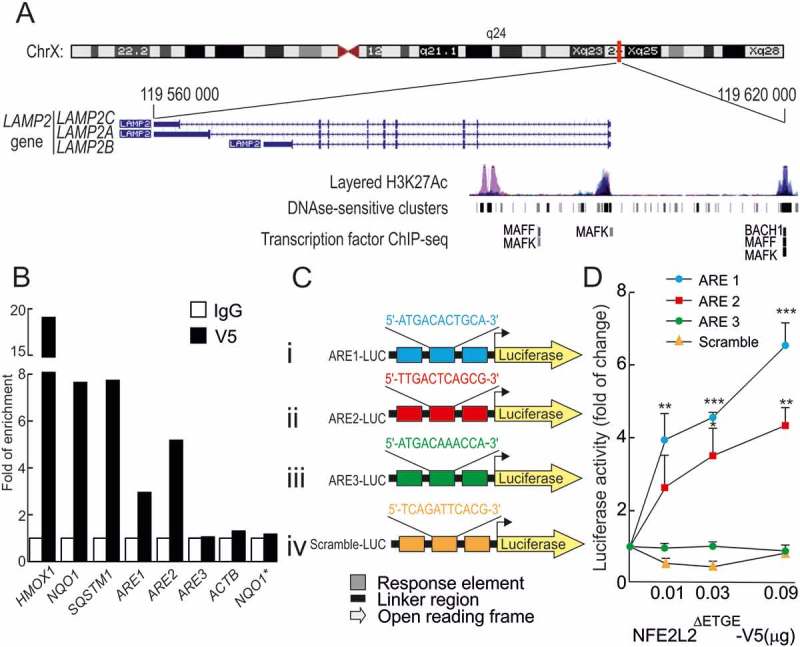
NFE2L2 binds to 2 functional antioxidant response elements (AREs) in the LAMP2 gene. (a) Scheme of the LAMP2 gene showing the 3 splice variants: LAMP2A, LAMP2B and LAMP2C, from The Encyclopedia of DNA Elements at UCSC (ENCODE) for human genome. Putative AREs in the LAMP2 gene were identified taking as reference the available information from ChIP of ARE-binding factors MAFK, MAFF and BACH1. These regions were localized in 200–400 base-pair-long DNAse-sensitive and H3K27Ac-rich regions, i.e. most likely regulatory promoter regions. (b) HEK293T cells were transfected with an expression vector for NFE2L2ΔETGE-V5 (lacking the KEAP1 regulatory domain). ChIP analysis was performed with anti-IgG or anti-V5 antibodies and the potential AREs with the highest score, termed ARE1, ARE2 and ARE3 (Table 1), were analyzed by qRT-PCR. The figure shows representative data normalized as the fold of enrichment with the anti-V5 antibody vs. the IgG antibody. The presence of already known AREs in HMOX1, NQO1 and SQSTM1 was analyzed as positive control and ACTB, and a region of NQO1 that does not contain an ARE (NQO1*) were amplified as negative controls. (c), Luciferase reporter constructs carrying 3 tandem putative ARE sequences from the LAMP2 gene (i-iii) or a scramble sequence as negative control (iv) controlling the expression of luciferase. (d) nfe2l2-KO mouse embryo fibroblasts were co-transfected with the reporters in C and increasing amounts of NFE2L2ΔETGE-V5 construct. Values were normalized to pTK-Renilla activity and presented as fold of change. Data are mean ± SD (n = 3). Statistical analysis was performed using Student’s t test. *p < 0.05, ** p < 0.01 and *** p < 0.001 vs. basal levels.
Table 1.
Putative antioxidant response elements (AREs) with a relative score higher than the 85% in the LAMP2 gene.
| ARE | Localization of the potential ARE | Max Score | Relative score | Putative binding sequence | Region of the gene |
|---|---|---|---|---|---|
| ARE1 | chrX:119,596,430–119,596,441 | 15.83 | 0.921 | ATGACACTGCA | Intron 1 |
| ARE2 | chrX:119 619,599–119,619,610 | 13.24 | 0.859 | TTGACTCAGCG | 5ʹ UTR (−16,500) |
| ARE3 | chrX:119,619,560–119,619,571 | 13.07 | 0.855 | ATGACAAACCA | 5ʹ UTR (−16,500) |
To validate the AREs with the highest score (termed ARE1, ARE2 and ARE3), we next performed ChIP analysis with NFE2L2. HEK293T cells were transfected with an expression vector for V5-tagged NFE2L2ΔETGE. This construct lacks the KEAP1 regulatory domain (ETGE) in order to facilitate NFE2L2 stabilization, translocation to the nucleus and binding to target genes. NFE2L2 was immunoprecipitated with an anti-V5 antibody and anti-IgG as a negative control. NFE2L2 binding was analyzed by qRT-PCR employing as template the immunoprecipitated DNA and primers designed to specifically amplify ARE1, ARE2 or ARE3 (Table S2). NFE2L2 bound to well-known positive control AREs in HMOX1, NQO1 and SQSTM1/p62, whereas NFE2L2 did not bind to ACTB or to a region of NQO1 that does not contain any ARE (NQO1*). Interestingly, NFE2L2 bound to ARE1 and ARE2 in LAMP2 but not to ARE3 (Figure 1b and Table S3).
We next analyzed the functionality of these putative AREs. Three tandem nucleotide sequences of each ARE or a control scramble sequence were cloned in the promoter region of a luciferase reporter as shown in Figure 1c. Nfe2l2-deficient cells were transiently co-transfected with these constructs and increasing amounts of NFE2L2-ΔETGE-V5. We found that NFE2L2 expression induced luciferase expression in reporters carrying ARE1 and ARE2 but not ARE3 or the scramble negative control (Figure 1d). As an additional control, change of a highly conserved G to A in the putative ARE1 and ARE2 sequences greatly reduced luciferase expression (Figure S1). Altogether, these results indicate that NFE2L2 binds and activates, at least, 2 AREs in the LAMP2 gene, termed here ARE1 and ARE2.
LAMP2A levels are regulated by NFE2L2
To determine if, in fact, NFE2L2 has an impact on LAMP2A expression, HEK293T cells were infected with a lentiviral vector expressing GFP as negative control or NFE2L2 ΔETGE-V5. As shown in Figure 2a-2c, NFE2L2 overexpression led to a modest but consistent increase in LAMP2A levels as well as in the positive controls HMOX1 and SQSTM1. In addition, we used a knockdown strategy to reduce NFE2L2 activity in the lung carcinoma cell line A549, which lacks KEAP1 and therefore exhibits high NFE2L2 protein levels. NFE2L2 knockdown reduced mRNA and protein levels of HMOX1, SQSTM1 and also LAMP2A (Figure 2d-2f).
Figure 2.
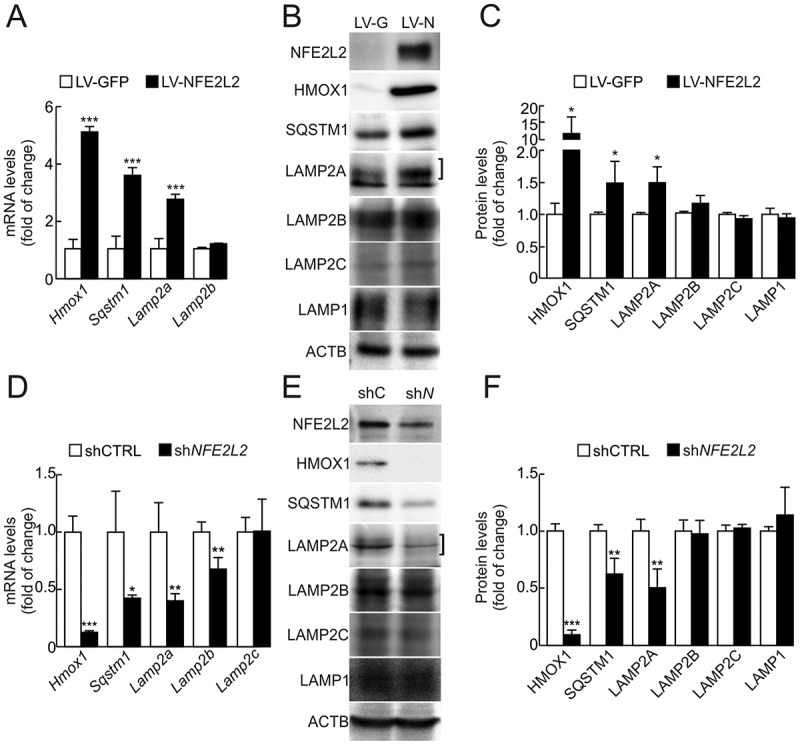
LAMP2A levels are modified upon genetic manipulation of NFE2L2. (a) HEK293T cells were transduced with GFP- or NFE2L2ΔETGE-V5-expressing lentivirus (LV-GFP or LV-NFE2L2, respectively). Expression levels of Hmox1, Sqstm1, Lamp2a, Lamp2b and Lamp2c were determined 3 days post-infection by qRT-PCR and normalized by Actb levels. Lamp2c levels were barely detectable in this cell type (data not shown). Data are mean ± SD (n = 4). Statistical analysis was performed with Student’s t test. ***p < 0.001 vs. LV-GFP-infected cells. (b) Representative immunoblots for the indicated proteins in cell lysates from cells transduced as in A. The bracket indicates the band corresponding to LAMP2A. (c) Densitometric quantification of representative immunoblots from B relative to ACTB protein levels. Data are mean ± SD (n = 4). Statistical analysis was performed using Student’s t test. *p < 0.05 vs. LV-GFP-infected cells. (d) A549 cells were transduced with lentivirus carrying shRNA against a scramble sequence (shCTRL) or against NFE2L2 (shNFE2L2). Expression levels of Hmox1, Sqstm1, Lamp2a, Lamp2b and Lamp2c were determined 3 days post-infection by qRT-PCR and normalized to Actb levels. Data are mean ± SD (n = 4). Statistical analysis was performed with Student’s t test. *p < 0.05, **p < 0.01 and ***p < 0.001 vs. shCTRL infected cells. (e) Representative immunoblots for the indicated proteins in cell lysates from cells transduced as in D. (f) Densitometric quantification of representative immunoblots from E relative to ACTB protein levels. Data are mean ± SD (n = 4). Statistical analysis was performed using Student’s t test. **p < 0.01 and ***p < 0.001 vs. shCTRL infected cells.
Because CMA and NFE2L2 activity have been separately well characterized in liver, we focused on the regulation of LAMP2A by NFE2L2 in mouse hepatocytes. Messenger RNA and protein levels of HMOX1, SQSTM1 and LAMP2A were lower in nfe2l2-knockout (nfe2l2-KO) than in Nfe2l2-wild type (Nfe2l2-WT) cells (Figure 3a, 3c and 3d). Moreover, lentivirus-induced overexpression of NFE2L2ΔETGE-V5 partially rescued the mRNA expression of these 3 genes in nfe2l2-KO hepatocytes (Figure 3b). Lamp2b and Lamp2c isoforms were not significantly changed upon overexpression or knockdown of NFE2L2 (Fig. S2), in agreement with the reported regulation- and cell-specific expression of these 2 isoforms, which do not participate in CMA. Moreover, reduction of LAMP2A/Lamp2a levels in the absence of NFE2L2 was also confirmed in human astrocytes, mouse hippocampal cells, embryo fibroblasts and cortical neurons from Nfe2l2-WT vs nfe2l2-KO mice (Fig. S3).
Figure 3.
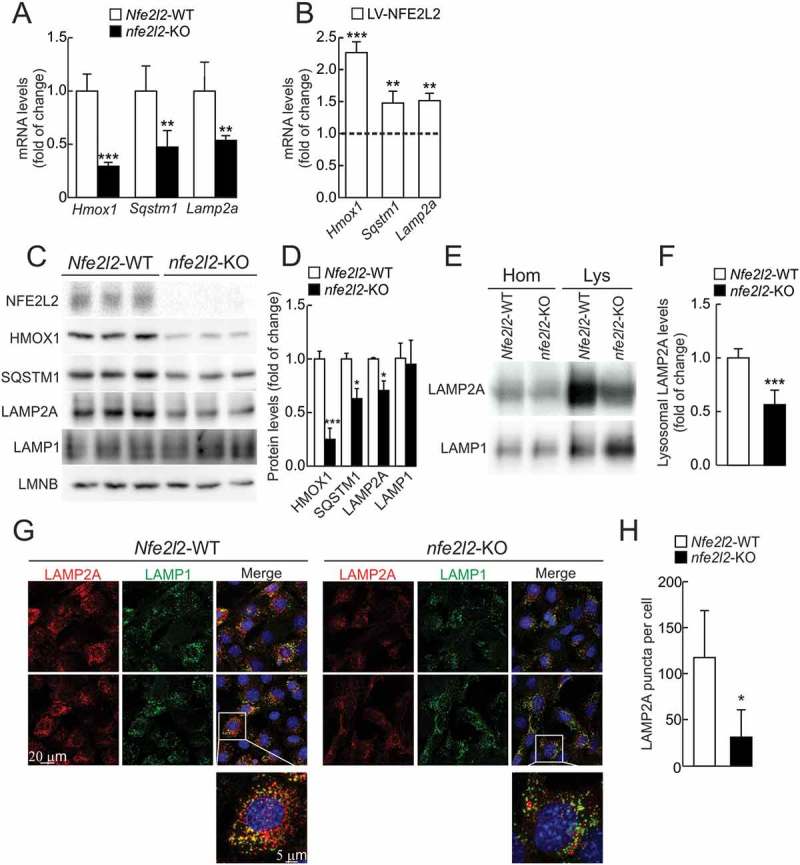
LAMP2A levels are reduced in the absence of NFE2L2. (a) Expression levels of Hmox1, Sqstm1 and Lamp2a from Nfe2l2-WT and nfe2l2-KO immortalized hepatocytes were determined by qRT-PCR and normalized to Actb levels. Data are mean ± SD (n = 4). Statistical analysis was performed with Student’s t test. **p < 0.01 and ***p < 0.001 vs. Nfe2l2-WT cells. (b) nfe2l2-KO immortalized hepatocytes were rescued with an NFE2L2ΔETGE-V5-expressing lentivirus (LV-NFE2L2). Expression levels of Hmox1, Sqstm1 and Lamp2a was analyzed 3 days post-infection by qRT-PCR and normalized to Tbp levels. The dotted line represents expression levels of nfe2l2-KO hepatocytes transduced with a control lentivirus (LV-GFP). Data are mean ± SD (n = 4). Statistical analysis was performed with Student’s t test. **p < 0.01 and ***p < 0.001 vs. LV-CTRL infected cells. (c) Representative immunoblots for the indicated proteins in cell lysates from Nfe2l2-WT and nfe2l2-KO immortalized hepatocytes. (d) Densitometric quantification of representative immunoblots from C relative to LMNB (lamin B) protein levels. Data are mean ± SD (n = 3). Statistical analysis was performed using Student’s t test. **p < 0.01 and ***p < 0.001 vs. Nfe2l2-WT cells. (e) Representative immunoblots of LAMP2A and LAMP1 in homogenates (Hom) and isolated lysosomes (Lys) from Nfe2l2-WT and nfe2l2-KO immortalized hepatocytes. (f) Densitometric quantification of representative immunoblots from E relative to LAMP1 levels. Data are mean ± SD (n = 6). Statistical analysis was performed using Student’s t test. ***p < 0.001 vs. Nfe2l2-WT levels. (g) Confocal analysis of double immunofluorescence with anti-LAMP2A (red) and anti-LAMP1 (green) antibodies in immortalized Nfe2l2-WT and nfe2l2-KO hepatocytes. The insert shows a higher magnification of the indicated cell. (h) Quantification of the total number of LAMP2A-positive puncta per cell. Values are mean ± SD (n = 3, with > 50 cells counted per experiment). Statistical analysis was performed using Student’s t test. *p < 0.05 vs. Nfe2l2-WT cells.
A modest reduction in LAMP2A levels was detected in whole cell lysates of nfe2l2-KO when compared to Nfe2l2-WT lysates (Figures 3c and 3d), and this reduction was more evident when we compared isolated lysosomes (Figures 3e and 3f), taking LAMP1 as a control lysosomal protein that is not involved in CMA. We also analyzed LAMP2A levels in lysosomes by immunofluorescence and, as shown in Figures 3g and 3h, the overall intensity and the number of puncta per cell of LAMP2A (red), which colocalized with LAMP1 (green), were dramatically reduced in nfe2l2-KO hepatocytes compared with Nfe2l2-WT hepatocytes. Altogether, NFE2L2 absence results in parallel changes in mRNA and protein levels of LAMP2A, especially in the lysosomal fraction.
NFE2L2 controls CMA under oxidant conditions
We characterized the redox status of immortalized hepatocytes derived from Nfe2l2-WT or nfe2l2-KO mice. As shown in Figures 4a and 4b, dihydroethidium (DHE) staining did not reflect major differences in ROS levels between these cell types under basal conditions. However, upon treatment for 16 h with the oxidative stress-inducing drug paraquat (PQ), nfe2l2-KO hepatocytes exhibited increased DHE fluorescence compared to Nfe2l2-WT cells. As NFE2L2 controls the production of the universal reducing agent NADPH, we analyzed the ratio between oxidized NADP and reduced NADPH in these cells. nfe2l2-KO hepatocytes showed an augmented NADP+:NADPH ratio, which may be sufficient to handle ROS under basal conditions but not in the presence of oxidants such as PQ (Figure 4c). Importantly, despite having higher ROS levels, nfe2l2-KO hepatocytes failed to increase LAMP2A levels upon PQ exposure (Figures 4d and 4e). Similar results were also observed with hydrogen peroxide (Fig. S4A and S4B).
Figure 4.
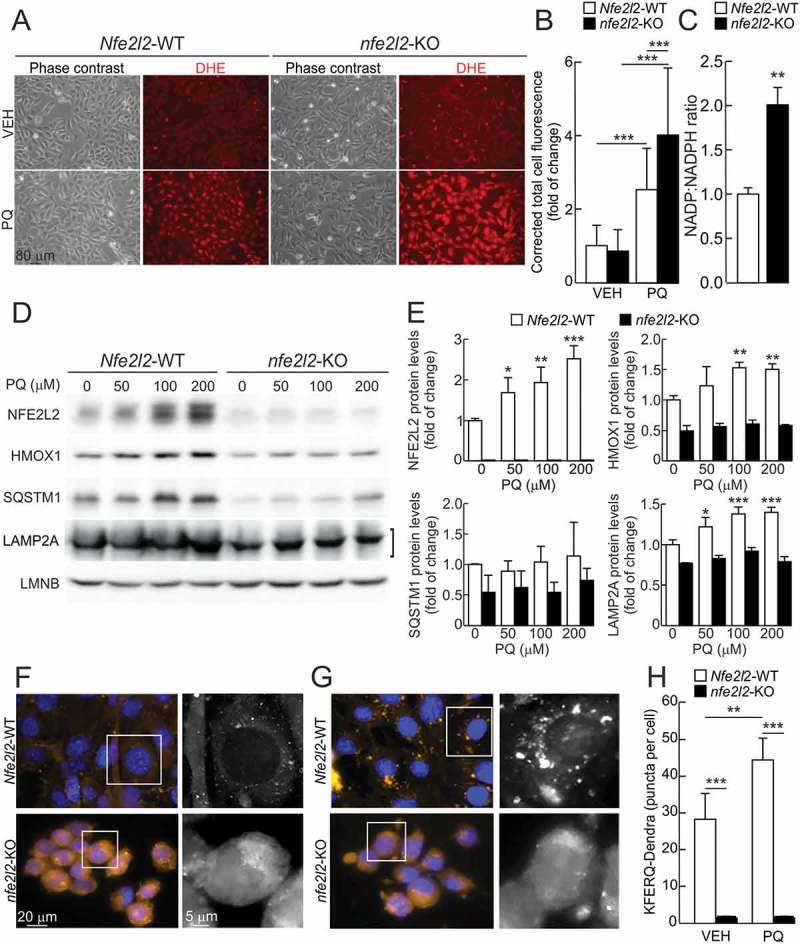
The oxidant agent paraquat (PQ) induces LAMP2A expression and CMA activity in an NFE2L2-dependent manner. (a) Representative images of ROS levels determined with the dihydroethidium probe (DHE) in immortalized Nfe2l2-WT and nfe2l2-KO hepatocytes in basal conditions or after treatment with 200 µM PQ for 16 h. The probe was added to a final concentration of 5 µM in the culture medium 1 h before in vivo imaging of the cells. (b) Corrected total cell fluorescence of representative images from A. Values are mean ± SD (n = 100 cells, derived from at least 3 different fields). Statistical analysis was performed using Student t test. ***p < 0.001 vs. untreated conditions. (c) NADP:NADPH ratio in immortalized Nfe2l2-WT and nfe2l2-KO hepatocytes. Data are mean ± SD (n = 4). Statistical analysis was performed using Student’s t test. **p < 0.01 vs. Nfe2l2-WT cells. (d) Immortalized hepatocytes from Nfe2l2-WT and nfe2l2-KO mice were submitted to the indicated concentrations of PQ for 16 h. Representative immunoblots for the indicated proteins in cell lysates. Note that anti-NFE2L2 antibody recognizes a nonspecific band in nfe2l2-KO cells. (e) Densitometric quantification of representative immunoblots from D relative to LMNB protein levels. Data are mean ± SD (n = 3). Statistical analysis was performed using Student’s t test. *p < 0.05, **p < 0.01 and ***p < 0.001 vs. untreated conditions. (f and g) Nfe2l2-WT and nfe2l2-KO hepatocytes were transduced with lentivirus carrying the CMA reporter KFERQ-PS-Dendra and, after photoswitching, were cultured without additions (f) or in the presence of 100 µM paraquat (PQ) (g) for 16 h. CMA was analyzed as the number of fluorescent puncta per cell at the end of the incubation time. Representative full-field images and insert showing black and white high magnification of the boxed regions in the Dendra channel. (h) Quantification of the number of puncta per cell after the indicated treatment. Values are mean ± SD (n = 3, with > 75 cells per experiment). Statistical analysis was performed using Student’s t test. **p < 0.01 and ***p < 0.001 vs. Nfe2l2-WT cells or untreated conditions.
The modulation of CMA activity by NFE2L2 was further analyzed in Nfe2l2-WT vs. nfe2l2-KO hepatocytes transduced with a lentivirus expressing the photoactivable CMA-specific fluorescent substrate KFERQ-PS-Dendra (modified from Koga et al. [30]). Upon 405 nm light exposure, Dendra protein is modified to emit red fluorescence, making it possible to track its delivery to lysosomes over newly synthesized protein. CMA activity is detected as a change from diffuse (cytosolic) fluorescence to a punctate (lysosomal) pattern, because CMA-active lysosomes are highlighted as red fluorescent puncta when the fluorescence substrate is delivered to this compartment. As shown in Figures 4f and 4h, Nfe2l2-WT cells exhibited a low cytosolic signal and some Dendra-positive fluorescent puncta, reflecting basal CMA activity. However, a more intense and diffuse fluorescent pattern throughout the cytoplasm, together with an extremely reduced number of Dendra-positive puncta per cell, were observed in nfe2l2-KO cells, consistent with impaired basal CMA activity (the KFERQ-PS-Dendra reporter is not being properly degraded by CMA and accumulates in the cytosol). When cells were treated for 16 h with PQ, a well-characterized stimulus for CMA [14], we found a significant increase in Dendra-positive puncta per cell in Nfe2l2-WT but not nfe2l2-KO hepatocytes, indicating impaired induction of CMA by PQ in the absence of NFE2L2 (Figures 4g and 4h).
Pharmacological activation of NFE2L2 induces LAMP2A expression and CMA
We next used a well-established activator of NFE2L2, sulforaphane (SFN), to test if pharmacological activation of NFE2L2 could lead to upregulation of LAMP2A levels and CMA activity. Treatment of Nfe2l2-WT hepatocytes with increasing doses of SFN for 16 h led to increased protein levels of NFE2L2, HMOX1, SQSTM1 and also LAMP2A, and this response was abolished in nfe2l2-KO cells (Figures 5a and 5b). Similar results were obtained with the NFE2L2 activator dimethyl fumarate (Fig. S4C and S4D). We further extended these observations to other cell types such as mouse embryonic fibroblasts, N2A neuroblastoma cells, mouse hippocampus-derived HT22 and human kidney HEK293T cells, hence corroborating again the conservation of this NFE2L2-LAMP2A axis among different cell types (Fig. S5A and S5B). Moreover, SFN-treated cells displayed mobilization of lysosomes (labeled either with LAMP2A or LAMP1) toward the perinuclear region, a typical characteristic of CMA activation [6]. Perinuclear LAMP2A fluorescence was much lower in nfe2l2-KO hepatocytes (Figures 5c and 5d).
Figure 5.
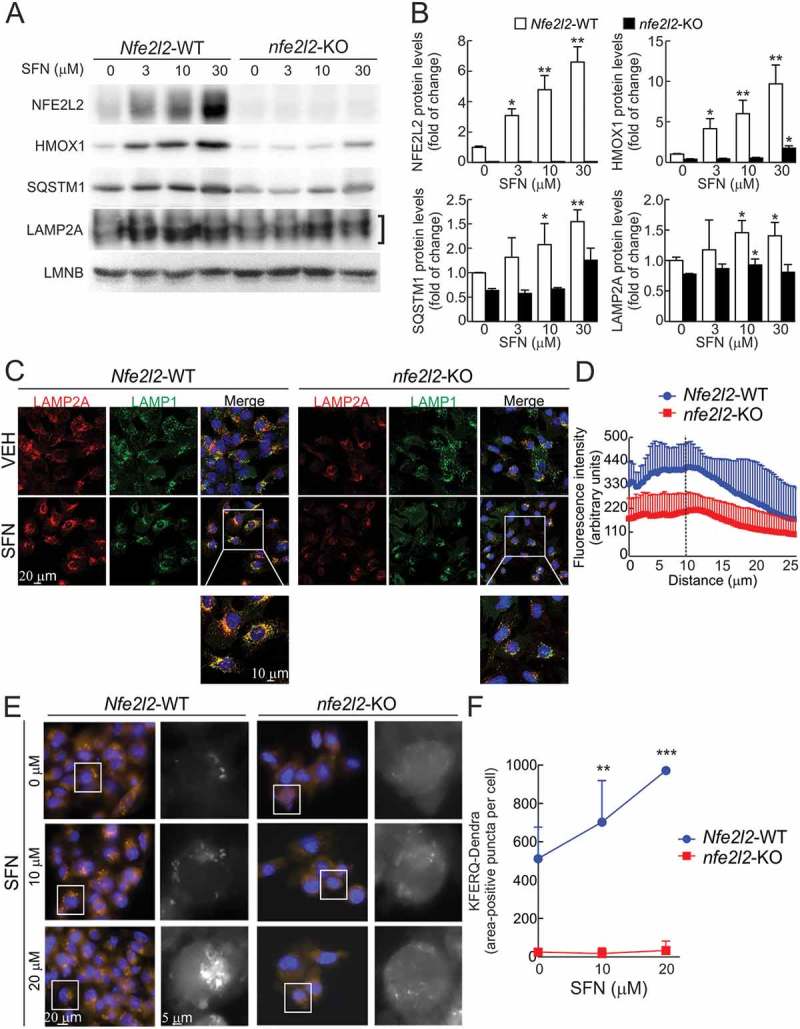
Pharmacological activation of NFE2L2 induces LAMP2A levels and CMA activation. (a) Immortalized hepatocytes from Nfe2l2-WT and nfe2l2-KO mice were submitted to the indicated concentrations of sulforaphane (SFN) for 16 h. Representative immunoblots for the indicated proteins in cell lysates. Note that anti-NFE2L2 antibody recognizes a nonspecific band in nfe2l2-KO cells. (b) Densitometric quantification of representative immunoblots from A relative to LMNB protein levels. Data are mean ± SD (n = 3). Statistical analysis was performed using Student’s t test. *p < 0.05, **p < 0.01 and ***p < 0.001 vs. untreated conditions. (c) Confocal analysis of double immunofluorescence with anti-LAMP2A (red) and anti-LAMP1 (green) antibodies in immortalized Nfe2l2-WT and nfe2l2-KO hepatocytes treated with vehicle (VEH) or 15 µM SFN for 16 h. The insert shows a higher magnification of the indicated cells. (d) Radial profile of LAMP2A fluorescence in cells treated with SFN in A. The graph shows the total mean fluorescence as a function of radial distance to the center of the cell. The dashed line represents the nuclear limits. Values are mean ± SD (n = 3, with > 50 cells counted per experiment). (e) Nfe2l2-WT and nfe2l2-KO hepatocytes were transduced with lentivirus carrying the CMA reporter KFERQ-PS-Dendra and, after photoswitching, were cultured without additions or in the presence of 10 µM or 20 µM SFN for 16 h. CMA was analyzed as the number of fluorescent puncta per cell at the end of the incubation time. Representative full-field images and insert showing black and white high magnification of the boxed regions in the Dendra channel. (f) Quantification of the KFERQ-Dendra-positive area per cell. Values are mean ± SD (n = 3, with > 75 cells per experiment). Statistical analysis was performed using Student t test. **p < 0.01 and ***p < 0.001 vs. untreated conditions.
Furthermore, to directly confirm CMA activation, we transduced cells with the KFERQ-PS-Dendra reporter. Compared to untreated cells, SFN induced a significant dose-dependent increase in Dendra-positive puncta per cell in Nfe2l2-WT cells (note that because perinuclear relocation made individual puncta indiscernible, data are presented as punctate KFERQ-Dendra positive area per cell) (Figures 5e and 5f). By contrast, SFN treatment failed to upregulate CMA in the absence of NFE2L2. Previous studies have shown compensatory cross-talk between CMA and macroautophagy. Because we have shown before that NFE2L2 also exerts a modulatory effect on macroautophagy, we next set out to investigate if CMA activation upon SFN treatment was primary or reactive to changes in macroautophagy. In macroautophagy-deficient atg5-KO cells (Fig. S5C-F) we found that SFN was still able to induce the expression of LAMP2A and subsequent increase in CMA activity. These findings indicate that the regulation of CMA by NFE2L2 is, for the most part, independent of changes in macroautophagy activity.
Impaired CMA in lysosomes from livers of nfe2l2-KO mice
To analyze the functional role of NFE2L2 in the modulation of CMA in vivo, we used livers of Nfe2l2-WT and nfe2l2-KO mice. Consistent with our in vitro results with cultured hepatocytes, mRNA levels of Hmox1, Sqstm1 and Lamp2a were reduced in livers from nfe2l2-KO mice (Figure 6a). Analysis of lysosomes isolated from these livers revealed a discrete decrease in LAMP2A levels in the nfe2l2-KO group (Figures 6b and 6c). The discrete differences in lysosomal LAMP2A levels are likely due to an overall increase in the ratio between lysosomal components and cargo in the nfe2l2-KO mice because of their lower CMA activity. In fact, contrary to the decrease in LAMP2A, levels of other lysosomal resident proteins such as HSPA8 and GBA were relatively higher in the lysosomes of nfe2l2-KO mice (Figures 6b and 6c).
Figure 6.
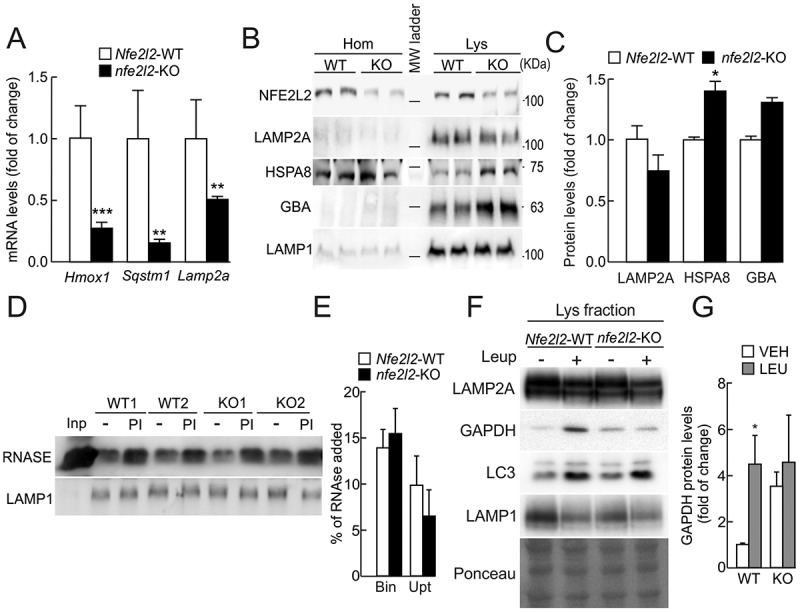
Role of NFE2L2 in the modulation of CMA in vivo. (a) Expression levels of Hmox1, Sqstm1 and Lamp2a in livers from Nfe2l2-WT and nfe2l2-KO mice were determined by qRT-PCR and normalized by the geometric mean between Actb and Tbp levels. Data are mean ± SEM (n = 4). Statistical analysis was performed using Student’s t test. **p < 0.01 and ***p < 0.001 vs. Nfe2l2-WT mice. (b) Immunoblots for the indicated proteins in homogenates and isolated lysosomes from livers of Nfe2l2-WT or nfe2l2-KO mice. Note that anti-NFE2L2 antibody recognizes a nonspecific band in nfe2l2-KO cells. (c) Densitometric quantification of lysosomal proteins in B relative to LAMP1 levels. Data are mean ± SD (n = 3 livers per mouse genotype). Statistical analysis was performed using Student’s t test. *p < 0.05 vs. Nfe2l2-WT mice. (d) Lysosomes isolated from livers of Nfe2l2-WT or nfe2l2-KO mice were pretreated or not with protease inhibitors (PI) to inhibit lysosomal proteolysis and incubated with purified RNASE for 20 min at 37ºC. At the end of the incubation, lysosomes were recovered by centrifugation and subjected to SDS-PAGE and immunoblot for the indicated proteins. LAMP1 is shown as control of similar lysosomal recovery after the incubation. (e) Densitometric quantification of RNASE binding (Bin) and uptake (Upt) in Nfe2l2-WT or nfe2l2-KO isolated lysosomes. Data are mean ± SD (n = 3 livers per mouse genotype). Statistical analysis was performed using Student’s t test. (f) Nfe2l2-WT and nfe2l2-KO mice were starved for 24 h. Lysosomal proteolysis was inhibited in vivo with 2 intraperitoneal injections of leupeptin (2 mg/100g body weight) 16 and 2 h before sacrifice. Representative immunoblots of the indicated endogenous proteins in lysosomal-enriched fractions from livers of Nfe2l2-WT (WT) and nfe2l2-KO (KO) mice. (g) Densitometric quantification of representative immunoblots from F relative to total protein levels stained with Ponceau Red. Data are mean ± SD (n = 3). Statistical analysis was performed using Student’s t test. *p < 0.05 vs. Nfe2l2-WT vehicle-treated mice.
To directly measure CMA activity in the isolated lysosomes, we incubated them with purified recombinant RNASE/RNAse A, a bona fide substrate of CMA [31], in the absence or presence of protease inhibitors (PI). The amount of RNASE recovered in untreated lysosomes represents the fraction bound at the lysosomal membrane, because the protein internalized during the incubation is rapidly degraded. Pretreatment with PI allows recovering lysosomes with both the RNASE bound and internalized. Thus, uptake was calculated as the difference in RNASE levels between non-treated and PI-treated conditions. As shown in Figures 6d and 6e, lysosomes from nfe2l2-KO mice showed a trend toward reduced RNASE uptake in comparison with Nfe2l2-WT littermates, although it did not reach statistical significance. We confirmed that the observed differences were not due to loss of lysosomal membrane integrity (Fig. S6A) or reduced proteolytic activity, which was in fact higher in the absence of NFE2L2 (Fig. S6B).
To gain further insight into the role of NFE2L2 in the modulation of CMA in vivo we directly measured the degradation of the endogenous CMA substrate GAPDH in lysosomes from Nfe2l2-WT and nfe2l2-KO mice. Mice were starved for 24 h to maximally induce CMA, and then injected intraperitoneally with vehicle or leupeptin to block proteolysis inside lysosomes. Because LAMP2A and LAMP1 levels may oscillate with leupeptin due to changes in the ratio between cargo and lysosomal components, in this case we normalized lysosomal GAPDH with total lysosomal protein as stained with Ponceau Red. As shown in Figures 6f and 6g, GAPDH accumulated in lysosomes from leupeptin-treated Nfe2l2-WT mice but not nfe2l2-KO mice, although these animals exhibited higher basal levels of lysosomal GAPDH. These results suggest that NFE2L2 deficiency impairs basal CMA at the level of substrate internalization.
Discussion
In this study, we report a molecular link between the transcription factor NFE2L2 and CMA, which occurs through the control of LAMP2A transcription. Knockout of Lamp2a or Nfe2l2 makes cells more susceptible to different stressors [8,32], indicating a crucial role in the maintenance of cell homeostasis under different circumstances. In fact, previous work supports the notion that redox status tightly controls CMA activity, presumably to eliminate proteins oxidized during mild oxidative damage [14]. Interestingly, CMA induction upon oxidative stress appears to occur through increased Lamp2a transcription [14], whereas CMA activation by nutrient deprivation relies on diminished degradation of LAMP2A and relocation of this receptor at the lysosomal membrane [33], pointing to the existence of several mechanisms of CMA regulation.
Several transcription factors might regulate LAMP2 expression. AP-1 and SP1 binding sites were found in the LAMP2 promoter [34], although they were not confirmed to be functional enhancers. MAF proteins, the other bZIP partner of the NFE2L2 heterodimer, heterodimerize also with NFAT (nuclear factor of activated T cells), which upon activation modulates Lamp2a transcription in T lymphocytes [16]. We have identified 2 functional AREs in the LAMP2 gene that lose activity when a highly conserved G is replaced to A. This is the binding site for MAF [35] and consequently we suggest that MAFs participate in a dual regulation of LAMP2 by both NFAT and NFE2L2. Of note, NFE2L2 would control not only inducible but also Lamp2a basal expression, as Nfe2l2-deficient cells exhibited reduced Lamp2a mRNA and protein levels. It is thus possible that different transcription factors co-exist to assure proteostasis under several circumstances and in distinct cell types. In fact, levels of basal CMA activity and the amplitude of the induction varies among different cell types [12,16,36,37]. Importantly, this novel NFE2L2-LAMP2A axis is conserved in different cell types.
Despite being encoded by the same gene, different transmembrane and cytosolic regions in each LAMP2 spliced variant enable specific functions. In fact, several roles have been proposed for LAMP2B and LAMP2C [10,38–40], but LAMP2A is the only isoform required for CMA [4,8,25]. LAMP2 isoforms display tissue-specific expression patterns [10,41]. While LAMP2A and LAMP2B are highly expressed in most tissues, LAMP2C appears to be very restricted [10], pointing to a different splicing regulation of the LAMP2 gene. Although we could not detect differences in the expression levels of LAMP2B or LAMP2C upon genetic manipulation of NFE2L2 in human HEK293T cells, mouse hepatocytes or HT22 cells, reduced levels of LAMP2B mRNA were found in A549 cells upon NFE2L2 silencing, and NFE2L2-deficient MEFs displayed reduced levels of the 3 isoforms. Differential regulation of the 3 isoforms has been reported previously [10,15,42]. For example, paraquat transcriptionally induces Lamp2a expression without affecting the other 2 isoforms [43]. Altogether, these data suggest the existence of additional post-transcriptional regulatory mechanisms of the Lamp2 gene that need to be unveiled in future studies.
Chemical and genetic manipulation of NFE2L2 results in modest changes in cellular LAMP2A mRNA and protein levels, albeit similar to those observed for the NFE2L2-responsive SQSTM1 gene. LAMP2A has a slow turnover, with a half-life of approximately 46 h for total LAMP2A and 57 h for lysosomal LAMP2A [33]. Hence, small changes in LAMP2A mRNA levels may have a marked impact on protein levels and CMA activity. Other studies show similar oscillations, further supporting this notion [4,16,44,45]. In contrast with the discrete changes in total cellular LAMP2A levels, the effects of NFE2L2-deficiency are more dramatic in isolated lysosomes, where LAMP2A is required for CMA. In fact, it has been extensively shown before that levels of LAMP2A at the lysosomal membrane are the ones correlating with changes in the rate of CMA [15]. As a result, CMA was barely detectable in NFE2L2-deficient hepatocytes, suggesting a crucial role for NFE2L2 in modulation of CMA.
While we have described the transcriptional regulation of Lamp2a by NFE2L2, other layers of CMA regulation are still possible. For example, lysosomes from nfe2l2-KO livers exhibited higher levels of HSPA8 compared to Nfe2l2-WT livers. Although it is possible that higher levels of HSPA8 are a result of the relative decrease in lysosomal cargoes, we cannot discard the possibility that the increase in lysosomal HSPA8 may represent a compensatory mechanism to counteract the decrease in LAMP2A levels. In fact, increased HSPA8 levels have been previously reported in other conditions with reduced lysosomal levels of LAMP2A, such as livers from old mice and in lamp2a-KO mice [25,46].
The impairment of CMA in nfe2l2-KO mice was evidenced by a deficient uptake in the in vitro reconstituted system of RNASE but more importantly by the reduced degradation in vivo of the endogenous bona fide CMA substrate GAPDH. The fact that the differences in uptake in vivo of the endogenous substrate are more pronounced than for the exogenously added protein in the isolated in vitro system could be attributed to the lack of other competing substrates in the latter. Interestingly, a previous report showed that astrocyte-specific NFE2L2 overexpression reduced the levels of another CMA substrate, MEF2D (myocyte enhancer factor 2D) [47]. However, this study was just correlative and ours is the first one to show that changes in CMA substrates are at the level of lysosomal degradation by CMA.
NFE2L2 has been reported to transcriptionally regulate macroautophagy [22]. Crosstalk between both autophagic pathways has been extensively reported both in vivo and in vitro, whereby one reacts in a compensatory manner to the loss of activity in the other [8,46,48]. However, in this work we show that the functional connection between NFE2L2 and CMA is independent of macroautophagy. Therefore, NFE2L2 might act as a regulatory node in the proteolytic network represented by macroautophagy and CMA.
Both CMA [25,46] and NFE2L2 [49,50] activities decline with age, probably favoring the accumulation of oxidized substrates. A wide variety of NFE2L2 inducers have been reported, and in fact, in this study we successfully used SFN to increase Lamp2a mRNA and protein levels and increase CMA activity. Considering the positive effect that genetic restoration of CMA had in proteostasis of the livers of old mice [44], it is reasonable to think that the pharmacological reinforcement of NFE2L2 and consequently CMA would be capable of a similar delay in the accumulation of oxidized protein aggregates during aging.
Materials and methods
Cell culture and reagents
Adenocarcinoma human alveolar A549 cells and human embryonic kidney HEK293T cells (ATCC, CCL-185 and CRL-3216, respectively) were grown in Dulbecco’s modified Eagle’s medium (Sigma-Aldrich, D5648) supplemented with 10% fetal bovine serum (HyClone, CH30160.03) and 80 μg/ml gentamicin (Laboratorios Normon, 763,011.1). Immortalized mouse hepatocytes were grown in the same medium supplemented with 10% fetal bovine serum, 2 mM glutamine (Gibco/Life Technologies, 25,030–024), 1 mM sodium pyruvate (Gibco/Life Technologies, 11,360–039), 5 mM HEPES, pH 7.4, 0.5 U/ml penicillin, 0.5 μg/ml streptomycin (Gibco/Life Technologies, 15,140–122). Other reagents include: Dihidroethiudium (Invitrogen, D11347). R,S-sulforaphane (SFN; LKT Laboratories, Inc., S8044). Sucrose (Panreac, 131,621). Leupeptin hemisulfate salt (> 95%; L5793) and paraquat (M2254) were purchased from Sigma-Aldrich. Transient transfections were performed with GenJET™ In Vitro Transfection Reagent from SignaGen (SL100488).
Bioinformatics analysis
Putative AREs in the LAMP2 gene were identified in ENCODE [26] for the human genome taking as reference the available information from ChIP of ARE binding factors MAFK, MAFF and BACH1. The putative MAFK, MAFF and BACH1 binding regions were localized in 200–400 base-pair-long DNase-sensitive and H3K27Ac-rich regions. In addition, a frequency matrix of the consensus ARE sequence based on the JASPAR database [27] was converted to a position-specific scoring matrix and a script was generated with the Python 3.4 program to scan the promoter sequences with candidate AREs as previously described [22].
Chromatin immunoprecipitation (chip) assays
HEK293T cells were grown on 10-cm plates until they reached 85% confluence and transfected with an NFE2L2 expression plasmid that lacks the high affinity binding site for KEAP1 and contains a V5 tag (NFE2L2ΔETGE-V5) [51]. Briefly, cells were cross-linked with 1% formaldehyde (Fluka, 47,630) and the reaction was stopped with 125 mM glycine (Bio-Rad, 161–0718). Cells were then washed twice with cold phosphate-buffered saline (PBS; 10 mM PO43−, 137 mM NaCl, 2.7 mM KCl, pH 7.4), lysed and sonicated in order to obtain adequate fragment sizes of DNA. Supernatant was diluted 10 fold with ChIP dilution buffer (0.01% sodium dodecyl sulfate [Merck Millipore, 817,034], 1.1% Triton X-100 [Sigma-Aldrich, T8787], 1.2 mM ethylenediaminetetraacetic acid, 16.7 mM Tris-HCl, pH 8.1, 167 mM NaCl, 1 mM phenylmethylsulfonyl fluoride [Sigma-Aldrich, P7626], 1 µg/ml leupeptin [Sigma-Aldrich, L5793]) and pre-cleared with protein G Sepharose (GE Healthcare, 17–0618-01). ChIP was carried out with anti-V5 antibody (Life Technologies, 37–7500) or anti-IgG (Abcam, ab18413). DNA was eluted and purified, analyzing the presence of previously identified putative AREs by quantitative real-time polymerase chain reaction (qRT-PCR) with specific primers (Table S2). Samples from at least 3 independent immunoprecipitations were analyzed.
Luciferase reporter generation
Artificial oligonucleotides with 3 tandem repetitions of the putative AREs identified separated with a BamHI sequence and a random separating-sequence, as well as the scramble sequence used as negative control, are described in Table S4. These oligonucleotides were subcloned into NheI and XhoI sites of the minimal Sod1 promoter pGL3basic-sod1-29 [52], removing the NheI site to facilitate religated vector exclusion. Scramble-LUC included also a SalI site instead of the separating-sequence to facilitate differentiation between the constructs.
Luciferase activity
Transient transfections of nfe2l2-KO cells were performed with the expression vectors ARE-LUC or SCRAMBLE-LUC as indicated. pTK-Renilla was used as an internal control vector. Luciferase assays were performed with the Dual-Luciferase Reporter Assay System (Promega, E1910) as described previously [53].
Production of lentiviral stocks and infection
Recombinant lentiviral stocks were produced in HEK293T/17 cells by co-transfecting 10 μg of transfer vector (GFP or NFE2L2ΔEGTE-V5), 6 μg of envelope plasmid pMD2.G (Addgene, 12,259; deposited by Didier Trono) and 6 μg of packaging plasmid pSPAX2 (Addgene, 12,260; deposited by Didier Trono), using Lipofectamine® 2000 Reagent (Invitrogen by Life Technologies, 116,668–019). After 12 h at 37°C the medium was replaced with fresh Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum and virus particles were harvested at 24 h and 48 h post-transfection. Cells were incubated in the presence of 2 µg/ml polybrene (Sigma-Aldrich, TR-1003-G) with the lentivirus during 24 h and mRNA was extracted 3 days after lentiviral transduction. Lentivirus carrying the KFERQ-PS-Dendra reporter were prepared as described previously [30] and added to hepatocytes in the presence of 2 µg/ml polybrene 24 h before the beginning of the CMA measurement.
Analysis of mrna levels
Total RNA extraction, reverse transcription and quantitative PCR were carried out as detailed elsewhere [54]. Primer sequences are shown in Tables S5 and S6. To ensure that equal amounts of cDNA were added to the PCR, the housekeeping genes Actb and Tbp were amplified. Data analysis was based on the ΔΔCT method with normalization of the raw data to housekeeping genes (Applied Biosystems, CA, USA). All PCRs were performed at least in triplicate.
Immunoblotting
Immunoblots were performed as described elsewhere [55]. The primary antibodies used are presented in Table 2. Membranes were analyzed using the appropriate peroxidase-conjugated secondary antibodies (Life Technologies, A15999; Amersham, NA931 and NA934; Merck Millipore, AP136P). Proteins were detected by enhanced chemiluminescence (GE Healthcare, RPN2232).
Table 2.
Antibodies used in immunoblots, immunofluorescence and ChIP.
| Antibody | Source | Catalog number | Dilution |
|---|---|---|---|
| NFE2L2/NRF2 | Cuadrado lab | - | 1:2000 (WB) |
| HMOX1 | Enzo Life Sciences | OSA110 | 1:2000 (WB) |
| SQSTM1/p62 | Sigma Aldrich | P0067 | 1:2000 (WB) |
| MmLAMP2A/Igp96 | Zymed/Invitrogen | 51–2200 | 1:1000 (WB). 1:100 (ICF) |
| HsLAMP2A | Abcam | ab18528 | 1:1000 (WB) |
| LAMP2B | Cuervo lab | - | 1:1000 (WB) |
| LAMP2C | Cuervo lab | - | 1:1000 (WB) |
| LAMP1 (1D4B) | Hybridoma Bank | 1D4B | 1:1000 (WB). 1:250 (ICF) |
| RNASE | Rockland Immunochemicals | 100–188 | 1:5000 (WB) |
| HSPA8/HSC70 (13D3) | Novus Biologicals | NB1202788 | 1:5000 (WB) |
| ACTB | Santa Cruz Biotechnology | sc-1616 | 1:5000 (WB) |
| LMNB/lamin B | Santa Cruz Biotechnology | sc-6217 | 1:5000 (WB) |
| GAPDH | Calbiochem | CB1001 | 1:5000 (WB) |
| LC3B | Cell Signaling Technology | 2775 | 1:2000 (WB) |
| V5 | Life Technologies | 37–7500 | 1:250 (ChIP) |
| IgG2a | Abcam | ab18413 | 1:250 (ChIP) |
ICF, immunocytofluorescence. WB, western blot. ChIP, chromatin immunoprecipitation.
Animals and treatments
Animals were housed under a 12-h light-dark cycle. Food and water were provided ad libitum and mice were cared for according to protocols approved by the Ethics Committee for Research of the Universidad Autónoma de Madrid following institutional, Spanish and European guidelines (Boletín Oficial del Estado of 18 March 1988; and 86/609/EEC, 2003/65/EC European Council Directives). Where indicated, food was removed for 24 h but water was still provided ad libitum. Where stated, mice received intraperitoneal injections of vehicle (0.9% saline buffer) or leupeptin (2 mg/100 g body weight) 16 h and 2 h before sacrifice.
Lysosomal isolation
For in vivo analysis, lysosomal-enriched fractions or CMA-active lysosomes from Nfe2l2-WT and nfe2l2-KO mouse livers were obtained as previously described [56]. Briefly, livers were mechanically homogenized in 0.25 M sucrose, filtered through double gauze and centrifuged at 6800 g. The supernatant was then centrifuged at 17,000 g and the pellet was resuspended with a ‘cold finger’ (dry glass test tube filled with ice). This step was repeated twice to wash the lysosomal-enriched fraction. Lysosomes active for CMA were isolated by centrifugation of the lysosomal-enriched fraction (light mitochondria and lysosomes) in a discontinuous metrizamide (AK Scientific, 69,696) density gradient [37,56]. The fraction of broken lysosomes at the moment of isolation was measured using the HEX/β-hexosaminidase latency assay [57]. In some experiments, a crude lysosomal fraction was prepared from hepatocytes derived from wild-type and nfe2l2-KO mice using the Lysosome Isolation Kit from BioVision (K235-50) according to the manufacturer’s instructions.
Immunocytofluorescence
Hepatocytes were seeded on sterile cover slips in 24-well plates (75,000 cells per well), cultured for 16 h and treated as indicated. Cells were washed with cold PBS and fixed in 4% paraformaldehyde for 15 min at room temperature. After 3 5-min washes with PBS, cells were incubated with blocking solution (0.2% powdered milk, 2% normal goat serum [Merck Millipore, NS20L], 0.1M glycine, 1% bovine serum albumin [Sigma-Aldrich, A7906] and 0.01% Triton X-100) for 30 min at room temperature. After briefly washing twice with PBS, the slides were incubated with the indicated primary antibodies (Table 2) for 90 min at room temperature in a humidified chamber. Then cells were washed 3 times with PBS and incubated with secondary antibodies for 45 min under the same conditions. To visualize the nuclei, cells were stained with DAPI (4,6-diamidino-2-phenylindole). The fluorescence images were captured using appropriate filters in a Leica DMIRE2TCS SP5 confocal microscope (Nussloch, Germany).
Measurement of CMA activity in intact cells
To measure CMA activity in intact cells the photoswitchable KFERQ-PSDendra2 reporter was transduced into cells using lentiviral delivery [30]. Cells were photoactivated with a 405-nm light-emitting diode (LED: Norlux) for 4 min with the intensity of 3.5 mA (current constant). After 16 h, cells were fixed and slides prepared using mounting medium containing DAPI (Abcam, ab104139) to highlight the cell nucleus. All images were acquired with an Axiovert 200 fluorescence microscope (Carl Zeiss Microscopy) with a 100x objective and 1.4 numerical aperture, mounted with an ApoTome.2 slider. Quantification was performed in individual frames after deconvolution and thresholding using ImageJ software (NIH) in a minimum of 20 cells per slide. In all cases, focal plane thickness was set at 0.17 μm and sections with maximal nucleus diameter were selected for quantification. Values are presented as number of puncta per cell section that in our acquisition conditions represents 10–20% of the total puncta per cell. CMA activation was detected as changes in the fluorescence pattern from diffuse/cytosolic to punctate/lysosomal and quantified as the average number of fluorescent puncta per cell or as the cellular area positive for KFERQ-Dendra puncta [30]. Where indicated, high-content microscopy was used instead to better determine dose-dependence effects in a larger number of cells. Briefly, cells were plated in glass-bottom 96-well plates, treated for the indicated times and, after fixation, images were acquired using a high-content microscope (Operetta, Perkin Elmer). Images of 9 different fields per well were captured, resulting in an average of 2,500–3,000 cells per condition. Nuclei and puncta were identified using the manufacturer’s software. The number of particles/puncta per cell was quantified using the ‘particle identifier’ function in the cytosolic region after thresholding in non-saturated images [37].
Measurement of CMA in vitro
CMA activity in vitro was measured using isolated intact lysosomes incubated with purified proteins and subjected to immunoblot [37,56]. Binding was calculated as the amount of substrate protein bound to the lysosomal membrane in the absence of protease inhibitors, and uptake by subtracting the amount of protein associated with lysosomes in the presence (protein bound to the lysosomal membrane and taken up by lysosomes) and absence (protein bound to the lysosomal membrane) of protease inhibitors [37]. Proteolytic activity was determined by incubating lysosomes with a cocktail of long-lived radiolabeled proteins (3H-pool) previously demonstrated to be enriched in CMA substrates [37,56] in the presence of 0.1% Triton X-100 to disrupt the lysosomal membrane. Samples were incubated in 20 mM MOPS, 1 mM DTT, 5.4 µM cysteine (Sigma-Aldrich, 168,149), pH 7.3, 0.25 M sucrose (Sigma-Aldrich, S0389) for 20 min at 37ºC. At the end of the incubation, 10% TCA was added to all the samples and they were subjected to filtration through a 0.2 μm filter, as previously described. Proteolysis was calculated as the percentage of acid precipitable radioactivity (protein) at the begging of the incubation that became acid soluble (amino acids) at the end of the incubation [37].
Image analysis
Different band intensities corresponding to immunoblot detection of protein samples were quantified using ImageJ software. The number of puncta per cell was analyzed with the ‘Analyze particles’ function of ImageJ after thresholding in non-saturated images [30]. DHE fluorescence intensity was quantified with ImageJ and is presented as the corrected total cell fluorescence (CTCF; CTCF = integrated density – [area of selected cell x mean fluorescence of background readings]). Perinuclear LAMP2A fluorescence was measured with the ‘Radial profile angle’ plugin of ImageJ after properly calibrating the image. A circular mask with a radius of 100 pixels (25 µm, approximately) was used to analyze fluorescence intensity per cell. For each cell, the algorithm measured the mean signal intensity in a series of concentric circles drawn along the established radius, starting from the nucleus and towards the cell periphery. Quantifications were performed in 50–75 cells per condition in 3 independent experiments.
Statistical analysis
Unless otherwise indicated, all experiments were performed at least 3 times and all data presented in the graphs are the mean of at least 3 independent samples ± standard error (SD). Student’s t-test was used to assess differences between groups. p < 0.05 was considered significant.
Funding Statement
This paper was funded by the Spanish Ministry of Economy and Competitiveness under the grant SAF2016-76520-R and by grants from the National Institute of Health/National Institute on Aging P01 AG031782. M.P. is recipient of a FPU fellowship of Autonomous University of Madrid. E.A. was supported by NIH/NIA AG038072 P&F.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Chiang HL, Terlecky SR, Plant CP, et al. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science. 1989. October 20;246(4928):382–385. PubMed PMID: 2799391. [DOI] [PubMed] [Google Scholar]
- [2].Chiang HL, Dice JF.. Peptide sequences that target proteins for enhanced degradation during serum withdrawal. J Biol Chem. 1988. May 15;263(14):6797–6805. PubMed PMID: 3360807. [PubMed] [Google Scholar]
- [3].Terlecky SR, Dice JF.. Polypeptide import and degradation by isolated lysosomes. J Biol Chem. 1993. November 05;268(31):23490–23495. PubMed PMID: 8226876. [PubMed] [Google Scholar]
- [4].Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996. July 26;273(5274):501–503. PubMed PMID: 8662539. [DOI] [PubMed] [Google Scholar]
- [5].Bandyopadhyay U, Kaushik S, Varticovski L, et al. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol. 2008. September;28(18):5747–5763. PubMed PMID: 18644871; PubMed Central PMCID: PMCPMC2546938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Agarraberes FA, Terlecky SR, Dice JF. An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J Cell Biol. 1997. May 19;137(4):825–834. PubMed PMID: 9151685; PubMed Central PMCID: PMCPMC2139836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cuervo AM, Dice JF, Knecht E. A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J Biol Chem. 1997. February 28;272(9):5606–5615. PubMed PMID: 9038169. [DOI] [PubMed] [Google Scholar]
- [8].Massey AC, Kaushik S, Sovak G, et al. Consequences of the selective blockage of chaperone-mediated autophagy. Proc Natl Acad Sci U S A. 2006. April 11;103(15):5805–5810. PubMed PMID: 16585521; PubMed Central PMCID: PMCPMC1458654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Eskelinen EL, Cuervo AM, Taylor MR, et al. Unifying nomenclature for the isoforms of the lysosomal membrane protein LAMP-2. Traffic. 2005. November;6(11):1058–1061. PubMed PMID: 16190986. [DOI] [PubMed] [Google Scholar]
- [10].Perez L, McLetchie S, Gardiner GJ, et al. LAMP-2C Inhibits MHC Class II Presentation of Cytoplasmic Antigens by Disrupting Chaperone-Mediated Autophagy. J Immunol. 2016. March 15;196(6):2457–2465. PubMed PMID: 26856698; PubMed Central PMCID: PMCPMC4779666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gough NR, Hatem CL, Fambrough DM. The family of LAMP-2 proteins arises by alternative splicing from a single gene: characterization of the avian LAMP-2 gene and identification of mammalian homologs of LAMP-2b and LAMP-2c. DNA Cell Biol. 1995. October;14(10):863–867. . PubMed PMID: 7546292. [DOI] [PubMed] [Google Scholar]
- [12].Massey A, Kiffin R, Cuervo AM. Pathophysiology of chaperone-mediated autophagy. Int J Biochem Cell Biol. 2004. December;36(12):2420–2434. . PubMed PMID: 15325582. [DOI] [PubMed] [Google Scholar]
- [13].Cuervo AM, Knecht E, Terlecky SR, et al. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am J Physiol. 1995. November;269(5 Pt 1):C1200–8. PubMed PMID: 7491910. [DOI] [PubMed] [Google Scholar]
- [14].Kiffin R, Christian C, Knecht E, et al. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell. 2004. November;15(11):4829–4840. PubMed PMID: 15331765; PubMed Central PMCID: PMCPMC524731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cuervo AM, Dice JF. Unique properties of lamp2a compared to other lamp2 isoforms. J Cell Sci. 2000. December;113(Pt 24):4441–4450. PubMed PMID: 11082038. [DOI] [PubMed] [Google Scholar]
- [16].Valdor R, Mocholi E, Botbol Y, et al. Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat Immunol. 2014. November;15(11):1046–1054. PubMed PMID: 25263126; PubMed Central PMCID: PMCPMC4208273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wang X, Tomso DJ, Chorley BN, et al. Identification of polymorphic antioxidant response elements in the human genome. Hum Mol Genet. 2007. May 15;16(10):1188–1200. PubMed PMID: 17409198; PubMed Central PMCID: PMCPMC2805149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci. 2014. April;39(4):199–218. . PubMed PMID: 24647116. [DOI] [PubMed] [Google Scholar]
- [19].Jain A, Lamark T, Sjottem E, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010. July 16;285(29):22576–22591. PubMed PMID: 20452972; PubMed Central PMCID: PMC2903417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jo C, Gundemir S, Pritchard S, et al. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat Commun. 2014. March 25;5:3496 PubMed PMID: 24667209; PubMed Central PMCID: PMCPMC3990284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chesser AS, Ganeshan V, Yang J, et al. Epigallocatechin-3-gallate enhances clearance of phosphorylated tau in primary neurons. Nutr Neurosci. 2016;19(1):21–31. PubMed PMID: 26207957. [DOI] [PubMed] [Google Scholar]
- [22].Pajares M, Jimenez-Moreno N, Garcia-Yague AJ, et al. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy. 2016. October 02;12(10):1902–1916. PubMed PMID: 27427974; PubMed Central PMCID: PMCPMC5079676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Pajares M, Jimenez-Moreno N, Dias IH, et al. Redox control of protein degradation. Redox Biol. 2015. December;6:409–420. PubMed PMID: 26381917; PubMed Central PMCID: PMCPMC4576413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Schneider JL, Cuervo AM. Autophagy and human disease: emerging themes. Curr Opin Genet Dev. 2014. June;26:16–23. PubMed PMID: 24907664; PubMed Central PMCID: PMCPMC4253630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cuervo AM, Dice JF. Age-related decline in chaperone-mediated autophagy. J Biol Chem. 2000. October 06;275(40):31505–31513. PubMed PMID: 10806201. [DOI] [PubMed] [Google Scholar]
- [26].Ep C. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012. September 06;489(7414):57–74. . PubMed PMID: 22955616; PubMed Central PMCID: PMCPMC3439153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mathelier A, Zhao X, Zhang AW, et al. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 2014. January;42(Database issue):D142–7. PubMed PMID: 24194598; PubMed Central PMCID: PMCPMC3965086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kwon AT, Arenillas DJ, Worsley Hunt R, et al. oPOSSUM-3: advanced analysis of regulatory motif over-representation across genes or ChIP-Seq datasets. G3 (Bethesda). 2012. September;2(9):987–1002. PubMed PMID: 22973536; PubMed Central PMCID: PMCPMC3429929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Andersen MC, Engstrom PG, Lithwick S, et al. In silico detection of sequence variations modifying transcriptional regulation. PLoS Comput Biol. 2008. January;4(1):e5 PubMed PMID: 18208319; PubMed Central PMCID: PMCPMC2211530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Koga H, Martinez-Vicente M, Macian F, et al. A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat Commun. 2011. July 12;2:386 PubMed PMID: 21750540; PubMed Central PMCID: PMCPMC3529934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Cuervo AM, Terlecky SR, Dice JF, et al. Selective binding and uptake of ribonuclease A and glyceraldehyde-3-phosphate dehydrogenase by isolated rat liver lysosomes. J Biol Chem. 1994. October 21;269(42):26374–26380. PubMed PMID: 7929357. [PubMed] [Google Scholar]
- [32].Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–426. PubMed PMID: 23294312; PubMed Central PMCID: PMCPMC4680839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cuervo AM, Dice JF. Regulation of lamp2a levels in the lysosomal membrane. Traffic. 2000. July;1(7):570–583. PubMed PMID: 11208145. [DOI] [PubMed] [Google Scholar]
- [34].Sawada R, Jardine KA, Fukuda M. The genes of major lysosomal membrane glycoproteins, lamp-1 and lamp-2. 5ʹ-flanking sequence of lamp-2 gene and comparison of exon organization in two genes. J Biol Chem. 1993. April 25;268(12):9014–9022. PubMed PMID: 8517882. [PubMed] [Google Scholar]
- [35].Kimura M, Yamamoto T, Zhang J, et al. Molecular basis distinguishing the DNA binding profile of Nrf2-Maf heterodimer from that of Maf homodimer. J Biol Chem. 2007. November 16;282(46):33681–33690. PubMed PMID: 17875642. [DOI] [PubMed] [Google Scholar]
- [36].Kon M, Kiffin R, Koga H, et al. Chaperone-mediated autophagy is required for tumor growth. Sci Transl Med. 2011. November 16;3(109):109ra117 PubMed PMID: 22089453; PubMed Central PMCID: PMCPMC4000261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Arias E, Koga H, Diaz A, et al. Lysosomal mTORC2/PHLPP1/Akt Regulate Chaperone-Mediated Autophagy. Mol Cell. 2015. July 16;59(2):270–284. PubMed PMID: 26118642; PubMed Central PMCID: PMCPMC4506737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Nishino I, Fu J, Tanji K, et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature. 2000. August 24;406(6798):906–910. PubMed PMID: 10972294. [DOI] [PubMed] [Google Scholar]
- [39].Tanaka Y, Guhde G, Suter A, et al. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature. 2000. August 24;406(6798):902–906. PubMed PMID: 10972293. [DOI] [PubMed] [Google Scholar]
- [40].Fujiwara Y, Kikuchi H, Aizawa S, et al. Direct uptake and degradation of DNA by lysosomes. Autophagy. 2013. August;9(8):1167–1171. PubMed PMID: 23839276; PubMed Central PMCID: PMCPMC3748189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Konecki DS, Foetisch K, Zimmer KP, et al. An alternatively spliced form of the human lysosome-associated membrane protein-2 gene is expressed in a tissue-specific manner. Biochem Biophys Res Commun. 1995. October 13;215(2):757–767. PubMed PMID: 7488019. [DOI] [PubMed] [Google Scholar]
- [42].Murphy KE, Gysbers AM, Abbott SK, et al. Lysosomal-associated membrane protein 2 isoforms are differentially affected in early Parkinson’s disease. Mov Disord. 2015. October;30(12):1639–1647. PubMed PMID: 25594542. [DOI] [PubMed] [Google Scholar]
- [43].Kiffin R, Kaushik S, Zeng M, et al. Altered dynamics of the lysosomal receptor for chaperone-mediated autophagy with age. J Cell Sci. 2007. March 1;120(Pt 5):782–791. PubMed PMID: 17284523. [DOI] [PubMed] [Google Scholar]
- [44].Zhang C, Cuervo AM. Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat Med. 2008. September;14(9):959–965. . PubMed PMID: 18690243; PubMed Central PMCID: PMCPMC2722716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Park C, Suh Y, Cuervo AM. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage [Article]. Nat Commun. 2015. April 16;6: 6823. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Schneider JL, Villarroya J, Diaz-Carretero A, et al. Loss of hepatic chaperone-mediated autophagy accelerates proteostasis failure in aging. Aging Cell. 2015. April;14(2):249–264. PubMed PMID: 25620427; PubMed Central PMCID: PMCPMC4364837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gan L, Vargas MR, Johnson DA, et al. Astrocyte-specific overexpression of Nrf2 delays motor pathology and synuclein aggregation throughout the CNS in the alpha-synuclein mutant (A53T) mouse model. J Neurosci. 2012. December 5;32(49):17775–17787. PubMed PMID: 23223297; PubMed Central PMCID: PMCPMC3539799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kaushik S, Massey AC, Mizushima N, et al. Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol Biol Cell. 2008. May;19(5):2179–2192. PubMed PMID: 18337468; PubMed Central PMCID: PMCPMC2366850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Rahman MM, Sykiotis GP, Nishimura M, et al. Declining signal dependence of Nrf2-MafS-regulated gene expression correlates with aging phenotypes. Aging Cell. 2013. August;12(4):554–562. PubMed PMID: 23521918; PubMed Central PMCID: PMC3714369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Suh JH, Shenvi SV, Dixon BM, et al. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc Natl Acad Sci U S A. 2004. March 09;101(10):3381–3386. PubMed PMID: 14985508; PubMed Central PMCID: PMC373470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].McMahon M, Itoh K, Yamamoto M, et al. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J Biol Chem. 2003. June 13;278(24):21592–21600. PubMed PMID: 12682069. [DOI] [PubMed] [Google Scholar]
- [52].Rojo AI, Salinas M, Martin D, et al. Regulation of Cu/Zn-superoxide dismutase expression via the phosphatidylinositol 3 kinase/Akt pathway and nuclear factor-kappaB. J Neurosci. 2004. August 18;24(33):7324–7334. PubMed PMID: 15317858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Rada P, Rojo AI, Evrard-Todeschi N, et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/beta-TrCP axis. Mol Cell Biol. 2012. September;32(17):3486–3499. PubMed PMID: 22751928; PubMed Central PMCID: PMC3422007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Rojo AI, Innamorato NG, Martin-Moreno AM, et al. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia. 2010. April;58(5):588–598. PubMed PMID: 19908287. [DOI] [PubMed] [Google Scholar]
- [55].Rojo AI, Medina-Campos ON, Rada P, et al. Signaling pathways activated by the phytochemical nordihydroguaiaretic acid contribute to a Keap1-independent regulation of Nrf2 stability: role of glycogen synthase kinase-3. Free Radic Biol Med. 2012. January 15;52(2):473–487. PubMed PMID: 22142471. [DOI] [PubMed] [Google Scholar]
- [56].Arias E. Methods to study chaperone-mediated autophagy. Methods Enzymol. 2017;588:283–305. PubMed PMID: 28237106; PubMed Central PMCID: PMC4300957. [DOI] [PubMed] [Google Scholar]
- [57].Storrie B, Madden EA. Isolation of subcellular organelles. Methods Enzymol. 1990;182: 203–225. PubMed PMID: 2156127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.