

Abstract
Purpose
Conventional therapy for malignant glioma fails to specifically target tumor cells. In contrast, substantial evidence indicates that if appropriately redirected, T cells can precisely eradicate tumors. Here we report the rational development of a fully human bispecific antibody (hEGFRvIII-CD3 bi-scFv) that redirects human T cells to lyse malignant glioma expressing a tumor-specific mutation of the EGFR (EGFRvIII).
Experimental Design
We generated a panel of bispecific single-chain variable fragments and optimized design through successive rounds of screening and refinement. We tested the ability of our lead construct to redirect naïve T cells and induce target cell–specific lysis. To test for efficacy, we evaluated tumor growth and survival in xenogeneic and syngeneic models of glioma. Tumor penetrance following intravenous drug administration was assessed in highly invasive, orthotopic glioma models.
Results
A highly expressed bispecific antibody with specificity to CD3 and EGFRvIII was generated (hEGFRvIII-CD3 bi-scFv). Antibody-induced T-cell activation, secretion of proinflammatory cytokines, and proliferation was robust and occurred exclusively in the presence of target antigen. hEGFRvIII-CD3 bi-scFv was potent and target-specific, mediating significant lysis of multiple malignant glioma cell lines and patient-derived malignant glioma samples that heterogeneously express EGFRvIII. In both subcutaneous and orthotopic models, well-engrafted, patient-derived malignant glioma was effectively treated despite heterogeneity of EGFRvIII expression; intravenous hEGFRvIII-CD3 bi-scFv administration caused significant regression of tumor burden (P < 0.0001) and significantly extended survival (P < 0.0001). Similar efficacy was obtained in highly infiltrative, syngeneic glioma models, and intravenously administered hEGFRvIII-CD3 bi-scFv localized to these orthotopic tumors.
Conclusions
We have developed a clinically translatable bispecific antibody that redirects human T cells to safely and effectively treat malignant glioma. On the basis of these results, we have developed a clinical study of hEGFRvIII-CD3 bi-scFv for patients with EGFRvIII-positive malignant glioma.
Introduction
Current therapy for malignant glioma is incapacitating (1) as a result of nonspecific, dose-limiting toxicity. In contrast, immunotherapy promises an exquisitely precise approach, and evidence now exists that adoptively transferred T cells expressing modified T-cell receptors (TCR) or chimeric antigen receptors (CAR) can eradicate large tumors in the central nervous system (CNS) in both preclinical and clinical studies (2–8). Although promising, these approaches rely on ex vivo expanded and genetically manipulated T cells, processes that are laborious, inconsistent, and often require complex viral transductions (9, 10). In addition, these T cells are almost always targeted to antigens shared with normal tissues, which has led to lethal autoimmune toxicity (11–13).
In contrast, using a combination of two single-chain variable fragments (scFv) with different specificities, we have developed a novel, "off-the-shelf," fully human bispecific antibody to redirect human CD3+ T cells to lyse tumor cells expressing the tumor-specific EGFR mutation, EGFRvIII. A similar CD19-targeted bispecific single-chain variable fragment (bi-scFv), blinatumomab, was recently approved by the FDA for the treatment of Philadelphia chromosome–negative relapsed or refractory precursor B-cell acute lymphoblastic leukemia (R/R ALL; ref. 14). Treatment, however, leads to the expected depletion of normal CD19- expressing B cells. Thus, a significant limitation of this promising therapeutic platform is the lack of tumor-specific targets.
EGFRvIII, however, is an entirely tumor-specific, constitutively activated, cell surface tyrosine kinase receptor that enhances cell growth and migration (15, 16) and confers radiation (17) and chemotherapeutic (18, 19) resistance. As EGFRvIII is completely absent from normal tissues but expressed on the surface of glioblastoma (GBM; ref. 20) and other common neoplasms, it offers an ideal immunotherapy target (21). Moreover, recent evidence indicates that treatment with antibody-redirected T cells produces long-lasting immunity against tumor cells lacking the target antigen (7, 22), suggesting that this approach may be superior to EGFRvIII-targeted vaccines that are limited by antigen escape (23, 24).
Previously, we described a murine bispecific antibody that extended survival in mice when challenged with EGFRvIII-positive glioma (25). We demonstrated that using this approach, which is agnostic to T-cell specificity, even typically suppressive regulatory T cells (Tregs) can be subverted to induce granzyme-mediated, antitumor cytotoxicity (26). Given the potential benefits of T-cell–based anti-EGFRvIII therapy, here we report the rational development and analysis of a fully human, EGFRvIII: CD3–targeted bispecific antibody suitable for clinical translation.
Through the use of fully human antibody fragments, we constructed a therapeutic with reduced potential for immunogenicity and increased clinical safety (27, 28). In this setting, murine antibody–associated complications, including cytokine release syndrome (28, 29) and human anti-mouse antibody (HAMA) formation leading to rapid clearance from patient serum (30), unpredictable dose–response relationships (27, 28) and an acute, potentially severe influenza-like syndrome (27, 28, 31, 32) are entirely averted.
As bi-scFv expression characteristics, physical properties, and target affinities are dependent on factors such as antibody fragment arrangement and linker composition (33, 34), we began by generating a panel of several different recombinant bi-scFv candidates. Each construct was transiently expressed and the panel progressively narrowed and refined on the basis of expression characteristics, ease of purification, and target cell specificity. Given that many antibodies to EGFRvIII cross-react with wild-type EGFR (EGFRwt), including cetuximab (35, 36), in addition to taking into account feasibility parameters such as expression and purification characteristics, validation was focused on determining the degree of specificity to human CD3 and EGFRvIII and the lack of cross-reactivity with EGFRwt.
Following this process, we tested our lead, fully human, target-cell–specific bi-scFv (hEGFRvIII-CD3 bi-scFv) in vitro for the ability to redirect naïve human T cells to generate proinflammatory, antitumor immune responses in an antigen-specific fashion. Tumor cell–specific lysis was assessed using multiple EGFRvIII-expressing glioma lines as well as multiple patient-derived malignant glioma samples that allowed for an assessment of cytotoxicity in the context of endogenous drivers, levels, and heterogeneity of EGFRvIII expression. Indeed, in all cases, hEGFRvIII-CD3 bi-scFv generated potent and target cell–specific antitumor responses. We tested efficacy as a single agent in vivo in both subcutaneous and orthotopic settings, utilizing glioma cell lines, patient-derived xenografts and highly invasive syngeneic models of glioma. We found that in all cases, intravenous administration of hEGFRvIII-CD3 bi-scFv cured well-established tumors, with the antibody accumulating to significant levels within the brain even in the context of highly infiltrative, syngeneic glioma. On the basis of these data, we have developed a phase I clinical trial to treat patients with EGFRvIII-positive glioma.
Materials and Methods
Study design
The purpose of this study was to rationally design, produce, and test a fully human anti-EGFRvIII:CD3 bispecific antibody for use as a therapeutic agent in patients with EGFRvIII-expressing tumors, including glioblastoma. We generated a panel of different fully human bi-scFvs with different variable fragment arrangements and linker compositions and tested for robustness of expression in transiently transfected, suspension adapted CHO cells (Cricetulus griseus), ease of purification using a tag-free, two-step chromatography process, and target specificity. We verified the specificity of the bi-scFvs using surface-bound antigen and multiple antigen-positive and antigen-negative cell lines. A stably transfected CHO cell master cell bank (MCB) was developed for mammalian cell culture–based production of the lead recombinant protein. T-cell responses were assessed in vitro and against both target-positive and target-negative glioma cell lines. Cytotoxicity was assessed using multiple glioma cell lines as well as multiple patient-derived malignant glioma samples with endogenous drivers, levels and heterogeneity of EGFRvIII expression. We tested efficacy in four different xenogeneic models and two different highly-invasive syngeneic models, including four different orthotropic models and two patient-derived malignant glioma models. To examine efficacy against well-established tumors, patient-derived xenografts were allowed to establish without treatment until they were grossly visible and easily palpable in the subcutaneous setting (cohort mean tumor volume > 120 mm3) or one-third median untreated survival times were reached in the orthotropic setting. Each experiment was performed multiple times, with T cells derived from various donors.
Library generation, expression, and screening
cDNA fragments encoding for the variable segments, linker sequences, and signal peptides tested were synthesized (Gen-Script) and used to generate a library of bi-scFv encoding mammalian expression vectors. Each of the sequences tested were generated using overlapping PCR and inserted into the multiple cloning site (MCS) of the pcDNA3.1+ mammalian expression vector (Invitrogen). Suspension-adapted CHO-S cells (Thermo Fisher Scientific) were transiently transfected with sequence-verified, endotoxin-free plasmid preparations (Qiagen) using FreestyleMax transfection reagent (Invitrogen) according to the manufacturer's instructions. Transfected cells were grown for 6 days. On each day posttransfection, CHO cell density and viability was assessed and supernatant was sampled to allow for determination of target protein concentration using flow cytometry as described below. The terminal cysteine residue of the antigenic peptide spanning the EGFRvIII-mutant fusion junction (PEPvIII, LEEKKGNYVVTDHC) was coupled using haloacetyl activation chemistry to Irreversible Thiol-coupling SepFast 4HF Chromatography Resin (BioToolomics) as per manufacturer's instructions. This custom-made affinity resin was used to capture EGFR-vIII-binding antibodies from clarified cell culture supernatant. A gel filtration, TSKgel SuperSW3000 chromatography column (Tosoh Bioscience) was then used to separate protein based on size. Resulting proteins were dialyzed (3 × 24 hours) against PBS at pH 7.4, sterile-filtered, and placed in septum vials for analysis and storage. Total protein yield was assessed following each processing step. Target specificity (EGFRvIII, CD3) and lack of cross reactivity (EGFRwt) was assessed using surface-bound antigen (surface plasmon resonance) and multiple antigen-positive and antigen-negative cell lines (flow cytometry) as described below. A similar production and purification scheme was used to generate and isolate control bi-scFvs.
Recombinant protein expression, purification, and analysis
hEGFRvIII-CD3 bi-scFv was produced for studies using a stably transfected, suspension-adapted CHO cell line. CHO cell codon–optimized cDNA was synthesized (GenScript) and cloned in to the pcDNA3.1+ MCS. High-quality, endotoxin-free, transfection-grade plasmid was prepared and sequence verified using standard procedures. Linearized plasmid (Sca1 restriction site) was used to transfect CHO-S cells. Transfected cells were allowed to grow for 48 hours (at 37°C and 8% CO2 on an orbital shaker) and then seeded in fresh media containing 1,000 μg/mL geneticin and grown under static conditions. Cell density and viability was monitored every three to four days. Transfected cells were seeded in shaker flasks when viability exceeded 30% and stable pools of cells were cryopreserved when viability exceeded 95%. Productivity was assessed and the highest titer–producing stable pools were selected for limiting-dilution single-cell cloning. Single-cell colonies were screened for protein expression, positive clones were scaled up, and in a series of three rounds of screening the highest titer–producing clones were identified and cryopreserved. 14-day fed-batch experiments were performed and the highest titer–producing clone was selected for second round of single-cell cloning and additional screening. A current Good Manufacturing Protocols (cGMP) MCB was generated and certified (BioReliance).
To produce hEGFRvIII-CD3 bi-scFv for experiments, cells were grown in shaker flasks or a WAVE Bioreactor System (GE Health-care). A two-step chromatography protocol was developed and optimized. Using an AKTA system and UNICORN software (GE Healthcare), affinity chromatography was performed using either custom-made affinity resin (described above) or commercially available Capto L BioProcess chromatography resin (GE Healthcare). A subsequent negative-selection chromatography step using Capto Q BioProcess anion exchange medium (GE Healthcare) allowed for further purification as well as robust viral clearance capacity important for clinical translatability. Resulting purified hEGFRvIII-CD3 bi-scFv was concentrated and formulated in PBS.
Purified protein was assessed by SDS-PAGE, Western blot, and analytic size-exclusion chromatography. For the former, 1 μg of protein was loaded on a NuPAGE Novex 4%–12% Bis-Tris Protein gel, electrophoresis was performed using MOPS running buffer, and protein was visualized using a Coomassie G-250 based Colloidal Blue Staining Kit (Life Technologies) as per manufacturer's instructions. Western blot analysis was performed using HRP-Protein L (GenScript) to detect bi-scFvs. One microgram of bi-scFv protein was loaded on a NuPAGE Novex 4%–12% Bis-Tris Protein gel and electrophoresis was performed in MOPS running buffer. Electrophoresed protein was transferred to a nitrocellulose membrane using an iBlot Blotting System (Invitrogen). The membrane was blocked, incubated with HRP-Protein L (1:5,000 dilution) overnight at 4°C on an orbital shaking platform, and developed using a WesternDot Chemiluminescent Detection Kit (Life Technologies) according to manufacturer's instructions. Gels were imaged using a Bio-Rad ChemiDoc MP Imaging System and analyzed using associated Image Lab 5.0 software. For analytic size-exclusion chromatography, 5 μg of purified protein was loaded on to a Super SW3000 TSK gel column (Tosoh Bioscience) connected to a high-pressure liquid chromatography (HPLC) system. The resulting 280 nm absorbance chromatogram, representing protein separated on the basis of molecular size, was used to determine percent purity and detect aggregates.
Flow cytometry
Bi-scFv was detected biding to the surface of cells using fluorescently labeled streptavidin tetramers. To generate these reagents, fluorescently labeled streptavidin (Life Technologies) was incubated with either biotinylated recombinant protein L (Pierce) or biotinylated PEPvIII (GenScript). After incubation, free, unbound peptide or protein was removed using a centrifugal molecular weight cut-off device (GE Healthcare). Protein L tetramer allowed for the detection of bi-scFv on the surface of cells, regardless of specificity, while PEPvIII tetramer allowed for simultaneous detection of bi-scFv cell surface binding (via CD3) and target tumor antigen binding (via tetramer peptide). For experiments detecting bi-scFv on the surface of cells, 1 μg of bi-scFv was incubated with 1 × 106 cells for 30 minutes. The reaction was washed and then incubated with tetramer. All tetramers and antibodies were titrated before use and used with appropriate isotype controls. Lymphocyte surface markers were stained per manufacturer's instructions using combinations of fluorescently labeled antibodies specific to CD4 (clone L200), CD8 (clone RPA-T8), CD25 (clone M-A251), and CD69 (clone L78) purchased from BD Biosciences. Tumor cells were stained for EGFRvIII using 1 μg of chimeric anti-EGFRvIII mAb ch-L8A4 (37) per 1 × 106 tumor cells. Briefly, ch-L8A4–expressing hybridoma cells were grown in a hollow-fiber bioreactor using Hybridoma-Serum Free Media (Life Technologies), and anti-EGFRvIII antibody was isolated from cell culture supernatant using protein A chromatography. ch-L8A4 was detected binding to the surface of cells using a fluorescently labeled goat anti-human IgG (Jackson ImmunoResearch). Cells were stained for EGFRwt using fluorescently labeled antibody (BD Biosciences, clone EGFR.1) as per manufacturer's instructions. All flow cytometry data were acquired with a BD Biosciences FACSCalibur flow cytometer and analyzed using FlowJo analysis software (FlowJo, LLC). All experiments were repeated.
Surface plasmon resonance
We assessed bi-scFv–binding affinities and kinetics to EGFRvIII, EGFRwt, and CD3 using a BIAcore 3000 Surface Plasmon Resonance System (GE Healthcare). The antigenic peptide spanning the EGFRvIII-mutant fusion junction (PEPvIII) was coupled to a CM5 sensor chip using thiol coupling chemistry. Peptide was coupled in a 10 mmol/L sodium acetate, pH 5.0 solution. Recombinant human EGFRwt protein and human CD3 epsilon protein (Novoprotein) were coupled to CM5 sensor chips using amine coupling chemistry. EGFRwt protein and human CD3 epsilon protein were coupled in 10 mmol/L sodium acetate solutions at pH 5.5 and pH 4.0, respectively. Optimized regeneration conditions were obtained at a flow rate of 30 μL/minute for 30 seconds with 10 mmol/L glycine pH 1.7 for EGFRvIII and CD3 chips and pH 2.5 for the EGFRwt chip. Bi-scFv–containing solutions (0, 100, 250, 500, 1,000 nmol/L) were analyzed on each chip. Positive control mAbs and negative control isotypes were used for each experiment. Association and dissociation rates were monitored for 180 and 300 seconds, respectively, with a 120-minute stabilization time and a 30 μL/minute flow rate. Samples were run in triplicate and compared with a blank reference flow cell prepared without target antigen. A global fit analysis using simultaneous ka/kd calculations was performed using BIAevaluation software (GE Healthcare). All experiments were repeated.
Cell lines and culture
All cell lines used were submitted for cell line authentication (Cell Check) and pathogen testing including mycoplasma testing (IMPACT) at IDEXX BioResearch prior to use. We confirmed species of origin, cell line–specific markers and assessed for possible cross-contamination with other cell lines or pathogens. Where applicable, microsatellite markers for parental and EGFR-vIII-transduced cell lines were compared to confirm lineage. Cells were grown in improved MEM zinc option media (Thermo Fisher Scientific) with 10% FBS (v/v) for a maximum of one week and subpassaged no more than once using 0.05% trypsin-EDTA (Thermo Fisher Scientific) before being used for experiments. All cell lines used were obtained from the ATCC. Notably, the U87-MG cell line obtained from the ATCC that was used in our study as well as that of others has recently been found to be of different genetic origin from the originally reported U87-MG cell line. On the basis of this genetic analysis, however, these currently used U87-MG cells are still thought to be bona flde human glioblastoma cells (38).
Disaggregation of patient-derived tumor samples
All studies involving patient samples were approved by the Duke University Medical Center Institutional Review Board. Tumor tissue derived from GBM samples were mechanically dissociated in a petri dish using presterilized scissors and forceps. For every 2 g of minced tumor tissue obtained, a 50 mL improved MEM zinc option media solution containing 2 mg/mL collage-nase and 20 U/mL of deoxyribonuclease (Worthington Biochemical Corporation) was stirred at 37°C for 1.5 hours. The solution was passed through a 70-μm strainer and then centrifuged at 500 × g for 5 minutes at room temperature. Resulting pellets were washed twice in PBS and carefully layered over a 15-mL solution of Lymphocyte Separation Medium (Corning). Differential migration following centrifugation (at room temperature for 20 minutes and 2,000 × g without a brake) allowed for isolation of glioma cells. Cells were harvested, washed twice in PBS, and assessed for viability and density (Trypan blue). Cells were then immediately used for xenograft studies or for in vitro experiments placed in cell culture as described below. Prior to experiments requiring cell culture, pilot studies were conducted to assess for EGFRvIII expression status over the cell culture period.
T-cell activation and cytokine secretion analysis
T cells were isolated from peripheral blood mononuclear cells (PBMC) using a negative selection–based Pan T Cell Isolation Kit (Miltenyi Biotec). Negatively selected T cells were incubated with target cells at a effector:target (E:T) ratio of 20:1 for 44 hours at 37°C. In some cases, hEGFRvIII-CD3 bi-scFv was added to the reaction at a concentration of 1 μg/mL. Following incubation, supernatant and cells were harvested for analysis. Cellular surface markers CD4, CD8, CD69, and CD25 were analyzed by flow cytometryand cytokine secretionwas quantified in thesupernatant using a Human Cytometric Bead Array Human Kit (BD Biosciences) according to the manufacturer's instructions. All reactions were performed in triplicate and all experiments were repeated.
T-cell proliferation
T cells isolated from PBMCs as described above were labeled with carboxyfluorescein succinimidyl ester (CFSE) using a Cell-Trace CFSE Cell Proliferation Kit (Invitrogen) as per manufacturer's instructions. CFSE-labeled T cells and target cells were incubated at a E:T ratio of 20:1 for 92 hours at 37°C. In some cases, hEGFRvIII-CD3 bi-scFv was added to the reaction at a concentration of 1 μg/mL. After incubation, cells were isolated, stained for CD4 and CD8 surface markers, and analyzed by flow cytometry for T-cell subsets and CFSE dilution. All reactions were performed in triplicate and all experiments were repeated.
Cytotoxicity assays
We assessed for cytotoxicity using chromium release assays. 51Cr (200 μCi; Perkin Elmer) was used to label 4 × 106 target cells. Target cells were incubated with 51Cr for 1 hour at 37°C, and the reaction mixture was resuspended every 15 minutes. Labeled cells were washed a total of three times, resuspended in cell culture media, and allowed to rest at room temperature for 20 minutes. Cells were washed an additional time to remove free 51Cr and then incubated with various combinations of T cells isolated from PBMCs as described above and or bi-scFvs at various concentrations. T cells were plated at an E:T ratio of 20:1 when present. Maximal lysis was induced using a 1.5% solution (v/v) of Triton X-100 (Sigma Aldrich). Cells were incubated at 37°C for 20 hours after which 50 μL of supernatant from each well was collected and combined with 150 μL of OptiPhase Supermix Scintilation Cocktail (Perkin Elmer). Radioactivity released in the culture medium was measuring using a 1450 MicroBeta TriLux Microplate Scintillation and Luminescence Counter (Perkin Elmer).
Subcutaneous and orthotopic glioma models
All animal experiments were performed according to protocols (A283-15-11) approved by the Duke University Institutional Animal Care and Use Committee (IACUC). For orthotopic U87-MG, U87-MG-EGFRvIII, or CT2A-EGFRvIII models, cells grown as described above were collected in logarithmic growth phase, washed twice in PBS, and mixed with an equal volume of 10% methyl cellulose (v/v). For orthotopic patient-derived malignant glioma models, cells were disaggregated, isolated, and washed as described above prior to being mixed with an equal volume of 10% methyl cellulose (v/v). Cell mixtures were loaded into a 250 μL Hamilton syringe and a 25-guage needle was attached. Using a stereotactic frame, 5 μL of the cell mixture was injected 2 mm to the right of the bregma and 4 mm below the surface of the skull at the coronal suture. A total of 5 × 104 U87-MG or U87-MG-EGFRvIII cells or 1 × 105 patient-derived glioma cells were implanted into 8- to 12-week-old female NOD-scid gamma (NSG) mice (Jackson Laboratory), with 10 mice per group base on pilot data and anticipated statistical power. A total of 3 × 104 CT2A-EGFRvIII cells were implanted into 8- to 12-week-old female heterozygous human CD3 transgenic mice (tgε600, Jackson Laboratory, strain number 20456 crossed with wild-type C57/BL6). Heterozygous mice were used, as the human CD3 transgene is known to induce lymphocyte apoptosis (39, 40) and these heterozygous mice were found to have less of a reduction in the total number of lymphocytes. Prior to the initiation of therapy, mice were randomized to groups using a random number generator. Tumor cell doses were based on pilot tumorigenicity studies.
For subcutaneous patient-derived glioma models, dissociated patient-derived malignant glioma cells were loaded into a repeating Hamilton syringe dispenser and a 19-gauge needle was attached. Fifty microliters of the tumor cell homogenate was injected subcutaneously in the right flank of 8- to 12-week-old female NSG mice (Jackson Labs), with 10 mice per group based on pilot data and anticipated statistical power. For subcutaneous syngeneic glioma models, CT2A-EGFRvIII cells were grown as described above, collected in logarithmic growth phase, washed twice in PBS, and loaded into a repeating Hamilton syringe dispenser. A 19-gauge needle was attached and 50 μL of the tumor cell preparation was injected subcutaneously in the right flank of 8- to 12-week-old female heterozygous human CD3 transgenic mice. Ten mice per group were used in these studies based on pilot data and anticipated statistical power. Tumors were allowed to engraft for 10 days before mice were randomized to different groups using a random number generator. Tumor progression was evaluated with calipers for the duration of the experiment. Tumor volume (millimeters cubed) was calculated (length × width2 × 0.52) in a perpendicular fashion. Mice were euthanized when tumors reached a volume of 2,000 mm3 or upon evidence of ulceration. Tumor measurements were included in the analyses up to the day of euthanasia.
Where indicated, immunodeficient mice (NSG) received a single intravenous injection of 1 × 107 in vitro expanded human T cells. Briefly, rapidly thawed PBMCs were washed twice in AIM V medium CTS (Thermo Fisher Scientific) containing 5% Human AB Serum (v/v; Valley Biomedical), assessed for viability and density (trypan blue) and resuspended at 1 × 106 viable cells per mL in AIM V growth media containing 5% (v/v) human AB serum, 300 IU/mL IL2 (Preometheus Laboratories), and 50 ng/mL OKT3 (Miltenyi Biotec). Cells were grown horizontally in a T150 cell culture flask at 37°C in a 5% CO2 incubator. After each two days of cell culture, cells were harvested and reseeded in growth media without OKT3 at a concentration of 1 × 106 viable cells per mL. When a sufficient number of cells were obtained, after 10–14 days of cell culture, cells were washed twice in PBS and where indicated 200 μL of either the cell mixture or vehicle (PBS) was administered via a single intravenous tail injection.
Where indicated, heterozygous human CD3 transgenic mice received a single intravenous injection of 1 × 107in vitro expanded autologous T cells. Autologous T cells were administered as heterozygous human CD3 transgenic mice have significantly reduced numbers of lymphocytes due to toxicity of the human CD3 transgene (39, 40). To prepare the expanded human CD3 transgenic lymphocytes, splenocytes were isolated from heterozygous human CD3 transgenic mice and expanded to sufficient numbers in RPMI 1640 cell culture medium (Thermo Fisher Scientific) containing 2 μg/mL conalbumin and 50 IU/mL IL2. A total of 2 × 106 cells/mL were seeded in 24-well plate cell culture wells containing 2 mL of cell solution per well. Cells were split at a ratio of 1 to 2 after 48 and 72 hours and, after five days of expansion, cells were washed twice in PBS and administered by a single intravenous tail injection in a 200 μL total volume.
Where indicated, hEGFRvIII-CD3 bi-scFv, control bi-scFvs, or vehicle (PBS) was administered in a total volume of up to 200 μL via intravenous tail vein injection.
Biodistribution studies
We assessed for hEGFRvIII-CD3 bi-scFv tumor accumulation using radiolabeled hEGFRvIII-CD3 bi-scFv and PET/CT imaging followed by autoradiography.
hEGFRvIII-CD3 bi-scFv (100 μg in 200 μl PBS, pH 7.4) was added to a glass vial containing 10 μg of Pierce IodoGen Reagent (Thermo Fisher Scientific). Iodine-124 (I-124; 5 μL; 1 mCi/μL, Zevacor Pharma) was added to the IodoGen vial, vortexed, and left to stand at room temperature for 5 minutes. The reaction mixture was transferred to a PD10 column (GE Healthcare) preconditioned with 5% human serum albumin (Grifols Biologicals). After adding the reaction mixture to the column, the column contents were eluted with PBS and the eluate was collected as 0.25-mL fractions after discarding the first 2 mL. Fractions with the highest radioactivity (typically fractions 5 to 10) were combined. Trichloroacetic acid (TCA) precipitation and iTLC was performed to determine the protein bound versus free I-124. Typical radiolabeling yield was in excess of 80% and final radiochemical purity was typically greater than 98%. Solutions were prepared for intravenous administration by diluting in PBS to give a final volume of 100 μL per mouse. Surface plasmon resonance utilizing I-124–labeled hEGFRvIII-CD3 bi-scFv and paired unlabeled hEGFRvIII-CD3 bi-scFv was used to ensure the maintenance of target binding following radioactive labeling.
Mice received 100 μCi of I-124–labeled hEGFRvIII-CD3 bi-scFv in 100 μL of PBS via intravenous tail injection. Three hours after administration, mice were anesthetized with 2% isoflurane (Abbott Laboratories) in 100% oxygen. The animals were then placed on a heated PET/CT animal holder that provided anesthesia through a nose cone, and transferred for dynamic list-mode PET imaging over 10 minutes. Images were reconstructed with an iterative reconstruction algorithm into an image pixel size of 0.4 × 0.4 × 0.4 mm. The image reconstruction software (Inveon Acquisition Workplace 1.5SP1; Siemens Medical Solutions USA, Inc.) provided for correction of radioactivity decay, random coincidences, dead-time losses, and photon attenuation. Regions of interest (ROI) were drawn on the coregistered CT image for large organs and over the region of the brain where tumor cells had been stereotactically injected to determine the uptake and washout of the radioactivity.
After PET/CT imaging, mice were humanly sacrificed and the cerebrum was removed from the cranium and rapidly frozen. Sections (20 μm) through the tumor were obtained with a cryotome (Thermo Fisher Scientific), mounted on adhesive microscope slides (Superfrost Plus, Thermo Fisher Scientific), and exposed to a phosphor storage screen film (super-resolution screen, Perkin Elmer) for 24 hours. Screens were read using a Cyclone Plus system (Perkin Elmer) and analyzed using Optiquant software (Perkin Elmer), as reported previously (41).
Statistical analysis
All experiments were designed and performed with consideration for statistical power and analysis. For mechanistic studies assessing the differences among groups in %CD25+, %CD69+, cytokine secretion and T-cell proliferation, an ANOVA model was used to assess whether any differences exist among the groups for both CD4+ and CD8+ cells separately. Upon a significant result for both CD4+ and CD8+ cells (F-test P < 0.0001), a t test was used to assess the difference between the hEGFRvIII-CD3 bi-scFv group and the group with the next highest mean.
Where indicated, ED50 concentrations were estimated in specific lysis experiments. The following four-parameter logistic model (i.e., an Emax model) was fit for the expected specific lysis given a drug concentration:
Where θ3 and θ4 are the minimum and maximum lysis %, θ1 is the drug ED50, and θ2 is the slope parameter. Parameters were estimated using the Gauss–Newton iterative method. For experiments assessing the effect of T cells, drug, and or control drug on specific lysis, a t test was used to assess the difference in specific lysis between the groups.
For subcutaneous tumor growth experiments, after tumors were allowed to establish, mice were randomized using a random number generator and the mean tumor volume for each group was assessed to ensure similarity between groups. Differences in the pattern of change in tumor volume among the groups were assessed from the initiation of therapy onward. As the data appear to show a linear trend for the time interval observed, a linear trend model with an interaction between time and group was fit to the raw tumor volume values. A table was generated to estimate slopes for each group from the model with P values testing whether each slope value is different from 0. A high P value for the group of interest indicates the change in tumor volume was not significantly different from 0. In addition, the difference in slope between the treated group and each of the other groups was assessed.
For orthotopic survival experiments, mice were randomized to different groups using a random number generator prior to administration of T cells or initiation of therapy. Given the nature typical of preclinical mouse survival data usually consisting of a plateau followed by a series of events, a generalized Wilcoxon test was used to assess whether a difference in survival pattern exists among the groups. All experiments were repeated.
Results
Generation and selection of a fully human bi-scFv
As a general rule, bi-scFv N- to C-terminal segment arrangement and linker composition govern bi-scFv antigen-binding kinetics and physical characteristics altering expression yield (33, 34, 42). Not only are these factors critical to eliciting effective, precisely targeted immunotherapeutic responses, but also these physical characteristics govern bispecific antibody production capacity, directly affecting clinical translatability. To determine an optimal bi-scFv design for the treatment of GBM, we began by generating a library of different bi-scFv constructs.
We started with protein sequences for mAbs specific for EGFR-vIII and CD3. We selected fully human, anti-human mAb clones 139 (anti-EGFRvIII) and 28F11 (anti-CD3), both of which have previously been used safely in the clinic (43–45). Using variable light- and heavy-chain sequences from these mAbs, we generated a panel of different N- to C-terminal combinations of variable heavy- and light-chain sequences joined by flexible glycine–serine linkers.
We expressed each of these constructs using a mammalian CHO expression system. This system was selected given our previous experience as well as that of others who have reported bi-scFvs expressed in CHO cells as fully functional protein, while expression of the same construct in other systems resulted in nonfunctional protein only (34). The use of CHO cells furthermore allows for straightforward integration with commonly used clinical protein–manufacturing infrastructure and regulatory pathways. Each construct was transiently expressed, purified using a two-step, tag-free chromatography process, and tested for specificity to CD3 and EGFRvIII, as well as lack of cross-reactivity with EGFRwt. The panel of constructs was progressively narrowed on the basis of antigen specificity and binding kinetics tested using surface plasmon resonance as well as flow cytometry using multiple cell lines.
Top constructs were then selected for further optimization and analysis. The effects of linker composition and length on binding affinity and physical characteristics have previously been reported and the 218 linker (GSTSGSGKPGSGEGSTKG) was found to increase binding affinity of scFv constructs (42). Accordingly, we generated additional constructs from our top bispecific antibodies, replacing glycine–serine linkers with 218 linkers. These constructs along with corresponding glycine–serine linker constructs were then expressed and purified, allowing us to assess for target specificity, lack of cross-reactivity, and protein yield. Through this sequential process, our library of bi-scFv constructs was progressively narrowed to a single target-specific, high-expressing construct (Fig. 1A). A table containing each of the constructs tested along with the corresponding target-binding kinetics, expression parameters, and purification yield is shown in Supplementary Table S1.
Figure 1.

Design and analysis of purified, lead bispecific construct, hEGFRvIII-CD3 bi-scFv. A, Vector map of fully human bispecific construct, hEGFRvIII-CD3 bi-scFv, with optimized variable segment arrangement and linker composition. SDS-PAGE (B) and Western blot (C) analysis of 1 μg of purified hEGFRvIII-CD3 bi-scFv. D, Elution spectrum following analytical size-exclusion chromatography. Five micrograms of purified hEGFRvIII-CD3 bi-scFv protein was used for the analysis.
We then generated a stable CHO cell line to produce our lead recombinant protein, hEGFRvIII-CD3 bi-scFv. CHO cell codon–optimized cDNA encoding for our lead construct was cloned into a mammalian expression vector under control of the cyto-megalovirus (CMV) enhancer–promoter. The sequence coding for the mature recombinant antibody was inserted downstream of the human serum albumin preproprotein signal peptide (MKWVTFISLLFLFSSAYS). This signal peptide was selected to improve secretion efficiency based on analysis of 16 difference signal peptides (46). In shaker flasks, the cell bank was capable producing greater than 25 mg of hEGFRvIII-CD3 bi-scFv per liter of cell culture. We developed a two-step, tag-free chromatography process for viral clearance and purification. The process generated a highly purified, approximately 50-kDa protein (Fig. 1B) capable of being identified on Western blot analysis via affinity to protein L (Fig. 1C) and of >98% purity (Fig. 1D).
hEGFRvIII-CD3 bi-scFv binds to human CD3 and EGFRvIII but not EGFRwt
We used flow cytometry to confirm dual specificity of hEGFR-vIII-CD3 bi-scFv to EGFRvIII and CD3-positive cells, as well as lack of cross reactivity with EGFRvIII-negative cells given that many antibodies to EGFRvIII can cross react with EGFRwt (Fig. 2). Our analyses revealed that hEGFRvIII-CD3 bi-scFv binds to both CD4+ and CD8+ lymphocytes known to express CD3 (Fig. 2A and B). As our recombinant protein was expressed without purifica-tion tags and there are no known antibodies to the drug, we developed a PEPvIII-Biotin/SA-PE tetramer to detect hEGFRvIII-CD3 bi-scFv on the surface of cells. The tetramer was constructed with the EGFRvIII antigenic-determining peptide, PEPvIII, allowing for simultaneous detection of hEGFRvIII-CD3 bi-scFv binding to the surface of lymphocytes and tumor antigen (Fig. 2A and B). Indeed, simultaneous binding to both CD3 and EGFRvIII mutation–containing peptide was detected. In addition, we tested hEGFRvIII-CD3 bi-scFv binding to both EGFRvIII-positive and EGFRvIII-negative cell lines. Using a similarly developed Protein L-Biotin/SA-Alexa Fluor 647 tetramer, we were unable to detect binding of hEGFRvIII-CD3 bi-scFv to the surface of EGFRvIII-negative U87-MG cells (Fig. 2C), but found robust binding to EGFRvIII-positive U87-MG cells. Similar results were obtained with multiple PBMC donors and against multiple EGFRvIII-positive and EGFRvIII-negative cell lines.
Figure 2.

hEGFRvIII-CD3 bi-scFv binds to T lymphocytes and EGFRvIII-positive glioma cells but not to EGFRvIII-negative cells. hEGFRvIII-CD3 bi-scFv binds to CD4+ (A) and CD8+ (B) T lymphocytes. hEGFRvIII-CD3 bi-scFv was detected on the surface of lymphocytes using PEPvIII-Bio/SA-PE tetramer, demonstrating simultaneous lymphocyte and tumor-antigen binding. No hEGFRvIII-CD3 bi-scFv antibody was detected on the surface of EGFRvIII-negative U87-MG cells (C), while hEGFRvIII-CD3 bi-scFv was detected on the surface of U87-MG-EGFRvIII cells (D). hEGFRvIII-CD3 bi-scFv was detected binding to the surface of tumor cells using Prot L-Bio/ SA-Alexa Fluor 647 tetramer. Dotted lines represent baseline tetramer signal in the absence of hEGFRvIII-CD3 bi-scFv while solid lines represent fluorescence signal in the presence of hEGFRvIII-CD3 bi-scFv.
We further investigated hEGFRvIII-CD3 bi-scFv binding kinetics to specific individual antigens using surface plasmon resonance. No binding to EGFRwt protein was detected, while binding to human CD3 epsilon and EGFRvIII antigen allowed for determination of binding kinetics. The association rate constant (ka), dissociation rate constant (kd), and equilibrium dissociation constant (KD) for EGFRvIII were 4.45 × 104 M−1s−1, 1.24 × 10−3 s−1, and 2.78 × 10−8 M, respectively. The ka, kd, and KD for CD3 epsilon were 5.35 × 104 mol/L−1s−1, 8.36 × 10−4 s−1, and 1.56 × 10−8 mol/L, respectively.
hEGFRvIII-CD3 bi-scFv activates T cells in an antigen-restricted fashion
We next tested for the ability of hEGFRvIII-CD3 bi-scFv to activate naïve T cells in vitro in an antigen-specific fashion. When T cells were incubated with both EGFRvIII-expressing glioma and hEGFRvIII-CD3 bi-scFv, 86.6% ± 1.2% of CD8+ T cells and 78.1 ± 1.65% of CD4+ T cells expressed the early activation marker, CD69. This is in direct contrast to T cells incubated with EGFRvIII-expressing glioma alone, where only 4.03% ± 1.50% of CD8+ T cells and 1.20% ± 0.29% of CD4+T cells expressed CD69 (Fig. 3A). Other combinations of T cells alone, T cells plus hEGFRvIII-CD3 bi-scFv, T cells plus either EGFRvIII-positive or EGFRvIII-negative cells, and T cells plus hEGFRvIII-CD3 bi-scFv and EGFRvIII-negative glioma were tested, and no differences were observed among these groups (Fig. 3A). An ANOVA model was used to assess whether any differences exist among the six groups in % CD69+ T cells for both CD4+ and CD8+ cells separately. Upon a significant result for both CD4+ and CD8+ cells (F-test P < 0.0001 in both cases), a t test was used to assess the difference between the hEGFRvIII-CD3 bi-scFv plus EGFRvIII-positive glioma group and the group with the next highest mean. For both CD4+ and CD8+ cells, the percent of T cells positive for CD69 was significantly higher in the hEGFRvIII-CD3 bi-scFv plus EGFRvIII-positive tumor group (P < 0.0001 in both cases).
Figure 3.

In the presence of EGFRvIII antigen, hEGFRvIII-CD3 bi-scFv activates T lymphocytes and redirects them to secrete proinflammatory cytokines and proliferate. T cells were incubated with various combinations of glioma cells and or hEGFRvIII-CD3 bi-scFv. Following incubation, T-cell surface activation markers CD69 (A) and CD25 (B) were assessed by flow cytometry. Data are reported as percent positive for both CD8+ and CD4+ T-cell subsets. For both CD8+ and CD4+ T cells, the percent CD69+ and percent CD25+ was significantly higher in the EGFRvIII-positive glioma and hEGFRvIII-CD3 bi-scFv group (P < 0.0001 in all cases). Resulting cell culture supernatants were analyzed for cytokine content (C) and indicated amounts of IFNγ, TNF, and IL2 were detected in the presence of both drug and target antigen, while no corresponding cytokines were detected in all other cases. T-cell proliferation in response to various combinations of glioma cells and or hEGFRvIII-CD3 bi-scFv was assessed and significant proliferation of both CD8+ and CD4+ T cells was observed in the presence of both drug and target antigen (P < 0.0002; D). Of those proliferating T cells, multiple rounds of proliferation were observed (E).
An equivalent analysis was conducted for the late activation marker, CD25, and a similar trend was observed, again highlighting the antigen specificity of the approach. When T cells were incubated with both EGFRvIII-expressing glioma and hEGFRvIII-CD3 bi-scFv, 72.63% ± 2.90% of CD8+ T cells and 84.43% ± 0.64% of CD4+ T cells expressed the late activation marker, CD25 (Fig. 3B). This is in direct contrast to T cells incubated with EGFRvIII-expressing glioma alone, where only 12.63% ± 1.19% of CD8+ T cells and 23.43% ± 0.06% of CD4+ T cells expressed CD25. Again, other combinations were tested and no differences were observed among these groups. (Fig. 3B) An ANOVA model was used to assess whether any differences exist among the six groups, as described above, and for both CD4 and CD8 cells, the % CD25+ was significantly higher in the hEGFRvIII-CD3 bi-scFv plus EGFRvIII-positive tumor group (P < 0.0001 in both cases).
These data demonstrating T-cell activation exclusively in the context of EGFRvIII-positive glioma and hEGFRvIII-CD3 bi-scFv demonstrate the exquisite antigen specificity of our approach, critical to safe and effective, tumor cell–specific immunotherapy.
hEGFRvIII-CD3 bi-scFv induces antitumor T-cell responses in an antigen-specific fashion
We next assessed for the ability of hEGFRvIII-CD3 bi-scFv to induce T cells to secrete proinflammatory, Th1-polarizing cytokines and to proliferate. These factors are associated with effective, T-cell–based, antitumor immune responses. T cells were incubated with various combinations of hEGFRvIII-CD3 bi-scFv and or EGFRvIII-positive or negative glioma cells in vitro and resulting cell culture supernatants were analyzed for cytokine content (Fig. 3C). Indeed, Th1-polarizing, proinflammatory cytokines, IFNγ, TNF, and IL2, were detected in copious amounts when hEGFRvIII-CD3 bi-scFv and EGFRvIII-positive glioma cells were present, while no detectable amounts of these cytokines were present in all other conditions tested (Fig. 3C). These data again highlight the antigen specificity of this approach.
We also assessed for both CD4+ and CD8+ T-cell proliferation in response to various combinations of hEGFRvIII-CD3 bi-scFv and or effector targets (Fig. 3D). When T cells were incubated with both EGFRvIII-expressing glioma and hEGFRvIII-CD3 bi-scFv, 77.57% ± 1.69% of CD8+ T cells and 64.07% ± 1.15% of CD4+ T cells underwent one or more cycles of division. This is in direct contrast to T cells incubated with EGFRvIII-expressing glioma alone, where only 0.51% ± 0.46% of CD8+ T cells and 0.67% ± 0.48% of CD4+ T cells replicated one or more times (Fig. 3D). Other combinations of T cells alone, T cells plus hEGFRvIII-CD3 bi-scFv, T cells plus either EGFRvIII-positive or EGFRvIII-negative cells, and T cells plus hEGFRvIII-CD3 bi-scFv and EGFRvIII-negative glioma were tested and no differences were observed among these groups (Fig. 3D). An ANOVA model was used to assess whether any differences exist among the six groups in % of T cells undergoing one or more cycles of division for both CD4 and CD8 cells separately. Upon a significant result for both CD4+ and CD8+ T cells (F-test P < 0.0001 in both cases), a t-test was used to assess the difference between the hEGFRvIII-CD3 bi-scFv plus EGFRvIII-positive glioma group and the group with the next highest mean. For both CD4+ and CD8+ T cells, the % of T cells undergoing one or more cycles of division was significantly higher in the hEGFRvIII-CD3 bi-scFv plus EGFRvIII-positive glioma group (CD4: P < 0.0001; CD8: P = 0.0002).
Of those proliferating T cells, multiple rounds of proliferation were observed, with 83.83% ± 2.70% of CD8+ T cells and 83.9% ± 1.8% of CD4+ T cells replicating two or more times (Fig. 3E). These data demonstrate robust, antigen-specific T-cell responses important to safe and effective tumor cell–specific hEGFRvIII-CD3 bi-scFv–induced T-cell responses.
hEGFRvIII-CD3 bi-scFv redirects T cells to lyse EGFRvIII-expressing glioma in vitro
We next sought to assess for hEGFRvIII-CD3 bi-scFv–induced functional, antigen-specific, cytotoxic responses in vitro. Using 51Cr release assays, we assessed for specific lysis of two different malignant glioma cell lines. Through the use of malignant glioma cell lines engineered to express EGFRvIII (U87-MG and D54-MG), we were able to assess for cytotoxic responses against EGFRvIII-expressing tumor cells as well as responses against parental cells lacking the target antigen. These cell lines were beneficial as they allowed for direct comparison between cells with identical genetic profiles but that differ only in EGFRvIII status.
Indeed, potent, antigen-specific, cytotoxic responses were observed in all cases (Fig. 4). Our results demonstrated exquisite antigen specificity even at the highest doses of hEGFRvIII-CD3 bi-scFv tested. A dose-response–based increase in tumor cell lysis was observed against U87-MG-EGFRvIII and D54-MG-EGFRvIII glioma cells, while no increase in specific lysis of parental U87-MG or D54-MG was observed (Fig. 4A and D).
Figure 4.

hEGFRvIII-CD3 bi-scFv redirects T cells to specifically lyse EGFRvIII-positive glioma. A, Tumor cell lysis was assessed against two different glioma cell lines (EGFRvIII-positive and -negative for both). A dose-response–based increase in tumor cell lysis was observed against U87-MG-EGFRvIII glioma cells while no increase in lysis of U87-MG was observed with increasing bispecific antibody concentrations. B, The ED50 for the U87-MG-EGFRvIII cell line was 15,881 pg/mL. Both T cells and drug were necessary for significant specific lysis of U87-MG-EGFRvIII cells (P = 0.0033 and P = 0.0032, respectively) while the combination failed to induce any significant increase in lysis of target-negative cells compared to groups without drug or target antigen (P = 0.391 and 0.195, respectively). C, Significant drug-induced specific lysis of EGFRvIII-positive glioma was also observed when compared with that induced by control bi-scFv that binds to EGFRvIII but not CD3 (P = 0.0033). D–F, The same trends were observed against a second malignant glioma cell line both with and without EGFRvIII (D54-MG). The estimated ED50 for the D54-MG-EGFRvIII cell line was 10,595 pg/mL. Likewise, both T cells and drug were necessary for significant specific lysis of D54-MG-EGFRvIII cells (P < 0.0001 in both cases) while the combination failed to induce any significant increase in lysis of target-negative cells compared with groups without drug or target antigen (P = 0.1011 and 0.4659, respectively). Significant drug-induced specific lysis was observed against the D54-MG-EGFRvIII cell line when compared with that induced by control bi-scFv (P = 0.0001). Among each of the EGFRvIII-positive cell lines tested, the number of EGFRvIII receptors per cell was either similar to or an order of magnitude less than the number of EGFRvIII receptors per cell found among the patient-derived cells used in this study that endogenously express the target antigen (see Supplementary Table S2).
We next sought to assess for the cytotoxic impact of T cells alone or hEGFRvIII-CD3 bi-scFv alone, against both EGFRvIII-positive and EGFRvIII-negative glioma cell lines (Figs. 4B and E). Both T cells and drug were necessary for significant specific lysis of U87-MG-EGFRvIII cells (P = 0.0033 and P = 0.0032, respectively) and D54-MG-EGFRvIII cells (P < 0.0001 in both cases), while the combination failed to induce any significant increase in lysis of target-negative cells compared with groups without drug or target antigen (P = 0.391 and 0.195, respectively, for U87-MG and P = 0.1011 and 0.4659, respectively, for D54-MG). These important controls demonstrate the lack of cytotoxicity when T cells or hEGFRvIII-CD3 bi-scFv are paired alone against glioma cells.
Having assessed for the effect of CD3 binding alone through the use of parental, antigen-negative glioma cell lines, we next sought to control for EGFRvIII binding alone. To do this, we generated a control bi-scFv with identical EGFRvIII-binding segments and capacity, but lack of reactivity, with CD3. Indeed, control bi-scFv failed to induce any significant specific lysis of U87-MG-EGFRvIII or D54-MG-EGFRvIII cells, while significant specific lysis was observed with hEGFRvIII-CD3 bi-scFv in both cases (P = 0.0033 and P = 0.0001, respectively; Fig. 4C and F).
To place results using these cell lines into context, we have quantified the number of EGFRvIII and EGFRwt receptors per cell for each of the cell lines used in our study along with for three different patient-derived tumor samples (Supplementary Table S2). These results demonstrate that the number of EGFRvIII receptors per cell in the induced expression glioma cell lines (U87-MG-EGFRvIII and D54-MG-EGFRvIII) are equivalent to or in one case tested an order of magnitude lower than the number of EGFRvIII receptors per cell found in EGFRvIII-positive patient-derived glioma samples. These results demonstrate that significant immune responses can be induced even when the target receptor expression level is an order of magnitude lower than that which can be expected to be found in patient tumor samples with endogenous levels and drivers of target antigen expression.
hEGFRvIII-CD3 bi-scFv redirects T cells to lyses patient derived malignant glioma in vitro
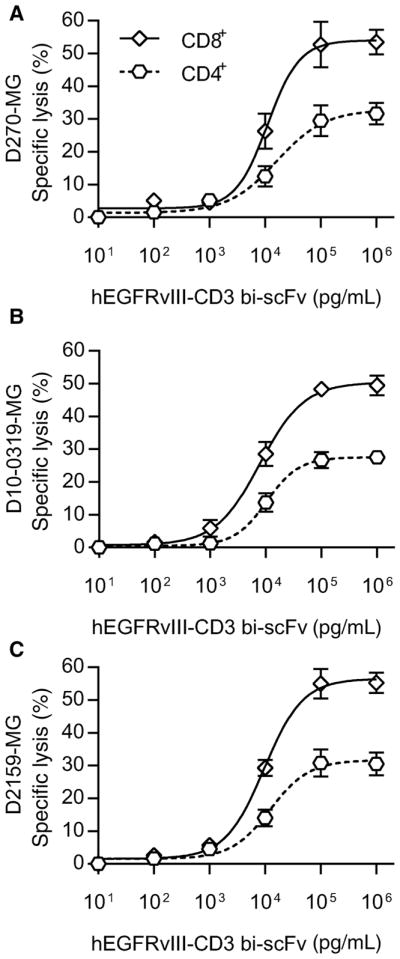
We next assessed for specific lysis of three different patient-derived glioma samples (D270-MG, D10-0319-MG, and D2159-MG) with endogenous drivers, levels, and heterogeneity of EGFR-vIII expression (Supplementary Table S2). As opposed to U87-MG and D54-MG cell lines that are uniformly modified to stably express EGFRvIII, the use of patient-derived samples allowed for an assessment of hEGFRvIII-CD3 bi-scFv–induced cytotoxicity in the context of EGFRvIII expression patterns that can be expected to be found among patients. In each of the samples tested, EGFRvIII expression was heterogeneous, with some cells that express EGFR-vIII and others that do not.
We assessed for specific lysis of D270-MG at various drug concentrations and indeed observed potent cytotoxic responses (Fig. 5A). Both T cells and hEGFRvIII-CD3 bi-scFv were necessary for induction of specific lysis (P = 0.0003 and P < 0.0001, respectively; Fig. 5B) and hEGFRvIII-CD3 bi-scFv–induced significant specific lysis when compared with control bi-scFv (P = 0.0003; Fig. 5C). Similar results were obtained when assessing for specific lysis of D10-0319-MG and D2159-MG. A dose-response–based increase in tumor cell lysis was observed against both D10-0319-MG and D2159-MG (Fig. 5D and E) and both T cells and hEGFRvIII-CD3 bi-scFv were necessary for induction of specific lysis of either patient-derived tumor sample (P < 0.0001 in all cases; Fig. 5E and H). Furthermore, similar to that observed with D270-MG, control bi-scFv failed to induce any significant specific lysis of either D10-0319-MG or D2159-MG, while significant specific lysis was observed with hEGFRvIII-CD3 bi-scFv in both cases (P = 0.0002 and P = 0.0003, respectively; Fig. F and I).
Figure 5.

hEGFRvIII-CD3 bi-scFv redirects T cells to lyse patient-derived glioma samples with endogenous drivers, levels, and heterogeneity of EGFRvIII expression. Tumor cell lysis was assessed among three different patient-derived tumor samples with endogenous drivers, levels, and heterogeneity of EGFRvIII expression (D270-MG, D10-0319-MG, and D2159-MG; see Supplementary Table S2). The proportion of cells expressing EGFRvIII in D270-MG, D10-0319-MG, and D2159-MG was 77.8 ± 3.61%, 73.6 ± 1.58%, and 83.6 ± 2.18%, respectively. Despite this heterogeneity, a dose-response–based increase in tumor cell lysis was observed in all cases. Increased D270-MG–specific lysis was observed with increasing bispecific antibody concentrations (A). The estimated ED50 was 12,511 pg/mL. Both T cells and drug were necessary for induction of specific lysis (P = 0.0003 and P < 0.0001, respectively; B) and hEGFRvIII-CD3 bi-scFv induced significant specific lysis when compared with control bi-scFv (P = 0.0003; C). Similar trends were observed against the two additional patient-derived malignant glioma samples tested, D10-0319-MG (D–F) and D2159-MG (G–I). The estimated ED50 for the D10-0319-MG cells and the D2159-MG cells was 6,916 pg/mL and 13,292 pg/mL, respectively. Both T cells and drug were necessary for significant specific lysis of either D10-0319-MG or D2159-MG to occur (P < 0.0001 in all cases) and significant drug-induced specific lysis was observed against both the D10-0319-MG and D2159-MG patient-derived glioma cells when compared with that induced against each of these tumors by control bi-scFv (P = 0.0002 and P = 0.0003, respectively).
These results demonstrate that despite heterogeneity of target–receptor expression, hEGFRvIII-CD3 is able to mediate significant specific lysis. This was evident in each of the patient-derived samples tested. Together, our in vitro cytotoxicity results utilizing multiple glioma cell lines and multiple patient-derived malignant glioma samples demonstrate that hEGFRvIII-CD3 bi-scFv is both potent and antigen specific, capable of mediating significant specific lysis of EGFRvIII-positive glioma, even in the context of heterogeneity of target receptor expression as is expected to be found among patients.
Both CD4+ and CD8+ T-cell subsets contribute directly to hEGFRvIII-CD3 bi-scFv–induced specific lysis of patient-derived malignant glioma
We next assessed for the ability of hEGFRvIII-CD3 bi-scFv to independently redirect both CD4+ and CD8+ T-cell subsets to lyse each of the patient-derived malignant glioma samples described previously (D270-MG, D10-0319-MG, and D2159-MG). Indeed, a dose-response–based increase in tumor cell lysis was observed in all cases, with both CD4+ and CD8+ T-cell subsets capable of inducing independent cytotoxic responses (Fig. 6). For the D270-MG patient-derived glioma cells, CD8+ T cells induced significantly more lysis compared with CD4+ T cells (P = 0.0123), although CD4+ T cells were still capable of inducing significant amounts of specific lysis (P = 0.0007; Fig. 6A). Similar trends were observed in each of the other two patient-derived malignant glioma samples tested (Fig. 6B and C), with CD8+ T cells inducing significantly more specific lysis compared with the CD4+ T-cell subset (P = 0.0031 and P = 0.0061 for D10-0319-MG and D2159-MG, respectively), although CD4+ T cells were able to contribute directly to significant amounts of specific lysis (P < 0.0001 and P = 0.0009 for D10-0319-MG and D2159-MG, respectively). These results further highlight the ability of hEGFR-vIII-CD3 bi-scFv to redirect any T cell to achieve tumor-specific lysis, regardless of endogenous T cell specificity or T cell subset, and are in agreement with the results reported by others investigating CD3 engaging bi-scFv therapy (47, 48).
Figure 6.
hEGFRvIII-CD3 bi-scFv redirects both CD4+ and CD8+ T-cell subsets to lyse patient-derived glioma samples. The ability of hEGFRvIII-CD3 bi-scFv to redirect both CD4+ and CD8+ T-cell subsets to independently lyse malignant glioma was assessed among three separate patient-derived glioma samples (D270-MG, D10-0319-MG, and D2159-MG). CD4+ or CD8+ T-cell subsets were isolated from human peripheral blood mononuclear cells (PBMC) and paired independently with patient-derived glioma cells at increasing concentrations of hEGFRvIII-CD3 bi-scFv. A dose-response–based increase in tumor cell lysis was observed in all cases, with both CD4+ and CD8+ T-cell subsets capable of inducing cytotoxic responses. For the D270-MG patient-derived glioma cells, at the maximum dose of hEGFRvIII-CD3 bi-scFv tested (106 pg/mL), CD8+ T cells induced significantly more cytotoxicity compared with CD4+ T cells (P = 0.0123) with the CD8+ T-cell subset inducing 53.52% ± 3.788% specific lysis compared with the CD4+ T-cell subset that induced specific lysis of 31.67% ± 3.327% of tumor cells (A). Similar trends were observed for the D10-0319-MG (B) and D2159-MG (C) patient-derived glioma cells. At a dose of 106 pg/mL of hEGFRvIII-CD3, the CD8+ T-cell subset induced a specific lysis rate of 49.50% ± 3.064% and 55.27% ± 3.138% compared with the CD4+ T-cell subset that induced a specific lysis rate of 27.55% ± 1.555% and 30.56% ± 3.464% when paired against the D10-0319-MG and D2159-MG patient-derived glioma samples, respectively. The CD8+ T-cell subset–specific lysis rate was significantly higher compared with the CD4+ T-cell subset–specific lysis rate for both D10-0319-MG and D2159-MG (P = 0.0031 and P = 0.0061, respectively). In all cases, however, the CD4+ T-cell subset induced significant cytotoxic responses (P = 0.0007; P < 0.0001; and P = 0.0009 for D270-MG, D10-0319-MG, and D2159-MG, respectively).
hEGFRvIII-CD3 bi-scFv requires engagement with both CD3 and EGFRvIII-target antigen to extend survival in vivo
To investigate efficacy in vivo, we tested the activity of hEGFR-vIII-CD3 bi-scFv in four different xenogenic models, including three different orthotropic models, a subcutaneous model, and two patient-derived malignant glioma models (Fig. 7).
Figure 7.

Intravenous administration of hEGFRvIII-CD3 bi-scFv requires engagement with both CD3 and EGFRvIII to extend survival in vivo and cures well-established patient-derived glioma with endogenous drivers, levels, and heterogeneity of EGFRvIII expression in both subcutaneous and orthotopic xenograft models. Antitumor responses produced by hEGFRvIII-CD3 bi-scFv were assessed using three different glioma models and both subcutaneous and orthotopic glioma models. The effect of both EGFRvIII and CD3 target binding on survival was assessed using the U87-MG-EGFRvIII and U87-MG cell lines. Both EGFRvIII and CD3 binding were necessary to induce significant increases in survival. NSG mice (n = 10) were implanted orthotopically with U87-MG-EGFRvIII (A) or U87-MG cells (B). Where indicated, these immunodeficient mice were reconstituted with 1 × 107 intravenously administered T cells one day post tumor implant (arrow) and 50 μg of hEGFRvIII-CD3 bi-scFv, 50 μg of control bi-scFv, or an equivalent volume of vehicle was administered daily by tail vein injection on days 2–6 post tumor implant (bar). hEGFRvIII-CD3 bi-scFv induced a significant increase in survival (P < 0.0001) compared with the other groups, including the group receiving control bi-scFv capable of binding to EGFRvIII-positive tumor cells, but not CD3 (A). Mice challenged with EGFRvIII-negative U87-MG were used to assess the impact of T-cell binding without tumor antigen binding. The difference in the survival among the three groups was assessed and no significant difference was observed (P = 0.5655; B). To assess for tumor burden and survival in mice with well-established patient-derived glioma with endogenous drivers, levels, and heterogeneity of EGFRvIII expression, NSG mice (n = 10) were implanted subcutaneously (C) or orthotopically (D) with patient-derived malignant glioma (D270-MG). The proportion of cells expressing EGFRvIII in this patient-derived tumor sample was 77.8% ± 3.61% (see Supplementary Table S2). Significant antitumor responses were observed despite this heterogeneity in target antigen expression. In the subcutaneous setting, tumors were allowed to establish for 10 days. On day 10 post tumor implant, mice were randomized and where indicated immunodeficient mice were reconstituted with 1 × 107 intravenously administered T cells (arrow). Fifty micrograms of hEGFRvIII-CD3 bi-scFv, 50 μg of control bi-scFv, or an equivalent volume of vehicle was administered daily by tail vein injection on days 11–20 post tumor implant (bar). The difference in tumor growth over time was assessed and a significant difference was observed in the hEGFRvIII-CD3 bi-scFv group compared with all other groups (P < 0.0001), with 9 of 10 mice in the hEGFRvIII-CD3 bi-scFv–treated group having an undetectable tumor burden at the completion of the study (C). In the orthotopic setting, tumors were allowed to establish for 13 days (1/3 median untreated survival). On day 13 post tumor implant, mice were randomized and where indicated immunodeficient mice were reconstituted with 1 × 107 intravenously administered T cells (arrow). Fifty micrograms of hEGFRvIII-CD3 bi-scFv, 50 μg of control bi-scFv, or an equivalent volume of vehicle was administered daily by tail vein injection on days 14–23 post tumor implant (bar). A significant difference in survival (P < 0.0001) was observed in the hEGFRvIII-CD3 bi-scFv–treated group, with 8 of 10 mice still alive at the termination of the study greater than 100 days post tumor implant. A schematic illustrating the sequence of events for these studies is shown in Supplementary Fig. S1.
We first sought to assess for the impact of both CD3 and EGFRvIII-target antigens on the survival advantage provided by hEGFRvIII-CD3 bi-scFv in vivo. Using the U87-MG model, we were able to control for the presence and absence of EGFRvIII-target antigen in an otherwise genetically identical glioma model. We evaluated for a survival advantage following intravenous administration of hEGFRvIII-CD3 bi-scFv, control bi-scFv, or vehicle in mice with orthotopic U87-MG-EGFRvIII or U87-MG (Fig. 7A and B). Immunodeficient NSG mice (n = 10) were implanted orthotopically with U87-MG-EGFRvIII or U87-MG cells. One day post tumor implant, mice were given human T cells (in vitro expanded from human PBMCs) or vehicle intravenously and, on days 2–6, post tumor implant, groups of mice were treated with daily intravenous injections of hEGFRvIII-CD3 bi-scFv, control bi-scFv, or vehicle. A schematic illustrating the timeline of events is shown in Supplementary Fig. S1. This early treatment period was selected to enhance the chance of observing any differences in survival among the hEGFRvIII-CD3 bi-scFv–treated cohort with EGFRvIII-negative tumors. Notably, the highly aggressive U87-MG tumor line also requires a short engraftment period (the 1/3 median U87-MG-EGFRvIII untreated survival time is 4.8 days). For both orthotopic U87-MG-EGFRvIII and U87-MG, we assessed for differences in survival that may occur via administration of T cells alone and found no significant difference between vehicle-treated groups with or without administration of T cells (P = 0.494 and P = 0.464, respectively; Fig. 7A and B). In the U87-MG-EGFRvIII orthotopic model, we assessed for differences between T-cell–replete groups receiving hEGFRvIII-CD3 bi-scFv, control bi-scFv or vehicle. Indeed, hEGFRvIII-CD3 bi-scFv induced a significant increase in survival (P < 0.0001) compared with the other groups, including the group receiving control bi-scFv capable of binding to EGFRvIII-positive tumor cells, but not CD3 (Fig. 7A). In the U87-MG orthotopic model, we assess for differences between groups treated with hEGFRvIII-CD3 bi-scFv or vehicle, allowing for determination of the impact of hEGFRvIII-CD3 bi-scFv binding to T cells without tumor antigen being present. Despite early treatment, the difference in the survival among the three groups was assessed and no significant difference was observed (P = 0.5655; Fig. 7B). Taken together, these data indicate that administration of T cells alone is not sufficient to induce a significant survival advantage and both EGFRvIII- and CD3-binding are necessary to induce significant increases in survival.
hEGFRvIII-CD3 bi-scFv cures well-established patient-derived malignant glioma in vivo
We next sought to assess for efficacy in both subcutaneous and orthotopic models of well-engrafted, patient-derived malignant glioma with endogenous drivers, levels, and heterogeneity of EGFRvIII-expression. NSG mice (n = 10) were implanted subcutaneously or orthotopically with patient-derived malignant glioma (D270-MG). In this patient-derived tumor sample, 77.8% ± 3.6% of cells were positive for EGFRvIII (see Supplementary Table S2). Tumors were allowed to establish (10 days in the subcutaneous setting allowing for an average tumor burden per group >120 mm3 and 13 days or 1/3 median untreated survival time in the orthotopic setting) at which point groups of mice were either reconstituted with human T cells (in vitro expanded from human PBMCs) or administered vehicle. The following day, groups began receiving treatment intravenously with hEGFRvIII-CD3 bi-scFv, control bi-scFv, or vehicle. A schematic illustrating the timeline of events is shown in Supplementary Fig. S1. In the subcutaneous setting, the difference in tumor growth over time was assessed and a significant difference was observed in the hEGFRvIII-CD3 bi-scFv group compared with all other groups (P < 0.0001; Fig. 7C). The mean tumor volume prior to the initiation of treatment was similar for each of the four groups, allowing for differences in the pattern of change in tumor volume among the four groups to be assessed from the initiation of treatment onward. As the data show a linear trend for the time interval observed, a linear trend model with an interaction between time and group was fit to the raw tumor volume values. This then allowed us to estimate the slopes for each group from the model with P values testing whether each slope value is different from 0. Indeed, groups receiving vehicle (no T cells), vehicle (T cells), and control bi-scFv (T cells) had an increase in tumor volume over time, with slopes of 125.38, 122.62, and 140.48, respectively. The tumor volume in the hEGFRvIII-CD3 bi-scFv–receiving group, however, decreased—data showing a negative slope (−11.74). At the termination of the experiment, 9 out of 10 mice in the hEGFRvIII-CD3 bi-scFv–treated group had an undetectable tumor burden that could not be palpated or measured with a caliper. In the orthotopic setting, survival was assessed over time and a significant difference in survival was observed in the hEGFRvIII-CD3 bi-scFv–treated group compared with all other groups (P < 0.0001), with 8 of 10 mice still alive at the termination of the study greater than 100 days post tumor implant (Fig. 7D). These data demonstrate that despite heterogeneity of EGFRvIII expression, hEGFRvIII-CD3 bi-scFv is capable of effectively treating both subcutaneous and orthotopic patient-derived malignant glioma.
When examined histologically following 5 days of treatment with either hEGFRvIII-EGFRvIII bi-scFv or control bi-scFv, we found significant D270-MG necrosis and T-cell infiltration in the hEGFRvIII-CD3 bi-scFv–treated cohorts, while a dense tumor xenograft with a lobular growth pattern and no obvious T cells in the tumor parenchyma was found in the control bi-scFv–treated cohorts. Representative histopathology is shown in Fig. 8. Taken together, these data indicate that intravenous administration of hEGFRvIII-CD3 bi-scFv is capable of mediating significant levels of T-cell infiltration to the tumor parenchyma, tumor cell necrosis, and regression of tumor burden resulting in an increase in overall survival in mice with well-engrafted, patient-derived malignant glioma.
Figure 8.

Treatment with hEGFRvIII-CD3 bi-scFv results in tumor cell necrosis and a dense T-cell infiltrate to the tumor parenchyma. The ability of hEGFRvIII-CD3 bi-scFv to produce tumor cell necrosis and induce T-cell infiltration to the tumor parenchyma was assessed using hematoxylin and eosin (H&E) staining and IHC for human CD3. Cohorts of NSG mice (n = 10) were implanted with patient-derived malignant glioma (D270-MG) that was allowed to establish for 10 days. Following this tumor engraftment period, all mice were reconstituted with 1 × 107 intravenously administered human T cells and then randomized to either treatment or control groups. Fifty micrograms of hEGFRvIII-CD3 bi-scFv or 50 μg of control bi-scFv was administered daily by tail vein injection on days 11–15. Following 5 days of treatment with either hEGFRvIII-CD3 bi-scFv or control bi-scFv, mice were humanely sacrificed, allowing tumors to be dissected, formalin fixed, and paraffin embedded. A representative H&E-stained section (10× magnification) from the hEGFRvIII-CD3 bi-scFv receiving cohort demonstrates tumor cell necrosis (A). A corresponding serial section stained with human CD3 IHC (10× magnification) shows a dense infiltrate of T cells in the tumor parenchyma (B). On the other hand, a representative H&E section (10× magnification) from the control bi-scFv–treated cohort, shows a dense tumor xenograft with a lobular growth pattern (C) and no obvious T cells in the tumor parenchyma when a corresponding serial section is stained with human CD3 IHC (10× magnification; D).
Intravenously administered hEGFRvIII-CD3 bi-scFv cures well-established, highly invasive, syngeneic malignant glioma
We next sought to evaluate the ability of hEGFRvIII-CD3 bi-scFv to treat highly invasive and aggressive syngeneic malignant glioma. Given that the CD3-binding portion of hEGFRvIII-CD3 bi-scFv binds only to human CD3 (49), a human CD3 transgenic mouse model (tgε600) with pharmacologically responsive, physiologically distributed human CD3 receptors (39, 40, 50) was used to assess for antitumor responses. This model provides an endogenous immune system that is pharmacologically responsive to the anti-human bi-scFv construct to be used in the clinic, drastically increasing the validity and clinical translatability of these preclinical studies. When heterozygous in a C57BL/6 background, these human CD3 transgenic mice possess low levels of peripheral T cells that respond to stimulation through both murine and human CD3 (40, 50).
The highly aggressive and infiltrative chemically induced murine glioma model CT-2A is syngeneic in the human CD3 transgenic C57/Bl6 background (51, 52). This tumor infiltrates throughout the brain and recapitulates the invasive nature of human glioblastoma (Fig. 9A). We transfected the CT-2A tumor line to stably express a murine homolog of EGFRvIII, providing a target for hEGFRvIII-CD3 bi-scFv that is antigenically indistinct from the human EGFRvIII.
Figure 9.

Intravenously administered hEGFRvIII-CD3 bi-scFv accumulates in highly invasive, syngeneic, orthotopic malignant glioma, curing well-established tumors. A representative H&E section (10× magnification; A) shows invasive CT-2A-EGFRvIII cells 6 days post orthotopic implantation, at which point mice have reached one-third the median survival time. To assess for tumor burden and survival in this model, heterozygous human CD3 transgenic mice (n = 10) were implanted subcutaneously (B) or orthotopically (C) with CT-2A-EGFRvIII glioma. In the subcutaneous setting, tumors were allowed to establish for 10 days. Mice with palpable tumor burden were then reconstituted with 1 × 107 intravenously administered autologous human CD3 transgenic T cells (arrow), given that these transgenic mice have diminished levels of T cells due to toxicity of the human CD3 transgene, and randomized to treatment groups. Fifty micrograms of hEGFRvIII-CD3 bi-scFv, 50 μg of control bi-scFv or, an equivalent volume of vehicle was administered daily by tail vein injection on days 11–20 post tumor implant (bar). The difference in tumor growth over time was assessed and a significant difference was observed in the hEGFRvIII-CD3 bi-scFv treated group compared with all other groups (P < 0.0001; B). In the orthotopic setting, tumors were allowed to establish for 6 days (1/3 median untreated survival). On day 6 post tumor implant, mice were reconstituted with 1 × 107 intravenously administered human CD3 transgenic T cells (arrow) and randomized to treatment groups. Fifty micrograms of hEGFRvIII-CD3 bi-scFv, 50 μg of control bi-scFv or, an equivalent volume of vehicle was administered daily by tail vein injection on days 7–16 post tumor implant (bar). A significant difference in survival (P < 0.0001) was observed in the hEGFRvIII-CD3 bi-scFv–treated group with, 8 of 10 mice still alive at the termination of the study greater than 70 days post tumor implant (C). Biodistribution studies were performed to quantify the extent to which intravenously administered hEGFRvIII-CD3 bi-scFv accumulates within invasive tumors within the brain. Human CD3 transgenic mice (n = 5) bearing 12-day established EGFRvIII-positive CT-2A glioma were injected with 100 μCi of I-124–labeled hEGFRvIII-CD3 bi-scFv. Micro-PET/CT imaging was performed three hours post intravenous injection, where 1.3% ± 0.07% of the injected dose per gram of tissue was found to be localized to the region of the tumor within the brain (D). A representative PET/CT image is shown in E demonstrating a region of significantly increased radioactive signal over background localized to coordinates within the brain where tumors were stereotactically injected. At the termination of the study, mice were humanely sacrificed, allowing for CNS tissue to be cryopreserved and microsectioned. Sections of CNS tissue were then subjected to autoradiography, allowing for further confirmation of the presence of radioactivity within the brain. A representative autoradiography image is shown in F, with regions of increased radioactivity uptake that corresponded to regions of grossly visible infiltrative tumor.
We used this pharmacologically responsive syngeneic model of highly invasive and aggressive malignant glioma to assess for tumor burden and survival following hEGFRvIII-CD3 bi-scFv therapy. To assess for efficacy in the subcutaneous setting, groups of heterozygous human CD3 transgenic mice (n = 10) were implanted subcutaneously with CT-2A-EGFRvIII cells. Tumors were allowed to establish for 10 days, at which point mice were reconstituted with autologous human CD3 transgenic T cells and randomized to treatment groups. The following day, groups began receiving daily intravenous treatments with either hEGFRvIII-CD3 bi-scFv, control bi-scFv, or an equal volume of vehicle. The difference in tumor growth over time was assessed and a significant difference was observed in the hEGFRvIII-CD3 bi-scFv–treated group compared with all other groups (P < 0.0001), with 8 of 10 mice in the hEGFRvIII-CD3 bi-scFv–treated group having no detectable tumor burden at the end of the study, while mice from all other groups had reached humane endpoints (Fig. 9B). In the orthotopic setting, CT-2A-EGFRvIII tumors were implanted in heterozygous human CD3 transgenic mice and allowed to establish for 6 days (1/3 the median untreated survival time), at which point mice were reconstituted with autologous human CD3 transgenic T cells and randomized to treatment groups. The following day, mice began receiving daily intravenous injections with either hEGFRvIII-CD3 bi-scFv, control bi-scFv, or an equal volume of vehicle for a period of 10 days. Survival was assessed over time and a significant difference in survival was observed in the hEGFRvIII-CD3 bi-scFv–treated group compared with all other groups (P < 0.0001), with 8 of 10 mice still alive at the termination of the study greater than 70 days post tumor implant (Fig. 9C). These data demonstrate the hEGFRvIII-CD3 bi-scFv is able to mediate significant efficacy following intravenous administration in both subcutaneous and orthotopic models of highly aggressive, invasive, syngeneic glioma.
Radiolabeled hEGFRvIII-CD3 bi-scFv accumulates in highly invasive, syngeneic, orthotopic malignant glioma following intravenous administration
To quantify the extent to which intravenously administered hEGFRvIII-CD3 bi-scFv accumulates within highly infiltrative tumors within the brain, we performed biodistribution studies using I-124–labeled hEGFRvIII-CD3 bi-scFv. PET/CT imaging and autoradiography were used to detect antibody accumulation within tumors in the brain. Heterozygous human CD3 transgenic mice (n = 5) were implanted orthotopically with CT-2A-EGFRvIII glioma. Tumors were allowed to establish for 12 days (cohorts of orthotopic CT-2A-EGFRvIII bearing heterozygous human CD3 transgenic mice received intravenous hEGFRvIII-CD3 bi-scFv injections at the same time point in separate efficacy studies) at which point mice were administered 100 μCi of intravenous I-124–labeled hEGFRvIII-CD3 bi-scFv. Micro PET/CT imaging was then performed three hours post intravenous injection. Indeed, in all cases, radioactive signal was found to localize to the right cerebral quadrant in the location where tumor cells were stereo-tactically injected, with 1.3% ± 0.07% of the injected dose per gram of tissue (%ID/g) localizing to tumors within the brain (Fig. 9D). Significant levels of radioactivity localized to ROIs where intracerebral tumor growth was expected, with a representative PET/CT image shown in Fig. 7E. To further confirm these findings, we then humanely sacrificed animals, allowing for CNS tissue to be cryopreserved, microsectioned, and subject to autoradiography. In all cases, areas of increased radioactivity were detected in the right cerebral hemisphere where tumors were grossly visible. A representative autoradiography figure demonstrating increased radioactive counts in regions of infiltrative glioma is shown in Fig. 9E.
Discussion
The application of bi-scFv therapy for the treatment of cancer has been successful, marked by the recent FDA approval of blinatumomab. The extension of this therapy for the treatment of other forms of cancer, including solid tumors, however, has yet to be realized. The lack of tumor-specific targets has resulted in targeting of healthy cells and tissue and barriers related to the effective production and scale-up of clinical-grade, therapeutic bi-scFvs has hampered progress. Here, we report the rational design, development and analysis of a fully human, anti-human bispecific antibody suitable for clinical translation and the safe and effective treatment of GBM.
To our knowledge, this is the first report of the rational, systematic development of a bi-scFv. We have implemented a three-step, systematic approach where we have progressively assessed for the effect of different arrangements of variable segments, linkers sequences, and signal peptides. Through this process, we have eliminated unfavorable constructs early in the development process and obtained a target-specific bi-scFv with favorable antigen-binding kinetics and expression and purification characteristics. We believe that this systematic approach can be extended to the development of other bi-scFvs to identify promising candidates early in the development process and can lead to overcoming barriers that have historically limited clinical translatability.
We tested our optimized construct, hEGFRvIII-CD3 bi-scFv, and demonstrated robust T-cell responses including upregulation of activation markers, secretion of proinflammatory cytokines, and proliferation. Importantly, these T-cell–mediated effects occur in an EGFRvIII antigen–restricted fashion. We have also tested for cytotoxicity against multiple malignant glioma cell lines, controlling for EGFRvIII expression and finding that cytotoxicity is restricted to cell populations where the EGFRvIII antigen is present, critical to the precise targeting and elimination of cancer cells for safe and effective anticancer immunotherapy.
Still we have found that in patient-derived samples with heterogeneous EGFRvIII expression patterns, we are able to induce specific lysis and effectively treat both subcutaneous and orthotopic xenografts. Validation of cytotoxicity and efficacy against these patient-derived malignant glioma samples with endogenous drivers, levels, and heterogeneity of target antigen expression is critical to gaining an understanding of potential effectiveness in the clinic and overall has enhanced rationale for continued investment in clinical translation.
In those cases where we have effectively eradicated patient-derived xenografts in vivo despite heterogeneity of target antigen expression, it may be that we have induced lysis of only EGFRvIII-positive tumor cells and that this may have been sufficient to obtain these results in the context of our preclinical in vitro and in vivo studies. Indeed, EGFRvIII is known to enhance tumor cell growth and invasion while conferring radiation (17) and chemo-therapeutic (18, 19) resistance. Among patients with malignant glioma, expression of EGFRvIII is an independent negative prognostic indicator (53). EGFRvIII also enhances the growth of neighboring EGFRvIII-negative tumor cells via cytokine-mediated paracrine signaling (54) and by transferring a functionally active oncogenic EGFRvIII receptor to EGFRvIII-negative cells through the release of lipid-raft–related microvesicles (55). Recent research has also found that EGFRvIII is expressed in glioma stem cells (GSC; ref. 56), an important consideration given the paradigm that tumor stem cells represent a subpopulation of cells that give rise to all cells in a differentiated tumor (57). Alternatively, hEGFRvIII-CD3 bi-scFv may be capable of mounting an immune response against cells that lack the target antigen. Indeed, others have already demonstrated this phenomenon with other CD3 engaging bi-scFv therapies and have attributed this to an activation and expansion of existing T-cell clones that are reactive toward tumor antigens other than that initially targeted (22). Another potential mechanism by which this can occur is through the induction of secondary immune responses following the release of cryptic tumor antigens. We look forward to future studies by our group and others where these important principles will be further investigated.
We also tested our antibody in vivo in a highly invasive and infiltrative, orthotopic, syngeneic model, finding that intravenously administered hEGFRvIII-CD3 bi-scFv accumulates within the tumor to significant levels and mediates specific and effective antitumor responses, curing even well-established orthotopic tumors. These findings are encouraging and suggest that straightforward intravenous administration of hEGFRvIII-CD3 bi-scFv may be an effective route of administration in the clinic. Indeed, future clinical studies will be necessary to further explore this route of administration and potential mechanisms by which biodistribution to the tumor can be enhanced.
Our method is also agnostic to endogenous T-cell specificity and potentially capable of redirecting any T cell. This makes a given patient's entire repertoire of T cells available for redirection. This concept is demonstrated in our studies examining both CD4+ and CD8+ T cells independently. Indeed, hEGFRvIII-CD3 bi-scFv is capable of inducing both CD4+ and CD8+ T-cell subsets to upregulate activation markers, secrete proinflammatory cytokines, proliferate, and lyse malignant glioma cells in a target–antigen–restricted fashion. We and others have demonstrated that this can result in subversion of even typically suppressive Tregs for anticancer immunotherapy (26) and expansion of preexisting T-cell clones that can have secondary specificity and reactivity toward tumor antigens other than those initially targeted (22). This method, therefore, in addition to entirely circumventing the need for ex vivo T-cell expansion and viral transduction, overcomes issues related to the limited T-cell repertoire and T-cell exhaustion that has been associated with and can limit the effectiveness of engineered adoptive T-cell therapy.
We have produced our construct from fully human antibodies. To our knowledge, this is the first report of a fully human, anti-human bi-scFv. The use of fully human antibody segments has the benefit of avoiding drug immunogenicity and potential HAMA responses that can lead to rapid drug clearance and other adverse reactions. The generation of a therapeutic, fully human bi-scFv antibody represents a significant advance.
To facilitate anticipated clinical studies, we have produced the bi-scFv antibody in a fashion suitable for clinical translational and compatible with clinical biologic manufacturing infrastructure. This has included generating and certifying a MCB and developing a scalable expression and tag-free purification and formulation process suitable for clinical translation. We believe that the assessment and consideration of such factors early in the development process is critical to successful translation of bi-scFv therapy. On the basis of our data and development work reported here, we have produced clinical grade hEGFRvIII-CD3 bi-scFv protein at Duke University Medical Center's Molecular Products and Cellular Therapies (MPACT) clinical manufacturing suite and are currently pursuing Investigational New Drug (IND) enabling studies that will allow us to initiate a phase I clinical trial to treat patients with EGFRvIII-positive GBM.
The primary goal of this study was to rationally develop and evaluate a fully human, anti-human bi-scFv to redirect patients' own T cells for safe and effective anticancer responses. Through further investigation in clinical studies, we will determine whether the therapeutic benefits observed here can be replicated in patients. If so, we believe this therapeutic approach has significant potential to enhance the standard of care for patients with GBM and other cancers.
Supplementary Material
Translational Relevance.
We have developed a clinically translatable method to specifically target malignant glioma using a tumor-specific, fully human bispecific antibody that redirects patients' own T cells to recognize and destroy tumors. Our work highlights the antigen specificity of our approach, critical to precisely eliminating cancer without the risk of toxicity and collateral damage to healthy cells and tissue. We have demonstrated robust, antitumor immune responses capable of curing well-established, patient-derived malignant glioma that heterogeneously expresses the target antigen. This translatable, off-the-shelf, fully human therapeutic is produced in a fashion compatible with existing clinical antibody manufacturing infrastructure and has significant potential to improve public health and quality of life for patients affected by malignant glioma and other cancers.
Acknowledgments
We would like to acknowledge each of the patients we've encountered who offer an endless source of inspiration; our family and friends for their continuous support; and Mark W. Dewhirst, D.V.M., Ph.D., Charles A. Gersbach, Ph.D., Kent J. Weinhold, Ph.D. and Michael R. Zalutsky, Ph.D., M.A. for their generous support and advice. This work was supported by funding from the NIH: F30CA196199 (to P.C. Gedeon), R01NS08541 (to J.H. Sampson), U01NS090284 (to J.H. Sampson), R01CA177476 (to J.H. Sampson), R01NS086943 (to J.H. Sampson), and R01NS099463 (to J.H. Sampson).
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
P.C. Gedeon, B.D. Choi, C.-T. Kuan, D.D. Bigner, and J.H. Sampson are listed as coinventors of patents regarding the use of fully human bispecific antibodies targeting EGFRvlll which belong to Duke University. J. H. Sampson reports receiving commercial research grants from Annias and Istari; holds ownership interest (including patents) in Annias, Neuronium, Duke University, and Istari; is a consultant/advisory board member for Bristol Myers Squibb, Medicenna, Insera Health, and Annias. No potential conflicts of interest were disclosed by the other authors.
Disclaimer
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Authors' Contributions
Conception and design: P.C. Gedeon, B.D. Choi, C.-T. Kuan, G.E. Archer, J.H. Sampson
Development of methodology: P.C. Gedeon, T.H. Schaller, S.K. Chitneni, C.-T. Kuan, G.E. Archer
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): P.C. Gedeon, T.H. Schaller, S.K. Chitneni, C.M. Suryadevara, D.J. Snyder, R.J. Schmittling, S.E. Szafranski, R.E. McLendon, S.T. Keir, L. Sanchez-Perez
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): P.C. Gedeon, T.H. Schaller, S.K. Chitneni, C.M. Suryadevara, R.J. Schmittling, S.E. Szafranski, P. Healy, J.E. Herndon, R.E. McLendon, E. Reap, L. Sanchez-Perez, J.H. Sampson
Writing, review, and/or revision of the manuscript: P.C. Gedeon, B.D. Choi, C.-T. Kuan, C.M. Suryadevara, S.E. Szafranski, P. Healy, J.E. Herndon, R.E. McLendon, S.T. Keir, E. Reap, L. Sanchez-Perez, J.H. Sampson
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): P.C. Gedeon, D.J. Snyder, S.E. Szafranski, X. Cui Study supervision: P.C. Gedeon, D.D. Bigner, J.H. Sampson
References
- 1.Imperato JP, Paleologos NA, Vick NA. Effects of treatment on long-term survivors with malignant astrocytomas. Ann Neurol. 1990;28:818–22. doi: 10.1002/ana.410280614. [DOI] [PubMed] [Google Scholar]
- 2.Brenner MK, Heslop HE. Adoptive T cell therapy of cancer. Curr Opin Immunol. 2010;22:251–7. doi: 10.1016/j.coi.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenberg SA. Cell transfer immunotherapy for metastatic solid cancer–what clinicians need to know. Nat Rev Clin Oncol. 2011;8:577–85. doi: 10.1038/nrclinonc.2011.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–46. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29:917–24. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hong JJ, Rosenberg SA, Dudley ME, Yang JC, White DE, Butman JA, et al. Successful treatment of melanoma brain metastases with adoptive cell therapy. Clin Cancer Res. 2010;16:4892–8. doi: 10.1158/1078-0432.CCR-10-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sampson JH, Choi BD, Sanchez-Perez L, Suryadevara CM, Snyder DJ, Flores CT, et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin Cancer Res. 2014;20:972–84. doi: 10.1158/1078-0432.CCR-13-0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi BD, Suryadevara CM, Gedeon PC, Herndon JE, 2nd, Sanchez-Perez L, Bigner DD, et al. Intracerebral delivery of a third generation EGFRvIII-specific chimeric antigen receptor is efficacious against human glioma. J Clin Neurosci. 2014;21:189–90. doi: 10.1016/j.jocn.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–4. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–9. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bargou R, Leo E, Zugmaier G, Klinger M, Goebeler M, Knop S, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science. 2008;321:974–7. doi: 10.1126/science.1158545. [DOI] [PubMed] [Google Scholar]
- 12.Amann M, Brischwein K, Lutterbuese P, Parr L, Petersen L, Lorenczewski G, et al. Therapeutic window of MuS110, a single-chain antibody construct bispecific for murine EpCAM and murine CD3. Cancer Res. 2008;68:143–51. doi: 10.1158/0008-5472.CAN-07-2182. [DOI] [PubMed] [Google Scholar]
- 13.Lutterbuese R, Raum T, Kischel R, Hoffmann P, Mangold S, Rattel B, et al. T cell-engaging BiTE antibodies specific for EGFR potently eliminate KRAS-and BRAF-mutated colorectal cancer cells. Proc Natl Acad Sci U S A. 2010;107:12605–10. doi: 10.1073/pnas.1000976107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Przepiorka D, Ko CW, Deisseroth A, Yancey CL, Candau-Chacon R, Chiu HJ, et al. FDA approval: blinatumomab. Clin Cancer Res. 2015;21:4035–9. doi: 10.1158/1078-0432.CCR-15-0612. [DOI] [PubMed] [Google Scholar]
- 15.Boockvar JA, Kapitonov D, Kapoor G, Schouten J, Counelis GJ, Bogler O, et al. Constitutive EGFR signaling confers a motile phenotype to neural stem cells. Mol Cell Neurosci. 2003;24:1116–30. doi: 10.1016/j.mcn.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 16.Pedersen MW, Tkach V, Pedersen N, Berezin V, Poulsen HS. Expression of a naturally occurring constitutively active variant of the epidermal growth factor receptor in mouse fibroblasts increases motility. Int J Cancer. 2004;108:643–53. doi: 10.1002/ijc.11566. [DOI] [PubMed] [Google Scholar]
- 17.Lammering G, Hewit TH, Holmes M, Valerie K, Hawkins W, Lin PS, et al. Inhibition of the type III epidermal growth factor receptor variant mutant receptor by dominant-negative EGFR-CD533 enhances malignant glioma cell radiosensitivity. Clin Cancer Res. 2004;10:6732–43. doi: 10.1158/1078-0432.CCR-04-0393. [DOI] [PubMed] [Google Scholar]
- 18.Montgomery RB, Guzman J, O'Rourke DM, Stahl WL. Expression of oncogenic epidermal growth factor receptor family kinases induces paclitaxel resistance and alters beta-tubulin isotype expression. J Biol Chem. 2000;275:17358–63. doi: 10.1074/jbc.M000966200. [DOI] [PubMed] [Google Scholar]
- 19.Nagane M, Narita Y, Mishima K, Levitzki A, Burgess AW, Cavenee WK, et al. Human glioblastoma xenografts overexpressing a tumor-specific mutant epidermal growth factor receptor sensitized to cisplatin by the AG1478 tyrosine kinase inhibitor. J Neurosurg. 2001;95:472–9. doi: 10.3171/jns.2001.95.3.0472. [DOI] [PubMed] [Google Scholar]
- 20.Moscatello DK, Holgado-Madruga M, Godwin AK, Ramirez G, Gunn G, Zoltick PW, et al. Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Res. 1995;55:5536–9. [PubMed] [Google Scholar]
- 21.Wikstrand CJ, Hale LP, Batra SK, Hill ML, Humphrey PA, Kurpad SN, et al. Monoclonal antibodies against EGFRvIII are tumor specific and react with breast and lung carcinomas and malignant gliomas. Cancer Res. 1995;55:3140–8. [PubMed] [Google Scholar]
- 22.Dao T, Pankov D, Scott A, Korontsvit T, Zakhaleva V, Xu Y, et al. Therapeutic bispecific T-cell engager antibody targeting the intracellular oncoprotein WT1. Nat Biotechnol. 2015;33:1079–86. doi: 10.1038/nbt.3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28:4722–9. doi: 10.1200/JCO.2010.28.6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gedeon PC, Choi BD, Sampson JH, Bigner DD. RINDOPEPIMUT Anti-EGFRvIII peptide vaccine oncolytic. Drug Future. 2013;38:147–55. doi: 10.1358/dof.2013.038.03.1933992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi BD, Kuan CT, Cai M, Archer GE, Mitchell DA, Gedeon PC, et al. Systemic administration of a bispecific antibody targeting EGFRvIII successfully treats intracerebral glioma. Proc Natl Acad Sci U S A. 2013;110:270–5. doi: 10.1073/pnas.1219817110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi BD, Gedeon PC, Herndon JE, II, Archer GE, Reap EA, Sanchez-Perez L, et al. Human regulatory T cells kill tumor cells through granzyme-dependent cytotoxicity upon retargeting with a bispecific antibody. Cancer Immunol Res. 2013;1:163–7. doi: 10.1158/2326-6066.CIR-13-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carter PJ. Potent antibody therapeutics by design. Nat Rev Immunol. 2006;6:343–57. doi: 10.1038/nri1837. [DOI] [PubMed] [Google Scholar]
- 28.Hansel TT, Kropshofer H, Singer T, Mitchell JA, George AJ. The safety and side effects of monoclonal antibodies. Nat Rev Drug Discov. 2010;9:325–38. doi: 10.1038/nrd3003. [DOI] [PubMed] [Google Scholar]
- 29.Gaston RS, Deierhoi MH, Patterson T, Prasthofer E, Julian BA, Barber WH, et al. OKT3 first-dose reaction: association with T cell subsets and cytokine release. Kidney Int. 1991;39:141–8. doi: 10.1038/ki.1991.18. [DOI] [PubMed] [Google Scholar]
- 30.Reynolds JC, Del Vecchio S, Sakahara H, Lora ME, Carrasquillo JA, Neumann RD, et al. Anti-murine antibody response to mouse monoclonal antibodies: clinical findings and implications. International journal of radiation applications and instrumentation Part B, Nucl Med Biol. 1989;16:121–5. doi: 10.1016/0883-2897(89)90182-7. [DOI] [PubMed] [Google Scholar]
- 31.Kuus-Reichel K, Grauer LS, Karavodin LM, Knott C, Krusemeier M, Kay NE. Will immunogenicity limit the use, efficacy, and future development of therapeutic monoclonal antibodies? Clin Diagn Lab Immunol. 1994;1:365–72. doi: 10.1128/cdli.1.4.365-372.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mascelli MA, Zhou H, Sweet R, Getsy J, Davis HM, Graham M, et al. Molecular, biologic, and pharmacokinetic properties of monoclonal antibodies: impact of these parameters on early clinical development. J Clin Pharmacol. 2007;47:553–65. doi: 10.1177/0091270006298360. [DOI] [PubMed] [Google Scholar]
- 33.Kipriyanov SM, Moldenhauer G, Braunagel M, Reusch U, Cochlovius B, Le Gall F, et al. Effect of domain order on the activity of bacterially produced bispecific single-chain Fv antibodies. J Mol Biol. 2003;330:99–111. doi: 10.1016/s0022-2836(03)00526-6. [DOI] [PubMed] [Google Scholar]
- 34.Mack M, Riethmuller G, Kufer P. A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc Natl Acad Sci U S A. 1995;92:7021–5. doi: 10.1073/pnas.92.15.7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jungbluth AA, Stockert E, Huang HJ, Collins VP, Coplan K, Iversen K, et al. A monoclonal antibody recognizing human cancers with amplification/ overexpression of the human epidermal growth factor receptor. Proc Natl Acad Sci U S A. 2003;100:639–44. doi: 10.1073/pnas.232686499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patel D, Lahiji A, Patel S, Franklin M, Jimenez X, Hicklin DJ, et al. Monoclonal antibody cetuximab binds to and down-regulates constitutively activated epidermal growth factor receptor vIII on the cell surface. Anticancer Res. 2007;27:3355–66. [PubMed] [Google Scholar]
- 37.Reist CJ, Batra SK, Pegram CN, Bigner DD, Zalutsky MR. In vitro and in vivo behavior of radiolabeled chimeric anti-EGFRvIII monoclonal antibody: comparison with its murine parent. Nucl Med Biol. 1997;24:639–47. doi: 10.1016/s0969-8051(97)00080-2. [DOI] [PubMed] [Google Scholar]
- 38.Allen M, Bjerke M, Edlund H, Nelander S, Westermark B. Origin of the U87MG glioma cell line: Good news and bad news. Sci Transl Med. 2016;8:354re3. doi: 10.1126/scitranslmed.aaf6853. [DOI] [PubMed] [Google Scholar]
- 39.Wang B, Biron C, She J, Higgins K, Sunshine MJ, Lacy E, et al. A block in both early T lymphocyte and natural killer cell development in transgenic mice with high-copy numbers of the human CD3E gene. Proc Natl Acad Sci U S A. 1994;91:9402–6. doi: 10.1073/pnas.91.20.9402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang N, Wang B, Salio M, Allen D, She J, Terhorst C. Expression of a CD3 epsilon transgene in CD3 epsilon(null) mice does not restore CD3 gamma and delta expression but efficiently rescues T cell development from a subpopulation of prothymocytes. Int Immunol. 1998;10:1777–88. doi: 10.1093/intimm/10.12.1777. [DOI] [PubMed] [Google Scholar]
- 41.Celen S, Koole M, De Angelis M, Sannen I, Chitneni SK, Alcazar J, et al. Preclinical evaluation of 18F-JNJ41510417 as a radioligand for PET imaging of phosphodiesterase-10A in the brain. J Nucl Med. 2010;51:1584–91. doi: 10.2967/jnumed.110.077040. [DOI] [PubMed] [Google Scholar]
- 42.Whitlow M, Bell BA, Feng SL, Filpula D, Hardman KD, Hubert SL, et al. An improved linker for single-chain Fv with reduced aggregation and enhanced proteolytic stability. Protein Eng. 1993;6:989–95. doi: 10.1093/protein/6.8.989. [DOI] [PubMed] [Google Scholar]
- 43.van der Woude CJ, Stokkers P, van Bodegraven AA, Van Assche G, Hebzda Z, Paradowski L, et al. Phase I, double-blind, randomized, placebo-controlled, dose-escalation study of NI-0401 (a fully human anti-CD3 mono-clonal antibody) in patients with moderate to severe active Crohn's disease. Inflamm Bowel Dis. 2010;16:1708–16. doi: 10.1002/ibd.21252. [DOI] [PubMed] [Google Scholar]
- 44.Dean Y, Depis F, Kosco-Vilbois M. Combination therapies in the context of anti-CD3 antibodies for the treatment of autoimmune diseases. Swiss Medical Weekly. 2012;142:w13711. doi: 10.4414/smw.2012.13711. [DOI] [PubMed] [Google Scholar]
- 45.Weber R, inventor. Amgen Fremont Inc, assignee. Antibodies directed to the deletion mutants of epidermal growth factor receptor and uses thereof. 2005012479A2 United States patent WO.
- 46.Kober L, Zehe C, Bode J. Optimized signal peptides for the development of high expressing CHO cell lines. Biotechnol Bioeng. 2013;110:1164–73. doi: 10.1002/bit.24776. [DOI] [PubMed] [Google Scholar]
- 47.Mack M, Gruber R, Schmidt S, Riethmuller G, Kufer P. Biologic properties of a bispecific single-chain antibody directed against 17–1A (EpCAM) and CD3: tumor cell-dependent T cell stimulation and cytotoxic activity. J Immunol. 1997;158:3965–70. [PubMed] [Google Scholar]
- 48.Brischwein K, Schlereth B, Guller B, Steiger C, Wolf A, Lutterbuese R, et al. MT110: a novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors. Mol Immunol. 2006;43:1129–43. doi: 10.1016/j.molimm.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 49.Elson G, inventor; Novimmune SA, assignee. Anti-CD3 antibody formulations. 20070065437A1. United States patent US. 2007 Mar 22;
- 50.Weetall M, Digan ME, Hugo R, Mathew S, Hopf C, Tart-Risher N, et al. T-cell depletion and graft survival induced by anti-human CD3 immunotoxins in human CD3epsilon transgenic mice. Transplantation. 2002;73:1658–66. doi: 10.1097/00007890-200205270-00023. [DOI] [PubMed] [Google Scholar]
- 51.Ranes MK, El-Abbadi M, Manfredi MG, Mukherjee P, Platt FM, Seyfried TN. N -butyldeoxynojirimycin reduces growth and ganglioside content of experimental mouse brain tumours. Br J Cancer. 2001;84:1107–14. doi: 10.1054/bjoc.2000.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ecsedy JA, Manfredi MG, Yohe HC, Seyfried TN. Ganglioside biosynthetic gene expression in experimental mouse brain tumors. Cancer Res. 1997;57:1580–3. [PubMed] [Google Scholar]
- 53.Heimberger AB, Hlatky R, Suki D, Yang D, Weinberg J, Gilbert M, et al. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin Cancer Res. 2005;11:1462–6. doi: 10.1158/1078-0432.CCR-04-1737. [DOI] [PubMed] [Google Scholar]
- 54.Inda MM, Bonavia R, Mukasa A, Narita Y, Sah DW, Vandenberg S, et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev. 2010;24:1731–45. doi: 10.1101/gad.1890510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, et al. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol. 2008;10:619–24. doi: 10.1038/ncb1725. [DOI] [PubMed] [Google Scholar]
- 56.Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther. 2012;23:1043–53. doi: 10.1089/hum.2012.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dirks PB. Cancer: stem cells and brain tumours. Nature. 2006;444:687–8. doi: 10.1038/444687a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.