

Abstract
Stimulation of the cytosolic NAD+ dependent deacetylase SIRT1 is cardioprotective against ischemia-reperfusion (IR) injury. NAD+ precursors including nicotinamide mononucleotide (NMN) are thought to induce cardioprotection via SIRT1. Herein, while NMN protected perfused hearts against IR (functional recovery: NMN 42±7% vs. vehicle 11±3%), this protection was insensitive to the SIRT1 inhibitor splitomicin (recovery 47±8%). Although NMN-induced cardioprotection was absent in Sirt3−/− hearts (recovery 9±5%), this was likely due to enhanced baseline injury in Sirt3−/− (recovery 6±2%), since similar injury levels in WT hearts also blunted the protective efficacy of NMN. Considering alternative cardiac effects of NMN, and the requirement of glycolysis for NAD+, we hypothesized NMN may confer protection in part via direct stimulation of cardiac glycolysis. In primary cardiomyocytes, NMN induced cytosolic and extracellular acidification and elevated lactate. In addition, [U-13C]glucose tracing in intact hearts revealed that NMN stimulated glycolytic flux. Consistent with a role for glycolysis in NMN-induced protection, hearts perfused without glucose (palmitate as fuel source), or hearts perfused with galactose (no ATP from glycolysis) exhibited no benefit from NMN (recovery 11±4% and 15±2% respectively). Acidosis during early reperfusion is known to be cardioprotective (i.e., acid post-conditioning), and we also found that NMN was cardioprotective when delivered acutely at reperfusion (recovery 39±8%). This effect of NMN was not additive with acidosis, suggesting overlapping mechanisms. We conclude that the acute cardioprotective benefits of NMN are mediated in part via glycolytic stimulation, with the downstream protective mechanism involving enhanced ATP synthesis during ischemia and/or enhanced acidosis during reperfusion.
Keywords: Lactate, Acidosis, Ischemia, NAD+, Glycolysis, NMN
1. Introduction
The SIRT family of NAD+ dependent lysine deacylases are important regulators of metabolic health [1,2]. SIRT activity is known to decline with age [3], and as such a number of NAD+ biosynthetic precursors are under investigation as nutriceuticals aimed at ameliorating diseases of aging [4,5]. Among these compounds, nicotinamide riboside (NR) and nicotinamide mononucleotide (NMN) are orally bioavailable and are currently the subject of clinical trials (NCT02812238, NCT02835664, NCT02303483, NCT02678611, etc.) [4,6].
Cardiac SIRT1 is important in several cardioprotective paradigms, including ischemic preconditioning (IPC) [7–9]. Recently, it was shown that NMN confers cardioprotection in a mouse model of ischemia-reperfusion (IR) injury, and this protection was lost in cardiac specific Sir1−/− mice (CS.Sir1−/−) [10]. Although thus far the biological activity of NMN has been mostly attributed to SIRT1 stimulation [11], the role of other mechanisms including other SIRTs has received less attention. Furthermore, the NAD+/NADH redox couple is critically important for metabolic pathways including glycolysis and mitochondrial oxidative phosphorylation [12]. The acute effects of boosting cellular NAD+ levels on these metabolic pathways are poorly understood, and the role that such metabolic perturbations may have in the protective effects of NMN are unknown. Herein, we investigated alternative cardioprotective mechanisms of NMN, beyond SIRT1.
2. Methods
2.1. Animals & reagents
Chemicals and reagents were from Sigma (St. Louis MO). Wild-type C57BL6/J mice and Sirt3−/− mice [13] were bred in-house, handled according to the “NIH Guide” with food and water ad libitum, and used at age 8–12 weeks.
2.2. Langendorff perfused mouse heart IR injury model
Following tribromoethanol anesthesia (200mg/kg ip), hearts were excised and perfused in Langendorff constant flow mode (4 ml/min.) as previously described [9]. Krebs Henseleit (KH) perfusion buffer was supplemented with either (i) 5 mM glucose plus 100 μΜ palmitate-BSA, (ii) palmitate-BSA alone (glucose-free), or (iii) 5 mM galactose plus 100 μΜ palmitate-BSA [9]. Drugs (vehicle, NMN, or splitomicin (Sp)) were delivered via a port above the perfusion cannula from a 100x stock in KH for 20 min., either immediately before ischemia or at the beginning of reperfusion. Cardiac function was monitored digitally at 1 kHz (DATAQ Instruments, Akron OH) via a pressure transducer linked to a fluid-filled left ventricular balloon [9]. Following 25 or 35 min. global no-flow ischemia and 60 min. reperfusion, hearts were stained with tetrazolium chloride for infarct determination by planimetry [9]. The concentration of Sp used (10 μΜ) is known to block cardioprotection by IPC, and the metabolic remodeling and cytosolic lysine deacetylation that accompany it [14]. This level of Sp also acutely blocks cardioprotection afforded by SIRT1 overexpression [15].
2.3. Cardiac metabolomics
Following 20 min. stable normoxic Langendoff perfusion accompanied by either vehicle or NMN delivery, hearts were freeze-clamped in liquid N2 and cardiac metabolites extracted in 80% MeOH for LC-MS/MS based metabolomics as previously described [16]. For glycolytic flux measurements, following 20 min. stable normoxic perfusion (with vehicle or NMN), glucose in KH buffer was replaced with [U-13C]glucose for 5 min. Hearts were then freeze-clamped in liquid N2 and the fractional saturation of selected cardiac metabolites with 13C label was determined by LC-MS/MS as previously described [16].
2.4. Cardiomyocytes
Primary cardiomyocytes were isolated by collagenase digestion, and intracellular pH was measured using fluorescence microscopy with the pH sensitive probe BCECF, as previously described [17,18]. A high K+ nigericin calibration from pHe 5.6–8.4 was used to derive a Boltzmann curve fit (Origin 9, OriginLab Corp.) which was used to convert BCECF emission ratios (dual excitation 440/490 nm) to pHi values. Cardiomyocyte oxygen consumption and extracellular acidification were measured in a Seahorse™ XF24 analyzer (Agilent, Santa Clara CA) either before or 10 min. after addition of NMN, as previously described [17]. Simulated IR (sIR) injury was performed as previously described, by exposing cells to 1 hr. anoxia in glucose free media at pH 6.5, followed by return to baseline conditions for 1 hr., and cell death assayed by LDH release [19,20].
2.5. Western blot
Vehicle or NMN treated hearts were fractionated by differential centrifugation as previously described [15], followed by separation by SDS-PAGE, transfer to nitrocellulose membranes and probing with anti-acetyl-lysine (K-Ac) or anti-SIRT3 antibodies (both from Cell Signaling Technology, Danvers MA), with enhanced chemiluminescence detection [9,13]. Purity of fractions was monitored by western blotting with antibodies against compartment-specific marker proteins.
2.6. Statistics
Statistical significance between groups was determined by ANOVA assuming a normal distribution followed by post-hoc Student’s t-test (significance threshold p<0.05). Numbers of replicates (n) per experimental group are listed in Figure legends.
3. Results
3.1. NMN-induced cardioprotection is insensitive to SIRT1 inhibition
It was previously reported that acute pre-ischemic delivery of NMN was cardioprotective in-vivo, with protection lost in mice harboring cardiac-specific deletion of SIRT1 [10]. However, the CS.Sirt1−/− mice exhibited enhanced IR injury levels at baseline (~50% greater infarct vs. wild-type), which could have over-ridden the ability of NMN to protect via other mechanisms. We previously showed that cardioprotection by ischemic preconditioning (IPC) is blocked in heterozygous whole body SIRT1 knockouts (Sirt1+/−). Furthermore, both IPC itself [9] and the metabolic remodeling that accompanies it [16] can be blocked by the pharmacologic SIRT1 inhibitor splitomicin (Sp). In addition, Sp acutely blocks cardioprotection afforded by SIRT1 over-expression [15]. As such, Sp is a useful tool to interrogate the role of SIRT1 in cardioprotective phenomena.
In this regard, Figures 1A and 1B show that pre-ischemic delivery of 1 mM NMN afforded significant protection against IR injury (post-IR functional recovery: NMN 42±7% vs. vehicle 11±3%; infarct size: NMN 34±4% vs. vehicle 66±4%; means ± SEM, n=6–7, p<0.01 for both parameters). However, in contrast with the CS.Sirt1−/− report mentioned above [10], we observed no effect of Sp on NMN-induced protection (p=0.71 vs. NMN alone).
Figure 1: NMN Induced Acute Cardioprotection is Independent of SIRT1.
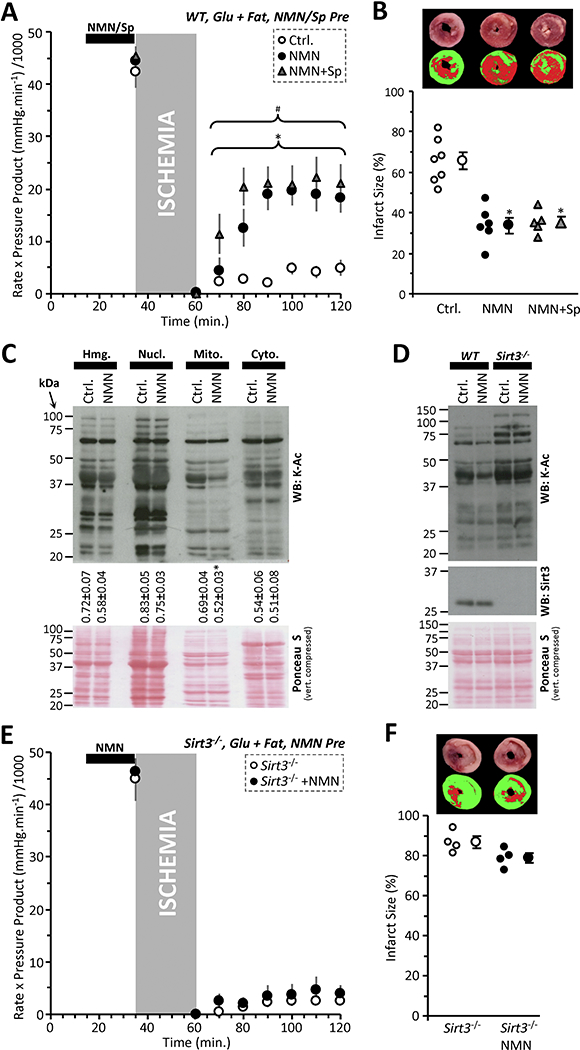
(A): Cardiac functional data (rate x pressure product) for mouse hearts subjected to 25 min. global ischemia and 60 min. reperfusion. Prior to the onset of ischemia hearts were perfused for 20 min. with vehicle (Ctrl, white circles), 1 mM NMN (black circles), or 1 mM NMN plus 10 μΜ splitomicin (Sp, gray triangles). Perfusion media contained both glucose (5 mM) and fat (palmitate-BSA 100 μΜ) as metabolic substrates. Data shown are means ± SEM, N=5–7 animals per group, *p<0.01 for all points indicated, between Ctrl. and NMN+Sp, #p<0.01 for all points indicated between Ctrl. and NMN.(B): Infarction data for the hearts from panel A, determined by tetrazolium chloride staining. Images above the graph show representative infarct photographs (upper), and pseudo-colored images (lower) used for quantitation by planimetry. In graph, individual data points are shown on the left, and means ± SEM on the right, for each treatment group. *p<0.001 vs. Ctrl. (C): Acetyl-lysine western blot on cardiac fractions from NMN treated hearts. Hearts were perfused for 20 min. with 1 mM NMN under normoxic conditions then separated into fractions (Hmg: homogenate, Nucl: nuclear, Mito: mitochondrial, Cyto: cytosolic) by differential centrifugation. Ponceau S stained membrane (loading control) is shown below compressed vertically to save space. Numbers below the blot indicate densitometric quantitation of acetyl-lysine intensity down whole lanes, normalized to protein loading, for N=4 independent experiments (means ± SEM). Fractionation controls are shown in Figure S1A. (D): Acetyl-lysine western blot on mitochondrial fraction from NMN treated hearts of WT and Sirt3−/− mice. Center image shows anti-SIRT3 blot, confirming status of knockout animals. Ponceau S stained membrane (loading control) is shown below compressed vertically to save space. (E): Cardiac functional data (rate x pressure product) for Sirt3−/− mouse hearts subjected to 25 min. global ischemia and 60 min. reperfusion. Prior to the onset of ischemia hearts were perfused for 20 min. with vehicle (Ctrl, white circles) or 1 mM NMN (black circles). Perfusion media contained both glucose (5 mM) and fat (palmitate-BSA 100 μΜ) as metabolic substrates. Data shown are means ± SEM, N=4 animals per group. (F): Infarction data for the hearts from panel E, determined by tetrazolium chloride staining. Images above the graph show representative infarct photographs (upper), and pseudo-colored images (lower) used for quantitation by planimetry. In graph, individual data points are shown on the left, and means ± SEM on the right, for each treatment group.
3.2. NMN effects on lysine acetylation
To interrogate the effects of NMN on sirtuin activity in general, we fractionated NMN-treated hearts and performed acetyl-lysine (K-Ac) western blotting. Purity of fractions is shown in Figure S1A. Figure 1C shows that NMN caused robust lysine deacetylation only in the mitochondrial fraction, not in the cytosol where cardiac SIRT1 is primarily located [21]. This result suggests that a mitochondrial sirtuin such as SIRT3 may be stimulated by NMN, and indeed Figure 1D shows that NMN-induced mitochondrial deacetylation was blunted in Sirt3−/− hearts.
3.3. NMN cardioprotection is lost in Sirt3−/− hearts, but baseline injury is greater
Consistent with a role for SIRT3 in acute NMN-induced cardioprotection, Figures 1E and 1F show that protection is lost in Sirt3−/− hearts. However, akin to the situation with CS.Sirt1−/− described above [10], hearts from Sirt3−/− mice also exhibited a significantly greater level of IR injury at baseline (infarct 87±3% in Sirt3−/− vs. 65±4% in WT, n=4, p<0.01). Such elevated IR injury at baseline may have precluded the ability of Sirt3−/− hearts to be protected at all. In support of this notion, extension of the ischemic time in WT hearts from 25 to 35 min. yielded a greater level of injury (9.4±2.2% functional recovery, 83.5.4±2.6% infarct, Figure S1B/C), and NMN pre-treatment failed to protect against this. As such, it is not possible to conclude that SIRT3 is required for NMN induced cardioprotection.
3.4. NMN stimulates cardiac glycolysis and cytosolic acidification
We recently showed that the SIRT1 inhibitor Sp causes mild cellular alkalinization in primary mouse cardiomyocytes [22]. Thus, we hypothesized that converse stimulation of SIRT1 activity might cause acidification. As Figures 2A and 2B show, 1 mM NMN induced significant cellular acidification. In addition this same concentration of NMN was capable of protecting myocytes against simulated IR injury (Figure 2C). Notably however, the degree of cytosolic acidification seen with NMN (−0.6 pH units) was almost double the opposing alkalinization we previously reported with Sp (+0.35 pH units) [22], suggesting additional (SIRT1-independent) mechanisms may be involved.
Figure 2: NMN Stimulates Cardiomyocyte Glycolysis Leading to Cell Acidification.
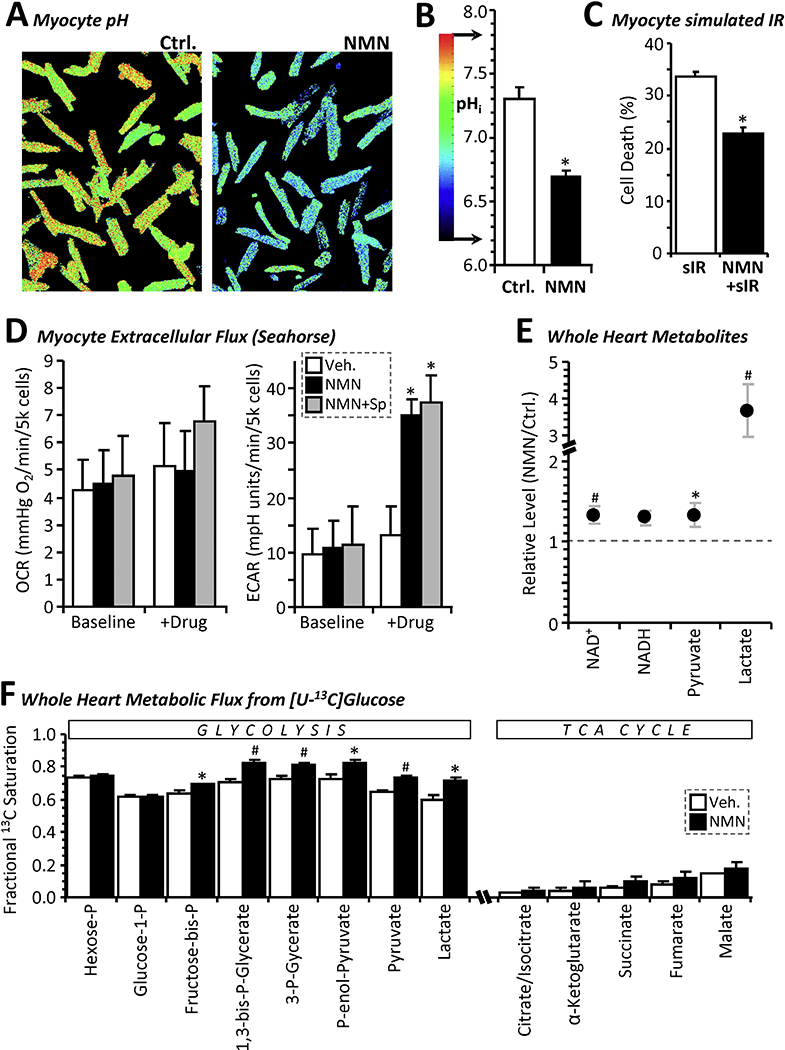
(A): Representative fluorescent microscope images of primary adult mouse cardiomyocytes stained with the pH sensitive vital dye BCECF and treated for 40 min. with 1 mM NMN or vehicle control. Images are pseudo-colored according to the pH scale in panel B. (B): Graph shows average pHi values from 9 independent plates of cells (means ± SEM, *p<0.001 between treatment groups). (C): Simulated IR injury. Cardiomyocytes were subjected to 60 min. similated ischemia followed by 60 min. simulated reperfusion (see methods), with 20 min. prior treatment with 1 mM NMN. Cell death (LDH release assay) is shown as mean ± SEM, N=5 independent cell preparations per group. (D): Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR) values measured via Seahorse™ XF analysis on primary adult mouse cardiomyocytes before and 10 min. after addition of vehicle (white bars) or 1 mM NMN (black bars) to the media. Where indicated, splitomicin (Sp, 10 μΜ) was present throughout (gray bars). Data are means ± SD for 7–8 wells per treatment group, on a single XF-24 plate. *p<0.001 between baseline and post drug treated, within a given treatment group. (E): Relative levels of selected metabolites in freeze-clamped perfused mouse hearts treated for 20 min. with 1 mM NMN or vehicle control. Note break in y-axis scale to accommodate lactate. Data are presented as the metabolite level in NMN treated hearts normalized to that in control hearts (means ± SEM, N=7 animals per group, *p<0.05, #p<0.01, between treatment groups). (F): Fractional saturation of metabolites from [U-13C]glucose infusion. Following vehicle control (white bars) or NMN treatment (black bars) for 20 min., glucose in KH buffer was replaced with [U-13C]glucose for 5 min., followed by freeze-clamp and isotopologue analysis by LC-MS/MS. Data are presented as fractional saturation - i.e., the fraction of the metabolite that is replaced by labeled metabolite within the allotted time, thus equating to metabolic flux from [U-13C]glucose to that point in metabolism (means ± SEM, N=5 animals per group, *p<0.05, #p<0.01 between vehicle and NMN groups).
Since NMN is a precursor for NAD+, and NAD+ is a substrate for gyceraldehyde-3-phosphate dehydrogenase (GAPDH) in glycolysis, we hypothesized that NMN-induced acidification may be partly due to direct glycolytic stimulation by mass action. Seahorse extracellular flux (XF) analysis (Figure 2D) revealed that 1 mM NMN caused a ~3-fold increase in cardiomyocyte extracellular acidification rate (ECAR) within 10 min., without significantly impacting oxygen consumption rate (OCR). Furthermore, this effect of NMN was not blocked by Sp, suggesting no role for SIRT1.
In support of NMN as a direct stimulator of glycolysis, metabolomics experiments (Figure 2E) showed that delivery of 1 mM NMN to perfused mouse hearts caused a significant elevation of NAD+ and a similar increase in NADH (the latter suggesting NAD+ reduction). NMN also drastically elevated cardiac lactate, with a moderate but significant rise in pyruvate. In addition, stable isotope resolved metabolomics with [U-13C]glucose (Figure 2F) showed that acute NMN delivery to perfused hearts significantly elevated the fractional labeling of key glycolytic intermediates, indicative of enhanced flux. No change in TCA cycle flux was seen, although this is likely due to low fractional saturation overall, consistent with glucose not being a preferred carbon source for the heart [16]. Together, these data indicate that NMN directly stimulates glycolysis, likely by increasing the availability of its key substrate NAD+. The lack of effect of NMN on cytosolic lysine acetylation (Figure 1C) may indicate an inability of SIRT1 to compete for NAD+ with the presumably more abundant enzymes of glycolysis.
3.5. Requirement for glycolysis in NMN cardioprotection
Although enhanced glucose utilization is typically associated with cardiac pathology and heart failure [23–27], more recently several parallels have been drawn between cardiac glycolysis and hypoxic survival [16,28]. Thus, we hypothesized glycolytic stimulation may play a partial role in the cardioprotective benefits of NMN [10]. To test this, hearts were perfused without glucose (i.e., with palmitate as sole metabolic substrate). Figures 3A and 3B show that such glucose restriction blocks the protective benefit of NMN.
Figure 3: NMN Induced Acute Cardioprotection Requires Glucose.
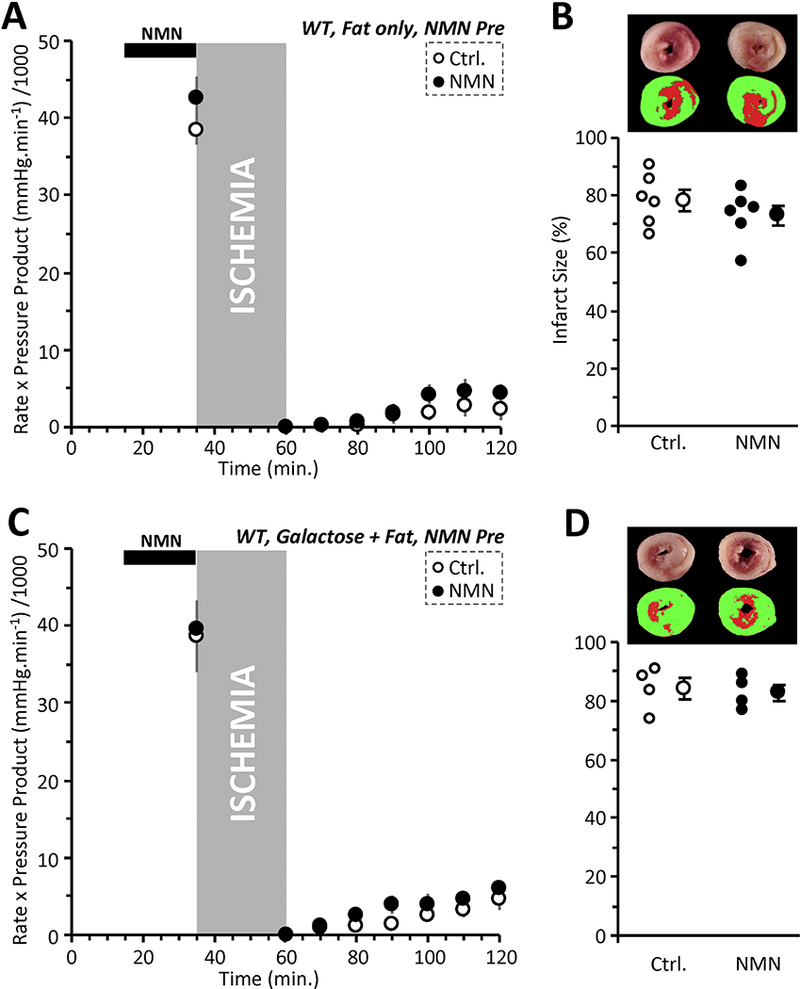
(A): Cardiac functional data (rate x pressure product) for mouse hearts subjected to 25 min. global ischemia and 60 min. reperfusion. Prior to the onset of ischemia (black bar at top), hearts were perfused for 20 min. with vehicle (Ctrl, white circles), or 1 mM NMN (black circles). Experiments were performed essentially as in Figure 1A, except that perfusion media contained zero glucose, such that palmitate was the sole metabolic substrate. Data shown are means ± SEM, N=6 animals per group. No significant differences were noted between Ctrl. and NMN groups. (B): Infarction data for the hearts from panel A, determined by tetrazolium chloride staining. Images above the graph show representative infarct photographs (upper), and pseudo-colored images (lower) used for quantitation by planimetry. In graph, individual data points are shown on the left, and means ± SEM on the right, for each treatment group. No significant differences were noted between Ctrl. and NMN groups. (C): Cardiac functional data (rate x pressure product) for mouse hearts subjected to 25 min. global ischemia and 60 min. reperfusion. Prior to the onset of ischemia (black bar at top), hearts were perfused for 20 min. with vehicle (Ctrl, white circles), or 1 mM NMN (black circles). Experiments were performed essentially as in Figure 1A, except that perfusion media contained 5 mM galactose instead of glucose. Palmitate-BSA was still present. Data shown are means ± SEM, N=4 animals per group. No significant differences were noted between Ctrl. and NMN groups. (D): Infarction data for the hearts from panel C, determined by tetrazolium chloride staining. Images above the graph show representative infarct photographs (upper), and pseu-docolored images (lower) used for quantitation by planimetry. In graph, individual data points are shown on the left, and means ± SEM on the right, for each treatment group. No significant differences were noted between Ctrl. and NMN groups.
Enhancing glycolysis could conceivably elicit cardioprotection via boosting either ischemic ATP synthesis or via acidification at reperfusion. To distinguish these phenomena, we performed experiments in which hearts were perfused with galactose, which yields no net glycolytic ATP, but still permits lactate generation. In this condition, NMN pre-delivery was not protective (Figure 3C/D). These data indicate that with NMN delivered prior to ischemia, the requirement for glycolysis is one of energetics rather than enhancing acidosis during ischemia.
3.6. NMN-induces acid post-conditioning
A key event in IR injury is the opening of the mitochondrial permeability transition (PT) pore. During ischemia, acid pH maintains the pore in a closed state, but pH rebound upon reperfusion triggers pore opening [29–31]. As such, reperfusion with acidic media (i.e., acid post conditioning) is cardioprotective via maintaining PT pore closure into early reperfusion [32–34]. Since acidosis is a key effect of NMN (Figure 2), we hypothesized that NMN delivery at reperfusion alone may be cardioprotective. Figures 4A and 4B show that, indeed, 1 mM NMN at reperfusion was significantly protective (recovery 39±8%, infarct 28±5%, means ± SEM, n=6, p<0.01 between groups). Using the isolated primary cardiomyocyte model of simulated IR injury (see Figure 2C), we also tested the additive effects of delivering acidic buffer and/or NMN at reperfusion. These experiments (Figure 4C) revealed that while acid or NMN alone were protective, no additive (synergistic) effect was seen with both interventions. These data suggest that NMN delivery at reperfusion likely protects via acidosis. Overall, our data suggest that NMN stimulates cardiac glycolysis, and this may contribute to its cardioprotective effects, with the mechanism (energetics or acid) dependent on timing of NMN delivery relative to ischemia. The concurrent involvement of previously outlined mechanisms for NMN-induced cardioprotection (i.e .SIRT1 or SIRT3) cannot be ruled out.
Figure 4: NMN is Protective at Reperfusion.
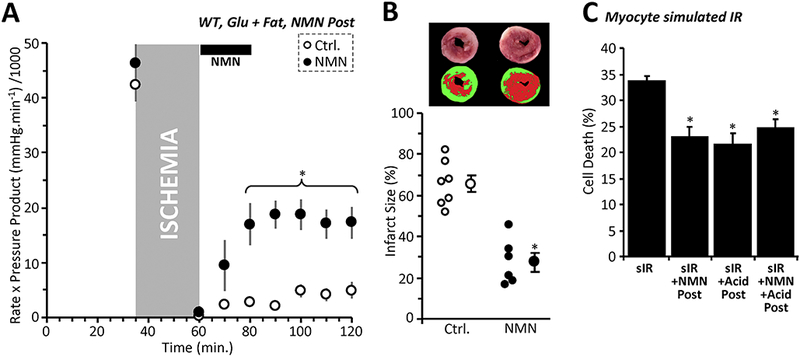
(A): Cardiac functional data (rate x pressure product) for mouse hearts subjected to 25 min. global ischemia and 60 min. reperfusion. At the onset of reperfusion hearts were perfused for 20 min. with vehicle (Ctrl, white circles), or 1 mM NMN (black circles). Perfusion media contained both glucose (5 mM) and fat (palmitate-BSA, 100 μΜ) as metabolic substrates. N.B. Control data in this panel and panel B are the same as those in Figures 1A and B. Data shown are means ± SEM, N=6–7 animals per group. *p<0.01 for all points indicated, between Ctrl. and NMN groups. (B): Infarction data for the hearts from panel A, determined by tetrazolium chloride staining. Images above the graph show representative infarct photographs (upper), and pseudo-colored images (lower) used for quantitation by planimetry. In graph, individual data points are shown on the left, and means ± SEM on the right, for each treatment group. *p<0.001 between Ctrl. and NMN groups. (C): Simulated IR (sIR) injury. Cardiomyocytes were subjected to sIR as in Figure 2C, with delivery of either acidic media, 1 mM NMN, or both at reperfusion. Cell death (LDH release assay) is shown as mean ± SEM, N=5 independent cell preparations per group. N.B. sIR alone data in this panel are the same as those in Figure 2C. *p<0.01 vs. sIR alone. No significant differences noted between acid/NMN/both groups.
4. Discussion
The heart is conventionally viewed as a “metabolic omnivore”, but under normal conditions the bulk of cardiac ATP requirements are furnished by mitochondrial ß-oxidation of fatty acids [23]. As such, a switch to favor glucose utilization is typically associated with cardiac pathology and heart failure [23–27]. However, several studies have also linked glycolysis to cardioprotection. A screen for compounds that enhance glycolysis yielded hits that were protective in models of cardiac IR injury [28]. Glycolysis is also up-regulated in IPC [16,35] and the presence of glucose is necessary for IPC [16]. Furthermore, the pH hypothesis of ischemic post conditioning (IPoC) posits that protection by IPoC is afforded by extending ischemic acidosis into early reperfusion, maintaining PT pore closure [32]. Together with our data showing NMN stimulates glycolysis, these findings suggest that NMN-induced cardioprotection may proceed in part via enhancement of glycolysis. With NMN delivery prior to ischemia, enhanced glycolysis appears to protect via enhancing bioenergetics (no protection when glycolytic ATP synthesis is nullified with galactose). On the other hand, NMN delivery at reperfusion appears to protect via acidosis (no additive effect with acidic reperfusion).
An important caveat in this study is the lack of information regarding the subcellular compartmentalization of the effects of NMN. Namely, acidification induced by NMN was cytosolic (consistent with stimulation of glycolysis), but lysine deacetylation induced by NMN was largely mitochondrial. It is not yet known whether NMN induces compartment-specific alterations in NAD+ or NADH pools, but the advent of novel NADH redox sensors (e.g., SoNar) may be useful in future studies aimed at determining which NAD+ pools are more sensitive to manipulation by compounds such as NMN [36]. A further caveat is the possibility that the lack of effect of Sp seen herein may be due to a low baseline activity of SIRT1. Sp has been shown to be effective under conditions of SIRT1 activation such as IPC [9], but it is unclear if the degree to which NMN may stimulate SIRT1 would confer Sp-sensitivity.
While acidosis is a response of all eukaryotes to hypoxia/ischemia, the potential of acidic pH to drive protective molecular signaling events has only recently become appreciated. For example, acidic pH imparts de-novo activities to several metabolic enzymes, resulting in generation of unique metabolites [18,37]. In particular, the oncometabolite 2-hydroxyglutarate (2-HG) is generated by lactate and malate dehydrogenases under acidic conditions [18,37]. 2-HG is a competitive inhibitor of the α-ketoglutarate dependent dioxygenase family of epigenetic regulators, which includes the JmjC domain-containing histone demethylases, the TET 5-methylcytosine hydroxylases and the EGLN prolyl-hydroxylases that regulate hypoxia inducible factor (HIF) [38,39]. As such, acid pH alone can induce HIF-1α under normoxic conditions via a pathway that requires 2-HG generation [18]. The role of such a metabolic/epigenetic signaling axis in the protective effects of NMN is currently unclear.
The observation that NMN rapidly induces metabolic acidosis raises the possibility that at least some of the benefits reported for dietary NMN supplementation [4–6,12] may be attributed to such effects. A wide variety of health benefits are reported for NMN and NR supplementation, including neurological, cardiac, obesity/diabetes-related and anti-aging [12]. In many cases, such benefits may be linked to enhancing glycolysis. For example, NMN treatment has been shown to increase performance in whole body glucose tolerance tests [40], which may simply be due to enhanced glucose uptake to fuel NMN-stimulated glycolysis. Similarly, NR enhances stem cell function, with stem-ness being generally associated with a glycolytic metabolic state [5].
Despite the reported benefits of dietary NMN/NR supplementation, our results also urge caution regarding the widespread human use of these nutriceuticals. For example, the loss of SIRT1 activity in aging is associated with HIF stabilization and a pseudo-hypoxic metabolic state [41]. As such, acute acidosis resulting from NMN supplementation may activate HIF, worsening pseudo-hypoxia. Similarly, HIF activation and metabolic acidosis are well-known hallmarks of cancer [42], and indeed the same screen that identified glycolytic stimulators as protective against hypoxic injury also found numerous anti-cancer drugs as glycolytic suppressors [28]. Acid pH is also known to promote the reverse reaction of isocitrate dehydrogenase (i.e., reductive carboxylation of α-ketoglutarate to citrate), which is an important driver of lipid biosynthesis in cancer. Together with the apparent ability of NR to promote stem-ness [5], these findings suggest that NAD+ boosting supplements may be beneficial to cancer cells.
5. Conclusions
Overall, our results suggest that the nutriceutical use of NMN and NR should not overlook the classical role that NAD+ plays in glycolysis. GAPDH is one of the most highly and stably expressed proteins in the cell, and is often used as a “housekeeping protein” for experimental loading controls. The levels of GAPDH are likely several orders of magnitude higher than those of NAD+ consuming signaling enzymes such as SIRTs, PARPs, and CD38. Thus, the biological effects of large-scale acute elevations in [NAD+] are likely to involve glycolytic stimulation. It remains to be determined whether long-term beneficial effects of NAD+ precursors are mediated by repetitive transient metabolic acidosis. Although we previously reported that SIRT1 inhibition (Sp) can alkalinize cells [22], this does not appear to be sufficient to override acidification caused by NMN (Figure 2D). This effect highlights the complex nature of overlapping mechanisms that regulate cell pH and survival, including both SIRT1 signaling and glycolytic metabolism.
Supplementary Material
HIGHLIGHTS.
The NAD+ precursor nicotinamide mononucleotide (NMN) acutely stimulates cardiac glycolysis.
During ischemia, NMN is cardioprotective via stimulating glycolytic ATP synthesis.
When delivered at reperfusion, NMN is cardioprotective via enhancing acidosis.
Acidosis induced by NMN may have implications for cancer and other phenotypes.
6. Acknowledgments
This work was funded by grants from the US National Institutes of Health: R01 HL-071158 (to PSB) and R01-HL127891 (to PSB and KWN). We thank Xenia Schafer (URMC Biochemistry) for technical support.
Footnotes
conflict disclosure
The authors declare no financial or other conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Correia M, Perestrelo T, Rodrigues AS, Ribeiro MF, Pereira SL, Sousa MI, Ramalho-Santos J, Sirtuins in metabolism, stemness and differentiation, Biochim. Biophys. Acta - Gen. Subj 1861 (2017) 3444–3455. [DOI] [PubMed] [Google Scholar]
- [2].Anderson KA, Green MF, Huynh FK, Wagner GR, Hirschey MD, Snapshot: Mammalian Sirtuins, Cell. 159 (2014) 956–956.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].van de Ven RAH, Santos D, Haigis MC, Mitochondrial Sirtuins and Molecular Mechanisms of Aging, Trends Mol. Med 23 (2017) 320–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Fang EF, Lautrup S, Hou Y, Demarest TG, Croteau DL, Mattson MP, Bohr VA, NAD + in Aging: Molecular Mechanisms and Translational Implications, Trends Mol. Med. 23 (2017) 899–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, D’Amico D, Ropelle ER, Lutolf MP, Aebersold R, Schoonjans K, Menzies KJ, Auwerx J, NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice., Science. 352 (2016) 1436–43. [DOI] [PubMed] [Google Scholar]
- [6].Trammell SAJ, Schmidt MS, Weidemann BJ, Redpath P, Jaksch F, Dellinger RW, Li Z, Abel ED, Migaud ME, Brenner C, Nicotinamide riboside is uniquely and orally bioavailable in mice and humans., Nat. Commun 7 (2016) 12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hsu C-P, Zhai P, Yamamoto T, Maejima Y, Matsushima S, Hariharan N, Shao D, Takagi H, Oka S, Sadoshima J, Silent Information Regulator 1 Protects the Heart From Ischemia/Reperfusion, Circulation. 122 (2010) 2170–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Rane S, He M, Sayed D, Vashistha H, Malhotra A, Sadoshima J, Vatner DE, Vatner SF, Abdellatif M, Downregulation of MiR-199a Derepresses Hypoxia-Inducible Factor-1 and Sirtuin 1 and Recapitulates Hypoxia Preconditioning in Cardiac Myocytes, Circ. Res 104 (2009)879–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Nadtochiy SM, Redman E, Rahman I, Brookes PS, Lysine deacetylation in ischaemic preconditioning: the role of SIRT1, Cardiovasc. Res. 89 (2011) 643–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yamamoto T, Byun J, Zhai P, Ikeda Y, Oka S, Sadoshima J, Nicotinamide Mononucleotide, an Intermediate of NAD+ Synthesis, Protects the Heart from Ischemia and Reperfusion, PLoS One. 9 (2014) e98972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Imai S, Guarente L, NAD+ and sirtuins in aging and disease, Trends Cell Biol. 24 (2014) 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Katsyuba E, Auwerx J, Modulating NAD + metabolism, from bench to bedside, EMBO J. 36 (2017) 2670–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Porter GA, Urciuoli WR, Brookes PS, Nadtochiy SM, SIRT3 deficiency exacerbates ischemia-reperfusion injury: implication for aged hearts, Am. J. Physiol. - Hear. Circ. Physiol 306 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Parker BL, Shepherd NE, Trefely S, Hoffman NJ, White MY, Engholm-Keller K, Hambly BD, Larsen MR, James DE, Cordwell SJ, Structural basis for phosphorylation and lysine acetylation cross-talk in a kinase motif associated with myocardial ischemia and cardioprotection., J. Biol. Chem 289 (2014) 25890–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nadtochiy SM, Yao H, McBurney MW, Gu W, Guarente L, Rahman I, Brookes PS, SIRT1-mediated acute cardioprotection, Am. J. Physiol. Circ. Physiol. 301 (2011) H1506–H1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Nadtochiy SM, Urciuoli W, Zhang J, Schafer X, Munger J, Brookes PS, Metabolomic profiling of the heart during acute ischemic preconditioning reveals a role for SIRT1 in rapid cardioprotective metabolic adaptation, J. Mol. Cell. Cardiol 88 (2015) 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nadtochiy SM, Madukwe J, Hagen F, Brookes PS, Mitochondrially targeted nitro-linoleate: a new tool for the study of cardioprotection, Br. J. Pharmacol 171 (2014) 2091– 2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Nadtochiy SM, Schafer X, Fu D, Nehrke K, Munger J, Brookes PS, Acidic pH Is a Metabolic Switch for 2-Hydroxyglutarate Generation and Signaling, J. Biol. Chem 291 (2016) 20188–20197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Nadtochiy SM, Zhu Q, Urciuoli W, Rafikov R, Black SM, Brookes PS, Brookes PS, Nitroalkenes Confer Acute Cardioprotection via Adenine Nucleotide Translocase 1, J. Biol. Chem 287 (2012) 3573–3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Nadtochiy SM, Madukwe J, Hagen F, Brookes PS, Mitochondrially targeted nitro-linoleate: a new tool for the study of cardioprotection, Br. J. Pharmacol 171 (2014) 2091– 2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tanno M, Sakamoto J, Miura T, Shimamoto K, Horio Y, Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1., J. Biol. Chem 282 (2007) 6823–32. [DOI] [PubMed] [Google Scholar]
- [22].Nadtochiy SM, Wang YT, Zhang J, Nehrke K, Schafer X, Welle K, Ghaemmaghami S, Munger J, Brookes PS, Potential mechanisms linking SIRT activity and hypoxic 2-hydroxyglutarate generation: no role for direct enzyme (de)acetylation, Biochem. J 474 (2017)2829–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Stanley WC, Recchia FA, Lopaschuk GD, Myocardial Substrate Metabolism in the Normal and Failing Heart, Physiol. Rev 85 (2005) 1093–1129. [DOI] [PubMed] [Google Scholar]
- [24].Sansbury BE, DeMartino AM, Xie Z, Brooks AC, Brainard RE, Watson LJ, DeFilippis AP, Cummins TD, Harbeson MA, Brittian KR, Prabhu SD, Bhatnagar A, Jones SP, Hill BG, Metabolomic analysis of pressure-overloaded and infarcted mouse hearts., Circ. Heart Fail 7 (2014) 634–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Allard MF, Schönekess BO, Henning SL, English DR, Lopaschuk GD, Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts., Am. J. Physiol 267 (1994) H742–50. [DOI] [PubMed] [Google Scholar]
- [26].Abel ED, Doenst T, Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy., Cardiovasc. Res 90 (2011) 234–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Nascimben L, Ingwall JS, Lorell BH, Pinz I, Schultz V, Tornheim K, Tian R, Mechanisms for Increased Glycolysis in the Hypertrophied Rat Heart, Hypertension. 44 (2004) 662–667. [DOI] [PubMed] [Google Scholar]
- [28].Gohil VM, Sheth SA, Nilsson R, Wojtovich AP, Lee JH, Perocchi F, Chen W, Clish CB, Ayata C, Brookes PS, Mootha VK, Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis, Nat. Biotechnol 28 (2010) 249–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Griffiths EJ, Halestrap AP, Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion., Biochem. J 307 ( Pt 1) (1995) 93–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nicolli A, Petronilli V, Bernardi P, Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore by matrix pH. Evidence that the pore open-closed probability is regulated by reversible histidine protonation., Biochemistry. 32 (1993) 4461–5. [DOI] [PubMed] [Google Scholar]
- [31].Kim J-S, Jin Y, Lemasters JJ, Reactive oxygen species, but not Ca2+ overloading, trigger pH- and mitochondrial permeability transition-dependent death of adult rat myocytes after ischemia-reperfusion., Am. J. Physiol. Heart Circ. Physiol 290 (2006) H2024–34. [DOI] [PubMed] [Google Scholar]
- [32].Cohen MV, Yang X-M, Downey JM, The pH Hypothesis of Postconditioning: Staccato Reperfusion Reintroduces Oxygen and Perpetuates Myocardial Acidosis, Circulation. 115 (2007) 1895–1903. [DOI] [PubMed] [Google Scholar]
- [33].Cohen MV, Yang Χ-M, Downey JM, Acidosis, oxygen, and interference with mitochondrial permeability transition pore formation in the early minutes of reperfusion are critical to postconditioning’s success, Basic Res. Cardiol 103 (2008) 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Inserte J, Barba I, Poncelas-Nozal M, Hernando V, Agullo L, Ruiz-Meana M, Garcia-Dorado D, cGMP/PKG pathway mediates myocardial postconditioning protection in rat hearts by delaying normalization of intracellular acidosis during reperfusion., J. Mol. Cell. Cardiol 50 (2011) 903–9. [DOI] [PubMed] [Google Scholar]
- [35].Tong H, Chen W, London RE, Murphy E, Steenbergen C, Preconditioning enhanced glucose uptake is mediated by p38 MAP kinase not by phosphatidylinositol 3-kinase., J. Biol. Chem 275 (2000) 11981–6. [DOI] [PubMed] [Google Scholar]
- [36].Zhao Y, Hu Q, Cheng F, Su N, Wang A, Zou Y, Hu H, Chen X, Zhou H-M, Huang X, Yang K, Zhu Q, Wang X, Yi J, Zhu L, Qian X, Chen L, Tang Y, Loscalzo J, Yang Y, SoNar, a Highly Responsive NAD+/NADH Sensor, Allows High-Throughput Metabolic Screening of Anti-tumor Agents., Cell Metab 21 (2015) 777–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Intlekofer AM, Wang B, Liu H, Shah H, Carmona-Fontaine C, Rustenburg AS, Salah S, Gunner MR, Chodera JD, Cross JR, Thompson CB, L-2-Hydroxyglutarate production arises from noncanonical enzyme function at acidic pH, Nat. Chem. Biol 13 (2017) 494–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S, Travins J, Weiss S, Looper R, Ligon KL, Verhaak RGW, Yan H, Kaelin WG, Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation., Nature. 483 (2012) 484–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim S-H, Ito S, Yang C, Wang P, Xiao M-T, Liu L, Jiang W, Liu J, Zhang J, Wang B, Frye S, Zhang Y, Xu Y, Lei Q, Guan K-L, Zhao S, Xiong Y, Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases, Cancer Cell. 19 (2011) 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Yoshino J, Mills KF, Yoon MJ, Imai S, Nicotinamide Mononucleotide, a Key NAD+ Intermediate, Treats the Pathophysiology of Diet- and Age-Induced Diabetes in Mice, Cell Metab. 14 (2011) 528–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gomes AP, Price NL, Ling AJY, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, Mercken EM, Palmeira CM, de Cabo R, Rolo AP, Turner N, Bell EL, Sinclair DA, Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging., Cell. 155 (2013) 1624–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Warburg O, Wind F, Negelein E, THE METABOLISM OF TUMORS IN THE BODY., J. Gen. Physiol 8 (1927) 519–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.