

Abstract
Controlling immune responses in autoimmunity and to biotherapeutics is an unmet need. In hemophilia, for example, up to 1/3 of patients receiving therapeutic factor VIII (FVIII) infusions develop neutralizing antibodies termed “inhibitors”. To address this problem in a mouse model of hemophilia A (HA), we utilized an antigen-specific regulatory T cell (Treg) approach in which we created a novel B-cell targeting chimeric receptor comprised of a FVIII antigen domain, linked with the CD28-CD3ζ transmembrane and signaling domains. We termed these “BAR” for B-cell-targeting antibody receptor. CD4+CD25hiCD127low human Tregs were retrovirally transduced to express a BAR containing the immunodominant FVIII C2 or A2 domains (C2 and A2 BAR). Such BAR Tregs specifically suppressed the recall antibody response of spleen cultures from FVIII-immunized mice in vitro and completely prevented anti-FVIII antibody development in response to FVIII immunization. Mechanistic studies with purified B cells and T cells from tolerized or control recipients demonstrated that the FVIII-specific B cells were directly suppressed or anergized while the T-cell response remained intact. Taken together, we report here a successful proof of principle strategy utilizing antigen-expressing Tregs to directly target specific B cells, an approach which could be adapted to address other adverse immune responses as well.
Introduction
Antigen-specific immune tolerance induction is a goal for treatment of a variety of unwanted immune responses. Clinically, however, tolerogenic immunotherapy is currently not well developed, even when there is a clearly defined target antigen. A prime example is anti-factor VIII (FVIII) neutralizing antibody (“inhibitor”) development, which occurs in 25-30% of hemophilia A (HA) patients receiving therapeutic FVIII injections. Herein, we present a novel approach to induce specific tolerance using regulatory T cells expressing domains of this defined antigen.
Foxp3 expressing regulatory T cells (Tregs), a subset of CD4 T cells with suppressive activities over a variety of cell types, play a central role in suppressing autoimmunity and in maintaining self-tolerance and immune homeostasis (1). Adoptive transfer of polyclonal Tregs has now been tested in early clinical trials for transplantation and for autoimmune diseases (2–4). However, the efficacy of adoptive therapy using ex vivo expanded polyclonal Tregs may be limited due to the scarcity of any specific T cells among the polyclonal populations. In addition, if used in very large numbers, expanded polyclonal Tregs may cause general immune suppression with risk of viral reactivation (5) or cancer (6). In contrast, using antigen-specific Tregs has advantages since fewer cells are needed and there would be reduced risks of nonspecific immune suppression. Direct isolation of antigen-specific Tregs from polyclonal populations is currently challenging because of limited clonal diversity of Treg pool and challenging expansion ex vivo. Recent success with chimeric antigen receptor-expressing T cells (CAR-Ts) in oncology provides a technical clue to generate antigen-specific Tregs, which serve as a promising alternative to the drawbacks of polyclonal Tregs (7).
In order to induce FVIII antigen-specific immune tolerance using Tregs, previously we have rendered polyclonal Tregs specific by expressing either a recombinant T-cell receptor (TCR; 17195) derived from HA patient’s T cell clone(8, 9) or a single chain chimeric antigen receptor (scFv; ANS8 CAR) that recognizes FVIII domain T- or B-cell epitopes, respectively (10). FVIII-specific Tregs prepared by these methods suppressed recall antibody responses both in vitro and in vivo, confirming the prophylactic and therapeutic potential of FVIII-specific Tregs in HA patients with inhibitors. Despite the successful demonstration of this FVIII-specific suppression effect, TCR-transduced Tregs are MHC-restricted, which limits its general application in an HLA heterogeneous population. In addition, our single chain Fv (ANS8) CAR-engineered Tregs depend on binding exposed epitopes of FVIII on cell surfaces. Moreover, it was unclear whether engineered Tregs suppressed antibody formation by inhibiting T-cell “help” or could act on FVIII-specific B cells.
To directly engage and suppress FVIII-specific B cells, we developed an alternative concept for a CAR analog, called BAR for (chimeric) “B-cell-targeting Antibody Receptor”, in which the extracellular domain of the BAR contains the immunodominant FVIII A2 or C2 domain. We hypothesized that A2 and/or C2 BAR expression in Tregs renders them specific, and such BAR Tregs could engage and suppress FVIII-specific B cells and inhibitor formation. In this study, we evaluated the phenotype of BAR transduced and long-term in vitro maintained human Tregs, as well as the specific suppressive function of BAR Tregs in vitro and in vivo. Furthermore, we demonstrated that A2 and C2 BAR Tregs might directly target FVIII-specific antibody formation bypassing the suppression of FVIII-specific T effectors for the first time.
Methods
Mice
FVIII exon 16 knock-out mice (E16) on C57BL/6 background were used as the model for HA; these were maintained from the colony of Dr Leon Hoyer at the American Red Cross (11, 12). HLA humanized DR1× E16 mice were created by crossing DR1-transgenic mice (Dr. Chella David, Mayo Clinic) with E16 mice on C57BL/6 background. Animal procedures were approved by the Institutional Animal Care and Use Committee at the Uniformed Services University of the Health Sciences (USUHS).
Reagents and antibodies
Recombinant human interleukin (IL)-2 was provided by the National Cancer Institute Biological Resources Branch (Frederick, MD). Phosphorothioate-backboned oligodeoxynucleotides (ODN, 25-bp) with random base pairs were synthesized by Integrated DNA Technologies (Coralville, IA) and used during Treg expansion (13). Recombinant human FVIII (rFVIII) was kindly provided by Dr. Birgit Reipert (Baxalta, Vienna, Austria). Anti-FVIII A2 monoclonal antibody (4A4) was provided by Dr. Pete Lollar (Emory).Anti-human CD3ε (clone 64.1) and anti-FVIII C2 (2C11) was purified in-house from hybridoma supernatants. Fixable viability dye eFluor 780 was from eBioscience (San Diego, CA).
The following anti-human antibodies for T cell stimulation or for flow cytometry were from the commercial sources: CD28 (clone CD28.2), CD127-PE (clone A019D5), and Helios-PE (clone 22F6) were from BioLegend (San Diego, CA); CD8α-PE-Cy7 (clone RPA-T8), CD4-FITC or PE (clone RPA-T4), CD25-PE-Cy7, CD4-PE-Cy7, and CD45RA violet Fluor 450 were from TONBO Biosciences (San Diego, CA); Foxp3-APC (clone 236A/E7) was from eBioscience; purified anti-chicken ovalbumin (OVA) was from Zymed (San Francisco, CA).
Generation of BAR retroviral vectors
The cDNA sequence for human FVIII-A2 and -C2 domains, and chicken ovalbumin (OVA) were derived from GenBank. To construct FVIII C2- BAR, the scFv region of the ANS8 CAR(10) was replaced with FVIII C2 domain, and the resultant (leader sequence)-C2-(G4S linker)-CD28-CD3ζ cDNA was codon optimized and synthesized by GenScript USA (Piscataway, NJ), and ligated into pRetroX-IRES-ZsGreen1 (Clontech, Mountain View, CA) retroviral vector. The FVIII A2-BAR and the control OVA-BAR were generated by replacing the C2 sequence in the C2-BAR construct with A2 or OVA cDNA sequence, respectively (Supplemental Figure 1A). The retroviral particles were produced using a Phoenix-Ampho packaging system (Clontech, Mountain View, CA). Culture supernatants containing the retroviral particles were concentrated with Retro-X Concentrator (Clontech), and aliquots stored at −80°C until use.
Human blood samples and T cell isolation
Buffy-coat blood samples from healthy male or female donors between 26 to 72 years of age were obtained from the Department of Transfusion Medicine, Clinical Center, National Institutes of Health (NIH). The procedures were approved by the USUHS Institutional Review Board. The isolation of Treg cells (CD4+CD25hiCD127lo) and CD4+ conventional T cells (Tcon, CD4+CD25−CD127hiCD45RA+) from human peripheral blood were as previously described, with the combination use of magnetic-activated cell sorting (MACS, Miltenyi Biotec Inc., Auburn, CA) and fluorescence-activated cell sorting (FACS; Aria II cell sorter, BD, Franklin Lakes, NJ) (10).
Retroviral transduction and expansion of transduced human CD4+ Tcon and Tregs
Transduction and expansion of CD4 Tcon and Treg cells were as previously described (9, 10). Briefly, thawed T cells were pre-stimulated with plate-bound or soluble 5 μg/ml of anti-human CD3ε (clone 64.1) and 2 μg/ml of anti-human CD28 (clone CD28.2, eBioscience) in complete culture media in the presence of 100 U/ml of IL-2 and 2 mM of ODN for 48 to 72 hours. Note that ODN were previously shown to stabilize Treg function by Kim et al (13). Transduction was performed by first spinning the retroviral particle supernatant onto 10 μg/ml retronectin pretreated 24-well culture plate at 2000g and 32°C for 2 hours, followed by centrifugation of the activated T cells onto the viral particle coated plate at 1500g, 32°C for 15 minutes. Ten days after the pre-stimulation, the successfully BAR transduced human Tregs were FACS sorted based on GFP expression, and re-stimulated once with 0.5 μg/ml soluble anti-human CD3ε in the presence of 6000 rad gamma-irradiated autologous PBMC (PBMC: T cell ratio = 2:1), in the presence of ODN (2 mM) and IL-2 (100 U/ml). Expanded T cells were frozen in FBS containing 10% DMSO, and stored at −80°C until use. Each experiment was performed with transduced T cells generated from a single donor, and was repeated 2-3 times.
FACS staining
To examine the expression of BAR on the transduced and expanded human Tregs, the cells were stained with fixable viability dye eFluor 780, together with the indicated specific or the respective isotype control antibody, followed by fluorescent labeled second antibody staining. Stained cells were acquired on an LSRII instrument (BD) using BD FACSDiva software and analyzed using FlowJo software (TreeStar, Inc. Ashland, OR).
For analyzing intracellular Foxp3 and Helios expression in the expanded BAR Tregs, the cells were re-stimulated with 0.5 μg/ml anti-human CD3ε for 48 hours. The cells were first surfaced stained with CD4-PE-Cy7 and Fixable Viability Dye eFluor 780, and then fixed with 2% paraformaldehyde solution and permeabilized in permeabilization buffer (0.1% Triton X-100 in PBS containing bovine serum albumin) overnight at 4°C. Permeabilized cells were stained with the indicated antibodies for at least 3 hours at 4°C.
DNA methylation analysis on human Treg-specific de-methylation regions (TSDR)
Genomic DNAs were extracted from approximately 1 × 106 the human T cells using the Wizard Genomic DNA purification kit (Promega). Genomic DNA samples at 20 ng/μl were shipped frozen to EpigenDx Inc. (Hopkinton, MA), where the TSDR methylation analysis was performed. Briefly, the genomic DNAs were bisulfite treated, PCR amplified toward TSDR region, and Pyrosequenced. The analysis covered nine CpGs spanning from −2263 to −2330 (from ATG the translation start site) in the Foxp3 genome.
B-cell ELISPOT and ELISA assays
To measure the FVIII-specific B cell suppression by BAR Tregs in vitro, an Enzyme-Linked ImmunoSpot (ELISPOT) protocol was used (14). As a model for inhibitor development in HA patients, anti-FVIII antibody response were established in E16 mice by weekly intravenous injection of 1 μg recombinant human FVIII in PBS for 4 weeks in most experiments. Pooled splenocytes (2 × 106) from four FVIII-immunized E16 mice were co-cultured with 1 × 105 of either A2-, C2-, or the control OVA-BAR human Tregs (BAR hTregs) in complete culture media. Two hours after mixing the Tregs and splenocytes to allow for potential interaction of specific B cells with the BARs, human rFVIII protein (0.1 μg/ml) was added and culture was continued for 5 days. The cells were washed twice in culture medium and transferred to 2 μg/ml rFVIII coated 96-well ELISPOT plates (EMD Millipore) and cultured overnight. The number of spots by FVIII-specific antibody secretion B cells (ASCs) was visualized by incubation with second antibody HRP-rabbit anti-mouse IgG (H+L) (ThermoFisher Scientific) followed by AEC substrate (BD Biosciences).
For measuring anti-FVIII total IgG antibody levels, an enzyme-linked immunosorbent assay (ELISA) was used as previously reported (15).
Statistical analysis
Statistical analysis was performed using Prism software (v6.0, GraphPad Software, La Jolla, CA). Student’s t test and Mann Whitney U test were chosen to evaluate the significance of the in vitro and in vivo suppression effect by FVIII-BAR hTregs. A P value < 0.05 was considered statistically significant.
Results
Design of BAR receptors for directly targeting FVIII-specific B cells
FVIII is a large glycoprotein of about 300 KDa, consisting multiple domains in the order of A1-A2-B-A3-C1-C2 (Figure 1A) (16). Expressing a BAR containing the full length FVIII protein on the surface of Tregs would be challenging. It is known that the majority of inhibitors from HA patients are directed against the functional A2 and C2 domains of FVIII (17). Therefore, we chose a strategy to separately engineer A2-BAR and C2-BAR, respectively, as was done by Lei and Scott previously (Figure 1A) (18). An OVA-BAR was also generated to serve as a control for antigen-specificity. The expected size for A2-, C2-, and OVA-BAR transgenes was 1898, 1274, and 1952bp, respectively, as confirmed by restriction enzyme digestion (Supplemental Figure 1). The expression of BAR in human Tregs was mediated through transduction by concentrated retroviral supernatant, and the transduced Tregs were sorted based on GFP expression and further expanded in vitro as described (9, 10).
Figure 1. Generation of human CD4+ Tregs expressing the chimeric B-cell-targeting antibody receptor (BAR).

(A) Schematic illustration for the generation of retroviral constructs for BARs. The immunodominant FVIII A2 or C2 domain was engineered as the extracellular domain of the chimeric receptor. The cDNA sequences for a BAR were arranged in the following order: antigen-CD28-CD3ζ from N- to C-terminal. The resulting BAR expression cassettes were cloned into a retroviral vector, RetroX-IRES-Zsgreen1, which contains a GFP reporter gene under the control of the internal ribosome entry site (IRES). (B) Expression of BAR in the transduced human Tregs. FACS sorted human Tregs (CD4+CD25hiCD127low) were transduced with BAR and expanded in vitro. At the end of the expansion (21 days after the initial T cell stimulation), the Tregs were stained with specific antibodies for the extracellular domain of BAR Tregs (mAb 4A4 for A2; mAb 2C11 for C2, and rabbit anti-OVA IgG for OVA), respectively. In the histogram shown, the blue dotted line is for BAR, the green solid line for isotope control, and the grey filled for unstained. The data shown were gated on live singlets. (C) Foxp3 and Helios expression in the expanded human Tregs, compared to that of freshly isolated Tcon. Long-term in vitro expanded BAR Tregs and freshly isolated Tcon were re-stimulated with soluble anti-CD3ε in the presence of recombinant human IL-2 for 48 hrs, followed by intracellular staining for Foxp3 and Helios. The dot plots shown were gated on live CD4+ singlets. (D) The % TSDR DNA methylation in the long-term expanded BAR Tregs, compared to that of the freshly FACS sorted Tregs and Tcon cells. The heat map shows the % methylation of 9 CpGs in the intron 1 of human Foxp3 genome. The bar graph shows the summarized data of mean ± SEM.
Expression of BAR molecule in the prepared BAR Tregs
The in vitro generated and expanded BAR Tregs were typically > 95% GFP+, indicative of successful BAR expression. To directly confirm the proper BAR expression, A2-, C2-, and OVA-BAR Tregs were surface stained with the specific antibodies. FACS result in Figure 1B shows that the all three BAR Tregs could be specifically recognized by corresponding antibodies, compared to isotype control staining. BAR expression enables human T cells to be specifically recognized by B cell receptors or their respective antibodies. As an example, the OVA-BAR transduced Tcon cells proliferated robustly in response to the cognate polyclonal anti-OVA antibodies stimulation in vitro (Supplemental Figure 2). These results imply that BAR Tregs may be able to interact with B cells bearing cognate B-cell receptors (BCR).
BAR-expressing human Tregs maintain the typical human Treg phenotypes after long-term in vitro expansion
Our goal is to re-target polyclonal human Tregs to suppress FVIII-specific B cells, for example. To check the quality of the prepared BAR Tregs, we first stained the cells for both CD4 and CD8 in case there is significant contamination of the cytotoxic T cells. As shown in the supplemental figure 3A, following the long-term in vitro expansion, > 99% BAR Tregs were positive for CD4.
Importantly, these in vitro expanded BAR Tregs maintained the Treg-specific phenotypes. Approximately, 70-80% of freshly isolated human Tregs co-expressed Foxp3 and Helios (9), while activated human conventional CD4 T cells transiently express FoxP3 but do not express Helios. Intracellular FACS data showed that the majority of the BAR hTregs still retained Foxp3 stably. In addition, at least 75% BAR hTreg co-expressed Foxp3 and Helios, which is the typical phenotype of bona fide human Tregs (Fig. 1C). As expected, the majority of the expanded Tregs do not express either IFN-γ or IL-2 (Supplemental Figure 3D). These qualities of prepared BAR Tregs were re-confirmed before we utilized these cells for each of our later in vitro and in vivo suppression experiments.
In addition, we examined the status of TSDR de-methylation on prepared BAR Tregs. As shown in Figure 1D, the average % TSDR methylation of the freshly FACS sorted Tregs and Tcon cells were 38% ± 1.7% and 89 ± 3.5%, respectively. The level of TSDR methylation in the long-term in vitro expanded A2- (19.1 ± 1.4%), C2- (40.2 ± 3.5%), and OVA-BAR Tregs (19.9 ± 1.5%) were lower or close to that of fresh sorted Tregs, but distinct from that of the Tcon cells. Taken together, we concluded that the BAR Tregs generated using our long term in vitro expansion protocol with ODNs maintained the typical signatures of human Tregs.
FVIII-BAR hTregs suppress antibody formation from FVIII-specific mouse B cells in vitro
To examine whether BAR hTregs suppress recall the anti-FVIII antibody response by specific memory B cells, we next performed a xenogeneic in vitro B cell suppression assay. In lieu of using patient PBMC as responders, splenocytes isolated from FVIII-immunized E16 mice were used. The results in Figure 2 show that, compared to the non-specific OVA-BAR hTreg control, both A2- and C2-BAR hTregs significantly suppressed the FVIII-specific ASCs formation even in a xenogeneic setting, as previously observed with TCR-and scFv transduced Tregs (9, 10). A mixture containing both A2-BAR Tregs and C2-BAR Tregs was also suppressive, as expected (Fig. 2C & D). These results suggest the functional utility of the FVIII BAR Tregs and encourage further in vivo testing.
Figure 2. In vitro suppression of anti-FVIII antibody production by BAR hTregs.
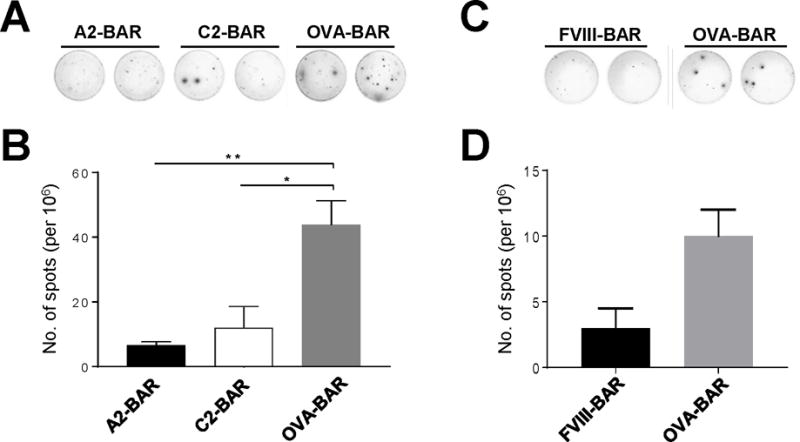
Splenocytes (2 × 106) from rFVIII immunized E16 mice were co-cultured with 1 × 105 different numbers of BAR hTregs in complete RPMI medium in the presence of 0.1 μg/ml human rFVIII. The cells were cultured for 5 days, and the suppressive activity of the BAR hTregs was evaluated by an anti-FVIII B-cell ELISPOT assay. (A) The in vitro suppressive effect of A2-BAR hTregs and C2-BAR hTregs on anti-FVIII antibody spot formation. (B) Summarized data for results shown in panel A. (C) The in vitro suppressive effect of FVIII-BAR hTregs (a mixture containing equal number of A2-BAR hTregs and C2-BAR hTregs). (D) Summarized data for results shown in panel C. Data are expressed as mean ± SEM. *p < 0.05 by student’s t test. The results shown are representatives of three independent experiments with similar results.
FVIII-BAR hTregs block anti-FVIII antibody formation in E16 mice in vivo
We next tested whether BAR hTregs could suppress the formation of anti-FVIII antibodies in vivo (Fig. 3). Considering both A2 and C2 of FVIII are major targets for inhibitors and choosing only one would lead to a partial effect, a mixture of equal numbers of A2-BAR hTregs and C2-BAR hTregs were used (called “FVIII-BAR hTregs” hereafter). Thus, E16 FVIII knockout mice were infused intravenously with 1 × 106 of BAR hTregs on day 0, and then immunized subcutaneously on day 1 with FVIII in incomplete Freund’s adjuvant. Anti-FVIII antibodies were detected by two weeks after immunization in control non-specific OVA-BAR hTreg-infused mice, and peaked at five weeks. However, in the FVIII-BAR hTreg infused group, no anti-FVIII antibody response was detected after immunization, an effect that was retained for at least 8 weeks in these FVIII-BAR hTreg infused mice (Figure 3A).
Figure 3. Prophylactic BAR Tregs therapy prevented the anti-FVIII antibody formation in vivo.
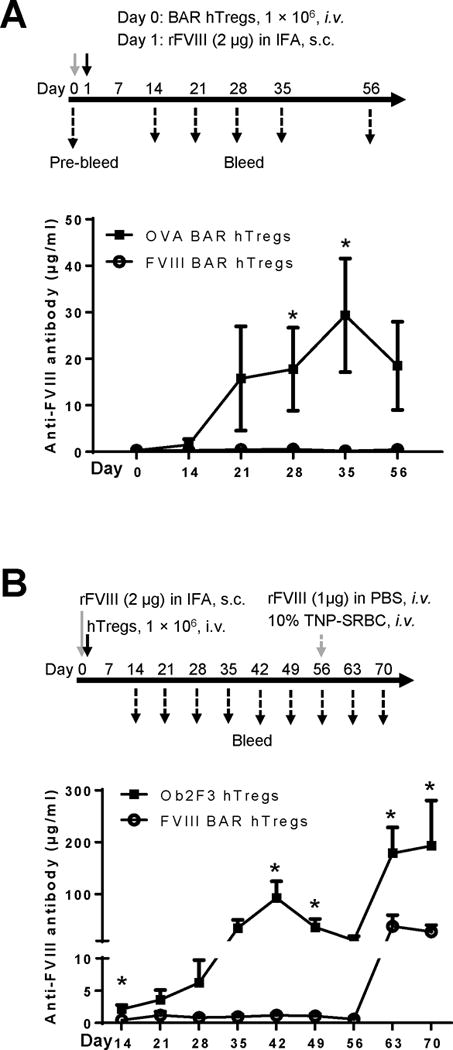
(A) Xenogeneic suppression of anti-FVIII antibody formation in E16 mice. The protocol of the in vivo experiment is shown at the top. Briefly, male E16 mice at 6-10 weeks of age (n=5) received 1 × 106 of FVIII-BAR hTregs on day 0 (circles). A control group of mice received OVA-BAR hTregs (squares). The mice were then immunized subcutaneously on day 1 with FVIII in incomplete Freund’s adjuvant (IFA). The serum anti-FVIII antibody levels at different time points were measured by an ELISA assay, and the result is shown in the lower part of panel A. (B) Xenogeneic suppression of anti-FVIII antibody formation in HLA humanized DR1×E16 mice. The protocol of the in vivo experiment is shown at the top. Briefly, age matched male or female DR1×E16 mice (n=5) were immunized subcutaneously with 2μg of rFVIII in IFA on day 0. Four hours later, the mice received 1 × 106 of FVIII-BAR hTregs. The control group of mice received similarly prepared non-relevant Ob2F3 hTregs, which express the TCR recognizing a myelin basic protein peptide. The serum anti-FVIII antibody levels at different time points were measured by an ELISA assay, and the result is shown in the lower part of panel B. The Tregs used in panel A and B were prepared from different donors. The data were expressed as mean ± SEM. *p < 0.05 between the FVIII-BAR hTregs group and the control Tregs group, by student’s t test.
To confirm the robust suppressive activity of FVIII BAR Tregs, we conducted another prophylactic in vivo experiment using a different strain of HA mice. As outlined in the upper panel of Figure 3B, age-matched male or female DR1×E16 mice (n=5) were immunized subcutaneously with 2μg of rFVIII in IFA on day 0. Four hours later, the mice received 1 × 106 of FVIII-BAR hTregs. A control group of mice received similarly prepared control non-relevant Ob2F3 hTregs, which express a TCR recognizing a human HLA-DR2 restricted myelin basic protein peptide. The treatment of FVIII BAR hTregs almost completely suppressed the total anti-FVIII antibody response compared to the control Ob2F3 hTregs, an effect that also lasted for as long as 8 weeks following the FVIII/IFA immunization. To examine the persistence of the FVIII tolerance effect, the mice were re-challenged with rFVIII at week 8, at which time the human Tregs would have been rejected. The mice in FVIII-BAR Treg group responded to the FVIII re-challenge, but the antibody levels were significantly lower compared to the control Ob2F3 Tregs group, presumably because of inhibition of the development of memory B cells after the initial treatment. On week 8, the mice also received an i.v. injection of 200 μl of 10% trinitrophenol-conjugated sheep red blood cells (TNP-SRBC) as an irrelevant antigen challenge to determine any potential general immune suppression by the BAR Tregs. As expected, the development of anti-TNP antibody was normal and was not significantly different in all groups (data not shown). From these in vivo suppression data, we conclude that infused FVIII-BAR hTregs can interact with specific B-cell precursors that produce anti-FVIII-antibodies, and suppress their antibody production in vivo.
Direct suppression of FVIII-specific B cells by FVIII-BAR hTregs in vivo
The anti-FVIII humoral immune response has been shown to be dependent on T-cell help in both humans and mice (19, 20). To examine the possible mechanism of action by FVIII-BAR hTregs, we did ex vivo co-culture of T and B cells from treated mice (Figure 4). At the end of the in vivo experiment described in Figure 3A, i.e. 8 weeks after immunization, we pooled splenocytes from “tolerant” E16 mice (recipients of FVIII BAR hTregs) and from control mice (recipients of OVA BAR hTregs). B (CD19+) and T cells (depleted of CD19+ cells) were purified from the pooled splenocytes in each group by MACS sorting. The B (1×106) and T cells (0.5 × 106), each from either the “tolerant” mice or the control mice, were then co-cultured for 6 days in the presence of 0.1 μg/ml of human rFVIII protein. As expected, when B and T cells that were both derived from OVA-BAR hTregs recipients, anti-FVIII antibody formation was detected. Interestingly, B cells from OVA-BAR hTregs-treated recipients could collaborate successfully with the T cells from the FVIII-BAR hTreg-treated recipients. In contrast, no recall antibody response to FVIII could be detected when the B cells were from the “tolerant” mice that received FVIII-BAR hTregs (Figure 4). These results suggest that a potential mechanism of FVIII-BAR hTregs in vivo was the direct suppression, anergy or killing of the FVIII-specific B cells.
Figure 4. Mechanism of suppression of FVIII-specific B cells by FVIII-BAR hTregs in vivo.

(A) Outline of the protocol. At the end of the in vivo experiment described in Figure 3 panel A, the mice were sacrificed and splenocytes were pooled from the FVIII-BAR hTreg or control OVA-BAR hTregs recipients. B and T cells, respectively, were purified from the pooled splenocytes by magnetic-activated cell sorting and co-cultured (B:T cell ratio = 2:1) in different combinations as indicated for 6 days in the presence of 0.1 μg/ml human rFVIII, followed by FVIII-specific B cell ELISPOT assay. (B) FVIII-specific ASC detected by the B cell ELISPOT assay. No anti-FVIII spot formation could be detected when the B cells were from the tolerant donors that received FVIII-BAR hTregs. (C) Summarized data from results shown in panel B. The data summarized from quadruplicate repeats (n = 4) were expressed as mean ± SEM. **p < 0.01, as compared to all other cell combinations except for condition #4 (the B and T cells were from the control mice and tolerant mice, respectively) or #1 (Both B and T cells were from the control mice group). The data was analyzed by student’s t test. N.D., none detected.
The therapeutic effect of FVIII-BAR hTregs on anti-FVIII antibody development in vivo
Finally, we tested the therapeutic effect of FVIII-BAR Tregs on primed mice with pre-existing anti-FVIII antibodies. The experimental protocol is outlined in Figure 5A. Age-matched male E16 mice (n = 4) were primed with two immunogenic doses of rFVIII (1μg in 100μl PBS) intravenously on day 0 and 7. The serum anti-FVIII antibody levels were (0.90 ± 0.06 μg/ml) on day 21; at the same time, none of the naïve E16 mice (n = 5) had detectable anti-FVIII titers (Fig. 5 and data not shown). These mice were randomly divided into three groups on day 28 and received a booster dose of rFVIII, followed by the i.v. injection of 2×106 FVIII-BAR hTregs, control OVA-BAR hTregs, or PBS, respectively. Interestingly, FVIII-BAR hTregs treatment significantly decreased the anti-FVIII antibody levels as compared to either the OVA-BAR Tregs or the PBS control (Fig. 5). These data support our main hypothesis that re-targeting polyclonal human Treg to FVIII-specific B cells through BAR receptor expression could block the B-cell differentiation pathway into ASCs or memory B cells, thereby efficiently control low titer anti-FVIII antibody formation (Figure 6).
Figure 5. The therapeutic effect of FVIII-BAR Tregs on anti-FVIII antibody formation in primed E16 mice.
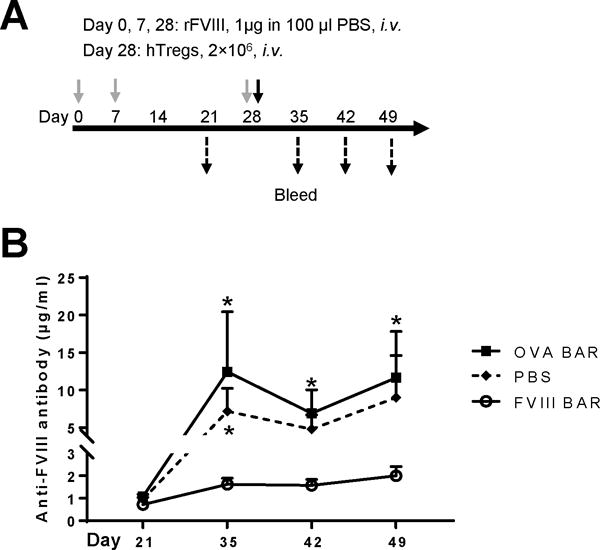
(A) Protocol of the in vivo experiment. Male E16 mice were primed with two immunogenic doses of rFVIII (1μg in 100μl PBS) intravenously on day 0 and 7. After the confirmation of detectible serum anti-FVIII antibody levels on day 21, the mice were randomly divided into three groups (n = 4 per group): OVA-BAR hTregs (squares), FVIII-BAR hTregs (circles), and PBS (diamonds). Each group of mice received a booster dose of rFVIII i.v. on day 28, followed by the i.v. injection of 2×106 FVIII-BAR hTregs, control OVA-BAR hTregs, or PBS, respectively. The mice were bled weekly after day 35, and the serum anti-FVIII antibody levels were measured by an ELISA assay as above. (B) Antibody titers before and after infusion of BAR Tregs. The antibody levels were measured by an ELISA assay. The data were expressed as mean ± SEM. *p < 0.05 between the FVIII-BAR hTregs group and the control OVA-BAR hTregs or PBS group. The non-normal distribution data was analyzed by Mann Whitney U test. Representative result from two independent experiments is shown.
Figure 6. Scheme of possible action of BAR hTregs.
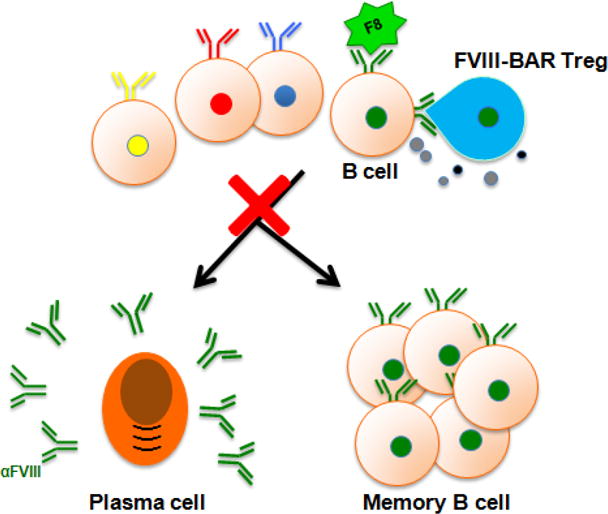
FVIII-BAR Tregs may specifically and directly engage FVIII-specific B cells, to anergize/kill or block their further differentiation into plasma cells and/or memory B cells.
Discussion
The antibody response to therapeutic FVIII (called inhibitors) blocks the efficacy of FVIII replacement therapy, and is a serious side effect in the treatment of hemophilia A patients. To prevent inhibitor formation and induce tolerance to FVIII, various experimental approaches have recently been reported, such as liver-specific or platelet-specific FVIII gene therapy (21–23), adoptive transfer of B cells engineered to express FVIII antigen-IgG fusion (24), or co-injection of FVIII with low dose non-depleting anti-CD3(25,26) or anti-CD4 mAb (27). Experimental FVIII tolerance induction has also been achieved through co-administration FVIII with rapamycin (28,29), or with IL-2/IL-2 mAb complexes (30). In addition, systemic tolerance to FVIII has been obtained in hemophilia A mice by oral feeding transgenic plant-expressing FVIII in chloroplasts (31). Interestingly, the tolerogenic effects induced by the above mentioned strategies were all dependent or associated with increased number or function of Tregs. This common pathway strongly suggests the feasibility of adoptive Treg cell therapy for FVIII tolerance induction. Instead of directly applying ex vivo expanded polyclonal Tregs (see ref. 32), we have been recently focused on developing an antigen-specific Treg approach with the aim to more efficiently address the anti-FVIII inhibitor problem (9, 10).
FVIII-specific B cells are responsible for the production of anti-FVIII inhibitors and memory formation. Thus, targeting FVIII-specific B cells without affecting normal protective immunity would be ideal. Current clinical protocols of long-term application of high doses of FVIII may work partially through suppressing FVIII-specific B cells (33), but it is extremely expensive and challenging, and it is not effective in many patients. In this study, we describe a novel BAR Treg approach, which targets FVIII-specific B cells directly and prevent the development of FVIII inhibitors.
Ellebrecht et al (34) recently reported the design of chimeric autoantibody receptor (CAAR) consisting of a major pemphigus vulgaris autoantigen, fused to CD137-CD3ζ signaling domains. We independently developed the BAR concept, similar to CAAR in terms of the antigen-based CAR design, in order to target FVIII-specific B cells. One major difference in our current report is that, instead of engineering cytotoxic T cells, we focused on developing antigen-specific human Tregs in order to increase the efficacy of polyclonal Treg therapy used in recent clinical trials (3). No significant contamination of CD8 cytotoxic T cells was found in our expanded BAR Tregs. More than 99% of the BAR Tregs after the long-term in vitro expansion were CD4+ (Supplemental Figure 3A). Due to the lack of purified FVIII-specific B cells, we could not directly examine potential cytotoxicity. However, the expression of cytotoxicity markers, Granzyme B, perforin, and the degranulation marker CD107a, was lower in the BAR Tregs than the BAR Tcon prepared from same donors and by identical treatment, and were distinctive from CD8 cytotoxic T cells (Supplemental Figure 3B&C).
In the Figure 4, we showed experimental evidence that BAR Tregs are capable of suppressing B-cell activation directly while not affecting FVIII-specific T cells. However, the detailed cellular and molecular mechanisms to explain these data still need to be determined. We consider two possibilities: the first is that FVIII-BAR Tregs directly suppress the differentiation and proliferation of FVIII-specific B cells. In this case, we need to determine whether cell-cell contact is a necessary event for the action of FVIII BAR Tregs, and if so, what are the critical molecular factors (e.g., TGFβ and CTLA-4, etc.). We checked LAP expression in BAR hTregs and did not find a significant expression of this TGF-β associated molecule, 48 hrs after receptor stimulation (data not shown). Similarly, no significant amount of TGF-β secretion was detected in the supernatants of cultured FVIII BAR hTregs 24 hrs or 48 hrs after anti-CD3-mediated re-stimulation using a CBA assay (data not shown). A second possible mechanism is the direct killing of the specific B cells by the FVIII-BAR hTregs. There are reports suggesting that activated antigen-specific Tregs could not only suppress but also kill cognate B cells in vitro and in vivo (35–37). However, as noted above, activated human Tregs do not produce significant amount of Granzyme B, a key cytotoxicity inducer compared to mouse Tregs (38,39). Consistently, we also found that the long-term in vitro expanded BAR Tregs were less potent in cytotoxic pathway markers, as noted above, compared to BAR Tcon (Supplemental Figure 3). Therefore, the effector molecules by BAR Tregs that mediate suppression, anergy or killing of cognate B cells will need to be further determined in future studies.
In this study, we directly tested BAR transduced hTregs in immunocompetent mouse recipients. This is because it is challenging at present to test HA patient’s antibody responses in vitro. However, it has been demonstrated that hTregs (CD4+CD25hiFoxp3+) suppress the activation of mouse responders as efficiently as murine Tregs (40). In addition, the properties of mouse and human Tregs in vitro and in vivo are not always correlated (41). Thus, a xenogeneic in vivo protocol was chosen. Despite xenogeneic recognition, the robust suppression of anti-FVIII antibody development by FVIII BAR Tregs was quite significant. Furthermore, we have also obtained evidence for a reciprocal effect in that OVA-BAR Tregs significantly suppressed anti-OVA antibody development (Supplemental Figure 4).
We realized that a critical issue in our design is the rejection of infused human cells by the host mice immune system. Indeed, anti-human lymphocyte antibody responses could be detected in recipient mice as early as one week after the injection of human cells, and GFP+ human cells could not be readily detected in the recipient peripheral blood two weeks following the injection of BAR Tregs (data not shown). Nonetheless, the anti-FVIII antibody response to FVIII/IFA immunization was almost completely blocked after prophylactic FVIII-BAR hTregs treatment (Figure 3), which provides evidence that the in vivo model of cross-species suppression by hTreg is still valuable, even though the human cell rejection is inevitable in an immunocompetent host.
A natural concern for the use of FVIII-BAR Tregs is whether it will be effective in patients with pre-existing anti-FVIII inhibitor antibodies. We can separate this concern into two questions: whether FVIII-BAR Tregs will have any effect on antibody-secreting plasma cells, and what are the potential adverse effects of pre-existing free antibodies on BAR Tregs. We think the fully differentiated plasma cells are not likely targets since they generally lack surface immunoglobulin. The effects of circulating free antibodies may have three possible effects. First, they would simply block the interaction between BAR Tregs and specific B cells. To test this, in our therapeutic experiment (Figure 5), we primed the host immune system with FVIII, and found that BAR Tregs were effective in mice with low titer anti-FVIII. We have not tested efficacy in mice with high anti-FVIII inhibitor titers. In such a scenario, removal of most of the circulating pathologic antibodies using the clinical available procedure of plasmapheresis should be helpful. A second possibility is that circulating antibodies have deleterious effects on BAR Tregs, through antibody-dependent cytotoxicity and/or antibody-mediated opsonization for phagocytosis. In this case, prophylactic application of BAR Tregs will still be useful, especially for high risk HA patients. An interesting third outcome for the free circulating antibodies may be that they stimulate the BAR Tregs and thereby enhance the efficacy. In the report by Ellebrecht et al., pre-existing polyclonal autoantibodies in an immunodeficient NSG mice recipient failed to neutralize or eliminate their cytotoxic CAAR T cells (34). Whether this could happen to BAR Tregs in an immunocompetent host needs to be confirmed in the future studies.
The BAR Treg approach takes advantage of the critical role of Tregs, which are natural suppressors of autoimmunity, as well as adverse or excessive immune responses. Severe side effects that commonly seen in the CAR therapy of cancer patients, such as the cytokine-release syndrome and neurologic toxicity(42), have not been reported in recent clinical trials for regulatory T-cell therapy (3). Nevertheless, the principle of advanced design, such as inducible CAR (43,44) or iC9 CAR (45), can be applied to BAR design as well if extra safety is warranted. In addition, gene editing tools like TALENs, and CRISPR/Cas9 can be used to remove the endogenous TCR and make universal BAR-Tregs available from a generic donor.
In summary, we report here a novel BAR Treg therapy approach for selectively targeting FVIII-specific B cells and suppression of an adverse immune response. Proof of principle xenogeneic experiments suggest that BAR Tregs are highly effective in preventing anti-FVIII antibody formation in response to active immunization. Successful translation of this approach may greatly benefit HA patients at high risk of inhibitor development. If successful, it can also be adapted to address other unwanted humoral immune responses.
Supplementary Material
Key points.
-
➢
Human regulatory T cells were engineered to express specific antigens and suppress B-cell responses.
-
➢
Human BAR-regulatory T-cell therapy specifically suppressed anti-FVIII antibody responses in vitro and in vivo by targeting B cells
Acknowledgments
The authors would like to thank Drs. Hong Wang and Si Hyug Jang for their technical support in this study. We also thank Dr. Birgit M. Reipert (Baxalta Innovation GmbH, Vienna, Austria) for providing recombinant FVIII and Dr. Pete Lollar for the anti-human FVIII monoclonals.
This work was funded in part by NIH grants RO1 HL126727 and R21 HL127495 to DWS, and the Bayer Hemophilia Special Project Award to AHZ.
References
- 1.Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nature reviews Immunology. 2008;8:523–532. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bluestone JA. Regulatory T-cell therapy: is it ready for the clinic? Nature reviews Immunology. 2005;5:343–349. doi: 10.1038/nri1574. [DOI] [PubMed] [Google Scholar]
- 3.Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, Herold KC, Lares A, Lee MR, Li K, Liu W, Long SA, Masiello LM, Nguyen V, Putnam AL, Rieck M, Sayre PH, Tang Q. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Science translational medicine. 2015;7:315ra189. doi: 10.1126/scitranslmed.aad4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang Q, Bluestone JA, Kang SM. CD4(+)Foxp3(+) regulatory T cell therapy in transplantation. Journal of molecular cell biology. 2012;4:11–21. doi: 10.1093/jmcb/mjr047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brunstein CG, Blazar BR, Miller JS, Cao Q, Hippen KL, McKenna DH, Curtsinger J, McGlave PB, Wagner JE. Adoptive transfer of umbilical cord blood-derived regulatory T cells and early viral reactivation. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2013;19:1271–1273. doi: 10.1016/j.bbmt.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brusko TM, Koya RC, Zhu S, Lee MR, Putnam AL, McClymont SA, Nishimura MI, Han S, Chang LJ, Atkinson MA, Ribas A, Bluestone JA. Human antigen-specific regulatory T cells generated by T cell receptor gene transfer. PloS one. 2010;5:e11726. doi: 10.1371/journal.pone.0011726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, Broady R, Levings MK. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. The Journal of clinical investigation. 2016;126:1413–1424. doi: 10.1172/JCI82771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ettinger RA, James EA, Kwok WW, Thompson AR, Pratt KP. Lineages of human T-cell clones, including T helper 17/T helper 1 cells, isolated at different stages of anti-factor VIII immune responses. Blood. 2009;114:1423–1428. doi: 10.1182/blood-2009-01-200725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim YC, Zhang AH, Su Y, Rieder SA, Rossi RJ, Ettinger RA, Pratt KP, Shevach EM, Scott DW. Engineered antigen-specific human regulatory T cells: immunosuppression of FVIII-specific T- and B-cell responses. Blood. 2015;125:1107–1115. doi: 10.1182/blood-2014-04-566786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoon J, Schmidt A, Zhang AH, Konigs C, Kim YC, Scott DW. FVIII-specific human chimeric antigen receptor T-regulatory cells suppress T- and B-cell responses to FVIII. Blood. 2017;129:238–245. doi: 10.1182/blood-2016-07-727834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qian J, Collins M, Sharpe AH, Hoyer LW. Prevention and treatment of factor VIII inhibitors in murine hemophilia A. Blood. 2000;95:1324–1329. [PubMed] [Google Scholar]
- 12.Bi L, Lawler AM, Antonarakis SE, High KA, Gearhart JD, Kazazian HH., Jr Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A. Nature genetics. 1995;10:119–121. doi: 10.1038/ng0595-119. [DOI] [PubMed] [Google Scholar]
- 13.Kim YC, Bhairavabhotla R, Yoon J, Golding A, Thornton AM, Tran DQ, Shevach EM. Oligodeoxynucleotides stabilize Helios-expressing Foxp3+ human T regulatory cells during in vitro expansion. Blood. 2012;119:2810–2818. doi: 10.1182/blood-2011-09-377895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hausl C, Ahmad RU, Schwarz HP, Muchitsch EM, Turecek PL, Dorner F, Reipert BM. Preventing restimulation of memory B cells in hemophilia A: a potential new strategy for the treatment of antibody-dependent immune disorders. Blood. 2004;104:115–122. doi: 10.1182/blood-2003-07-2456. [DOI] [PubMed] [Google Scholar]
- 15.Zhang AH, Skupsky J, Scott DW. Effect of B-cell depletion using anti-CD20 therapy on inhibitory antibody formation to human FVIII in hemophilia A mice. Blood. 2011;117:2223–2226. doi: 10.1182/blood-2010-06-293324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lollar P. Pathogenic antibodies to coagulation factors. Part one: factor VIII and factor IX. Journal of thrombosis and haemostasis : JTH. 2004;2:1082–1095. doi: 10.1111/j.1538-7836.2004.00802.x. [DOI] [PubMed] [Google Scholar]
- 17.Scandella D, Mahoney S DeGraaf, Mattingly M, Roeder D, Timmons L, Fulcher CA. Epitope mapping of human factor VIII inhibitor antibodies by deletion analysis of factor VIII fragments expressed in Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America. 1988;85:6152–6156. doi: 10.1073/pnas.85.16.6152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lei TC, Scott DW. Induction of tolerance to factor VIII inhibitors by gene therapy with immunodominant A2 and C2 domains presented by B cells as Ig fusion proteins. Blood. 2005;105:4865–4870. doi: 10.1182/blood-2004-11-4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bray GL, Kroner BL, Arkin S, Aledort LW, Hilgartner MW, Eyster ME, Ragni MV, Goedert JJ. Loss of high-responder inhibitors in patients with severe hemophilia A and human immunodeficiency virus type 1 infection: a report from the Multi-Center Hemophilia Cohort Study. American journal of hematology. 1993;42:375–379. doi: 10.1002/ajh.2830420408. [DOI] [PubMed] [Google Scholar]
- 20.Evans GD, Mendelson GM, Lever AM, Baglin TP. Development of autoantibodies and factor VIII inhibitor in an HIV-infected haemophiliac following treatment with combination anti-retroviral therapy. British journal of haematology. 1998;102:1382–1383. [PubMed] [Google Scholar]
- 21.Matsui H, Shibata M, Brown B, Labelle A, Hegadorn C, Andrews C, Chuah M, VandenDriessche T, Miao CH, Hough C, Lillicrap D. A murine model for induction of long-term immunologic tolerance to factor VIII does not require persistent detectable levels of plasma factor VIII and involves contributions from Foxp3+ T regulatory cells. Blood. 2009;114:677–685. doi: 10.1182/blood-2009-03-202267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sack BK, Merchant S, Markusic DM, Nathwani AC, Davidoff AM, Byrne BJ, Herzog RW. Transient B cell depletion or improved transgene expression by codon optimization promote tolerance to factor VIII in gene therapy. PloS one. 2012;7:e37671. doi: 10.1371/journal.pone.0037671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Du LM, Nurden P, Nurden AT, Nichols TC, Bellinger DA, Jensen ES, Haberichter SL, Merricks E, Raymer RA, Fang J, Koukouritaki SB, Jacobi PM, Hawkins TB, Cornetta K, Shi Q, Wilcox DA. Platelet-targeted gene therapy with human factor VIII establishes haemostasis in dogs with haemophilia A. Nat Commun. 2013;4:2773. doi: 10.1038/ncomms3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lei TC, Su Y, Scott DW. Tolerance induction via a B-cell delivered gene therapy-based protocol: optimization and role of the Ig scaffold. Cellular immunology. 2005;235:12–20. doi: 10.1016/j.cellimm.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 25.Peng B, Ye P, Rawlings DJ, Ochs HD, Miao CH. Anti-CD3 antibodies modulate anti-factor VIII immune responses in hemophilia A mice after factor VIII plasmid-mediated gene therapy. Blood. 2009;114:4373–4382. doi: 10.1182/blood-2009-05-217315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Waters B, Qadura M, Burnett E, Chegeni R, Labelle A, Thompson P, Hough C, Lillicrap D. Anti-CD3 prevents factor VIII inhibitor development in hemophilia A mice by a regulatory CD4+CD25+-dependent mechanism and by shifting cytokine production to favor a Th1 response. Blood. 2009;113:193–203. doi: 10.1182/blood-2008-04-151597. [DOI] [PubMed] [Google Scholar]
- 27.Oliveira VG, Agua-Doce A, Lafaille MA Curotto de, Lafaille JJ, Graca L. Adjuvant facilitates tolerance induction to factor VIII in hemophilic mice through a Foxp3-independent mechanism that relies on IL-10. Blood. 2013;121:3936–3945. S3931. doi: 10.1182/blood-2012-09-457135. [DOI] [PubMed] [Google Scholar]
- 28.Moghimi B, Sack BK, Nayak S, Markusic DM, Mah CS, Herzog RW. Induction of tolerance to factor VIII by transient co-administration with rapamycin. Journal of thrombosis and haemostasis : JTH. 2011;9:1524–1533. doi: 10.1111/j.1538-7836.2011.04351.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maldonado RA, LaMothe RA, Ferrari JD, Zhang AH, Rossi RJ, Kolte PN, Griset AP, O’Neil C, Altreuter DH, Browning E, Johnston L, Farokhzad OC, Langer R, Scott DW, Andrian UH von, Kishimoto TK. Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:E156–165. doi: 10.1073/pnas.1408686111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu CL, Ye P, Lin J, Djukovic D, Miao CH. Long-term tolerance to factor VIII is achieved by administration of interleukin-2/interleukin-2 monoclonal antibody complexes and low dosages of factor VIII. Journal of thrombosis and haemostasis : JTH. 2014;12:921–931. doi: 10.1111/jth.12576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Su J, Sherman A, Rogers GL, Liao G, Hoffman BE, Leong KW, Terhorst C, Daniell H, Herzog RW. Plant-based oral tolerance to hemophilia therapy employs a complex immune regulatory response including LAP+CD4+ T cells. Blood. 2015;125:2418–2427. doi: 10.1182/blood-2014-08-597070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sarkar D, Biswas M, Liao G, Seay HR, Perrin GQ, Markusic DM, Hoffman BE, Brusko TM, Terhorst C, Herzog RW. Ex Vivo Expanded Autologous Polyclonal Regulatory T Cells Suppress Inhibitor Formation in Hemophilia. Mol Ther Methods Clin Dev. 2014;1 doi: 10.1038/mtm.2014.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hausl C, Ahmad RU, Sasgary M, Doering CB, Lollar P, Richter G, Schwarz HP, Turecek PL, Reipert BM. High-dose factor VIII inhibits factor VIII-specific memory B cells in hemophilia A with factor VIII inhibitors. Blood. 2005;106:3415–3422. doi: 10.1182/blood-2005-03-1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, Di Zenzo G, Lanzavecchia A, Seykora JT, Cotsarelis G, Milone MC, Payne AS. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. 2016;353:179–184. doi: 10.1126/science.aaf6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bystry RS, Aluvihare V, Welch KA, Kallikourdis M, Betz AG. B cells and professional APCs recruit regulatory T cells via CCL4. Nature immunology. 2001;2:1126–1132. doi: 10.1038/ni735. [DOI] [PubMed] [Google Scholar]
- 36.Zhao DM, Thornton AM, DiPaolo RJ, Shevach EM. Activated CD4+CD25+ T cells selectively kill B lymphocytes. Blood. 2006;107:3925–3932. doi: 10.1182/blood-2005-11-4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gotot J, Gottschalk C, Leopold S, Knolle PA, Yagita H, Kurts C, Ludwig-Portugall I. Regulatory T cells use programmed death 1 ligands to directly suppress autoreactive B cells in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:10468–10473. doi: 10.1073/pnas.1201131109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J Immunol. 2005;174:1783–1786. doi: 10.4049/jimmunol.174.4.1783. [DOI] [PubMed] [Google Scholar]
- 39.Ashley CW, Baecher-Allan C. Cutting Edge: Responder T cells regulate human DR+ effector regulatory T cell activity via granzyme B. J Immunol. 2009;183:4843–4847. doi: 10.4049/jimmunol.0900845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tran DQ, Glass DD, Uzel G, Darnell DA, Spalding C, Holland SM, Shevach EM. Analysis of adhesion molecules, target cells, and role of IL-2 in human FOXP3+ regulatory T cell suppressor function. J Immunol. 2009;182:2929–2938. doi: 10.4049/jimmunol.0803827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klein L, Khazaie K, von Boehmer H. In vivo dynamics of antigen-specific regulatory T cells not predicted from behavior in vitro. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:8886–8891. doi: 10.1073/pnas.1533365100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127:3321–3330. doi: 10.1182/blood-2016-04-703751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sakemura R, Terakura S, Watanabe K, Julamanee J, Takagi E, Miyao K, Koyama D, Goto T, Hanajiri R, Nishida T, Murata M, Kiyoi H. A Tet-On Inducible System for Controlling CD19-Chimeric Antigen Receptor Expression upon Drug Administration. Cancer immunology research. 2016;4:658–668. doi: 10.1158/2326-6066.CIR-16-0043. [DOI] [PubMed] [Google Scholar]
- 44.Foster AE, Mahendravada A, Shinners NP, Chang WC, Crisostomo J, Lu A, Khalil M, Morschl E, Shaw JL, Saha S, Duong MT, Collinson-Pautz MR, Torres DL, Rodriguez T, Pentcheva-Hoang T, Bayle JH, Slawin KM, Spencer DM. Regulated Expansion and Survival of Chimeric Antigen Receptor-Modified T Cells Using Small Molecule-Dependent Inducible MyD88/CD40. Molecular therapy : the journal of the American Society of Gene Therapy. 2017;25:2176–2188. doi: 10.1016/j.ymthe.2017.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Straathof KC, Pule MA, Yotnda P, Dotti G, Vanin EF, Brenner MK, Heslop HE, Spencer DM, Rooney CM. An inducible caspase 9 safety switch for T-cell therapy. Blood. 2005;105:4247–4254. doi: 10.1182/blood-2004-11-4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.