

SUMMARY
The biogenesis of the RNA payload of mature sperm is of great interest, as RNAs delivered to the zygote at fertilization can affect early development. Here, we tested the hypothesis that small RNAs are trafficked to mammalian sperm during the process of post-testicular maturation in the epididymis. By characterizing small RNA dynamics during germ cell maturation in mice, we confirm and extend prior observations that sperm undergo a dramatic switch in RNA payload from piRNAs to tRNA fragments (tRFs) upon exiting the testis and entering the epididymis. Small RNA delivery to sperm could be recapitulated in vitro by incubating testicular spermatozoa with caput epididymosomes. Finally, tissue-specific metabolic labeling of RNAs in intact mice definitively shows that mature sperm carry RNAs that were originally synthesized in the epididymal epithelium. These data demonstrate that soma-germline RNA transfer occurs in male mammals, most likely via vesicular transport from the epididymis to maturing sperm.
In-Brief/eTOC Blurb
Recent studies suggest that sperm carry RNAs first synthesized in epididymal somatic cells. Sharma et al. test this hypothesis, characterizing small RNA population dynamics during sperm maturation. They show that caput epididymosomes can deliver RNAs to immature sperm in vitro and track RNAs in vivo from epididymis to sperm using metabolic labeling.
INTRODUCTION
Inheritance of phenotypic changes in the absence of any change in DNA is known as epigenetic inheritance, and is essential for cell state inheritance during mitosis. Epigenetic information is not only inherited during cell division, as at least some epigenetic information can be transmitted from one generation to the next in multicellular organisms, a mechanistically complex process that requires epigenetic information to be maintained throughout the disruptive processes of gametogenesis, fertilization, and early embryonic development. Although decades of study of cell state inheritance and other cases of epigenetic inheritance have identified several major molecular conduits of epigenetic information – chromatin structure, small RNAs, DNA modifications, prions, and transcription factors – much remains to be understood regarding the establishment and erasure of epigenetic information during gametogenesis and early development.
Small (<40 nt) RNAs are one of the best-established carriers of epigenetic information, and play key roles in epigenetic inheritance paradigms in C. elegans (Fire et al., 1998), A. thaliana (Arteaga-Vazquez and Chandler, 2010; Hamilton and Baulcombe, 1999; Zilberman et al., 2003), S. pombe (Volpe et al., 2002), D. melanogaster (Brennecke et al., 2008; de Vanssay et al., 2012), and mammals (Chen et al., 2016a; Gapp et al., 2014; Rassoulzadegan et al., 2006; Rodgers et al., 2015; Sharma et al., 2016). Multiple distinct families of small RNAs have been described which differ from one another in their biogenesis and in their regulatory functions, and include well-studied species such as microRNAs, endo-siRNAs, and the germline-enriched class of piRNAs which are crucial for self-nonself recognition during gametogenesis (Aravin et al., 2006; Batista et al., 2008; Brennecke et al., 2007; Mochizuki et al., 2002; Shirayama et al., 2012; Vagin et al., 2006). In addition, a number of less-studied small RNA species have been described, including cleavage products of various noncoding RNAs including tRNAs – these latter RNAs are commonly referred to as tRNA fragments, or “tRFs” (Anderson and Ivanov, 2014; Lee et al., 2009; Sobala and Hutvagner, 2011). While tRFs have been found in numerous small RNA sequencing datasets, they appear to be particularly abundant in contexts related to reproduction, as for example 3′ tRFs are the primary cargo of the Tetrahymena Piwi-related argonaute Tiwi12 (Couvillion et al., 2010), while 5′ tRFs comprise the vast majority (~80%) of readily-cloned small RNAs in mature mammalian sperm (Chen et al., 2016a; Garcia-Lopez et al., 2014; Peng et al., 2012; Rompala et al., 2018; Sharma et al., 2016).
The mechanisms responsible for the biogenesis of the small RNA payload of mammalian sperm are not completely understood. Small RNA dynamics have been extensively studied during the process of testicular spermatogenesis, where three Piwi-clade argonaute proteins (MIWI, MILI, and MIWI2) orchestrate two major waves of piRNA biosynthesis (Aravin et al., 2006; Aravin et al., 2007; Kuramochi-Miyagawa et al., 2004; Li et al., 2013). Initially, “pre-pachytene” piRNAs – primarily derived from repeat elements – are expressed beginning in spermatogonia (Aravin et al., 2007), followed by a wave of unique pachytene piRNAs which are processed from 214 long noncoding precursor transcripts (Li et al., 2013). Given that small RNAs found in testes are almost entirely comprised of piRNAs, it is curious that the vast majority of small RNAs present in mature fertilization-competent sperm are tRNA fragments. Indeed, tRFs are vanishingly scarce (~2% of all small RNAs) in intact testes (Peng et al., 2012; Sharma et al., 2016) despite the fact that testes are primarily comprised of gametes at varying stages of development, raising the broader question of how and when small RNAs are gained or lost during post-testicular maturation.
We recently uncovered a potential role for the epididymis – a long convoluted tubular structure where mammalian sperm undergo post-testicular maturation – in modulating the small RNA repertoire of maturing sperm (Sharma et al., 2016). We found that tRNA cleavage occurs robustly in the epididymis, and that small vesicles secreted by the epididymal epithelium, known as epididymosomes (Belleannee et al., 2013; Krapf et al., 2012; Sullivan et al., 2007; Sullivan and Saez, 2013), carry a very similar population of small RNAs to that gained by sperm during epididymal transit (Sharma et al., 2016). Moreover, as sperm successively pass from caput (proximal) to cauda (distal) epididymis, sperm gain specific small RNAs that are locally enriched in the underlying epithelium. Finally, we and others (Reilly et al., 2016) have shown that epididymosomes can deliver small RNAs to relatively immature caput epididymal sperm in vitro, suggesting that the sperm small RNA repertoire is shaped by soma-to-germline trafficking of small RNAs.
Here, we rigorously test this hypothesis, characterizing developmental dynamics of small RNAs across multiple purified sperm populations spanning testicular and post-testicular development. We detected very low levels of tRFs in all testicular germ cell populations, including mature testicular spermatozoa, confirming that tRFs are only gained upon entry into the epididymis. This maturation step could be partially recapitulated in vitro, as we show that epididymosomes from the proximal epididymis can deliver a wide variety of small RNAs, including both microRNAs and tRFs, to testicular spermatozoa. Finally, we use a chemogenetic approach to definitively identify the tissue of origin for small RNAs present in mature cauda epididymal sperm. Using epididymis-specific expression of UPRT to enable 4-thiouracil labeling of small RNAs synthesized in the caput epididymis, we show that mature cauda sperm carry small RNAs first synthesized in the epididymis epithelium. Taken together, these data demonstrate that small RNAs are trafficked from soma to germline in mammals, and provide further evidence that the small RNA payload of mature sperm is shaped by successive waves of epididymosomal delivery during the post-testicular maturation process.
RESULTS
Improved characterization of the small RNA contents of mouse sperm
Increasing interest in the potential for sperm RNAs to serve regulatory functions in the early embryo has motivated a number of groups to characterize the mammalian sperm RNA payload via deep sequencing methods. Although many studies yield similar findings – with 5′ tRNA fragments representing the vast majority (~80%) of sperm small RNAs (Chen et al., 2016a; Garcia-Lopez et al., 2014; Peng et al., 2012; Rompala et al., 2018; Sharma et al., 2016; Yang et al., 2016) – there is nonetheless substantial variation between studies, some of which report significantly lower tRF levels (de Castro Barbosa et al., 2016; Kawano et al., 2012). Beyond trivial protocol differences such as the size range of purified RNAs, RNA recovery depends on many additional factors, ranging from the purity of the sperm preparation to effects of nucleotide modifications on RNA adaptor ligation or cloning. We therefore sought to characterize the effects of several protocol steps – sperm washing, sperm head and tail separation, RNA isolation and enzyme treatments – on recovery of 18–40 nt small RNAs from murine sperm samples. As the sperm small RNA payload is dramatically remodeled at the transition from testicular spermatogenesis to post-testicular sperm maturation in the epididymis, we characterized the effects of key protocol steps not only on RNA recovery from the tRF-dominated payload of mature cauda sperm, but also on recovery of the piRNA-dominated cargo of selected testicular sperm populations. Overall, we assessed the RNA contents of six germ cell populations obtained from throughout the reproductive tract of sexually mature (8–12 week old) male mice, as well as intact testis and cauda epididymis. Sperm populations included mature sperm obtained from caput and cauda epididymis, primary spermatocytes, two fractions of round spermatids (Sharma et al., 2016), and a previously-unsequenced population of testicular spermatozoa purified using Percoll gradients (Methods, Supplemental Figure S1A).
First, given that sperm samples are typically washed following isolation from epididymal contents, we asked how different wash conditions affect small RNA recovery. 10–12 week old male mice were sacrificed, and the cauda epididymis was dissected with cuts to the corpus epididymis and vas deferens immediately adjacent to the bulk of the cauda. Epididymal contents were gently squeezed out into tissue culture media, and sperm from four animals were pooled, gently pelleted and washed with PBS, then split into four aliquots: one aliquot was immediately pelleted for RNA isolation, and the other three subjected to low, medium, or high stringency detergent washes (Supplemental Figures S1B–C, Methods). Total RNA was extracted, and 18–40 nt small RNAs were gel-purified and cloned using the Illumina Truseq protocol. Overall, we find similar RNAs captured consistently across all four samples, with 5′ tRFs representing the most abundant RNA species in all four datasets (Table S1). Relative to unwashed sperm, any detergent washing step resulted in a decreased overall abundance of microRNAs, resulting from loss of a specific subset of microRNAs such as let-7 family members (Supplemental Figures S1B–C). This presumably reflects the removal of excess adherent epididymosomes or ribonucleoprotein complexes from the surface of sperm – indeed, the specific microRNAs lost after washing include species that are particularly abundant in partially-purified epididymosome preparations relative to sperm (Sharma et al., 2016). After higher-stringency washing, we obtained lower levels of rRNA fragments and snoRNA fragments, potentially resulting from permeabilization of sperm and loss of cytoplasmic contents. Indeed, while sperm largely remained intact following the low stringency washes, we observed that sperm washed at the intermediate stringency were often broken into head and tail sections (not shown), consistent with these conditions at least partially disrupting the sperm membrane. Overall, these analyses demonstrate that overall RNA recovery is not greatly affected by the washing protocol, but suggest that a low stringency detergent wash should be used to remove adherent material from the sperm surface to provide the most reproducible results between different investigators.
We next investigated the subcellular localization of various small RNAs, separating sperm into heads and tails by sonication followed by sucrose gradient fractionation (Figure 1A). Small RNA profiles were broadly similar between heads and tails (Figure 1B), with a few notable differences for cauda sperm samples. Most dramatically, we find that piRNAs are highly enriched (~5-fold) in the tail preparations, consistent with the fact that remnants of the chromatoid body, central to piRNA biogenesis, localize to the sperm midpiece during sperm maturation (de Mateo and Sassone-Corsi, 2014; Meikar et al., 2011). More surprisingly, microRNAs are also generally enriched in tails relative to sperm heads, although this enrichment is less dramatic than that observed for piRNAs. Other RNA classes exhibited modest preferences for the head vs. tail – tRFs were overall moderately (~1.5-fold) enriched in sperm heads (Schuster et al., 2016), but a handful of specific tRFs (notably tRF-Gly-TCC) were relatively more abundant in tails (Table S2). As intracytoplasmic sperm injection (ICSI) – used in humans for a subset of assisted reproduction cases – typically involves injection of a sperm head only into the oocyte, the differences documented here should motivate further investigation into potential impacts of ICSI on preimplantation development.
Figure 1. Subcellular localization and 3′ end modifications of sperm RNAs.

A) Separation of sperm into head and tail-enriched fractions. Representative images of cauda epididymal sperm, showing intact sperm (top), along with head and tail-enriched fractions, as indicated. Note that head and tail fractions are highly enriched but are not completely pure, as occasional sperm tails can be appreciated in the head preparation. Red scale bar = 10 μm.
B) Subcellular fractionation of sperm small RNAs. Coarse differences between sperm head and tail preparations are shown for testicular sperm and cauda epididymal sperm. Pie charts show small RNA populations for sperm heads and tails, along with “intact sperm” which have been sonicated for head/tail separation but not separated. Notable here is the enrichment of piRNAs and microRNAs in the tails of cauda sperm, in contrast to the tRNA fragment enrichment in sperm heads.
C) Resolution of cyclic 2′-3′ phosphates by the phosphatase activity of polynucleotide kinase (PNK).
D) Effects of PNK treatment on recovery of major small RNA populations from cauda epididymis or cauda sperm.
E) PNK treatment results in a shift towards longer RNA inserts in small RNA-Seq data. Data are shown for cauda epididymis and cauda sperm.
F) Effects of PNK treatment on lengths of 5′ fragments of specific tRNAs, as indicated. Data are shown for cauda sperm RNAs – similar behavior was observed for cauda epididymis and caput sperm tRFs. For each tRNA, the three anticodon nucleotides are indicated on the x axis. See also Supplemental Figures S1–2.
Finally, we noted that in several biological systems, tRNAs are cleaved by nucleases of the RNase A and T families (Andersen and Collins, 2012; Emara et al., 2010; Lee and Collins, 2005; Yamasaki et al., 2009), which are known to leave either 2′-3′ cyclic phosphate or 3′ phosphate moieties at the end of the 5′ fragment, both of which can interfere with small RNA cloning protocols. We therefore repeated our characterization of sperm small RNAs following treatment with T4 Polynucleotide Kinase (PNK) in the absence of ATP, which resolves 2′-3′ cyclic phosphates left at the 3′ end of RNaseA and RNAseT family cleavage products (Figure 1C). PNK treatment of all samples tested – intact testis, cauda epididymis, caput sperm, and cauda sperm – resulted in a dramatic increase in rRNA-mapping reads (Supplemental Figure S2A, Table S3). In addition, for all samples with the exception of the piRNA-dominated testes, PNK treatment revealed a previously-unappreciated population of tRNA fragments, with 5′ tRFs becoming 2–4-fold more abundant (normalized relative to all non-rRNA reads) in PNK-treated RNA samples (Figure 1D and Supplemental Figure S2B). Furthermore, PNK treatment resulted in a shift in the RNA length distribution to longer species (Figure 1E). Closer inspection of tRNA-mapping reads in these libraries revealed two new entities, both mapping to tRNA 5′ ends. For most tRFs, PNK treatment revealed a population of slightly larger (~33–36 nt) tRNA cleavage products than the predominant ~28–32 nt species observed in no-PNK libraries, with this new population typically resulting from cleavage within or immediately downstream of the anticodon (Figure 1F). For a smaller subset of tRNAs, we observed ~19–22 nt RNAs representing D loop cleavage that are extremely scarce in no-PNK libraries (as shown for tRNA-Leu-CAG, Figure 1F). These data reveal what are presumably initial cleavage events for an RNAseA or RNAseT family nuclease at accessible tRNA loop structures, with the previously-described shorter 28–32 nt fragments representing secondary degradation or trimming products.
Together, our data provide a more detailed picture of small RNA populations in the male reproductive tract and in sperm, and provide experimental guidance for studies on environmental effects on the germline epigenome.
Small RNA dynamics during sperm development and maturation
Having a clearer picture of the small RNA populations present in mature sperm, we next turned to the dynamics of small RNA biogenesis during sperm development and maturation. As noted in the Introduction, sperm isolated from either the cauda (distal) or caput (proximal) epididymis carry a small RNA payload comprised primarily of tRNA fragments (tRFs). In contrast, tRNA cleavage as assayed both by small RNA sequencing and by Northern blot is nearly undetectable in intact testes, where the vast majority of small RNAs are piRNAs (Peng et al., 2012; Sharma et al., 2016). Although these data suggest that tRFs are gained upon entry into epididymis, it remains formally possible that tRFs are gained during the last stages of testicular spermiogenesis as round spermatids mature into testicular spermatozoa.
Analysis of the small RNA payload of six germ cell populations – primary spermatocytes, two round spermatid populations, testicular spermatozoa (Supplemental Figure S1A), and caput and cauda epididymal sperm – recapitulates prior findings, with piRNAs dominating the small RNA repertoire of spermatocytes and round spermatids, and tRFs being the primary small RNA population in both caput and cauda epididymal sperm (Figure 2A, Table S4). Turning to the previously-unexamined testicular spermatozoa, we find that even at this late stage of maturation these sperm resemble other testicular populations rather than epididymal sperm – they continue to carry high levels of piRNAs (~80% of small RNA reads), having yet to gain abundant tRFs (~5% of reads). The RNA payload of mammalian sperm therefore shifts from piRNAs to tRFs at some point after sperm exit the testis for the epididymis.
Figure 2. Bulk small RNA dynamics across sperm development.

A) Bulk changes to small RNA payload throughout testicular and post-testicular sperm development. Pie charts as in Figure 1B. For these six germ cell populations, small RNAs were cloned without PNK treatment, as PNK had essentially no effect on testicular RNA populations beyond increasing the fraction of rRNA-mapping reads (Supplemental Figure S2A).
B) Validation of deep sequencing data by q-RT-PCR. Using U6 ncRNA as a normalization control, we assayed the levels of the indicated small RNAs in triplicate samples of testicular sperm, caput sperm, and cauda sperm. Bars show U6-normalized changes in small RNA abundance (y axis, log2 scale) relative to testicular sperm. These data confirm the gain of tRFs observed in epididymal sperm relative to testicular sperm, as well as the strong cauda-specific enrichment for tRF-Val-CAC. miR-10b also exhibits the dramatic gain in the epididymis observed in deep sequencing data, while the surprising loss of miR-34c in caput sperm followed by its restoration in cauda sperm is also confirmed. In contrast, let-7f levels are relatively stable across sperm maturation.
In principle, the apparent gain of tRNA fragments in epididymal sperm could simply result from the massive loss of piRNAs, leaving behind only the previously rare population of tRNA fragments. To distinguish between “degradation only” and a “degradation plus tRF gain” models, we spiked in several exogenous small RNAs at defined concentrations per sperm and deep sequenced testicular and caput sperm small RNAs. We consistently find a ~2-fold decrease in overall RNA levels as testicular sperm enter the epididymis (Supplemental Figure S3), which cannot explain the ~10-fold enrichment of tRNA fragments in caput sperm. In addition, other sperm small RNAs (such as microRNAs) do no change in abundance at a similar scale as tRFs, again arguing that piRNA degradation alone cannot explain changes in RNA contents as sperm enter the epididymis. Finally, we confirm that cauda sperm gain tRFs relative to testicular sperm by q-RT-PCR, using U6 ncRNA (Nixon et al., 2015) as an internal control (Figure 2B). Together, our results are consistent with a “piRNA degradation plus tRF gain” model for the dramatic remodeling of sperm small RNA payload during epididymal maturation.
In addition to the bulk differences in abundance of broad classes of small RNAs, we also find marked differences in specific individual RNA species present in each germ cell population, confirming for example the previously-described gain in tRF-Val-CAC that occurs as sperm transit from caput to cauda epididymis (Sharma et al., 2016). In contrast to the highly tissue-restricted expression of piRNAs and tRFs, individual microRNAs exhibit more diverse behaviors during spermatogenesis and sperm maturation. To investigate microRNA dynamics in more detail, we renormalized microRNA abundances according to total microRNA levels for each sperm population, rather than to all small RNAs, to avoid the confounding normalization effects of bulk piRNA/tRF loss and gain. This analysis revealed a range of dynamic behaviors of individual microRNAs, ranging from relatively stable microRNAs such as let-7a and miR-181a/b to microRNAs exhibiting dramatic changes in abundance throughout development (Figure 3). Many microRNAs were relatively rare in testicular sperm populations, then gained upon entry into the epididymis, with miR-21a, miR-29c, miR-199a, miR-200b/c, miR-10a/b and many others dramatically increasing in relative abundance during epididymal maturation. This is consistent with these RNAs potentially being trafficked to epididymal sperm alongside the bulk delivery of tRFs (Reilly et al., 2016; Sharma et al., 2016).
Figure 3. MicroRNA dynamics across sperm development.
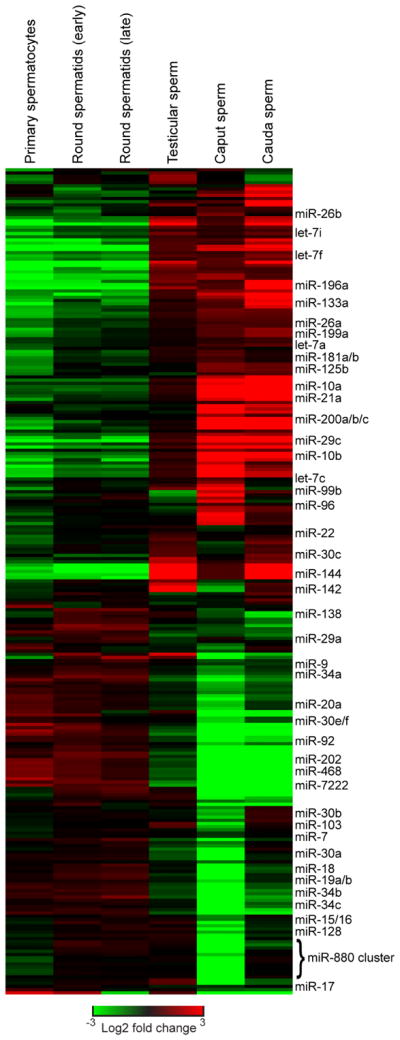
MicroRNA abundances for all six germ cell populations were re-normalized only to the total microRNAs at a given developmental stage, then zero-centered – re-normalized according to the median abundance across the six stages analyzed.
The contrasting behavior, with microRNAs expressed during testicular spermatogenesis being lost/diluted at later developmental stages, was also common. Curiously, for a surprisingly large subset of these microRNAs, the decrease in abundance observed in caput sperm was at least partially reversed later in the epididymis. In these cases, the microRNAs in question are abundant in testicular sperm as well as cauda sperm, but are scarce specifically in caput sperm. Interestingly, the majority of such microRNAs originate from genomic microRNA clusters of various sizes, ranging from small clusters (miR-15/16, the “oncomiR”17–92 cluster, etc.) up to the megabase-scale imprinted X-linked miR-880 cluster. The changes in abundance of these microRNAs can be quite dramatic – many members of the miR-880 cluster (miR-871, miR-465c) are present at ~500 reads per million (across all small RNAs, not just microRNAs) in all four testicular germ cell populations, dropping to ~25 ppm in caput sperm before rebounding to ~400 ppm in cauda sperm. These changes in abundance cannot be explained by global changes in sperm RNA contents, and U6-normalized q-RT-PCR (Figure 2B) confirmed the caput-specific loss of miR-34c observed in the deep sequencing dataset. Instead, this behavior is most consistent with these microRNAs initially being lost (by degradation or cytoplasmic elimination) upon epididymal entry, followed by a second load of these miRNAs either being generated in situ in cauda sperm from their precursor molecules, or shipped from epididymis epithelium to maturing sperm by epididymosomes. Consistent with the latter hypothesis, many of the microRNAs that are specifically scarce in caput sperm are abundant in cauda epididymosomes (Table S4). These data thus uncover surprisingly complex small RNA dynamics during sperm development, and further underscore the major role of epididymal transit in modulation of the sperm epigenome.
Caput epididymosomes deliver small RNAs to testicular spermatozoa
Our data confirm and extend prior reports showing major differences between the small RNAs generated during testicular spermatogenesis and those gained during epididymal transit. What is the mechanism by which tRFs and other small RNAs are gained as sperm enter the epididymis? We previously showed that epididymosomes secreted by the epithelium of cauda epididymis have a similar RNA payload to that of mature sperm, and we and others have shown that epididymosomes can deliver small RNAs to relatively “immature” caput sperm (Reilly et al., 2016; Sharma et al., 2016). However, as the RNA repertoire of caput sperm is not particularly distinct from that of cauda sperm – caput sperm already carry high levels of most tRFs, for example – these reconstitution studies could only focus on a small handful of fairly cauda-specific species such as tRF-Val-CAC. In contrast, the RNA payload of testicular spermatozoa, which is dominated by piRNAs, is highly distinct from that of either epididymal sperm population, providing us with a relatively blank slate to more broadly explore the ability of epididymosomes to deliver RNAs to immature sperm.
Testicular sperm were isolated, then either mock treated or incubated for two hours with caput epididymosomes, and resulting sperm were extensively washed to remove any contaminating vesicles (Figure 4A). We first used TaqMan qRT-PCR to assay two prominent tRFs present in epididymosomes but scarce in testicular sperm, finding that epididymosomal delivery results in a ~4–10-fold increase in abundance for these two tRFs (Figure 4B). To more broadly interrogate epididymosomal RNA delivery to sperm, we next carried out deep sequencing of small RNAs from mock and “reconstituted” sperm. Globally, we observed significantly higher levels of tRFs and microRNAs in reconstituted spermatozoa, relative to mock-treated sperm (Figures 4C, Table S5). Although two hours incubation is clearly insufficient to fully establish the caput small RNA payload, this is perhaps unsurprising given that purified caput sperm from intact animals have spent up to several days undergoing this delivery process.
Figure 4. Reconstitution of small RNA delivery to testicular sperm.

A) Experimental schematic. Purified testicular sperm, which carry extremely low levels of tRFs, were incubated with caput epididymosomes for 2 hours, followed by extensive washing. Small RNAs were purified from either mock-treated testicular sperm or reconstituted sperm, and analyzed either by q-RT-PCR or deep sequencing.
B) Delivery of two prominent tRFs to testicular sperm. Taqman q-RT-PCR for the indicated tRFs, with individual replicates plotted (on a log2 y axis) relative to the average level for mock-treated testicular sperm.
C) Deep sequencing of testicular sperm either mock-treated or incubated with caput epididymosomes. Pie charts show average levels of various small RNA classes, revealing increased levels of microRNAs and tRFs delivered to testicular sperm by epididymosomes.
D) Distribution of small RNA changes in sperm following epididymosome fusion. X axis shows the log2 fold difference between reconstituted and mock-treated sperm – positive values indicate delivery by epididymosomes. Importantly, because genome-wide normalization methods assume no global change in RNA abundance, RNAs that are in fact unaltered in the face of an overall gain of RNAs will exhibit a decrease in relative abundance, as observed here for the bulk of piRNAs (average log2 fold decrease of ~0.6). Values over −0.6 therefore indicate gain of RNA during the fusion protocol.
E) Scatterplot of small RNA abundances from deep sequencing data. The strong diagonal for piRNAs indicates RNAs present in testicular sperm that are not affected by the delivery process. Note that essentially all RNAs here either lie along this diagonal, indicating that they are either absent or nearly so in caput epididymosomes, or above the diagonal, indicating widespread delivery of many RNA species to testicular sperm during reconstitution.
More granular analysis of specific small RNA species revealed that the vast majority of piRNAs were unaffected by epididymosomal fusion (Figures 4D–E), consistent with the fact that piRNAs are expressed during testicular spermatogenesis and are largely absent from epididymosomes (and indicating that epididymosomal fusion does not induce piRNA degradation). This is clearly visualized in the scatterplot in Figure 4E, where piRNA abundance is strongly correlated between mock-treated and reconstituted sperm samples. Globally, nearly all small RNAs either fall on the same diagonal as the piRNAs – meaning that they are unaffected by epididymosomal fusion – or above this diagonal, consistent with delivery by epididymosomes. Individual tRFs and microRNAs exhibited a range of behaviors (Figures 4D–E), ranging from those unaffected by epididymosomal incubation and which are generally already abundant in testicular sperm and/or absent from caput epididymosomes, to efficiently-delivered small RNAs that are highly abundant in caput epididymosomes (such as miR-10a/b, miR-148, miR-143, tRF-Glu-CTC, tRF-Gly-GCC, and tRF-His-GTG – Table S5).
These data show that caput epididymosomes are capable of fusing with mature testicular spermatozoa to deliver their small RNA cargo in vitro, and together with the in vivo small RNA dynamics detailed above are most consistent with a mechanism of RNA biogenesis in mammalian sperm where small RNAs generated in the epididymis are trafficked to sperm in epididymosomes.
Chemogenetic tracking of small RNAs from epididymis to sperm
Although multiple observations here and elsewhere are most readily explained by the hypothesis that small RNAs in mature sperm are first generated in the epididymal epithelium, they do not formally rule out the possibility that the small RNAs gained by sperm during epididymal maturation in intact animals are instead processed in situ from precursor molecules first synthesized during testicular spermatogenesis. In order to distinguish between these possibilities, we made use of the “TU tracer” mouse (Gay et al., 2013), in which tissue-specific expression of Cre recombinase is used to drive expression of uracil phosphoribosyltransferase (UPRT) (Figure 5A). Expression of UPRT allows cells to incorporate 4-thiouracil (4-TU) into newly-synthesized RNAs, thereby enabling tissue-specific metabolic labelling of RNA.
Figure 5. Tissue-specific labeling and detection of small RNAs in intact animals.

A) Schematic of the TU tracer locus. In the absence of Cre, GFP is expressed from a ubiquitous promoter, with no UPRT expression. Cre drives LoxP recombination, eliminating GFP and juxtaposing the promoter with HA-UPRT.
B) Western blot showing GFP and HA-UPRT levels in livers and several reproductive tissues isolated from TU-tracer animals not expressing Cre (top) or expressing liver-specific Albumin promoter-driven Cre recombinase (bottom). In the absence of Cre, GFP is expressed from a ubiquitous promoter, with no UPRT expression. In mice expressing liver specific Cre recombinase, HA-UPRT expression is confined to the liver, while the concomitant near-complete elimination of GFP underlines the relatively homogeneous cellular makeup of this tissue. See Supplemental Figure S4A for uncropped Western blot images.
C) Dot blot for RNA isolated from TU-tracer animals not expressing Cre, injected with either 4-TU or vehicle alone. As shown, only positive control (Biotinylated DNA oligo) is detectable upon incubation with Streptavidin-HRP.
D) Dot blot for RNA isolated from Alb-Cre X TU tracer mice injected with two different doses of 4-TU. Upon 4-TU injection RNAs are specifically labeled, in a dose-dependent manner, in liver tissue and not in control tissue (cauda epididymis).
E) SLAM-Seq analysis of 4-TU-labeled RNAs. TU tracer X Alb-Cre animals were either injected with 4-TU (400 mg/kg body weight) or with vehicle (solvent only), and were sacrificed 5–6 hours after the last injection for tissue harvest. Small RNAs were isolated from either cauda epididymis or from liver, and subject to SLAM-Seq, and sequencing reads were mapped to microRNAs using an error-tolerant pipeline. Mismatches were identified for all reads mapping to a given microRNA, and boxplots (box: 25th/50th/75th percentile; whiskers: 10th/90th percentile) show the frequency of various mismatches for mock-injected and 4-TU-injected animals, as indicated.
To assess the efficiency and specificity of this system, we first generated mice expressing UPRT specifically in liver by mating Albumin-Cre (Alb-Cre) mice with TU-tracer mice. Cre-mediated recombination was extremely efficient, with nearly quantitative deletion of the GFP insert and concomitant expression of UPRT (Figure 5B, Supplemental Figure S4A) – remaining GFP likely reflects non-hepatocyte cell populations, such as blood cells, present in the liver. The resultant mice expressed UPRT specifically in liver, and injection of 4-TU in these mice resulted in liver-specific labeling of RNAs, with undetectable background incorporation in other tissues (Figures 5C–D). Northern blot of labeled RNAs revealed 4-TU labeling of a wide range of RNA species including microRNAs (see below), but tRNAs and rRNAs were not efficiently labeled (not shown), either due to poor use of 4-TU by PolI and PolIII, or due to checkpointing of these typically highly-modified RNAs. This protocol therefore enables us to characterize microRNA trafficking in vivo, but unfortunately cannot be used to track tRFs.
Although tracking TU-labeled RNAs in vivo could in principle be accomplished by biotinylation of 4-TU (Figures 5C–D) and deep sequencing of avidin-purified RNAs, we note that even in an abundant tissue such as the liver only a very small fraction of RNA was 4-TU-labeled, and the abundance of any RNAs trafficked between cells would of course be far lower. Along with the fact that sperm overall have very low levels of total RNA, we anticipated extreme difficulty in purification of rare biotinylated RNAs. We therefore turned instead to thiol(S)-linked alkylation for the metabolic sequencing of RNA (SLAM-Seq) (Alberti et al., 2018; Herzog et al., 2017) for characterization of 4-TU-labeled small RNAs. This method is based on chemically modifying the 4-thiouracil in RNA with iodoacetamide to generate a covalent adduct that causes nucleotide misincorporation during reverse transcription, with G misincorporation opposite the modified U (seen as a U->C mutation in downstream deep sequencing) occurring most frequently. This method enables quantitative analysis of 4-TU incorporation in any clonable population of small RNAs, sidestepping inevitable RNA losses during biotin purification. To characterize the signal-to-noise for this method, we first applied SLAM-Seq to small RNAs purified from the liver of Alb-Cre-expressing mice. Background mutation rates in the cloning process were characterized using Alb-Cre mice that had not been injected with 4-TU, while analysis of cauda epididymis in these animals controlled for both cloning errors and for 4-TU incorporation into non-UPRT-expressing cells. As expected, we find that 4-TU incorporation caused a significant increase in U->C misincorporation events (Figure 5E). This required 4-TU injection, was dose-dependent, and required tissue-specific expression of UPRT. Variation in the extent of U->C misincorporation for individual microRNAs reflects in vivo RNA synthesis and decay rates, as well as any effects of local sequence context on RT misincorporation. Together, these results demonstrate our ability to identify RNAs first synthesized in a given genetically-defined cell type.
We therefore applied this system to track microRNAs synthesized in the caput epididymis, using the Defb41 promoter (Bjorkgren et al., 2012) to drive Cre in the most proximal “initial segment” of the caput epididymis (Figure 6A). Mice carrying both the TU tracer cassette and Defb41-Cre exhibited the expected caput-specific expression of UPRT (Figure 6B, Supplemental Figure S4B). Injection of 4-TU into these animals resulted in caput-specific incorporation of 4-TU as assessed by biotinylation of total RNA (Supplemental Figure S4C), and SLAM-Seq analysis of microRNAs purified from uninjected and TU-injected animals confirmed the expected TU-dependent increase in U->C mutation rates in caput epididymis RNAs, along with no change in U->C mutation rates in the testis (Figures 6C–D, Table S6).
Figure 6. Chemogenetic tracking of RNAs from epididymis to sperm.

A) Experimental design. UPRT is expressed specifically in the initial segment of the caput epididymis due to expression of Defb41-Cre. Upon injection of 4-thiouracil (4-TU), RNAs synthesized in this tissue incorporate 4-TU. Animals are injected with 4-TU every 2 days for 9 days (5 injections), and 5 hours after injection on Day 9 sperm are isolated from the cauda epididymis and assayed for 4-TU labeling of small RNAs.
B) Western blots showing HA-UPRT expression specifically in the caput epididymis of Defb41-Cre X TU-tracer animals. Note that in contrast to the near-complete loss of GFP in Alb-Cre animals (Figure 5B), the continued expression of GFP in the caput epididymis is consistent with our expectation that only a limited subsection of the caput epididymis – the initial segment – expresses Defb41. See Supplemental Figure S4B for uncropped Western blot images.
C) SLAM-Seq induces miscloning of uracil in a Cre- and 4-TU-dependent manner. Defb41-Cre X TU-tracer animals were either injected with 4-TU (every other day for 9 days) or sham-injected, and testis, caput epididymis, and cauda epdidiymal sperm were dissected. Small RNAs were purified and subject to SLAM-Seq. Reads were mapped to microRNAs using a mismatch-tolerant custom pipeline (Methods) and nucleotide misreads were compiled for all microRNAs with at least 1000 reads (boxes show median and 25th/75th percentiles, whiskers show 10th and 90th percentiles). 4-TU induces misreading of uracils specifically in the caput epididymis, but not the testis, with U->C being the most common mutation observed (p = 1.3e–42, paired t test). Importantly, U->C mutations were also significantly (p = 7.4e–7, paired t test) elevated in a 4-TU-dependent manner in cauda sperm obtained from animals expressing Cre specifically in the caput epididymis epithelium (bottom panel).
D) Log2 ratio of U->C rates for 4-TU injected vs. uninjected animals, for all microRNAs with at least 1000 reads for the indicated tissues. Box plots show the distribution of log2 fold change in U->C “mutation” rates for the three indicated tissues, with two separate replicates shown. The modest increase in U->C mutation in mature sperm reflects multiple factors: 1) the signal to noise in caput epididymis is already modest (2-fold); 2) cauda sperm carry a wide range of unlabelled RNAs originating in the testis and other parts of the epididymis; 3) sperm in the cauda epididymis can survive for longer than the time frame of this experiment and so will include a population of unlabelled sperm that passed through the caput epididymis prior to 4-TU injections.
Confident that we could reliably detect small RNAs synthesized in the caput epididymis, we finally turn to SLAM-Seq analysis of small RNAs purified from cauda epididymal sperm. Consistent with the hypothesis that cauda sperm carry microRNAs originally synthesized in the caput epididymis, SLAM-Seq of cauda sperm microRNAs revealed significantly greater 4-TU labelling than testes (Figures 6C–D, Supplemental Figure S4D). Although this effect is quantitatively modest, it was significant in two biological replicates – Supplemental Figure S4D shows the cumulative distribution plots for U->C mutations across all microRNAs for these samples. Taken together, these data conclusively demonstrate that at least a subset of small RNAs in mature sperm are originally synthesized in the caput epididymis, and further support our hypothesis that small RNAs are trafficked from epididymis to sperm via epididymosomes.
DISCUSSION
Our data provide a detailed view of the biogenesis of the small RNA repertoire of maturing mammalian sperm. We characterized dynamics of small RNAs across sperm development, finding that multiple waves of small RNAs are synthesized/loaded into sperm over the course of testicular spermatogenesis and post-testicular maturation in the epididymis. Moreover, we show that caput epididymosomes can deliver small RNAs to testicular spermatozoa in vitro, and use in vivo chemogenetic tracking in intact animals to definitively identify epididymis-derived RNAs in mature sperm. Together, our data reveal a surprising role for soma-to-germline trafficking in sculpting the sperm RNA payload.
An improved characterization of sperm RNA payload
Increasing interest in functional roles for sperm RNAs in early development (see Conine et al, accompanying manuscript) has resulted in a number of studies reporting on the sperm RNA payload, with at times conflicting results. Here, we further refine our understanding of sperm small RNAs. Most notably, we find that a substantial additional population of 5′ tRFs are revealed following PNK treatment to resolve cyclic 2′-3′ phosphates. This is consistent with well-characterized roles for RNase A family members such as Angiogenin (Fu et al., 2009; Yamasaki et al., 2009), and RNAse T family members (Andersen and Collins, 2012; Lee and Collins, 2005; Thompson et al., 2008), in tRNA cleavage in various organisms. We note that although Angiogenin is not expressed at particularly high levels in the epididymis, several epididymis-specific paralogs of Ang – Rnase9–12 – are extremely highly expressed in this tissue, along with Rnaset2a (Castella et al., 2004; Johnston et al., 2005). These are therefore the likeliest nucleases responsible for the unusually high levels of tRNA cleavage observed in this tissue under unstressed conditions. Our data strongly suggest that RNaseA or T enzymes first cleave mature tRNAs at exposed loop regions, most notably the anticodon but in some cases also at the D or T loops, with exonuclease trimming or other degradation processes resulting in the slightly shorter 5′ tRFs previously reported in multiple studies.
What happens to the 3′ ends of cleaved tRNAs? Scrutiny of our small RNA datasets reveals low levels of reads (~10–100 ppm, compared to ~10–50,000 ppm for abundant 5′ tRFs) mapping to the 3′ ends of tRNAs, generally including the non-templated CCA found at the 3′ ends of mature tRNAs. Ongoing studies suggest that 3′ fragments resulting from tRNA cleavage are inherently challenging to clone, as we can robustly detect 3′ tRNA cleavage products by Northern blotting of epididymal RNAs (Supplemental Figure S5). Although recent studies have documented enhanced full-length tRNA cloning following RNA demethylation with an engineered AlkB RNA demethylase (Cozen et al., 2015; Zheng et al., 2015), we have been unsuccessful thus far in enhancing 3′ tRF capture using this enzyme (not shown), possibly owing to our inability to successfully generate deep sequencing libraries using the thermostable RT reported by Zheng et al. We anticipate that advances in cloning will enable more quantitative capture of 3′ tRNA fragments from mature sperm RNA populations, a key goal for future studies given the potential roles for 3′ tRFs in LTR element control (Martinez et al., 2017; Schorn et al., 2017).
Small RNA dynamics throughout sperm maturation
Analysis of small RNA repertoires of various purified sperm populations confirmed prior reports of a global loss of piRNAs and gain of tRFs that occurs over the course of post-testicular maturation, and further defined a narrow anatomical window during which this transition occurs. In addition to this large-scale remodeling of the sperm small RNA payload, we also identified more subtle dynamic behaviors for a subset of RNAs. Most interestingly, a large group of microRNAs known to be expressed during spermatogenesis were abundant, as expected, in testicular sperm populations, but were then surprisingly lost in caput epididymal sperm before recovering in abundance in cauda sperm (Figure 3). The majority of microRNAs exhibiting this behavior are encoded in genomic clusters, including the imprinted X-linked miR-880 cluster, the miR-17–92 “oncomiR” cluster, and many smaller clusters. Intriguingly, in the accompanying study (Conine et al), we find that embryos generated from caput sperm exhibit defects in preimplantation gene regulation, and that these gene regulatory defects appear to result from the transient loss of these clustered microRNAs in caput sperm.
Understanding the basis for this unusual behavior will be of great interest. The likeliest scenario is that these microRNAs (along with the bulk of piRNAs) are degraded, or ejected in the cytoplasmic droplet (Sprando and Russell, 1987), upon epididymal entry. Clustered microRNAs would then either be trafficked from the epididymis to transiting sperm in epididymosomes, or, less likely, generated in situ via processing of precursor transcripts. An extreme version of this model is that entry into the epididymis is accompanied by a global purge of all small RNAs including all microRNAs, with those microRNAs that appear to maintain abundance in caput epididymis (such as various let-7 family members) simply being those that are replenished earlier than the clustered microRNAs that are gained more distally. Whatever the mechanism for this process, the reason for the strong caput/cauda gradients in expression and trafficking of various small RNAs remains unclear, and will be the subject of future studies.
Soma-to-germline RNA trafficking in the epididymis
How do transcriptionally inactive sperm gain new small RNAs during epididymal transit? Multiple lines of evidence support the hypothesis that small RNAs synthesized in the epididymis epithelium are trafficked to maturing sperm in small extracellular vesicles, collectively known as epididymosomes: 1) Small RNAs that are gained as sperm enter the epididymis, such as tRFs, are highly abundant in the epididymis epithelium. 2) More locally, gradients in small RNA expression between the proximal and distal epididymis are manifest in sperm transiting the relevant region (as in, eg, the ~10-fold difference in tRF-Val-CAC between caput and cauda epididymis, sperm, and epididymosomes). 3) Using the TU tracer system, we show here that microRNAs first synthesized in the caput epididymis make their way to maturing sperm, thus demonstrating that soma-to-germline transfer of small RNAs occurs in intact animals. 4) The likely mechanism for this transfer is RNA trafficking in epididymosomes, as purified epididymosomes carry a very similar RNA payload to that gained by sperm during epididymal transit. 5) Epididymosomes can deliver small RNAs to sperm in vitro.
Our data thus identify a role for soma-to-germline trafficking in shaping the sperm RNA repertoire in mammalian sperm. This unites mammalian spermatogenesis with gametogenesis in a number of other species where RNAs produced in somatic support cells have been suggested or shown to be subsequently transferred into developing germ cells (Bourc’his and Voinnet, 2010). Previously-described or proposed examples of such soma-to-germline communication include: 1) communication between the somatic macronucleus and the germline micronucleus in ciliates such as Tetrahymena (Mochizuki et al., 2002) and Oxytricha (Fang et al., 2012), 2) piRNA communication from somatic follicle cells to the germline during Drosophila oogenesis (Ghildiyal and Zamore, 2009; Malone et al., 2009), and 3) release of transposon repression in vegetative nuclei in Arabidopsis pollen leading to generation of anti-transposon siRNAs which are transported into the sperm nuclei (Martinez et al., 2016; Slotkin et al., 2009).
Intriguingly, the process detailed here is quite distinct from most prior examples of soma-to-germline communication, as the proposed trafficking process involves a vesicle intermediate rather than direct cytoplasmic connections (Mahajan-Miklos and Cooley, 1994; McCue et al., 2011), and the RNAs being shipped are primarily microRNAs and tRFs as opposed to repeat element-derived piRNAs/siRNAs. Importantly, although a multitude of cell types secrete RNAs into extracellular vesicles in mammals (Tkach and Thery, 2016), there is as yet no evidence that circulating vesicles have direct access to the germline, which is protected by the blood-testis barrier – for example, we find no evidence in the Alb-Cre X TU-tracer experiments for liver to germline communication. Thus the epididymis, along with somatic cells of the testis such as Sertoli cells, occupy a privileged position in this regard.
Epigenetic remodeling during gametogenesis
Many potentially heritable “epigenetic marks” in the germline are known to be erased during key stages of development, as for example cytosine methylation patterns undergo two major waves of erasure and re-establishment during the mammalian life cycle (Feng et al., 2010) – once in primordial germ cells, and again shortly after fertilization. Our findings reveal novel epigenetic reprogramming events that occur during mammalian spermatogenesis, as we document a major transition from a piRNA-dominated stage of testicular spermatogenesis to the ultimately tRF-dominated payload of epididymal sperm, as well as a transient loss of clustered microRNAs that occurs in the caput epididymis.
The mechanism by which this epigenetic remodelling occurs is of significant interest given the mounting evidence that environmental signaling may be able to modulate specific epigenetic marks in germ cells and thereby potentially affect the phenotype in future generations (Chen et al., 2016b; Lane et al., 2014; Rando, 2012). While it is well appreciated that, say, an organism’s dietary conditions can influence the epigenome in somatic cells (Sharma and Rando, 2017), it has not been clear how this information is communicated to the developing gametes and thus to offspring. The origin of sperm small RNAs in the epididymis provides a focus for future studies on the mechanism by which environmental conditions might influence the sperm epigenome. Understanding the molecular basis underlying sorting and packaging of small RNAs into epididymosomes will also be of great interest (Gutierrez-Vazquez et al., 2013; Koppers-Lalic et al., 2014) as it will not only uncover a basic mechanism of communication between the somatic cells of epididymis and the maturing gametes, but will also shed light on how environmental information is signaled to sperm.
STAR METHODS
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Oliver Rando (oliver.rando@umassmed.edu).
Experimental Model and Subject Details
Mice
All animal care and use procedures were in accordance with guidelines of University of Massachusetts Medical School Institutional Animal Care and Use Committee. Mice were group housed (maximum of 5 per cage) with a 12-hour light-dark cycle (lights off at 7 pm) and free access to food and water ad libitum. Mice were weaned from mothers at 21 days of age. All animals used for experiments were 8–12 weeks old and fed either control diet (Bioserv AIN-93g) or normal chow diet (TU-tracer experiments).
Wild type FVB/NJ mice obtained from Jackson Laboratory were used for all experiments other than TU-tracer experiments (Figures 5–6, Supplemental Figure S4). For our breeding colonies, six week old FVB/NJ mice were purchased from Jackson Laboratory, and 24 hours after arrival at UMass Animal Facility mice were moved from chow to control diet (Bioserv AIN-93g). After being on the control diet for 2 weeks, mice were mated and the offspring were weaned on to control diet. The first three litters generated from the breeding pair were used for sperm and tissue isolations.
For TU-tracer experiments, B6(D2)-Tg(CAG-GFP,-Uprt)985Cdoe (CAGFPstopUPRT mice) and Albumin-cre (B6.Cg-Speer6-ps1Tg(Alb-cre)21Mgn/J) mice were purchased from Jackson Laboratory. Defb41 iCre KI (TUKO 10) mouse embryos were a gift from Dr. Petra Sipilä, Univeristy of Turku Finland. Defb41 iCre KI (TUKO 10) mouse line was regenerated at the Mouse Facility of UMass Medical School.
Method Details
Tissue and sperm isolation
Testes, epididymis, caput sperm, cauda sperm, spermatocytes and round spermatids were isolated as described previously (Sharma et al., 2016). Briefly, testes were dissected from 10–12 week mice, directly frozen in liquid nitrogen and stored at −80 °C until RNA extraction. Cauda and Caput epididymis were dissected from mice and placed in Whitten’s Media (100 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 5.5 mM Glucose, 1 mM Pyruvic acid, 4.8 mM Lactic acid (hemicalcium), and HEPES 20 mM) at 37 °C. To collect caput sperm, two small incisions were made at the proximal end of caput and using a 26G needle holes were poked in the rest of the tissue to let the caput epididymal fluid ooze out. For cauda sperm collection, cauda epididymis were gently squeezed to allow the caudal fluid to ooze out. Sperm-containing media was incubated for 15 minutes at 37 °C, then transferred to a fresh tube and the sperm-free epididymis tissues were directly frozen in liquid nitrogen and stored at −80 °C. The sperm were incubated for another 15 minutes at 37 °C, after incubation sperm were collected by centrifugation at 2000 × g for 2 minutes, followed by a 1X PBS wash, and a second wash with somatic lysis buffer (low or mid SLB, see below) for 10 minutes on ice to eliminate somatic cell contamination. Somatic lysis buffer treated sperm were collected by centrifugation at 3000×g for 5 minutes, and finally washed with 1X PBS before freezing down. For caput sperm collection, somatic lysis buffer concentration or incubation time was increased if the sperm pellet appeared red (due to contaminating red blood cells). Sperm sample purity was confirmed by microscopic examination of the samples.
For testicular spermatocyte and spermatid isolation, testes were acquired from one mouse at 10–12 weeks of age and incubated without their tunica albuginea in 5 ml elutriation buffer (100 mM NaCl, 45 mM KCl, 6 mM Na2HPO4, 0.6 mM KH2PO4, 0.23% Sodium DL-Lactate, 0.1% Glucose, 0.1% BSA, 0.011% Sodium Pyruvate, 1.2 mM MgSO4 and 1.2 mM CaCl2) containing 25 μg/ml liberase TM (Roche Diagnostics GmbH) for 30 min at 37 °C with gentle agitation every 5 m in to make cell suspension. The cell suspension was next pipetted 20 times with a 10-ml plastic pipette. After homogenization by pipetting 10 times through a P1000 pipette, the single cell suspension was filtered twice through a 40-μm cell strainer (Fisher Scientific) on ice and centrifuged at 1500 rpm at 4 °C for 10 min, and then the pellet was resuspended in 20 ml elutriation buffer. Next, testis cell populations were separated by centrifugal elutriation using a JE-5.0 elutriation system and a 4-ml standard elutriation chamber (Beckman Coulter). The system was assembled following the manufacturer’s instruction. The precise elutriation conditions are as follows: Fractions 1–3 were run at 3000 rpm and fractions 4–5 were run at 2000 rpm. The flow rate was 14, 18, 31, 23, and 40 ml/min for fractions 1–5, respectively. Tubes from fractions 3 to 5 were spun at 1500 rpm at 4 °C for 10 min. Pellets from the same fract ion were combined, resuspended in 200 μl of elutriation buffer and loaded onto 23–35% Percoll gradient, and centrifuged at 11,000 rpm at 4 °C for 15 min. The cells were then collected in 15-ml conical polypropylene tubes and spun at 1500 rpm at 4 °C for 15 min, and the pellets were stored at −80 °C. As described previously (Sharma et al., 2016), fraction 5 corresponds to primary spermatocytes and fractions 4 and 3 correspond to early and late round spermatids, respectively.
For mature testicular spermatozoa isolation, testes from one mouse (8–12 weeks old) were minced in a 35-mm Petri dish containing 1 ml 150 mM NaCl. Finely minced tissue slurry was then transferred to a 15ml conical tube and set aside for 3–5 minutes to allow tissue pieces to settle down. Next, the cell suspension was loaded onto 10.5 ml of 52% isotonic percoll (Sigma). The tubes were then centrifuged at 12000× g for 10 minutes at 10 °C. The pellet was resuspended in 10 ml of 150 mM NaCl and spun at 600× g at 4 °C for 10 minutes. The pellet was next washed with 75 mM NaCl solution 3 times, followed by treatment with somatic lysis buffer (0.01% SDS, 0.005% Triton-X) and one wash with 1X PBS to retrieve purified mature testicular spermatozoa. Spermatozoa pellets were then either flash frozen or used for fusion with epididymosomes.
Sperm washing
Cauda epididymis were dissected from 8–12 week old FVB mice, placed in Whitten’s media pH 7.4 (100 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 5.5 mM Glucose, 1 mM Pyruvic acid, 4.8 mM Lactic acid (hemicalcium), and HEPES 20 mM), and gently squeezed at the distal end to allow the caudal fluid to ooze out into the media. Sperm containing media was incubated for 15 minutes at 37 °C, then transferred to a fresh tube and incubated for another 15 minutes at 37 °C. To ensure homogeneity of sperm samples undergoing varying concentrations of somatic lysis buffer (SLB) treatment, cauda sperm from multiple animals were pooled, mixed thoroughly and then re-divided into four tubes. Sperm were collected by centrifugation at 2000×g for 2 minutes, then washed with 1X PBS and spun down. Three of these tubes were treated with three different concentrations of somatic lysis buffer, namely, low SLB (0.01% SDS, 0.005% Triton-X), mid SLB (0.1% SDS, 0.005% Triton-X) and high SLB (0.1% SDS, 0.5% Triton-X) respectively, for 10 minutes on ice followed by centrifugation at 3000×g for 5 minutes. After one final wash with 1X PBS, the sperm pellet was obtained by centrifugation at 3000×g for 5 minutes, and flash frozen in liquid Nitrogen. The fourth sample was flash frozen right after the first PBS wash, without any somatic lysis. All sperm pellets were stored at −80 °C until RNA extraction.
Separation of sperm head and tail
For the separation of cauda sperm head and tail, epididymides from one sexually mature mouse (12–15 weeks old) were slit open and sperm were gently squeezed out in in a dish containing PBS. The dish was then incubated at 37 °C for 15 minutes. Sperm slurry from five such dishes were pooled into a 15 ml conical tube and incubated at 37 °C for another 15 minutes. Next, the supernatant was centrifuged at 1500× g for 15 minutes at 4°C and the cell pellet was either washed with cold 75 mM NaCl three times, or with somatic lysis buffer (0.01% SDS and 0.005% Triton X-100), to remove somatic cells followed by centrifugation at 4000× g for 5 minutes at 4°C. Purity of sperm preparations was confirmed by microscopic examination. The sperm pellet was resuspended in 5 ml mitochondrial wash buffer (0.25 M sucrose, 0.01 M Tris, 0.001 M EDTA, pH 7.4) and sonicated for 10 seconds at 4°C at setting 4 using a Sonifier cell disruptor, Model W185 (Heat System-Ultrasonic, Inc., Plainview, NY). The sonicate was layered over a discontinuous sucrose gradient containing 10 ml 0.9 M sucrose and 15 ml 2.0 M sucrose. The gradient was centrifuged at 10,000 rpm in a SW 28 rotor, at 10°C for 30 minutes. Sperm tails were collected from the interface of 0.9 M sucrose and 2.0 M sucrose, and the heads from the bottom of the tube. Heads and tails were then washed with TE buffer and PBS, flash frozen and stored at −80°C for later analysis.
For the separation of heads and tails from testicular spermatozoa, testes from one sexually mature mouse (12–15 weeks old) were gently minced in a 35 mm Petri dish containing 1 ml modified Whitten’s media (22 mM HEPES, 1.2 mM MgCl2, 100 mM NaCl, 4.7 mM KCl, 1.0 mM pyruvic acid, 5.5 mM glucose, 4.8 mM lactic acid, pH 7.3) and 1 mM PMSF. Minced tissue slurry from 13 mice was pooled into one 50 ml tube and set aside for 3–5 minutes. The supernatant was layered over 30 ml 7.5% Percoll gradient and centrifuged at 1000× g for 20 minutes at 13 °C. The cell pellet was washed with cold 75 mM NaCl to remove somatic cells followed by centrifugation at 4000× g for 5 minutes at 4°C. The wash step was repeated three times. Next, the pellet was resuspended in 5 ml modified Whitten’s media containing 1 mM PMSF and layered over 52% Percoll gradient, followed by centrifugation at 15,000 rpm in SW 28 rotor for 15 minutes at 10 °C. The pellet which contains purified testicular spermatozoa, was washed with modified Whitten’s media and sperm purity was examined by microscopic examination. Next, purified testicular spermatozoa were resuspended in 5 ml of mitochondrial wash buffer, sonicated, washed and flash frozen as described above for cauda sperm head/tail separation.
Epididymosome isolation
Epididymosomes were isolated as previously described (Sharma et al., 2016). Briefly, caput epididymides were dissected from 8–12 weeks old male mice and placed in dishes containing 1 ml Whitten’s media (100 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 5.5 mM Glucose, 1 mM Pyruvic acid, 4.8 mM Lactic acid (hemicalcium), and HEPES 20 mM) pre-warmed at 37°C. The epididymides were then gently squeezed using forceps to isolate epididymal luminal content. The dishes containing epididymal luminal contents were then placed in an incubator set at 37°C with 5% CO2 for 15 minutes, to allow any remaining epididymal contents to release from the tissue. Next, the media containing epididymal luminal contents was transferred to a 1.5 ml tube and allowed to incubate for an additional 15 minutes. At the end of the 15 minutes, any tissue pieces or non-motile sperm settled down at the bottom of the tube and all the contents of the tube except for the bottom ~50 μl were transferred to a fresh tube. Next, the tube was spun in a tabletop centrifuge at 2000× g for 2 minutes to pellet down sperm. Supernatant, which contains epididymosomes, was then transferred to a fresh tube and centrifuged at 10000× g for 30 minutes at 4°C to get rid of any non-sperm cells and cellular debris. Supernatant from this spin was then transferred to a polycarbonate thick wall tube (13 × 56 mm, Beckman Coulter, Catalog number 362305) and centrifuged at 100000× g for 2 hours at 4°C in a tabletop ultracentrifuge (Beckman Optima TL) using TLA100.4 rotor. The pellet from this spin was then washed with 500 μl 1X PBS and centrifuged for another 2 hours at 100000× g at 4°C. Finally, the pellet containing epididymosomes was resuspended in 50 μl ice-cold 1X PBS, transferred to a 1.5 ml tube and flash frozen in liquid nitrogen or used for fusion with sperm.
Testicular spermatozoa and caput epididymosome fusion
After final wash with 1XPBS, caput epididymosomes were resuspended in 62.5 μl media out of which 12.5 μl was flash frozen and 50 μl was used for fusion with testicular spermatozoa. For fusion reaction, purified 100 μl of testicular spermatozoa were mixed with 50 μl of caput epididymosomes in presence of 1 mM ZnCl2 and incubated at 37°C for 2 hours. In parallel, a mock fusion reaction was also performed where no epididymosomes were added to the reaction. At the end of the incubation, additional 850 μl of 1X PBS was added to the reaction and sperm were spun down at 4000× g for 5 minutes. Next, the pellet was washed with 1X PBS three times and processed further for RNA extraction.
Two lines of evidence argue that gain of small RNAs in “reconstituted sperm” (Figure 4) results from epididymosomal RNA delivery to testicular sperm, rather than simple contamination by epididymosomes. First, initial reconstitution studies carried out with only one wash step resulted in a small RNA profile that almost perfectly matched the caput epididymosome RNA repertoire, demonstrating that the washes used in the final protocol removed excess epididymosomes. Second, the RNAs gained during reconstitution – dots above the diagonal in Figure 4E – are a subset of epididymosomal RNAs, consistent with a subset of RNAs being present in delivery-competent vesicles and again arguing against gross contamination of testicular sperm by the epididymosomal material.
PNK treatment
Total RNA from testis, cauda epididymis or sperm (caput and cauda) was extracted as previously described (Sharma et al., 2016). Following gel purification of 18–40 nucleotide small RNAs and isopropanol precipitation, small RNAs were treated with T4 Polynucleotide Kinase (PNK)(NEB M0201) + T4 PNK reaction buffer, incubated at 37°C for 30 minutes followed by heat inactivation at 65°C for 20 minutes. Following enzymatic treatment an equal volume of Acid Phenol:Chloroform (Ambion) was added to purify the RNA by aqueous phase separation. Finally, RNA was isopropanol precipitated prior to small RNA cloning.
Small RNA deep sequencing
Isolation of 18–40 nts small RNAs was carried out as previously described (Sharma et al., 2016) by purification from 15% polyacrylamide-7M urea denaturing gels. The resulting small RNA was used for preparing sequencing libraries using the Small RNA TruSeq kit from Illumina (as per the manual). Small RNA sequencing data was analyzed as described previously (Sharma et al., 2016).
TaqMan assays
Taqman assay quantitative RT-PCR were performed as described previously (Sharma et al., 2016). Custom small RNA assays for tRF-Val-CAC, tRF-GluCTC, tRF-GlyGCC and pre-designed microRNA assay for miR-34c, Let7c, Let7f and mir-10a were purchased from Life Technologies. U6 small nuclear RNA was used as an internal control to normalize small RNA levels in various sperm populations.
Note that values for cauda sperm in Figure 2B may be overestimated by ~4-fold, as Ct values for U6 were similar for testicular sperm and caput sperm, but were consistently 2 cycles higher in cauda sperm, suggesting that U6 decreases in abundance from caput to cauda sperm. In this case, using U6 for normalization would inflate the small RNA changes in cauda sperm, thereby accounting for the fact that all the RNAs assayed here appear to be ~4-fold more abundant in cauda sperm than expected from deep sequencing data, where for example miR-34c and let-7f levels are similar in cauda and testicular sperm. Regardless, our U6-normalized data are qualitatively highly concordant with our deep sequencing data, and strongly support the hypothesis that tRFs are indeed gained by sperm as they enter the epididymis.
Generation of mice expressing tissue-specific UPRT
To generate mice expressing tissue specific UPRT, we crossed homozygous CAGFPstopUPRT mice with either liver-specific Albumin-cre mice or caput epididymis specific Defb41-cre mice, to generate heterozygous [Uprt−/+, Alb-Cre] or [Uprt−/+, Defb41-Cre] mice, respectively. Since 4-thiouracil incorporation is sensitive to the levels of UPRT, we then backcrossed these mice with homozygous CAGFPstopUPRT mice to produce mice homozygous for Uprt gene. Mouse genotyping was performed using quantitative PCR (qPCR) with DNA isolated from ear punches. For genotyping CAGFPstopUPRT mice, following primers and probes were used: 5′-ATT CCA AGA TCT GTG GCG TC-3′ and 5′-CTT CTC GTA GAT CAG CTT AGG C-3′ for mutant allele; 5′-CAC GTG GGC TCC AGC ATT-3′ and 5′-TCA CCA GTC ATT TCT GCC TTT G-3′ for wildtype allele; probe 5′-/Cy5/CCA ATG GTC GGG CAC TGC TCA A/Black Hole Quencher 2/-3′ for wildtype allele and probe 5′-/6-FAM/CCG CAT CGG GAA AAT CCT CAT CCA/Black Hole Quencher 2/-3′ for mutant allele. Albumin–cre mice were genotyped using: 5′-TTG GCC CCT TAC CAT AAC TG-3′ for both wildtype and mutant allele, 5′-TGC AAA CAT CAC ATG CAC AC-3′ for wildtype allele, and 5′-GAA GCA GAA GCT TAG GAA GAT GG-3′ for mutant allele. Primers for Defb41-cre mice included: 5′-AGA TGC CAG GAC ATC AGG AAC CTG-3′ and 5′-ATC AGC CAC ACC AGA CAC AGA GAT C-3′ for transgene, 5′-CTA GGC CAC AGA ATT GAA AGA TCT-3′ and 5′-GTA GGT GGA AAT TCT AGC ATC ATC C-3′ for internal positive control. The qPCR conditions were as follows: 95°C for 3 min, followed by 10 cycles of 95°C for 30 sec, 65°C for15 sec, de crement of temperature by 0.5°C per cycle, 68°C for10 sec, and 28 cycles of 95°C for 20 sec, 60°C for 20 sec, and 72°C for 15 sec, with a final extension at 72°C for 2 min.
4-thiouracil (4-TU) treatment
4-TU was dissolved in DMSO at 200 mg/ml and stored at −20°C. Prior to use, the stock solution was diluted at 1:4 with corn oil with rigorous shaking and working solution was then kept in dark. 8–12 weeks old adult mice were intraperitoneally injected with either 4-TU (400 mg/kg body weight) or solvent alone (control) every other day for 5 times, as sperm transits from caput to cauda epididymis in approximately 10 days. Tissue and sperm samples were collected (as described above) 5 hours after the last injection, flash frozen in liquid nitrogen and stored at −80°C until RNA extraction.
Northern and Dot blot analysis
We followed published method for dot blot analysis with some modifications (Radle et al., 2013). In brief, equal amount of RNA (in binding buffer) from different experimental groups, was applied to Zeta membrane (Bio-Rad). A biotin-labeled DNA oligo was used as a positive control. The membrane was air-dried at room temperature for 5 minutes, followed by blocking at room temperature for 30 minutes with 10% SDS in PBS and 1mM EDTA. Next, the membrane was incubated in a solution containing 10% SDS in PBS and Streptavidin-HRP (Thermo Fisher Scientific) for 15 minutes at room temperature, followed by three washes with PBS containing SDS for 5 minutes each. After the final wash, biotinylated RNA were detected with Amersham ECL Western Blotting Reagent (GE Healthcare Life Sciences) using Amersham Imager 600 (GE Healthcare Life Sciences). For Northern blot analysis, 30 μg of RNA was denatured and run on a 15% acrylamide 7M urea gel, followed by transfer to a positively charged nylon membrane and UV-crosslinking. The membrane was next incubated with Streptavidin-HRP, washed and detected using ECL reagent, as above. For Northern blot analysis of tRNA fragments (Supplemental Figure S5), we used the following probes: 3′tRF GlyGCC 5′ TGCATTGGCCGGGAACCGAACCCGGGCCTCCCGCG 3′ and 3′tRF Val CAC 5′ TGTTTCCGCCCGGTTTCGAACCGGGGACCTTTCGCG 3′
Western blot
Protein extracts were prepared from mouse tissues using RIPA lysis buffer (150 mM sodium chloride, 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris, pH 8.0). Protein extracts were denatured with 2X Laemmli buffer (4% SDS, 10% 2-mercaptothanol, 20% glycerol, 0.004% bromophenol blue, 0.125 M Tris-HCl, pH 6.8) at 95°C for 5 min and run on a 10% SDS -PAGE, followed by transfer to PVDF membrane. The membrane was next probed with anti-GFP (Invitrogen), anti-HA (Thermo Scientific) or anti-Actin (Thermo Scientific) antibodies at 1:1000 dilution followed by secondary antibody treatment at 1:2000 dilution (anti-mouse HRP and anti-rabbit HRP, Cell Signaling).
SLAM-seq
4 μg of isolated total RNA was treated with 10 mM iodoacetamide under optimal reaction conditions (50 mM NaPi, pH8; 50% v/v DMSO; 15 min at 50°C). The reaction was quenched by addition of 10 mM DTT, followed by ethanol precipitation and small RNA library preparation as described previously (Jayaprakash et al., 2011). Briefly, total RNA was size selected (18–30 nt) by 15% denaturing polyacrylamide gel electrophoresis and subjected sequentially to 3′ and 5′ adapter ligation (employing linker oligonucleotides with four random nucleotides at the ligation interface to avoid ligation biases). Gel-purified ligation products were reverse transcribed using SuperScript III (Invitrogen) according to the manufacturer’s instructions, followed by PCR amplification and high-throughput sequencing on an Illumina HiSeq 2000 instrument (SR100 mode).
Quantitation and statistical analysis
For small RNA-Seq datasets, individual small RNA abundances were normalized to parts per million based on total number of reads mapping to microRNAs, tRNAs, piRNAs (repeatmasker and unique piRNAs), and mRNA (Refseq) – rRNA-mapping reads were excluded. Significant differences in small RNA abundance were calculated by t-test, adjusted for multiple hypothesis testing by multiplying by the number of RNAs above an average abundance of 10 ppm. Complete genome mapping results for all small RNA-Seq datasets, with sample numbers, are available in Tables S1–S5.
For the analysis of SLAM-seq datasets, small RNA reads were aligned using bowtie v.1.2 (Langmead et al., 2009), allowing for 3 mismatches, to all mouse pre-miRNA hairpins described in mirbase v.21 (Kozomara and Griffiths-Jones, 2014) and extended downstream by 20 basepairs. All reads mapping to 5p and 3p arms of a miRNA hairpin locus were filtered to only retain the most frequent 5′ isoform. Mutation rates were determined by investigating the occurrence of any given mutation within the miR body (nt 1–18) of the most abundant miRNA 5′ isoforms, normalized to the genome matching instances at this position and frequency of each nucleotide within the inspected region. Reads containing U>C conversions within the miR body were counted and normalized to U-content and total mapping reads to assess abundance in cpm. For any given sample, further analysis was restricted to microRNAs with at least 1000 reads.
Statistical significance for TU tracer experiments was assessed in two ways. For Figures 5E and 6C, significant changes in U->C conversion rate were assessed across the entire set of detected microRNAs by paired two-tailed t-test, with Uninjected and TU-injected samples being paired for a given tissue. In addition, in Supplemental Figure S4D we used KS tests to assess significance for the distribution of microRNAs exhibiting a given change in U->C conversion upon TU injection, comparing distributions in the testis vs. in cauda epididymal sperm. For TU tracer studies, n=2 animals, with each replicate set representing a sample of intact testis, intact caput epididymis, and cauda epididymal sperm obtained from the same animal.
Data and Software Availability
All raw fastq files and processed data files of deep-sequencing data can be found in GEO with the accession number GSE112990.
Supplementary Material
Small RNA-Seq data for cauda sperm pellets washed 1X in PBS and then either snap frozen or subject to one of three detergent washing protocols (Methods). RNAs were successively mapped to rRNAs, tRNAs, microRNAs, unique piRNA transcripts, repeatmasker (rmsk), and RefSeq (genes), as indicated in the “RNA Class” column. Total read numbers are provided here – unless otherwise specified, for the majority of analyses rRNA-mapping reads were discarded and remaining reads were normalized to reads per million.
As in Table S1, for sperm sonicated to separate heads from tails. Samples labeled “sperm” represent small RNAs cloned from sonicated sperm that were not further treated, while “heads” and “tails” show data for purified sperm fragments. Sperm samples include testicular spermatozoa and cauda epididymal sperm. In addition, cauda sperm samples include samples washed with either low stringency detergent conditions (0.01% SDS, 0.005% Triton X-100) or with hypotonic lysis buffer (75 mM NaCl), as indicated.
Small RNA levels are shown for small RNA cloning experiments using total RNA obtained from intact testis or cauda epididymis or from purified sperm. Note that the caput sperm libraries were low quality, requiring 5 extra PCR cycles to yield appreciable library material, and are undersequenced. We therefore do not discuss these data in detail in the body of the manuscript. Nonetheless, we include these data here as the same gross features observed in cauda sperm – dramatically increased levels of rRNA fragments revealed by PNK treatment, and increased levels of tRFs when excluding rRNA-mapping reads – are seen here. In addition, the specific tRF lengths revealed by PNK treatment (Figure 1F) are also observed in caput sperm (not shown).
All small RNA-Seq data for primary spermatocytes, round spermatids, testicular spermatozoa, and caput and cauda epididymal sperm, as indicated, as well as cauda epididymosomes. All libraries here were cloned without PNK treatment.
As in Table S1, for testicular sperm either mock-treated or fused with caput epididymosomes. Note that 2 columns here (mock-treated sperm) are reproduced from Table S4.
Sheets show analysis of each mutation type (C>A, etc.). In each sheet, data for individual microRNAs are shown as rows, with six columns showing raw read counts for the six libraries (testis, caput epididymis, and cauda sperm, all with or without 4-TU injection), and the following six columns showing the average mutation rate for all relevant nucleotides (eg all cytosines for the C>A sheet) in the microRNA’s first 18 nt. For the T>C sheet, columns are also included with data normalized to parts per million, and three columns are included showing the 4-TU/no TU ratio for the relevant mutation rates.
Research Highlights.
Detailed characterization of small RNA dynamics in mammalian sperm maturation
Caput epididymosomes can deliver small RNAs to testicular spermatozoa in vitro
Metabolic labeling of RNAs in intact animals tracks RNAs from epididymis to sperm
Acknowledgments
We thank members of the Rando lab for critical reading of the manuscript and helpful comments. We thank P. Sipilä for the generous gift of the Defb41:Cre mouse strain. This work was supported by NIH grants R01HD080224 and DP1ES025458 (OJR), and the Austrian Academy of Sciences and the European Research Council ERC-StG-338252 (SLA). US is a Charles H. Hood postdoctoral Fellow. CCC is a Helen Hay Whitney Foundation postdoctoral Fellow.
Footnotes
AUTHOR CONTRIBUTIONS
The majority of experiments were designed by US and OJR. US carried out RNA characterizations in epididymosomes and various sperm populations, and carried out epididymosome fusion experiments. SK, FS, and CCC carried out analyses of sperm wash conditions and RNA treatments, FS purified testicular sperm populations, FS and US carried out TU tracer experiments, and SLAM-Seq was performed and analyzed by BR, VAH, and SLS. US and OJR carried out most data analysis, and wrote the manuscript, which was reviewed and edited by all authors.
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alberti C, Manzenreither RA, Sowemimo I, Burkard TR, Wang J, Mahofsky K, Ameres SL, Cochella L. Cell-type specific sequencing of microRNAs from complex animal tissues. Nature methods. 2018 doi: 10.1038/nmeth.4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen KL, Collins K. Several RNase T2 enzymes function in induced tRNA and rRNA turnover in the ciliate Tetrahymena. Mol Biol Cell. 2012;23:36–44. doi: 10.1091/mbc.E11-08-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P, Ivanov P. tRNA fragments in human health and disease. FEBS letters. 2014;588:4297–4304. doi: 10.1016/j.febslet.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravin A, Gaidatzis D, Pfeffer S, Lagos-Quintana M, Landgraf P, Iovino N, Morris P, Brownstein MJ, Kuramochi-Miyagawa S, Nakano T, et al. A novel class of small RNAs bind to MILI protein in mouse testes. Nature. 2006;442:203–207. doi: 10.1038/nature04916. [DOI] [PubMed] [Google Scholar]
- Aravin AA, Sachidanandam R, Girard A, Fejes-Toth K, Hannon GJ. Developmentally regulated piRNA clusters implicate MILI in transposon control. Science (New York, NY. 2007;316:744–747. doi: 10.1126/science.1142612. [DOI] [PubMed] [Google Scholar]
- Arteaga-Vazquez MA, Chandler VL. Paramutation in maize: RNA mediated transgenerational gene silencing. Curr Opin Genet Dev. 2010;20:156–163. doi: 10.1016/j.gde.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista PJ, Ruby JG, Claycomb JM, Chiang R, Fahlgren N, Kasschau KD, Chaves DA, Gu W, Vasale JJ, Duan S, et al. PRG-1 and 21U-RNAs interact to form the piRNA complex required for fertility in C. elegans. Molecular cell. 2008;31:67–78. doi: 10.1016/j.molcel.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belleannee C, Calvo E, Caballero J, Sullivan R. Epididymosomes convey different repertoires of microRNAs throughout the bovine epididymis. Biology of reproduction. 2013;89:30. doi: 10.1095/biolreprod.113.110486. [DOI] [PubMed] [Google Scholar]
- Bjorkgren I, Saastamoinen L, Krutskikh A, Huhtaniemi I, Poutanen M, Sipila P. Dicer1 ablation in the mouse epididymis causes dedifferentiation of the epithelium and imbalance in sex steroid signaling. PloS one. 2012;7:e38457. doi: 10.1371/journal.pone.0038457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourc’his D, Voinnet O. A small-RNA perspective on gametogenesis, fertilization, and early zygotic development. Science (New York, N Y. 2010;330:617–622. doi: 10.1126/science.1194776. [DOI] [PubMed] [Google Scholar]
- Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell. 2007;128:1089–1103. doi: 10.1016/j.cell.2007.01.043. [DOI] [PubMed] [Google Scholar]
- Brennecke J, Malone CD, Aravin AA, Sachidanandam R, Stark A, Hannon GJ. An epigenetic role for maternally inherited piRNAs in transposon silencing. Science (New York, N Y. 2008;322:1387–1392. doi: 10.1126/science.1165171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castella S, Benedetti H, de Llorens R, Dacheux JL, Dacheux F. Train A, an RNase A-like protein without RNase activity, is secreted and reabsorbed by the same epididymal cells under testicular control. Biology of reproduction. 2004;71:1677–1687. doi: 10.1095/biolreprod.104.031666. [DOI] [PubMed] [Google Scholar]
- Chen Q, Yan M, Cao Z, Li X, Zhang Y, Shi J, Feng GH, Peng H, Zhang X, Zhang Y, et al. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science (New York, N Y. 2016a;351:397–400. doi: 10.1126/science.aad7977. [DOI] [PubMed] [Google Scholar]
- Chen Q, Yan W, Duan E. Epigenetic inheritance of acquired traits through sperm RNAs and sperm RNA modifications. Nature reviews. Genetics. 2016b doi: 10.1038/nrg.2016.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couvillion MT, Sachidanandam R, Collins K. A growth-essential Tetrahymena Piwi protein carries tRNA fragment cargo. Genes & development. 2010;24:2742–2747. doi: 10.1101/gad.1996210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozen AE, Quartley E, Holmes AD, Hrabeta-Robinson E, Phizicky EM, Lowe TM. ARM-seq: AlkB-facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nature methods. 2015;12:879–884. doi: 10.1038/nmeth.3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Castro Barbosa T, Ingerslev LR, Alm PS, Versteyhe S, Massart J, Rasmussen M, Donkin I, Sjogren R, Mudry JM, Vetterli L, et al. High-fat diet reprograms the epigenome of rat spermatozoa and transgenerationally affects metabolism of the offspring. Mol Metab. 2016;5:184–197. doi: 10.1016/j.molmet.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Mateo S, Sassone-Corsi P. Regulation of spermatogenesis by small non-coding RNAs: role of the germ granule. Semin Cell Dev Biol. 2014;29:84–92. doi: 10.1016/j.semcdb.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vanssay A, Bouge AL, Boivin A, Hermant C, Teysset L, Delmarre V, Antoniewski C, Ronsseray S. Paramutation in Drosophila linked to emergence of a piRNA-producing locus. Nature. 2012;490:112–115. doi: 10.1038/nature11416. [DOI] [PubMed] [Google Scholar]
- Emara MM, Ivanov P, Hickman T, Dawra N, Tisdale S, Kedersha N, Hu GF, Anderson P. Angiogenin-induced tRNA-derived stress-induced RNAs promote stress-induced stress granule assembly. The Journal of biological chemistry. 2010;285:10959–10968. doi: 10.1074/jbc.M109.077560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang W, Wang X, Bracht JR, Nowacki M, Landweber LF. Piwi-interacting RNAs protect DNA against loss during Oxytricha genome rearrangement. Cell. 2012;151:1243–1255. doi: 10.1016/j.cell.2012.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Jacobsen SE, Reik W. Epigenetic reprogramming in plant and animal development. Science (New York, N Y. 2010;330:622–627. doi: 10.1126/science.1190614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Fu H, Feng J, Liu Q, Sun F, Tie Y, Zhu J, Xing R, Sun Z, Zheng X. Stress induces tRNA cleavage by angiogenin in mammalian cells. FEBS letters. 2009;583:437–442. doi: 10.1016/j.febslet.2008.12.043. [DOI] [PubMed] [Google Scholar]
- Gapp K, Jawaid A, Sarkies P, Bohacek J, Pelczar P, Prados J, Farinelli L, Miska E, Mansuy IM. Implication of sperm RNAs in transgenerational inheritance of the effects of early trauma in mice. Nature neuroscience. 2014;17:667–669. doi: 10.1038/nn.3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Lopez J, Hourcade JD, Alonso L, Cardenas DB, Del Mazo J. Global characterization and target identification of piRNAs and endo-siRNAs in mouse gametes and zygotes. Biochimica et biophysica acta. 2014;1839:463–475. doi: 10.1016/j.bbagrm.2014.04.006. [DOI] [PubMed] [Google Scholar]
- Gay L, Miller MR, Ventura PB, Devasthali V, Vue Z, Thompson HL, Temple S, Zong H, Cleary MD, Stankunas K, et al. Mouse TU tagging: a chemical/genetic intersectional method for purifying cell type-specific nascent RNA. Genes & development. 2013;27:98–115. doi: 10.1101/gad.205278.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghildiyal M, Zamore PD. Small silencing RNAs: an expanding universe. Nature reviews Genetics. 2009;10:94–108. doi: 10.1038/nrg2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez-Vazquez C, Villarroya-Beltri C, Mittelbrunn M, Sanchez-Madrid F. Transfer of extracellular vesicles during immune cell-cell interactions. Immunol Rev. 2013;251:125–142. doi: 10.1111/imr.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton AJ, Baulcombe DC. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science (New York, N Y. 1999;286:950–952. doi: 10.1126/science.286.5441.950. [DOI] [PubMed] [Google Scholar]
- Herzog VA, Reichholf B, Neumann T, Rescheneder P, Bhat P, Burkard TR, Wlotzka W, von Haeseler A, Zuber J, Ameres SL. Thiol-linked alkylation of RNA to assess expression dynamics. Nature methods. 2017;14:1198–1204. doi: 10.1038/nmeth.4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaprakash AD, Jabado O, Brown BD, Sachidanandam R. Identification and remediation of biases in the activity of RNA ligases in small-RNA deep sequencing. Nucleic acids research. 2011;39:e141. doi: 10.1093/nar/gkr693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston DS, Jelinsky SA, Bang HJ, DiCandeloro P, Wilson E, Kopf GS, Turner TT. The mouse epididymal transcriptome: transcriptional profiling of segmental gene expression in the epididymis. Biology of reproduction. 2005;73:404–413. doi: 10.1095/biolreprod.105.039719. [DOI] [PubMed] [Google Scholar]
- Kawano M, Kawaji H, Grandjean V, Kiani J, Rassoulzadegan M. Novel small noncoding RNAs in mouse spermatozoa, zygotes and early embryos. PloS one. 2012;7:e44542. doi: 10.1371/journal.pone.0044542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppers-Lalic D, Hackenberg M, Bijnsdorp IV, van Eijndhoven MA, Sadek P, Sie D, Zini N, Middeldorp JM, Ylstra B, de Menezes RX, et al. Nontemplated nucleotide additions distinguish the small RNA composition in cells from exosomes. Cell reports. 2014;8:1649–1658. doi: 10.1016/j.celrep.2014.08.027. [DOI] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic acids research. 2014;42:D68–73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapf D, Ruan YC, Wertheimer EV, Battistone MA, Pawlak JB, Sanjay A, Pilder SH, Cuasnicu P, Breton S, Visconti PE. cSrc is necessary for epididymal development and is incorporated into sperm during epididymal transit. Developmental biology. 2012;369:43–53. doi: 10.1016/j.ydbio.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramochi-Miyagawa S, Kimura T, Ijiri TW, Isobe T, Asada N, Fujita Y, Ikawa M, Iwai N, Okabe M, Deng W, et al. Mili, a mammalian member of piwi family gene, is essential for spermatogenesis. Development. 2004;131:839–849. doi: 10.1242/dev.00973. [DOI] [PubMed] [Google Scholar]
- Lane M, Robker RL, Robertson SA. Parenting from before conception. Science (New York, N Y. 2014;345:756–760. doi: 10.1126/science.1254400. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SR, Collins K. Starvation-induced cleavage of the tRNA anticodon loop in Tetrahymena thermophila. The Journal of biological chemistry. 2005;280:42744–42749. doi: 10.1074/jbc.M510356200. [DOI] [PubMed] [Google Scholar]
- Lee YS, Shibata Y, Malhotra A, Dutta A. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs) Genes & development. 2009;23:2639–2649. doi: 10.1101/gad.1837609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XZ, Roy CK, Dong X, Bolcun-Filas E, Wang J, Han BW, Xu J, Moore MJ, Schimenti JC, Weng Z, et al. An ancient transcription factor initiates the burst of piRNA production during early meiosis in mouse testes. Molecular cell. 2013;50:67–81. doi: 10.1016/j.molcel.2013.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan-Miklos S, Cooley L. Intercellular cytoplasm transport during Drosophila oogenesis. Developmental biology. 1994;165:336–351. doi: 10.1006/dbio.1994.1257. [DOI] [PubMed] [Google Scholar]
- Malone CD, Brennecke J, Dus M, Stark A, McCombie WR, Sachidanandam R, Hannon GJ. Specialized piRNA pathways act in germline and somatic tissues of the Drosophila ovary. Cell. 2009;137:522–535. doi: 10.1016/j.cell.2009.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez G, Choudury SG, Slotkin RK. tRNA-derived small RNAs target transposable element transcripts. Nucleic acids research. 2017;45:5142–5152. doi: 10.1093/nar/gkx103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez G, Panda K, Kohler C, Slotkin RK. Silencing in sperm cells is directed by RNA movement from the surrounding nurse cell. Nat Plants. 2016;2:16030. doi: 10.1038/nplants.2016.30. [DOI] [PubMed] [Google Scholar]
- McCue AD, Cresti M, Feijo JA, Slotkin RK. Cytoplasmic connection of sperm cells to the pollen vegetative cell nucleus: potential roles of the male germ unit revisited. J Exp Bot. 2011;62:1621–1631. doi: 10.1093/jxb/err032. [DOI] [PubMed] [Google Scholar]
- Meikar O, Da Ros M, Korhonen H, Kotaja N. Chromatoid body and small RNAs in male germ cells. Reproduction. 2011;142:195–209. doi: 10.1530/REP-11-0057. [DOI] [PubMed] [Google Scholar]
- Mochizuki K, Fine NA, Fujisawa T, Gorovsky MA. Analysis of a piwi-related gene implicates small RNAs in genome rearrangement in tetrahymena. Cell. 2002;110:689–699. doi: 10.1016/s0092-8674(02)00909-1. [DOI] [PubMed] [Google Scholar]
- Nixon B, Stanger SJ, Mihalas BP, Reilly JN, Anderson AL, Tyagi S, Holt JE, McLaughlin EA. The MicroRNA Signature of Mouse Spermatozoa Is Substantially Modified During Epididymal Maturation. Biology of reproduction. 2015 doi: 10.1095/biolreprod.115.132209. [DOI] [PubMed] [Google Scholar]
- Peng H, Shi J, Zhang Y, Zhang H, Liao S, Li W, Lei L, Han C, Ning L, Cao Y, et al. A novel class of tRNA-derived small RNAs extremely enriched in mature mouse sperm. Cell Res. 2012;22:1609–1612. doi: 10.1038/cr.2012.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radle B, Rutkowski AJ, Ruzsics Z, Friedel CC, Koszinowski UH, Dolken L. Metabolic labeling of newly transcribed RNA for high resolution gene expression profiling of RNA synthesis, processing and decay in cell culture. J Vis Exp. 2013 doi: 10.3791/50195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando OJ. Daddy issues: paternal effects on phenotype. Cell. 2012;151:702–708. doi: 10.1016/j.cell.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassoulzadegan M, Grandjean V, Gounon P, Vincent S, Gillot I, Cuzin F. RNA-mediated non-mendelian inheritance of an epigenetic change in the mouse. Nature. 2006;441:469–474. doi: 10.1038/nature04674. [DOI] [PubMed] [Google Scholar]
- Reilly JN, McLaughlin EA, Stanger SJ, Anderson AL, Hutcheon K, Church K, Mihalas BP, Tyagi S, Holt JE, Eamens AL, et al. Characterisation of mouse epididymosomes reveals a complex profile of microRNAs and a potential mechanism for modification of the sperm epigenome. Sci Rep. 2016;6:31794. doi: 10.1038/srep31794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers AB, Morgan CP, Leu NA, Bale TL. Transgenerational epigenetic programming via sperm microRNA recapitulates effects of paternal stress. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:13699–13704. doi: 10.1073/pnas.1508347112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rompala GR, Mounier A, Wolfe CM, Lin Q, Lefterov I, Homanics GE. Heavy Chronic Intermittent Ethanol Exposure Alters Small Noncoding RNAs in Mouse Sperm and Epididymosomes. Front Genet. 2018;9:32. doi: 10.3389/fgene.2018.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorn AJ, Gutbrod MJ, LeBlanc C, Martienssen R. LTR-Retrotransposon Control by tRNA-Derived Small RNAs. Cell. 2017;170:61–71.e11. doi: 10.1016/j.cell.2017.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster A, Tang C, Xie Y, Ortogero N, Yuan S, Yan W. SpermBase: A Database for Sperm-Borne RNA Contents. Biology of reproduction. 2016;95:99. doi: 10.1095/biolreprod.116.142190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma U, Conine CC, Shea JM, Boskovic A, Derr AG, Bing XY, Belleannee C, Kucukural A, Serra RW, Sun F, et al. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science (New York, N Y. 2016;351:391–396. doi: 10.1126/science.aad6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma U, Rando OJ. Metabolic Inputs into the Epigenome. Cell metabolism. 2017;25:544–558. doi: 10.1016/j.cmet.2017.02.003. [DOI] [PubMed] [Google Scholar]
- Shirayama M, Seth M, Lee HC, Gu W, Ishidate T, Conte D, Jr, Mello CC. piRNAs initiate an epigenetic memory of nonself RNA in the C. elegans germline. Cell. 2012;150:65–77. doi: 10.1016/j.cell.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin RK, Vaughn M, Borges F, Tanurdzic M, Becker JD, Feijo JA, Martienssen RA. Epigenetic reprogramming and small RNA silencing of transposable elements in pollen. Cell. 2009;136:461–472. doi: 10.1016/j.cell.2008.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobala A, Hutvagner G. Transfer RNA-derived fragments: origins, processing, and functions. Wiley Interdiscip Rev RNA. 2011;2:853–862. doi: 10.1002/wrna.96. [DOI] [PubMed] [Google Scholar]
- Sprando RL, Russell LD. Comparative study of cytoplasmic elimination in spermatids of selected mammalian species. Am J Anat. 1987;178:72–80. doi: 10.1002/aja.1001780109. [DOI] [PubMed] [Google Scholar]
- Sullivan R, Frenette G, Girouard J. Epididymosomes are involved in the acquisition of new sperm proteins during epididymal transit. Asian journal of andrology. 2007;9:483–491. doi: 10.1111/j.1745-7262.2007.00281.x. [DOI] [PubMed] [Google Scholar]
- Sullivan R, Saez F. Epididymosomes, prostasomes, and liposomes: their roles in mammalian male reproductive physiology. Reproduction. 2013;146:R21–35. doi: 10.1530/REP-13-0058. [DOI] [PubMed] [Google Scholar]
- Thompson DM, Lu C, Green PJ, Parker R. tRNA cleavage is a conserved response to oxidative stress in eukaryotes. RNA. 2008;14:2095–2103. doi: 10.1261/rna.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkach M, Thery C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell. 2016;164:1226–1232. doi: 10.1016/j.cell.2016.01.043. [DOI] [PubMed] [Google Scholar]
- Vagin VV, Sigova A, Li C, Seitz H, Gvozdev V, Zamore PD. A distinct small RNA pathway silences selfish genetic elements in the germline. Science (New York, N Y. 2006;313:320–324. doi: 10.1126/science.1129333. [DOI] [PubMed] [Google Scholar]
- Volpe TA, Kidner C, Hall IM, Teng G, Grewal SI, Martienssen RA. Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science (New York, N Y. 2002;297:1833–1837. doi: 10.1126/science.1074973. [DOI] [PubMed] [Google Scholar]
- Yamasaki S, Ivanov P, Hu GF, Anderson P. Angiogenin cleaves tRNA and promotes stress-induced translational repression. The Journal of cell biology. 2009;185:35–42. doi: 10.1083/jcb.200811106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A, Ha S, Ahn J, Kim R, Kim S, Lee Y, Kim J, Soll D, Lee HY, Park HS. A chemical biology route to site-specific authentic protein modifications. Science (New York, NY. 2016 doi: 10.1126/science.aah4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Qin Y, Clark WC, Dai Q, Yi C, He C, Lambowitz AM, Pan T. Efficient and quantitative high-throughput tRNA sequencing. Nature methods. 2015;12:835–837. doi: 10.1038/nmeth.3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberman D, Cao X, Jacobsen SE. ARGONAUTE4 control of locus-specific siRNA accumulation and DNA and histone methylation. Science (New York, N Y. 2003;299:716–719. doi: 10.1126/science.1079695. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Small RNA-Seq data for cauda sperm pellets washed 1X in PBS and then either snap frozen or subject to one of three detergent washing protocols (Methods). RNAs were successively mapped to rRNAs, tRNAs, microRNAs, unique piRNA transcripts, repeatmasker (rmsk), and RefSeq (genes), as indicated in the “RNA Class” column. Total read numbers are provided here – unless otherwise specified, for the majority of analyses rRNA-mapping reads were discarded and remaining reads were normalized to reads per million.
As in Table S1, for sperm sonicated to separate heads from tails. Samples labeled “sperm” represent small RNAs cloned from sonicated sperm that were not further treated, while “heads” and “tails” show data for purified sperm fragments. Sperm samples include testicular spermatozoa and cauda epididymal sperm. In addition, cauda sperm samples include samples washed with either low stringency detergent conditions (0.01% SDS, 0.005% Triton X-100) or with hypotonic lysis buffer (75 mM NaCl), as indicated.
Small RNA levels are shown for small RNA cloning experiments using total RNA obtained from intact testis or cauda epididymis or from purified sperm. Note that the caput sperm libraries were low quality, requiring 5 extra PCR cycles to yield appreciable library material, and are undersequenced. We therefore do not discuss these data in detail in the body of the manuscript. Nonetheless, we include these data here as the same gross features observed in cauda sperm – dramatically increased levels of rRNA fragments revealed by PNK treatment, and increased levels of tRFs when excluding rRNA-mapping reads – are seen here. In addition, the specific tRF lengths revealed by PNK treatment (Figure 1F) are also observed in caput sperm (not shown).
All small RNA-Seq data for primary spermatocytes, round spermatids, testicular spermatozoa, and caput and cauda epididymal sperm, as indicated, as well as cauda epididymosomes. All libraries here were cloned without PNK treatment.
As in Table S1, for testicular sperm either mock-treated or fused with caput epididymosomes. Note that 2 columns here (mock-treated sperm) are reproduced from Table S4.
Sheets show analysis of each mutation type (C>A, etc.). In each sheet, data for individual microRNAs are shown as rows, with six columns showing raw read counts for the six libraries (testis, caput epididymis, and cauda sperm, all with or without 4-TU injection), and the following six columns showing the average mutation rate for all relevant nucleotides (eg all cytosines for the C>A sheet) in the microRNA’s first 18 nt. For the T>C sheet, columns are also included with data normalized to parts per million, and three columns are included showing the 4-TU/no TU ratio for the relevant mutation rates.