

Abstract
Background
In patients with end-stage heart failure, the primary etiology often originates in the left ventricle, and eventually the contractile function of the right ventricle (RV) also becomes compromised. RV tissue-level deficits in contractile force and/or kinetics need quantification to understand involvement in ischemic and non-ischemic failing human myocardium.
Methods and Results
The human population suffering from heart failure is diverse, requiring many subjects to be studied in order to perform an adequately powered statistical analysis. From 2009-present we assessed live tissue-level contractile force and kinetics in isolated myocardial RV trabeculae from 44 non-failing and 41 failing human hearts. At 1 Hz stimulation rate (in vivo resting state) the developed active force was not different in non-failing compared to failing ischemic nor non-ischemic failing trabeculae. In sharp contrast, the kinetics of relaxation were significantly impacted by disease, with 50% relaxation time being significantly shorter in non-failing vs. non-ischemic failing, while the latter was still significantly shorter than ischemic failing. Gender did not significantly impact kinetics. Length-dependent activation was not impacted. Although baseline force was not impacted, contractile reserve was critically blunted. The force-frequency relation was positive in non-failing myocardium, but negative in both ischemic and non-ischemic myocardium, while the β-adrenergic response to isoproterenol was depressed in both pathologies.
Conclusions
Force development at resting heart rate is not impacted by cardiac pathology, but kinetics are impaired and the magnitude of the impairment depends on the underlying etiology. Focusing on restoration of myocardial kinetics will likely have greater therapeutic potential than targeting force of contraction.
Keywords: Heart Failure, Myocardial Biology, Excitation-Contraction Coupling
Introduction
End-stage heart failure impact over 6 million people in the US alone, and is a growing and very costly problem. End-stage heart failure is typically characterized by contractile dysfunction of the ventricles. In vivo, ventricular contractility is impaired resulting in overall reduced pump function. Ventricular contraction is mainly determined by two factors: the number of myocytes contributing to contraction and the average contractile force they produce. In addition to ventricular contraction, the rate (i.e. shortening and re-lengthening speed of the tissue) at which this contractile process takes place is critically important for function; when the contractile and relaxation speed is insufficient, a component of diastolic dysfunction arises and further deteriorates overall pump function.
Investigation of myocardial kinetics of viable human myocardium has been limited[1]. The majority of previous studies have focused on contractile strength, thus, contractile kinetics were not always assessed or reported. A second factor is that the diversity in the human population, especially when compared to inbred laboratory mice, increases the variability in assessed parameters. It is thus necessary to investigate a larger number of subjects to achieve the same statistical power as studies using animal-based models. Live contracting human myocardial tissue is logistically much harder to obtain than tissue from animal models of disease, specifically in large numbers required. Thus, most past studies were performed on a relatively small patient/sample population, allowing only for large differences to be detected as statistically significant. Another complicating factor is that to quantify parameters obtained in the failing heart, a comparison with healthy tissue parameters obtained in an identical manner to the failing tissue is paramount. Non-failing viable myocardium is often even more difficult to obtain than failing tissue, and most past studies, including our own, have typically included a very low sample size of non-failing controls, or none at all. Again, the vast differences even among healthy humans (due to size, race, age, gender, etc.) result in a broad spectrum of quantifiable myocardial contraction and kinetic parameters which severely limits statistical power. Although all these past studies[2–16] have made significant contributions to further understanding myocardial dysfunction at the tissue level, the low numbers of subjects, mixed etiologies, lack of non-failing controls, and gender-indifferent studies, have produced conflicting results regarding contractile function and kinetic behavior of end-stage failing myocardium. Moreover, although experimental conditions of most studies were chosen to be close to in vivo conditions, several studies were performed at non-physiological temperatures, or non-physiological pacing rates, possibly further contributing to conflicting results.
Consequently, we lack sufficiently-powered quantitative data on force development and myocardial kinetics of viable RV tissue, as it relates to end-stage failing hearts of ischemic versus non-ischemic etiologies. Moreover, little is known about the impact of gender on human myocardial contractility. Therefore, we set out to quantify the kinetics of relaxation in failing right ventricular myocardium, wherever possible stratified by non-ischemic and ischemic etiology and gender. We performed the study over 7.5 years on a large patient/sample group (44 non-failing hearts, 41 end-stage failing hearts) to allow for the detection of significant, functional differences that would be clinically relevant. Moreover, we investigated the impact of disease on contractile force and kinetics encompassing the three main mechanisms of contractile regulation[17]: the length-dependent activation[18, 19], frequency-dependent activation[20], and β-adrenergic stimulation[21].
Methods
Human Tissue Collection
All explanted hearts were obtained directly in the operating room and immediately flushed with cardioplegic solution after removal from donors/patients as described previously[2, 22]. The hearts were transferred to the laboratory (within 10–30 minutes) in cold cardioplegic solution containing (in mM): 110 NaCl, 16 KCL, 16 MgCl2, 10 NaHCO3, and 0.5 CaCl2. All hearts were procured and treated with identical protocols, solutions and timing regardless of their source. All human tissues were experimented on with approval from the Institutional Review Board (IRB) of The Ohio State University and conform to the declaration of Helsinki. Informed consents were acquired from cardiac transplant patients. All end-stage failing hearts (n = 41) were acquired from patients in the operating room undergoing cardiac transplantation at The Ohio State University Wexner Medical Center. Non-transplantable donor hearts (n = 44) were acquired in the operating room in collaboration with Lifeline of Ohio Organ Procurement. The biometric characteristics of these hearts are provided in Tables 1 and 3. Characteristics of the dimensions of the hearts, as well as wall-thickness is given in Tables 2 and 4. The age of the donors was 48 ± 15 (SD) years, the patients with failing ischemic hearts were 59 ± 7 years old, and the failing non-ischemic patients were 53 ± 10 years of age. Body mass index was also similar: 29.6 ± 8.0, 26.8 ± 4.3, and 29.4 ± 4.3 resp. Heart weight, on average, was 19.7% higher in failing hearts compared to the donor hearts, with no significant difference by etiology.
Table 1.
Characteristics of non-failing hearts
| ID # | Sex | Etiology | Age | Race | BMI | Heart Weight (g) |
|---|---|---|---|---|---|---|
| 118258 | M | Non-Failing | 38 | Caucasian | 29.9 | 575 |
| 147381 | M | Non-Failing | 58 | Caucasian | 32.1 | 512 |
| 156910 | F | Non-Failing | 62 | Caucasian | 35.5 | 478 |
| 168021 | F | Non-Failing | 62 | Caucasian | 26.0 | 896 |
| 219852 | F | Non-Failing | 30 | Caucasian | 24.2 | 299 |
| 240603 | F | Non-Failing | 51 | Caucasian | 23.9 | 320 |
| 294050 | M | Non-Failing | 23 | Caucasian | 33.9 | 462 |
| 313956 | F | Non-Failing | 38 | Caucasian | 31.0 | 406 |
| 331253 | F | Non-Failing | 42 | Hispanic | 29.6 | 390 |
| 364587 | M | Non-Failing | 19 | Caucasian | 24.5 | 300 |
| 380071 | F | Non-Failing | 43 | Caucasian | 60.9 | 603 |
| 394176 | M | Non-Failing | 57 | Caucasian | 32.0 | 748 |
| 402879 | M | Non-Failing | 54 | Caucasian | 30.9 | 474 |
| 415217 | M | Non-Failing | 42 | Caucasian | 28.6 | 508 |
| 435578 | M | Non-Failing | 20 | Caucasian | 25.9 | 324 |
| 442404 | M | Non-Failing | 69 | Caucasian | 40.3 | 659 |
| 452192 | F | Non-Failing | 55 | African American | 24.5 | 350 |
| 460025 | F | Non-Failing | 69 | Caucasian | 29.6 | 435 |
| 474083 | F | Non-Failing | 41 | African American | 37.7 | |
| 476074 | F | Non-Failing | 29 | Caucasian | 20.2 | 271 |
| 481041 | M | Non-Failing | 41 | African American | 20.3 | 455 |
| 481043 | F | Non-Failing | 65 | Caucasian | 26.0 | 451 |
| 488240 | F | Non-Failing | 51 | Caucasian | 39.7 | |
| 507207 | F | Non-Failing | 72 | Caucasian | 30.5 | 456 |
| 514489 | F | Non-Failing | 42 | Caucasian | 28.3 | 327 |
| 600245 | F | Non-Failing | 51 | Caucasian | 20.6 | 507 |
| 618200 | F | Non-Failing | 58 | Caucasian | 499 | |
| 632941 | F | Non-Failing | 68 | Caucasian | 31.1 | 402 |
| 685884 | M | Non-Failing | 36 | Caucasian | 25.2 | 415 |
| 694855 | F | Non-Failing | 46 | Caucasian | 22.3 | 356 |
| 712301 | M | Non-Failing | 67 | Caucasian | 29.2 | 527 |
| 749693 | M | Non-Failing | 65 | African American | 20.8 | 643 |
| 753820 | F | Non-Failing | 43 | African American | 20.9 | 605 |
| 785258 | F | Non-Failing | 51 | Caucasian | 23.0 | 335 |
| 809108 | M | Non-Failing | 60 | Caucasian | 33.3 | 842 |
| 845013 | M | Non-Failing | 26 | Caucasian | 24.1 | 497 |
| 872295 | M | Non-Failing | 56 | Caucasian | 31.5 | 539 |
| 921821 | M | Non-Failing | 22 | African American | 26.3 | 383 |
| 925852 | F | Non-Failing | 34 | African American | 50.4 | 672 |
| 947200 | F | Non-Failing | 63 | Caucasian | 34.5 | 608 |
| 958987 | M | Non-Failing | 40 | Caucasian | 33.7 | 615 |
| 984478 | F | Non-Failing | 54 | African American | 21.1 | 348 |
| 987692 | F | Non-Failing | 34 | Caucasian | 29.0 | 313 |
Table 3.
Characteristics of failing hearts
| ID # | Sex | Primary Etiology | Detailed Classification | Age | Race | Heart Weight (g) | BMI | Height (m) | Body Weight (kg) | Heart Weight:Body Weight (g/kg) |
|---|---|---|---|---|---|---|---|---|---|---|
| 100713 | M | Failing Ischemic | 64 | Caucasian | 27.4 | 1.80 | 88.7 | |||
| 110713 | M | Failing Ischemic | 50 | Caucasian | 714 | 21.6 | 1.75 | 66.0 | 10.8 | |
| 214010 | M | Failing Ischemic | 64 | Caucasian | 495 | 27.5 | 1.78 | 87.1 | 5.68 | |
| 233587 | M | Failing Ischemic | 65 | Caucasian | 692 | 29.2 | 1.78 | 92.5 | 7.48 | |
| 250585 | M | Failing Ischemic | 65 | Caucasian | 621 | 24.5 | 1.69 | 69.9 | 8.88 | |
| 328163 | M | Failing Ischemic | 63 | Caucasian | 506 | 24.3 | 1.78 | 77.1 | 6.56 | |
| 422358 | M | Failing Ischemic | 52 | Caucasian | 498 | 23.0 | 1.83 | 76.9 | 6.48 | |
| 479062 | M | Failing Ischemic | 50 | Caucasian | 636 | 22.3 | 1.83 | 74.8 | 8.50 | |
| 537263 | M | Failing Ischemic | 62 | African American | 427 | 23.0 | 1.83 | 76.8 | 5.56 | |
| 597750 | M | Failing Ischemic | 67 | Caucasian | 630 | 34.5 | 1.77 | 108.0 | 5.83 | |
| 611422 | M | Failing Ischemic | 68 | Caucasian | 576 | 24.5 | 1.83 | 82.1 | 7.02 | |
| 645444 | M | Failing Ischemic | 47 | African American | 411 | 27.2 | 1.78 | 86.2 | 4.77 | |
| 774694 | F | Failing Ischemic | 50 | Caucasian | 486 | 33.5 | 1.50 | 75.3 | 6.45 | |
| 777902 | M | Failing Ischemic | 59 | Caucasian | 530 | 32.4 | 1.83 | 108.6 | 4.88 | |
| 101230 | M | Failing Non-Ischemic | 36 | African American | 563 | 36.9 | 1.78 | 117.0 | 4.81 | |
| 102011 | M | Failing Non-Ischemic | Sarcoid Myocarditis | 62 | Caucasian | 527 | 31.8 | 1.73 | 95.3 | 5.53 |
| 110624 | F | Failing Non-Ischemic | Dilated | 51 | Caucasian | 508 | 25.7 | 1.65 | 70.0 | 7.26 |
| 203056 | M | Failing Non-Ischemic | 51 | African American | 704 | 27.4 | 1.85 | 93.9 | 7.50 | |
| 323104 | M | Failing Non-Ischemic | familial cardiomyopathy/systolic heart failure | 63 | Caucasian | 543 | 29.6 | 1.73 | 88.5 | 6.14 |
| 335581 | F | Failing Non-Ischemic | 40 | Caucasian | 715 | 30.6 | 1.70 | 88.5 | 8.08 | |
| 369452 | M | Failing Non-Ischemic | 61 | African American | 540 | 29.8 | 1.73 | 89.1 | 6.06 | |
| 390112 | F | Failing Non-Ischemic | 60 | Caucasian | 608 | 29.3 | 1.65 | 79.8 | 7.62 | |
| 437918 | M | Failing Non-Ischemic | 40 | Caucasian | 731 | 24.3 | 1.70 | 70.3 | 10.4 | |
| 450564 | M | Failing Non-Ischemic | Dilated | 30 | Caucasian | 482 | 25.8 | 1.78 | 81.6 | 5.90 |
| 465645 | M | Failing Non-Ischemic | 59 | Caucasian | 972 | |||||
| 497522 | F | Failing Non-Ischemic | 62 | Caucasian | 649 | 25.9 | 1.80 | 83.9 | 7.74 | |
| 522421 | F | Failing Non-Ischemic | Dilated with possible familial component | 56 | Caucasian | 470 | 33.9 | 1.55 | 81.4 | 5.77 |
| 537114 | M | Failing Non-Ischemic | 48 | Middle Eastern | 439 | 22.3 | 1.77 | 69.9 | 6.28 | |
| 588415 | M | Failing Non-Ischemic | 52 | Caucasian | 390 | 28.2 | 1.73 | 84.4 | 4.62 | |
| 599014 | M | Failing Non-Ischemic | 50 | Caucasian | 667 | 31.6 | 1.80 | 102.5 | 6.51 | |
| 602102 | M | Failing Non-Ischemic | 64 | Caucasian | 465 | 31.0 | 1.80 | 100.5 | 4.63 | |
| 622584 | M | Failing Non-Ischemic | Noncompaction | 54 | Caucasian | 606 | 39.6 | 1.80 | 128.2 | 4.73 |
| 679533 | M | Failing Non-Ischemic | 47 | Caucasian | 930 | 30.6 | 1.91 | 111.6 | 8.33 | |
| 728878 | M | Failing Non-Ischemic | 40 | Caucasian/African American | 747 | 25.5 | 1.96 | 98.0 | 7.62 | |
| 820447 | M | Failing Non-Ischemic | 68 | Caucasian | 567 | 31.8 | 1.80 | 103.0 | 5.50 | |
| 879242 | F | Failing Non-Ischemic | 58 | Vietnamese | 489 | 22.5 | 1.58 | 56.3 | 8.69 | |
| 897154 | F | Failing Non-Ischemic | 53 | African American | 428 | 30.2 | 1.60 | 77.2 | 5.54 | |
| 904475 | F | Failing Non-Ischemic | Post partum cardiomyopathy | 35 | Caucasian | 262 | 28.5 | 1.58 | 71.2 | 3.68 |
| 911614 | M | Failing Non-Ischemic | 65 | Caucasian | 716 | 33.5 | 1.83 | 112.3 | 6.38 | |
| 956256 | M | Failing Non-Ischemic | 64 | Caucasian | 714 | 34.1 | 1.79 | 109.3 | 6.53 | |
| 975654 | M | Failing Non-Ischemic | 60 | Caucasian | 483 | 24.3 | 1.80 | 78.6 | 6.15 |
Table 2.
Dimensions of non-failing hearts
| ID # | Overall Dimension (cm) | Wall Thickness (cm): RV | Wall Thickness (cm): LV | Wall Thickness (cm): Septum | ||
|---|---|---|---|---|---|---|
| 118258 | 13 | 11.5 | 7.5 | 0.9 | 2.4 | 2 |
| 147381 | 13.5 | 10 | 9.5 | 0.9 | 1.7 | 1.9 |
| 156910 | 14.5 | 10.5 | 9.5 | 0.7 | 1.6 | 1.7 |
| 168021 | ||||||
| 219852 | 14 | 9.5 | 10.5 | 0.5 | 1.5 | 1 |
| 240603 | 11 | 9 | 7.5 | 0.6 | 1.1 | 1.3 |
| 294050 | 14 | 9.5 | 9.5 | 0.7 | 1.8 | 1.6 |
| 313956 | 10 | 9.5 | 9.5 | 0.6 | 2.3 | 1.8 |
| 331253 | 12.4 | 12 | 11 | 1.8 | 1.8 | 1.3 |
| 364587 | 10.5 | 8.5 | 8 | 0.9 | 1.4 | 1.4 |
| 380071 | 14.5 | 13.2 | 11.5 | 1 | 1.8 | 2 |
| 394176 | 16 | 14 | 13 | 1.5 | 2 | 2 |
| 402879 | 14 | 10.5 | 10.2 | 0.5 | 2 | 1.5 |
| 415217 | 11 | 10 | 9.5 | 0.5 | 1.5 | 1.5 |
| 435578 | 13.3 | 9 | 8.5 | 1 | 1.8 | 1.4 |
| 442404 | 15 | 10.5 | 11 | 1.5 | 2 | |
| 452192 | 11.5 | 8.5 | 8.5 | 0.4 | 1.5 | 1.8 |
| 460025 | 11 | 9.5 | 10 | 0.7 | 1.7 | 1.7 |
| 474083 | ||||||
| 476074 | 10 | 7 | 7 | 0.5 | 1.3 | 2.1 |
| 481041 | 12 | 10 | 9 | 0.5 | 1.7 | 2.3 |
| 481043 | ||||||
| 488240 | 0.6 | 1.4 | 1.5 | |||
| 507207 | 15 | 10.9 | 9.8 | 0.5 | 1.4 | 1.4 |
| 514489 | 11 | 9 | 8.5 | 0.9 | 2 | 1.4 |
| 600245 | 13.5 | 10 | 11.5 | |||
| 618200 | 0.7 | 1.8 | 1.5 | |||
| 632941 | 12 | 9 | 8.5 | |||
| 685884 | 0.8 | 1.5 | 1.4 | |||
| 694855 | 12 | 10 | 9.5 | 0.8 | 2.2 | 2.9 |
| 712301 | 15.3 | 11.8 | 11 | 0.8 | 1.9 | 1.6 |
| 749693 | 14.5 | 9.5 | 9 | 0.8 | 2 | 2 |
| 753820 | 13.5 | 10.5 | 10 | 0.8 | 1.5 | |
| 785258 | 11 | 8.5 | 9 | 0.9 | 1.7 | 1.2 |
| 809108 | 16.5 | 14 | 12.5 | 0.6 | 2 | 2 |
| 845013 | 14.5 | 11 | 9.5 | 0.6 | 1.3 | 1.5 |
| 872295 | 13.5 | 10 | 9 | 0.6 | 2 | 1.8 |
| 921821 | ||||||
| 925852 | 14 | 10.5 | 10.5 | 0.5 | 2.3 | 2 |
| 947200 | 11 | 10 | 13 | |||
| 958987 | 14 | 11.5 | 10 | 0.6 | 1.5 | 1.5 |
| 984478 | 9.5 | 8.5 | 8 | 0.7 | 1.9 | 1.5 |
| 987692 | 12.5 | 9 | 7 | 0.7 | 1.5 | 1.6 |
Table 4.
Dimensions of failing hearts
| ID # | Overall Dimension (cm) | Wall Thickness (cm): RV | Wall Thickness (cm): LV | Wall Thickness (cm): Septum | ||
|---|---|---|---|---|---|---|
| 100713 | ||||||
| 110713 | 14 | 13 | 10 | 0.7 | 2 | 1.2 |
| 214010 | 15 | 14 | 9 | 0.9 | 1.2 | 1.5 |
| 233587 | 14 | 10.5 | 10.5 | 0.7 | 1 | 0.5 |
| 250585 | 12.5 | 11 | 8.5 | 0.9 | 1.9 | 0.8 |
| 328163 | 15 | 13 | 14 | 0.7 | 1.9 | 2.3 |
| 422358 | 14 | 12 | 11 | 0.7 | 1.5 | 1.3 |
| 479062 | 0.7 | 1.4 | 1 | |||
| 537263 | 11 | 10 | 8.5 | 0.9 | 2 | 1.2 |
| 597750 | 14 | 14 | 12 | 0.7 | 1.7 | 1.6 |
| 611422 | 13 | 11.5 | 10 | 0.5 | 2 | 1.5 |
| 645444 | 14 | 11 | 10 | 0.7 | 1.5 | 1 |
| 774694 | 14 | 13 | 11 | 0.6 | 1.2 | 1.8 |
| 777902 | 11 | 10.5 | 10.5 | 0.8 | 1.5 | 1.1 |
| 101230 | 14 | 14 | 11.5 | |||
| 102011 | 15 | 10 | 10.5 | 0.5 | 1.5 | 0.9 |
| 110624 | ||||||
| 203056 | 13 | 12.5 | 11 | 0.7 | 1.4 | 1.2 |
| 323104 | 14 | 13 | 13.5 | 0.8 | 1.1 | 1.7 |
| 335581 | 12 | 11.5 | 10.5 | 1.2 | 1.5 | 1.6 |
| 369452 | 14 | 12.5 | 11 | 0.6 | 1.5 | 2 |
| 390112 | 12 | 11.5 | 10.5 | 0.3 | 1.3 | 0.5 |
| 437918 | 14.5 | 13 | 10 | 0.5 | 1 | 0.6 |
| 450564 | 12 | 11 | 8 | 0.6 | 1.4 | 1.2 |
| 465645 | 16 | 14.5 | 14 | 0.5 | 1.5 | 1.7 |
| 497522 | 14.5 | 11.5 | 10.5 | 0.7 | 1 | 0.7 |
| 522421 | 10 | 10 | 8 | 1 | 1.3 | 1.3 |
| 537114 | 12 | 10 | 11 | 1.2 | 1.2 | 1 |
| 588415 | 10.5 | 10 | 8.5 | 0.7 | 1 | 1.3 |
| 599014 | 14.5 | 14 | 13.5 | 0.5 | 1.6 | 1.2 |
| 602102 | 11.5 | 10 | 9 | 0.8 | 1.4 | very thin |
| 622584 | 16 | 10.6 | 9.6 | 0.9 | 1.7 | 1.3 |
| 679533 | 17 | 15 | 13 | 1.1 | 1.8 | 1.5 |
| 728878 | 15 | 12 | 11 | 0.9 | 1.4 | 1.3 |
| 820447 | 14 | 10.5 | 11.5 | 0.7 | 1.3 | 1 |
| 879242 | 15 | 12.5 | 11.5 | 1.5 | 2 | 1.9 |
| 897154 | 11 | 10.5 | 10.5 | 0.9 | 2 | 1.5 |
| 904475 | 9 | 7.5 | 7 | 0.7 | 1.2 | 0.8 |
| 911614 | 15 | 12 | 11 | 0.9 | 1.5 | 1.2 |
| 956256 | 13 | 13 | 13 | 0.9 | 2.3 | |
| 975654 | 14.5 | 10.5 | 11.5 | 1 | 1.7 | 1.3 |
Trabeculae Isolation
The RV of each heart was transferred from the cardioplegic solution to a cold modified Krebs-Henseleit solution (K-H) bubbled with 95% O2-5% CO2 containing (in mM): 137 NaCl, 5 KCl, 0.25 CaCl2, 20 NaHCO3, 1.2 NaH2PO4, 1.2 MgSO4, 10 dextrose, and 20 BDM (2,3-butanedione monoxime) and pH of 7.4. Linear, small, and free-running trabeculae were isolated with the aid of a stereo dissection microscope, and kept in this solution at 0–4 °C until the time of the experiment. Thin, free running trabeculae were chosen since they contain all cell-types found in the heart, and their mechanical properties very closely match those of the ventricle, including inotropic properties of ejection[23], and they have a long experimental life[24], whereas papillary muscles (often used in studies using mice and rats), are way too big in humans to ensure a properly oxygenated preparation. Muscles were transferred into custom-made setups as previously described for animal models[25] and the perfusion solution was changed to another modified K-H without BDM. This solution was maintained at 37 °C and continuously bubbled with 95% O2-5% CO2 resulting in pH of 7.4. Stimulation was started at baseline frequency of 0.5 Hz while CaCl2 concentration of the solution was slowly raised to 2 mM over ~15 minutes. Muscles were gradually stretched (over a few minutes) until an increase in developed force was exceeded by an increase in resting tension. This length, designated as L100 or Lopt (optimal length), roughly corresponds to sarcomere length of ~2.2 μm, which is near or at the in vivo sarcomere length at end-diastole[26].
Baseline trabeculae function
Once muscles had stabilized in the set-up, at 1 Hz, baseline contractile force was assessed. This frequency reflects a normal resting in vivo heart rate of 60 bpm. After collecting contractile and kinetic data at this baseline frequency 5–10 steady state twitches were averaged to minimize mechanical noise. Under the stereo microscope, dimensions of the trabeculae were assessed (resolution of 10 μm), and all recorded forces were normalized to the cross-section area of the trabecula. Average dimensions of all trabeculae (n=85, 1 per heart included in baseline analysis) were 441 ± 18 μm in width, 296 ± 12 μm in thickness, and 2.9 ± 0.1 mm in length, with an average cross-section area of 0.116 ± 0.010 mm2. There were no significant differences between dimensions of muscles from non-failing or failing hearts, nor between muscles from males or females, all were selected to be comparable. The failing hearts themselves were significantly larger due to hypertrophy and/or dilatation.
Assessment of contractile regulation and contractile reserve
The three most physiologically relevant regulatory systems were investigated in subsets of muscles. First, the length-dependent activation mechanism was assessed by measuring contractile force and kinetics at 4 different muscle lengths. We assessed the muscle at optimal length (Lopt, at 0.5 Hz). Then, the muscle length was reduced to 85% of optimal length (L85%), resulting in a length slightly below the in vivo prevailing end-systolic sarcomere length [24]. Thereafter, it was assessed at 90% of Lopt (L90%), then at 95% of Lopt (L95%), and then measured at Lopt again. The force-frequency relationship was determined at Lopt using 6 frequencies ranging from 0.5 Hz to 3 Hz. This range encompasses the entire physiological range of a healthy adult human heart. At each frequency, the muscle was allowed to stabilize for ~2–5 minutes, and contractile force and kinetics were recorded. Lastly, the β-adrenergic response was determined by assessing a concentration-response protocol (1 nM – 1 μM) for isoproterenol at Lopt and 0.5 Hz.
Data analysis and statistics
All hearts at time of arrival in the lab received a 6-digit code that served as a de-identifier. At the time of measurement and analysis, for almost all cases the pathology/etiology and gender/age of the heart was not known to the experimenter. All tension and kinetic measurements were collected and analyzed using custom made programs in LabView (National Instruments). Muscle tensions were normalized to the cross-sectional area of the muscles and expressed as mN/mm2. Statistical analysis was performed with ANOVA, followed by post-Hoc tests where appropriate. Statistical significance was set at two-tailed P < 0.05 unless stated otherwise. All data is shown as mean ± S.E.M. unless stated otherwise.
Results
A total of 85 human hearts rendered at least 1 successful experiment in which baseline contractile force was assessed. The overall success rate of getting baseline data from an individual muscle is >60%, and the overall success rate of getting at least 1 successful experiment per heart is >90%. For RV trabeculae, these success rates did not appear to be impacted by the disease status of the heart.
Active baseline force development in failing versus non-failing trabeculae
Typical examples of twitch contractions under physiological conditions (1 Hz, 37 °C, and at optimal preload) are shown in Figure 1A. Without further stratification of either disease etiology or gender, baseline contractile force of all 85 trabeculae is shown in Figure 1B. On average, active developed force was not significantly different between muscles from non-failing and failing hearts (19.97 ± 2.11 vs. 18.62 ± 2.08 mN/mm2, resp., P=0.65). Diastolic (resting) tension likewise was not different (6.19 ± 0.44 vs. 6.50 ± 0.50 mN/mm2, resp., P=0.65), and there was no correlation between diastolic tension and active developed force at optimal length in either group (not shown).
Figure 1.

A: Original twitch contractions of a non-failing (#958987), failing ischemic (#597750), and failing non-ischemic (#110624) muscle. B: Active developed force in trabeculae isolated from n=44 non-failing and n=41 end-stage failing hearts was not different. C: Stratified by gender (34 female, 51 male), active developed force and diastolic force were not different in either non-failing or failing trabeculae.
About 77% of end-stage failing patients undergoing cardiac transplantation are male[27]. This was closely reflected in our cohort of 41 end-stage failing hearts where 32 (78%) were from males and 9 were from females. In addition, most non-failing donor hearts not used for transplantation are of female origin: in our data set of 44 non-failing hearts, 25 were female and 19 were male. If there were to be a gender-based difference in myocardial force development, it could potentially mask an impact of pathology. We thus stratified the data by gender, and analyzed whether a potential gender-based difference in active force development was present in our data. Two-way ANOVA on gender (male/female) and pathology (non-failing/failing) reported that there was no overt gender-based impact (P=0.11), nor pathology-based impact (P=0.76), nor an interaction between these two factors (P=0.51). Figure 1C shows the averages and standard errors for non-failing and failing males and females. Two-way ANOVA testing included two-tailed testing. According to many previous studies, it could be expected that males would be stronger, not potentially weaker. A one-sided t-test (to test whether males are specifically stronger than females) between non-failing females versus non-failing males indeed showed statistical significant higher force in males (24.20 ± 3.79 (n=19) vs. 16.76 ± 2.20 mN/mm2 (n=25), P=0.040). The difference between males and females was not significant in failing myocardium (P=0.32).
Baseline contraction and relaxation kinetics
Time from electrical stimulation to peak tension (time to peak, TTP, in ms) was not statistically different (P=0.11) between non-failing and failing trabeculae (Figure 2A). Time from peak tension to 50% relaxation (RT50, Figure 2B) was significantly longer in failing versus non-failing trabeculae (155 ± 4 ms vs. 129 ± 3 ms, P<0.0001). Total twitch time (Figure 2C), calculated as time from stimulation to 90% relaxation time (note, 100% relaxation time cannot be accurately assessed as force asymptotically approaches resting tension), was also significantly longer: 519 ± 12 ms in failing trabeculae vs. 451 ± 9 ms in non-failing trabeculae (P<0.0001). Stratification by gender revealed no gender-based effect on any of these parameters. For instance, RT50% was 131 ± 5 ms in non-failing males vs. 128 ± 4 ms in non-failing females (P=0.56), and 158 ± 5 ms in failing males vs. 146 ± 8 ms in failing females (P=0.25).
Figure 2.

Twitch timing parameters of 85 human hearts stratified between trabeculae from failing (n=41) and non-failing (n=44) hearts. A: Time to peak tension was not significantly different. B: Time from peak tension to 50% relaxation was significantly (*, P<0.00001) prolonged in trabeculae from failing hearts. C: Twitch duration, calculated as time from stimulation to 90% relaxation, was significantly (*, P<0.00001) prolonged in trabeculae from failing hearts. D: Data from figure 1A relative to active developed force shows the impact of etiology on twitch kinetics. E: Stratified by etiology (failing ischemic, n=14, failing non-ischemic, n=27), time to peak tension was not significantly different. F: Time from peak tension to 50% relaxation was significantly prolonged in both non-ischemic and ischemic failing muscles compared to trabeculae from non-failing hearts (*, P<0.0001), while this parameter in trabeculae from ischemic failing hearts was significantly slower than in trabeculae from non-ischemic failing hearts (**, P<0.001). G: Twitch duration, calculated as time from stimulation to 90% relaxation, was significantly prolonged in both non-ischemic and ischemic failing muscles compared to trabeculae from non-failing hearts (*, P<0.005).
Although gender-stratification did not reveal a gender-based difference in twitch kinetics, the underlying etiology of the failing hearts could be a discriminating factor. Based on clinical assessment of the patient at time of cardiac transplantation, end-stage failing hearts were divided into those with a primary diagnosis of ischemic etiology (n=14), and those with a non-ischemic etiology (n=27). We found that trabeculae isolated from ischemic failing hearts had a significantly longer TTP (Figure 2D) compared to non-failing trabeculae (ANOVA, P<0.05). 50% relaxation time (Figure 2E) was significantly prolonged in both etiologies (P<0.001). In addition to being prolonged compared to non-failing trabeculae, ischemic-failing trabeculae relaxed significantly slower than non-ischemic failing trabeculae (P<0.01). A similar observation was made for total twitch time (Figure 2F): non-failing preparations had a significantly shorter twitch time compared to both ischemic and non-ischemic failing ones (P<0.005), while ischemic- failing trabeculae had a significantly longer twitch time than non-ischemic failing trabeculae (P<0.05).
Next, we investigated the impact of etiology on the maximal speed of contraction and relaxation. dF/dt, analogous to dP/dt of the ventricle, reflects the maximal speed of force development, and was not different (ANOVA P=0.94) between the three groups (Figure 3A). Negative dF/dt, the maximal speed of relaxation, was also not different (ANOVA, P=0.67). The derivative of force (dF/dt) is however highly dependent on the level of force. In order to study pure kinetics of the twitch contraction and relaxation, we divide dF/dt by the active developed force, allowing us to obtain a pure kinetic rate (unit: s−1). The maximal kinetic rate of contraction (Figure 3B) was also not different between the groups (ANOVA, P=0.15), but the maximal kinetic rate of relaxation was significantly slower in both failing etiologies (ANOVA, P<0.0001). The kinetic rate of relaxation was not dependent on the level of developed force.
Figure 3.

A: Maximal speed of contraction and relaxation were not different between any groups. B: The maximal kinetic rate (s−1) of contraction (maximal positive dF/dt/Fdev) was not different (P=0.15) between the groups, but maximal kinetic rate of relaxation (maximal negative dF/dt/Fdev) was significantly slower in both ischemic and non-ischemic failing myocardium (ANOVA, P<0.0001), while the difference between the ischemic and non-ischemic groups was not significant (P=0.13).
Regulation of contraction and kinetics; impact of muscle length
The three main mechanisms that the heart uses to regulate cardiac output are length-dependent activation, frequency-dependent activation, and β-adrenergic stimulation. First, we assessed length-dependent activation in a subset of trabeculae. In Figure 4A, B, and C we show trabeculae from all three groups at 4 different muscle lengths, ranging from 85% of optimal length to optimal length (L100). On average in Figure 4D we show that forces in all groups responded virtually equally to a change in muscle length. Since the absolute forces of the stronger muscles dominate this data presentation mode, we also plotted the data in Figure 4E related to the individual maximal force of the muscle at optimal length to investigate the true length-dependency and weigh all trabeculae equally in the analysis. As can be seen, this relationship is fairly linear, and not different between the three groups. Regarding kinetics, we observed a slowing of time to peak tension as muscle length increased, in all groups (Figure 4F). Plotted to their individual maximum, Figure 4G show that the slowing down of kinetics was both qualitatively and quantitatively similar in all groups. Likewise, total twitch time also slowed as length increased (Figure 4H), while the difference between non-failing and both failing etiologies was significant. The length-dependent slowing down of total twitch time was similar in all groups (Figure 4I).
Figure 4.

A: Twitch contractions of a trabecula from a non-failing heart (#768159) at 85%, 90%, 95%, and 100% of optimal length. B: Twitch contractions of a trabecula from a failing ischemic heart (#328163) at 85%, 90%, 95%, and 100% of optimal length. C: Twitch contractions of a trabecula from a failing non-ischemic heart (#101230) at 85%, 90%, 95%, and 100% of optimal length. D: Length dependent force development was virtually identical in all three groups, * indicates P<0.0001 of the impact of length on force. E: Same data as panel D, plotted to each muscle’s individual maximum to allow equal weight of each muscle in the statistical analysis. * indicates P<0.0001 of the impact of length on force. F: Time to peak force increases with muscle length in all groups (*P<0.0001), while in general, this parameter was faster shorter in non-failing myocardium (n=20, **P<0.05). G: Normalized data from panel F shows a significant interaction between muscle length and time to peak tension in all three groups (*P<0.0001). H: Total twitch time increases with muscle length in all three groups (*P<0.0001), while this parameter is shorter at all muscle lengths in the non-failing versus failing ischemic (n=3, $, P<0.01) and failing non-ischemic group (n=8, #, P<0.01). I: Normalized data from panel H shows a significant interaction between muscle length and total twitch time in all three groups (*P<0.0001).
Regulation of contraction and kinetics; impact of frequency
Frequency-dependent activation was assessed to reflect heart rates encompassing the entire in vivo range of the adult heart. In Figure 5A, B, and C we show trabeculae from all three groups at 6 different frequencies, ranging from 0.5 Hz to 3 Hz, at optimal length. Averages from all muscles are shown in Figure 5D. In non-failing myocardium, force increase between 0.5 and 2 Hz (positive force-frequency relationship, FFR), while in both failing ischemic and failing non-ischemic myocardium force decayed (negative force-frequency relationship, FFR). To equally weigh each muscle in the analysis, and to investigate the average shape of the FFR curve, data were normalized to those obtained at 0.5 Hz in the same muscle (Figure 5E). This analysis shows a positive FFR in non-failing myocardium, while in failing ischemic myocardium this FFR is flatter between 0.5 and 2 Hz compared to failing non-ischemic myocardium. Diastolic force also changed with frequency (Figure 5F); at 1 and 1.5 Hz, diastolic force slightly declined in both non-failing and failing ischemic muscles. In all groups, diastolic tension was higher at 3 Hz than baseline of 0.5 Hz. Kinetics were also significantly different between non-failing and both failing ischemic and failing non-ischemic trabeculae (Figure 5G); the maximal kinetic rate of relaxation was significantly faster in non-failing myocardium over the entire frequency range. However, the acceleration of this kinetic rate (shape of the curve) was equal in all three groups.
Figure 5.

A: Twitch contractions of a trabecula from a non-failing heart (#118258) at 6 frequencies, spanning the in vivo range showing a positive force-frequency relationship. B: Twitch contractions of a trabecula from a failing ischemic heart (#100714) at 6 frequencies showing a blunted/negative force-frequency relationship. C: Twitch contractions of a trabecula from a failing non-ischemic heart (#437918) at 6 frequencies showing a blunted/negative force-frequency relationship. D: Frequency-dependent regulation of force was different between failing and non-failing myocardium. The interaction of force and frequency was significantly (P<0.01) more positive in non-failing myocardium (n=33). E: Same data as panel A, plotted to each muscle’s force at 0.5 Hz to allow equal weight of each muscle in the statistical analysis. At all frequencies, force in non-failing trabeculae exceeded the force at the 0.5 Hz baseline. $ P<0.05 vs. non-ischemic failing trabeculae (n=8); * P<0.05 vs. ischemic failing trabecular (n=13). F: Relative to diastolic force at 0.5 Hz, diastolic force decreases ($, P<0.05) in the non-failing and failing ischemic group. The relative increase in diastolic force was significantly higher (*, P<0.05) in the failing non-ischemic group. In all groups, at 3 Hz the diastolic force was significantly higher than the 0.5 Hz baseline (#, P<0.05). G: The maximal kinetic rate of relaxation was significantly impacted by frequency in all groups (#, P<0.0001) and in addition was significantly faster (*, P<0.05) in non-failing trabeculae versus both failing groups.
Regulation of contraction and kinetics; impact of β-adrenergic stimulation
Finally, we investigated the response to beta-adrenergic stimulation by treatment with increasing concentrations of isoproterenol (1 nM – 1 μM). Because frequency-dependent inotropy uses some of the same pathways as β-adrenergic stimulation, such as troponin I and myosin light chain-2 phosphorylation [28, 29], the experiments were performed at 0.5 Hz to minimize the impact of frequency. In non-failing myocardium, we observed a robust increase in force, significantly greater than the increase in both failing groups (Figure 6A). Relative to baseline (no isoproterenol), isoproterenol- induced force development was similarly depressed in ischemic and non-ischemic myocardium (Figure 6B), with a slight rightward-shift of the curve in failing non-ischemic myocardium compared to non-failing myocardium (P<0.05). Kinetics accelerated similarly in each group (curve shape is the same), as illustrated for TTP in Figure 6C, and for total twitch time in Figure 6D (non-failing trabeculae had a significantly shorter TTP over the entire concentration range), while maximal rate of tension decline (-dF/dt/F) showed a similar result (not shown).
Figure 6.

A: Force of contraction was increased in all groups upon addition of isoproterenol in a concentration-dependent manner (P<0.0001). B: Same data as panel A, plotted to each muscle’s force at baseline to allow equal weight of each muscle in the statistical analysis. $ P<0.05 vs. failing non-ischemic trabeculae (n=6); * P<0.05 vs. failing ischemic trabeculae (n=3). C: Time to peak tension was significantly accelerated in all three groups (#, P<0.001), while in non-failing trabeculae (n=17) it was shorter compared to both failing groups.
Averaged kinetic profile stratified by primary etiology
To depict the average baseline contractile and kinetic characteristics of our data, we generated a plot in which an original twitch tracing was recalibrated in both amplitude and timing kinetics to reflect the average assessed twitch per group. This data is included to be used a reference for modeling studies, since original examples of twitches are heavily impacted by the large spread in quantifying parameters. In Figure 7, we present twitch data calibrated on 5 parameters that reflect the average diastolic force, active developed force, TTP, RT50, and total twitch time.
Figure 7.
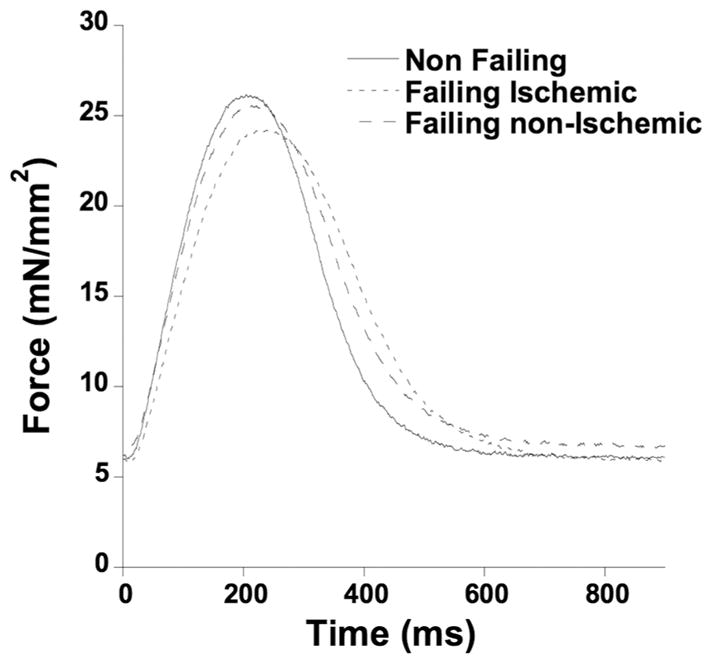
Single twitch contraction reflecting the average values of diastolic force, active developed force, time to peak tension, time to 50% relaxation, and total twitch time.
Discussion
The present study, shows several novel findings; in isolated RV myocardium 1) active force development at resting heart rate of the viable myocardium is not impaired, nor enhanced in heart failure, 2) Healthy male myocardium is stronger than healthy female myocardium, but gender does not have a major impact on contraction and relaxation in diseased myocardium, 3) kinetics of relaxation are impaired in end-stage failing myocardium while impairment is greater in myocardium with an ischemic etiology compared to a non-ischemic etiology. In addition, our study confirms several findings that previously were only shown in studies with significantly lower n-numbers, often preventing a sufficiently powered statistical analysis. We showed/statistically confirmed that 6) regulation of contraction via the length-dependent activation mechanism is not impaired in right ventricular trabeculae, 7) frequency-dependent regulation of contraction is significantly impaired, and 8) β-adrenergic regulation of contraction is impaired in myocardium from patients with end-stage heart failure.
Active force development at resting heart rate is not impaired
We found no significant difference in active force development between non-failing and failing human trabeculae. This is in agreement with several previous studies[2, 9, 13, 15], but in contrast to several other studies that observed either decreased developed force in failing preparations[5, 6, 14], or an increased level of developed force[12, 16]. Analysis of our data and past studies shows that there is a very large spread in the forces of isolated myocardium. As a result, when a small sample population is used, false-positives are more likely to occur. Additional reasons for discrepancies can be partially explained by baseline stimulation rate; when isolated myocardium is stimulated at sub-physiological rates, as has been done for baseline studies in several of these past studies, force developments may be more impacted in non-failing myocardium. Non-failing myocardium has a much more robust dependency on inter-beat duration than does failing myocardium, and the further the stimulation frequency is removed from the typical in vivo baseline of 60 beats per minute, the larger the differences are between non-failing and failing myocardium. Another factor that may have caused misinterpretations is that nearly all past studies combined non-ischemic and ischemic myocardium together as “end-stage failing myocardium”, or “myopathic myocardium”, and composition of the failing group may impact the outcome of the study. Temperature used during the experiments can also be a confounding factor, as it critically affects both the level of force development (typically higher at lower temperature), as well as slows down kinetics. Temperature-dependency of these factors can be potentially of different magnitude in failing versus non-failing myocardium, possibly adding to discrepancies in findings at low temperature compared to physiological temperature. Lastly, previous studies did not stratify between male or female tissue origin, which may have under-powered analyses, see below.
Changes in myofilament calcium sensitivity can alter force development capability. We did not measure this parameter in this study, as we focused on the twitch force and kinetics. However, further investigation into myofilament calcium sensitivity in the future would help understanding of the molecular mechanism of heart failure. Specifically, the assessment of myofilament calcium sensitivity in combination with intact muscle physiology would be needed to more completely understand the dynamic impact of altered myofilament responsiveness[30]. In addition, it is important to note that we assessed right ventricular trabeculae from left-sided failing hearts. Left-sided heart failure can lead to pressure overload on the right side through the pulmonary vasculature. This is seen in the clinic where vast majority of left-sided heart failure patients exhibit right-sided heart failure symptoms such ascites, jugular vein distension, hepatomegaly, and pitting edema.
Impact of gender on contraction and relaxation
Although we did not find a statistically significant difference in developed force (or any other parameter) between male and female myocardium in general (i.e. without disease stratification), a more detailed analysis showed a statistically larger force (average ~44%) in non-failing males compared to non-failing females. This was significant for 1-sided testing of the data, which was based on past studies that reported higher force in males. Thus, this sizeable difference in developed force (~44% higher in males) may have contributed to the outcome of past studies. Indeed, in our study, and several previous studies, failing myocardium was exclusively[11] or predominantly obtained from males[9, 14], with more than half of the non-failing groups obtained from females. Moreover, the majority of past studies did not disclose gender composition of the tested groups. Thus, findings in previous studies of failing myocardium being slightly stronger than non-failing myocardium may have been impacted by this potential gender bias. Kinetics, however, were not impacted by gender based on our results. Therefore current and past results on myocardial kinetics will be much less or more likely not at all impacted by a gender bias between groups. In addition, there was virtually no correlation between age of the patient/donor and contractile parameters (R=0.14 for developed force (at 1 Hz), and R=0.10 for 50% relaxation time).
Kinetics of relaxation are impaired in end-stage failing myocardium
Kinetics of relaxation were generally significantly slower in failing myocardium, with the most profound slowing down of kinetics observed in myocardium from patients with ischemic failing myocardium. This kinetic impairment is in close agreement with most previous studies, that either found a slowing of kinetics, or a trend in slower kinetics that was not statistically significant, the latter again likely due to low n-numbers in these studies. Moreover, the larger deficit in kinetics in ischemic myocardium, brought to light by our stratification of etiology, may have resulted in different conclusions depending on the etiology-composition in previous studies. In past studies in which most failing myocardium was of non-ischemic origin it would have been harder to detect statistical differences due to a milder kinetic phenotype. Like with gender, several past studies did not disclose or describe the etiology of the failing hearts.
None of the failing hearts used in this study have a primary diagnosed etiology of diastolic dysfunction. However, from the data, it can be derived that 13 of 14 failing ischemic hearts had kinetics that were slower than the average non-failing heart, while 21 of 27 failing non-ischemic hearts showed slower kinetics than the average non-failing heart. Thus, in our cohort, over 80% of the failing group had kinetics below the average of the non-failing group.
As expected in all failing hearts, the left ventricular performance was generally poor, with the LV EF in all failing hearts well under 25%. At the myocardial level, at least at resting heart rate, the RV myocardium tested in this study did not exhibit a contractile deficit. The combination of impaired kinetics with sufficient force development would lead us to conclude that improvement of cardiac contractile function may need to be more directed towards an improvement of kinetics rather than force per se. The still viable RV myocardium of the heart produces adequate force to sustain a cardiac output compatible with life, but does so with impaired kinetics. As demand for cardiac output increases with exercise, this kinetic impairment, rather than reduction in levels of developed force, may become prominent in limiting cardiac output.
Regulation of contraction via the length-dependent activation mechanism is not impaired
One of the main regulatory pathways that impacts cardiac force is volume-dependent changes in contractile strength, i.e. the length-dependent activation mechanism. In none of the tested parameters was this mechanism impaired in the right ventricular preparations we studied. With an increase in muscle length, all groups responded to a similar quantitative and qualitative increase in force, and decrease in kinetic rates. The changes in force upon an increase in muscle length are virtually instantaneous[31, 32]. Likewise, the changes in kinetics are virtually instantaneous[33, 34]. Although the complete underlying mechanism is currently still incompletely understood, lack of impact of etiology on this mechanism could indicate that future therapeutic interventions may be better directed at other regulatory mechanisms. On the other hand, we did not make a direct assessment of this length-dependent activation in left ventricular myocardium, so assessment of left-ventricular tissue as it pertains to length-dependent activation could potentially be a subject for future studies. In addition, length-dependent activation has not been studied as a function of β-adrenergic stimulation and/or activation frequencies in failing human hearts.
Frequency-dependent regulation of contraction is impaired in end-stage failing myocardium
The impaired regulation of frequency-dependent force development is a main hallmark of heart failure. Our results are in close agreement with previous studies that showed a similar frequency-dependence of force. In addition, we here for the first time detail the kinetic impairment in relaxation of failing hearts not shown in past studies. In virtually all past studies, relaxation was determined by assessment of RT50. Although this parameter is a good measure for relaxation properties at baseline frequencies when the muscle has time to fully relax, RT50 is not a reliable parameter to describe relaxation properties at higher frequencies when diastolic tension rises. This higher diastolic tension abbreviates the twitch contraction, and assessment of a 50% relaxation value is then greatly impacted by the elevated diastolic tension. As a result, RT50, and even more so RT90 are artificially abbreviated at high frequency. We analyzed the pure kinetic rate of relaxation, and showed that in failing myocardium the slower relaxation offset found at low frequency is maintained at high frequency. Despite that failing myocardium accelerates rather similarly to non-failing myocardium, its initial deficit in speed is not overcome, and becomes exceedingly impactful on relaxation as frequency increases. The failing non-ischemic group displayed a larger elevation in diastolic tension when frequency increased. This increase in diastolic tension strongly, or even solely, contributing to the more negative force-frequency relationship than observed in ischemic failing muscles. As frequency increased, there was an initial drop in the low end of the frequency range in diastolic tension in the non-failing and the failing ischemic group, possibly related to frequency- induced myofilament calcium desensitization[28]. In the non-ischemic failing group, this decrease did not occur, similar to animal studies where pressure-overload- induced, non-ischemic heart failure significantly impaired the frequency-dependent myofilament calcium desensitization[29].
β-adrenergic regulation of contraction is impaired in end-stage failing myocardium
In close agreement with previous studies[2, 21], the β-adrenergic response was blunted in failing myocardium. A significant acceleration of kinetics was observed upon stimulation; however, similar to our results in frequency-dependent activation, the baseline deficit in relaxation speed persists throughout this concentration-response protocol, thereby maintaining the impaired relaxation of failing myocardium. Therefore, the blunting β-adrenergic response is in large part due to the initial kinetic deficit that is not overcome even at the highest dose. It is likely that improvement of baseline kinetics will result in an improved β-adrenergic responsiveness as well.
Limitations of the study
Isolated muscle preparations can suffer from core-hypoxia, i.e. when a preparation is very think, oxygen diffusion to the center of the preparation is lower than oxygen consumption, leading to a hypoxic core. The size limits at which core hypoxia occurs critically highly depend on temperature, frequency of stimulation, and species, as investigated in past studies by us and others[35, 36]. Although the experiments in this study were performed on preparations that were typically half the size of those used in most previous human studies, they were still fairly large compared to studies on rats and mice that show core hypoxia can occur already at 150–200 μm in diameter[37]. The impact of potential core-hypoxia in this study was assessed by correlation analysis between muscle diameter contractile response. We observed no correlation between muscle size and contractile performance, including frequency response. Only at the very highest stimulation frequency 3 Hz (typically not attained in vivo given the patients age), could there have been a significant impact of core hypoxia. In addition, the muscle sizes were not different between the 3 groups, and thus would have been a factor equally impacting all groups.
We did not assess protein levels and phosphorylation levels in this study. Protein phosphorylation is a very rapid process, and can occur in less than a second (i.e. the fight/flight response is nearly instantaneous), and can subside/return to baseline in 10’s of seconds/few minutes, because of the constitutively active phosphatase activity. The phosphorylation status of procured quiescent tissue (in our studies frozen typically within 10–30 minute post-explantation, in most past studies typically several hours) may thus provide, at best, a weak substitute for actual experiments on tissue in contractile homeostasis. Although we have collected several muscles at homeostasis, we can unfortunately only freeze each muscle in this study under 1 condition at the very end of the study (typically in presence of maximal β-adrenergic stimulation). Hence, a correlative study on the impact of phosphorylation on force development and regulation needs samples at different contractile states (different lengths, frequencies, and beta-stimulation), and is currently lacking.
Summary
Under baseline conditions, kinetics (but not force development) are impaired in right ventricular myocardium from hearts in end-stage failure, with a more prominent slowing down of kinetics in myocardium from hearts with a primary ischemic failing etiology. Whereas the length-dependent activation mechanism in all etiologies is completely preserved, both in regulation of active force development and regulation of kinetics, frequency-dependent as well as β-adrenergic stimulation are markedly impaired. This impairment is mainly due to the inability to overcome the slower kinetics present at baseline. Since pharmacological treatment of end-stage failing myocardium can only target the remaining viable myocardium, improvement in the kinetics of contraction would likely have the biggest impact on restoring normal regulatory function in these pathological tissues. Therapies targeting improvement of baseline kinetics, perhaps rather than targeting improvement in contractile strength, are critically needed.
Acknowledgments
The authors would like to thank Susan Montgomery, Erin Bumgardner, and Emily Jarvis for their help in obtaining consent from the patients for this study. The authors would like to thank Abraham Zawodni (Lifeline of Ohio) for help with obtaining clinical correlates for the donor hearts.
Sources of Funding:
Funding for this project was supported by NIH RC1HL099538 (to PMLJ), NIH R01HL113084 (to PMLJ), AHA 16PRE33410549 (to JHC), AHA 16POST27760155 (to MTE), and AHA 12PRE11480008 (to NMN).
Footnotes
Discolsures:
The authors have no conflicts to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Janssen PM, Biesiadecki BJ, Ziolo MT, Davis JP. The Need for Speed: Mice, Men, and Myocardial Kinetic Reserve. Circ Res. 2016;119:418–21. doi: 10.1161/CIRCRESAHA.116.309126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Milani-Nejad N, Canan BD, Elnakish MT, Davis JP, Chung JH, Fedorov VV, et al. The Frank-Starling mechanism involves deceleration of cross-bridge kinetics and is preserved in failing human right ventricular myocardium. Am J Physiol Heart Circ Physiol. 2015;309:H2077–86. doi: 10.1152/ajpheart.00685.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pieske B, Maier LS, Bers DM, Hasenfuss G. Ca2+ handling and sarcoplasmic reticulum Ca2+ content in isolated failing and nonfailing human myocardium. Circ Res. 1999;85:38–46. doi: 10.1161/01.res.85.1.38. [DOI] [PubMed] [Google Scholar]
- 4.Hasenfuss G, Holubarsch C, Hermann HP, Astheimer K, Pieske B, Just H. Influence of the force-frequency relationship on haemodynamics and left ventricular function in patients with non-failing hearts and in patients with dilated cardiomyopathy. Eur Heart J. 1994;15:164–70. doi: 10.1093/oxfordjournals.eurheartj.a060471. [DOI] [PubMed] [Google Scholar]
- 5.Hasenfuss G, Mulieri LA, Blanchard EM, Holubarsch C, Leavitt BJ, Ittleman F, et al. Energetics of isometric force development in control and volume-overload human myocardium. Comparison with animal species. Circ Res. 1991;68:836–46. doi: 10.1161/01.res.68.3.836. [DOI] [PubMed] [Google Scholar]
- 6.Hasenfuss G, Mulieri LA, Leavitt BJ, Allen PD, Haeberle JR, Alpert NR. Alteration of contractile function and excitation-contraction coupling in dilated cardiomyopathy. Circ Res. 1992;70:1225–32. doi: 10.1161/01.res.70.6.1225. [DOI] [PubMed] [Google Scholar]
- 7.Gwathmey JK, Hajjar RJ. Relation between steady-state force and intracellular [Ca2+] in intact human myocardium. Index of myofibrillar responsiveness to Ca2+ Circulation. 1990;82:1266–78. doi: 10.1161/01.cir.82.4.1266. [DOI] [PubMed] [Google Scholar]
- 8.Gwathmey JK, Slawsky MT, Hajjar RJ, Briggs GM, Morgan JP. Role of intracellular calcium handling in force-interval relationships of human ventricular myocardium. J Clin Invest. 1990;85:1599–613. doi: 10.1172/JCI114611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pieske B, Sutterlin M, Schmidt-Schweda S, Minami K, Meyer M, Olschewski M, et al. Diminished post-rest potentiation of contractile force in human dilated cardiomyopathy. Functional evidence for alterations in intracellular Ca2+ handling. J Clin Invest. 1996;98:764–76. doi: 10.1172/JCI118849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hajjar RJ, DiSalvo TG, Schmidt U, Thaiyananthan G, Semigran MJ, Dec GW, et al. Clinical correlates of the myocardial force-frequency relationship in patients with end-stage heart failure. J Heart Lung Transplant. 1997;16:1157–67. [PubMed] [Google Scholar]
- 11.Rossman EI, Petre RE, Chaudhary KW, Piacentino V, 3rd, Janssen PM, Gaughan JP, et al. Abnormal frequency-dependent responses represent the pathophysiologic signature of contractile failure in human myocardium. J Mol Cell Cardiol. 2004;36:33–42. doi: 10.1016/j.yjmcc.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 12.Chaudhary KW, Rossman EI, Piacentino V, 3rd, Kenessey A, Weber C, Gaughan JP, et al. Altered myocardial Ca2+ cycling after left ventricular assist device support in the failing human heart. J Am Coll Cardiol. 2004;44:837–45. doi: 10.1016/j.jacc.2004.05.049. [DOI] [PubMed] [Google Scholar]
- 13.Gwathmey JK, Copelas L, MacKinnon R, Schoen FJ, Feldman MD, Grossman W, et al. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ Res. 1987;61:70–6. doi: 10.1161/01.res.61.1.70. [DOI] [PubMed] [Google Scholar]
- 14.Pieske B, Maier LS, Piacentino V, 3rd, Weisser J, Hasenfuss G, Houser S. Rate dependence of [Na+]i and contractility in nonfailing and failing human myocardium. Circulation. 2002;106:447–53. doi: 10.1161/01.cir.0000023042.50192.f4. [DOI] [PubMed] [Google Scholar]
- 15.Hajjar RJ, Schmidt U, Helm P, Gwathmey JK. Ca++ sensitizers impair cardiac relaxation in failing human myocardium. J Pharmacol Exp Ther. 1997;280:247–54. [PubMed] [Google Scholar]
- 16.Gwathmey JK, Warren SE, Briggs GM, Copelas L, Feldman MD, Phillips PJ, et al. Diastolic dysfunction in hypertrophic cardiomyopathy. Effect on active force generation during systole. J Clin Invest. 1991;87:1023–31. doi: 10.1172/JCI115061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biesiadecki BJ, Davis JP, Ziolo MT, Janssen PML. Tri-modal regulation of cardiac muscle relaxation; intracellular calcium decline, thin filament deactivation, and cross-bridge cycling kinetics. Biophysical Reviews. 2014;6:273–89. doi: 10.1007/s12551-014-0143-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frank O. Zur Dynamik des Herzmuskels. Z Biol. 1895;32:370–447. [Google Scholar]
- 19.Starling EH. The Lineacre lecture on the law of the heart. Longmans Green; London: 1918. [Google Scholar]
- 20.Bowditch HP. Über die Eigenthümlichkeiten der Reizbarkeit, welche die Muskelfasern des Herzens zeigen. Berichte über die Verhandlungen der Königlich-Sächsischen Gesellschaft der Wissenschaften zu Leipzig. Mathematisch Physische Klasse. 1871;23:652–89. [Google Scholar]
- 21.Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K, et al. Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N Engl J Med. 1982;307:205–11. doi: 10.1056/NEJM198207223070401. [DOI] [PubMed] [Google Scholar]
- 22.Elnakish MT, Canan BD, Kilic A, Mohler PJ, Janssen PM. Effects of zacopride, a moderate IK1 channel agonist, on triggered arrhythmia and contractility in human ventricular myocardium. Pharmacol Res. 2017;115:309–18. doi: 10.1016/j.phrs.2016.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Tombe PP, Little WC. Inotropic effects of ejection are myocardial properties. Am J Physiol Heart Circ Physiol. 1994;266:H1202–13. doi: 10.1152/ajpheart.1994.266.3.H1202. [DOI] [PubMed] [Google Scholar]
- 24.Janssen PML, Lehnart SE, Prestle J, Hasenfuss G. Preservation of contractile characteristics of human myocardium in multi-day cell culture. J Mol Cell Cardiol. 1999;31:1419–27. doi: 10.1006/jmcc.1999.0978. [DOI] [PubMed] [Google Scholar]
- 25.Janssen PML, Stull LB, Marban E. Myofilament properties comprise the rate-limiting step for cardiac relaxation at body temperature in the rat. Am J Physiol Heart Circ Physiol. 2002;282:H499–H507. doi: 10.1152/ajpheart.00595.2001. [DOI] [PubMed] [Google Scholar]
- 26.Rodriguez EK, Hunter WC, Royce MJ, Leppo MK, Douglas AS, Weisman HF. A method to reconstruct myocardial sarcomere lengths and orientations at transmural sites in beating canine hearts. Am J Physiol Heart Circ Physiol. 1992;263:H293–306. doi: 10.1152/ajpheart.1992.263.1.H293. [DOI] [PubMed] [Google Scholar]
- 27.Weiss ES, Allen JG, Patel ND, Russell SD, Baumgartner WA, Shah AS, et al. The impact of donor-recipient sex matching on survival after orthotopic heart transplantation: analysis of 18 000 transplants in the modern era. Circ Heart Fail. 2009;2:401–8. doi: 10.1161/CIRCHEARTFAILURE.108.844183. [DOI] [PubMed] [Google Scholar]
- 28.Varian KD, Janssen PML. Frequency-dependent acceleration of relaxation involves decreased myofilament calcium sensitivity. Am J Physiol Heart Circ Physiol. 2007;292:H2212–9. doi: 10.1152/ajpheart.00778.2006. [DOI] [PubMed] [Google Scholar]
- 29.Varian KD, Kijtawornrat A, Gupta SC, Torres CA, Monasky MM, Hiranandani N, et al. Impairment of diastolic function by lack of frequency-dependent myofilament desensitization rabbit right ventricular hypertrophy. Circ Heart Fail. 2009;2:472–81. doi: 10.1161/CIRCHEARTFAILURE.109.853200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chung JH, Biesiadecki BJ, Ziolo MT, Davis JP, Janssen PM. Myofilament Calcium Sensitivity: Role in Regulation of In vivo Cardiac Contraction and Relaxation. Front Physiol. 2016;7:562. doi: 10.3389/fphys.2016.00562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mateja RD, de Tombe PP. Myofilament length-dependent activation develops within 5 ms in guinea-pig myocardium. Biophys J. 2012;103:L13–5. doi: 10.1016/j.bpj.2012.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Milani-Nejad N, Chung JH, Canan BD, Davis JP, Fedorov VV, Higgins RS, et al. Insights into Length-Dependent Regulation of Cardiac Cross-Bridge Cycling kinetics in Human Myocardium. Arch Biochem Biophys. 2016 doi: 10.1016/j.abb.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Monasky MM, Varian KD, Davis JP, Janssen PML. Dissociation of force decline from calcium decline by preload in isolated rabbit myocardium. Pflugers Arch. 2008;456:267–76. doi: 10.1007/s00424-007-0394-0. [DOI] [PubMed] [Google Scholar]
- 34.Monasky MM, Taglieri DM, Jacobson AK, Haizlip KM, Solaro RJ, Janssen PM. Post-translational modifications of myofilament proteins involved in length-dependent prolongation of relaxation in rabbit right ventricular myocardium. Arch Biochem Biophys. 2013;535:22–9. doi: 10.1016/j.abb.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schouten VJ, ter Keurs HE. The force-frequency relationship in rat myocardium. The influence of muscle dimensions. Pflugers Arch. 1986;407:14–7. doi: 10.1007/BF00580714. [DOI] [PubMed] [Google Scholar]
- 36.Raman S, Kelley MA, Janssen PML. Effect of muscle dimensions on trabecular contractile performance under physiological conditions. Pflugers Arch. 2006;451:625–30. doi: 10.1007/s00424-005-1500-9. [DOI] [PubMed] [Google Scholar]
- 37.Stull LB, Leppo M, Marban E, Janssen PML. Physiological determinants of contractile force generation and calcium handling in mouse myocardium. J Mol Cell Cardiol. 2002;34:1367–76. doi: 10.1006/jmcc.2002.2065. [DOI] [PubMed] [Google Scholar]