

Abstract
Fatty acid induced hypothalamic inflammation (HI) is a potential cause of the obesity epidemic. It is unclear whether saturated or n-6 polyunsaturated fat is the primary driver of these effects. Premenopausal women are protected, in part, from obesity and associated comorbidities by circulating 17β-estradiol (E2). It is unknown how HI interacts with E2, because most studies of HI do not examine females despite the involvement of E2 in hypothalamic energy homeostasis. Our objective is to determine the effects of high fat diets with varying levels of linoleic acid (LA) and saturated fat on the energy and glucose homeostasis in female mice with and without E2. Female C57BL/6J mice were fed either a control diet or a 45% kilocalories from fat diet with varying levels of LA (1%, 15%, or 22.5% kilocalories from LA) with or without E2 (300 μg/kg/day orally). After 8 weeks, the oil-treated high fat groups gained more weight than control regardless of fat type. E2 reduced body fat accumulation in all high fat groups. Glucose clearance from glucose challenge was impaired by LA. Nighttime O2 consumption was increased by E2, regardless of diet and independent of activity. Neuropeptides and HI genes were not affected by LA or SFA content. These data show that fatty acid type does not affect body weight, but does affect glucose metabolism in females, and these effects are not associated with an induction in HI gene expression.
Keywords: estrogen, fatty acids, obesity, inflammation, saturated, polyunsaturated, linoleic acid
Introduction
Diet and lifestyle play a causative role in obesity1, but there is also an endocrinological aspect seen in the difference between sexes. Obesity and its comorbidities are more common in post- than premenopausal women, simultaneous with reduced circulating 17β-estradiol (E2, or steroid)2–5. Food intake follows the menstrual cycle, peaking during the luteal phase, with a nadir peri-ovulatory6–9. Unsurprisingly, the post-menopausal fall in circulating E2 is associated with weight gain10. Hormone replacement therapy can mitigate these effects11. A dietary treatment for obesity in women requires a greater understanding of the physiology of female obesity.
Originally considered a behavioral problem, the etiology of obesity is now thought to be primary physiological, especially after the discovery of leptin12 and genetic obesity. Hypothalamic inflammation (HI) can impair energy balance regulation13,14, including leptin signaling. Whether HI causes obesity is an ongoing question15, but it is suggestive that HI involves dysregulation of hormone and signaling pathways in genetic obesity. Dietary fatty acids (FA) can induce HI16, which may form a causal connection between the molecular mechanism of obesity and its increase in the population. Within the context of HI and energy balance, E2 is involved in these pathways17, compounding the case for greater experimental consideration.
The concept of obesity being simple over-nutrition has been challenged by the discovery of endocrinological action of specific nutrients. The caloric energy from fat, for example, cannot explain the current obesity epidemic, since over the relevant time period fat intake was stable18–20. FA profile, however, has changed; monounsaturated and saturated FAs (MUFA and SFA) have been replaced by polyunsaturated (PUFA), in particular linoleic acid (LA)21. Different FAs have demonstrated different metabolic effects22. In males, high-fat diets differing in dietary SFA and LA caused minor differences in obesity, but significant differences in glucose metabolism23.
Determining FA-specific effects is difficult because foods contain a mixture of FAs. Lard is commonly used, and has a highly variable FA profile. Coconut oil, an almost pure SFA source, has been used to compare obesity from SFA and PUFA feeding24–26. The metabolic effects of different FAs have been studied in females27, but the overwhelming majority of HFD and HI research is performed on male animals to avoid the complications of estrogen.
Our objective is to determine whether diet-induced obesity from HFDs high in SFA or LA can be linked to HI gene expression in E2-deficient and E2-treated ovariectomized (ovx) female mice. We hypothesize that mice fed LA will gain more weight, show impaired glucose metabolism, and exhibit higher expression of HI gene biomarkers compared to mice fed SFA. We expect these effects to be more pronounced in the oil treated than in E2 treated mice. To address this objective, ovx females were fed 3 different HFDs over 8 weeks, with weekly measurements of body weight and food intake followed by body composition assessment, indirect calorimetry, glucose and insulin tolerance tests (GTT and ITT), and tissue harvest for HI gene expression.
Materials and Methods
Animals
All animal treatments were in accordance with guidelines based on National Institutes of Health standards and were performed with Institutional Animal Care and Use Committee approval at Rutgers University. Female wild-type (WT C57BL/6J) mice were selectively bred in-house, and maintained under controlled temperature (23°C) and photoperiod conditions (12/12 h light/dark cycle) with food and water ad libitum.
Drugs and Diets
Estradiol benzoate (EB) and 4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF, 1 mg/mL) were purchased from Sigma-Aldrich. Ketamine, Marcaine®, and Rimadyl® were purchased from Henry Schein Animal Health (Columbus, OH, USA). EB was dissolved in 100% ethanol (Sigma) prior to dissolving in coconut oil (Nature’s Way). Diets were purchased from Research Diets (New Brunswick, NJ): CON (10% kcal fat; D12450B) and 3 HFDs based on Research Diets 45% kcal fat diet D12451, with either 1%, 15% or 22.5% of kcals from LA, the remainder consisting of coconut oil SFAs. See Table 1 for FA profile of experimental diets.
Table 1.
Composition of all diets fed to female mice.
| Diets | CON | 1% | 15% | 22.5% |
|---|---|---|---|---|
| kcals/g | 3.85 | 4.7 | 4.7 | 4.7 |
| Oils (g) | 45 | 202.5 | 202.5 | 202.5 |
| Coconut oil | 0 | 133 | 64.5 | 21.5 |
| Flaxseed oil | 0 | 10 | 10 | 10 |
| Lard | 20 | 0 | 0 | 0 |
| Safflower oil | 0 | 0 | 45 | 45 |
| Soybean oil | 25 | 2 | 2 | 2 |
| Sunflower oil | 0 | 57.5 | 81 | 124 |
| Carbohydrate (g) | 700 | 255.6 | 255.6 | 255.6 |
| Protein (g) | 203 | 203 | 203 | 203 |
| % energy from carbohydrate | 70 | 31 | 31 | 31 |
| % energy from protein | 20 | 24 | 24 | 24 |
| % energy from fat | 10 | 45 | 45 | 45 |
| % from SFA | 2.26 | 31 | 17 | 8 |
| % from LA | 4.22 | 1 | 15 | 22.5 |
| Fatty acids (g) | 43.3 | 199.8 | 199.9 | 199.6 |
| C6, Caproic | 0.0 | 0.8 | 0.4 | 0.1 |
| C8, Caprylic | 0.0 | 10.2 | 5.0 | 1.7 |
| C10, Capric | 0.0 | 7.8 | 3.8 | 1.3 |
| C12, Lauric | 0.0 | 63.3 | 30.7 | 10.2 |
| C14, Myristic | 0.2 | 23.9 | 11.6 | 3.9 |
| C16, Palmitic | 6.5 | 14.0 | 12.9 | 11.9 |
| C16:1, Palmitoleic | 0.3 | 0.0 | 0.0 | 0.0 |
| C18, Stearic | 3.1 | 16.9 | 11.0 | 7.4 |
| C18:1, Oleic | 12.6 | 52.4 | 51.0 | 55.8 |
| C18:2, Linoleic | 18.3 | 4.7 | 67.7 | 101.4 |
| C18:3, Linolenic | 2.2 | 5.8 | 5.8 | 5.9 |
| C20:4, Arachidonic | 0.1 | 0.0 | 0.0 | 0.0 |
| C20:5, Eicosapentaenoic | 0.0 | 0.0 | 0.0 | 0.0 |
| C22:6, Docosahexaenoic | 0.0 | 0.0 | 0.0 | 0.0 |
| SFA (%) | 22.7 | 69 | 37.7 | 18.3 |
| MUFA (%) | 29.9 | 26 | 25.5 | 28.0 |
| PUFA (%) | 47.4 | 5 | 36.8 | 53.7 |
SFA, saturated fatty acids; MUFA, monounsaturated fatty acids; PUFA, polyunsaturated fatty acids
Experimental Design
Adult females (10 weeks old) were ovariectomized (ovx) via a single ventral incision under isoflurane anesthesia (2% in O2:N2O (2:1)) delivered by face mask with a local injection of Marcaine® (2 mg/kg) followed by 48 h of pain management using an injection of Rimadyl® (4 mg/kg, every 24 h; Henry Schein). Females fed ad libitum CON; (n = 24) or one of 3 high-fat diets (1%, 15% or 22.5%; n = 24 for each diet) for 8 weeks. Half of each diet group was administered oil and the other half EB (300 μg/kg) daily perorally via a peanut butter carrier (all groups: n = 12 – CON-oil, CON-E2, 1%-oil, 1%-E2, 15%-oil, 15%-E2, 22.5%-oil, 22.5%-E2). We chose peroral E2 dosage to reduce the stress-inducing effects of repeated injection and to maintain a constant systemic level of E2, similar to hormone replacement therapy in postmenopausal women28. Females were housed in group-matched pairs and body weight and food intake were measured weekly. At the end of 8 weeks, body composition was measured using an EchoMRI 3-in-1 Body Composition Analyzer (Echo Medical Systems, Houston, TX, USA). Calorimetric and activity measurements (48 h run) were performed using a Columbus Instruments’ Comprehensive Lab Animal Monitoring System (CLAMS) (Columbus Instruments, Inc., Columbus, OH, USA). After suitable recovery, a glucose tolerance test (GTT) was performed on each female. Females were fasted overnight (1700h-900h) in a new cage. At the start of the test and 30 min after application of a local anesthetic (Lidocaine®; Henry Schein) to the tail, mice were placed in Plexiglass restrainers and tails were nicked to collect a baseline (time=0) glucose reading using a glucometer (AlphaTRAK2, Carlsbad, CA, USA). Immediately after baseline, females were given an intraperitoneal (i.p.) glucose injection (2.0 g/kg body weight) and placed back individually into clean cages. Tail blood samples were collected at 15, 30, 60, 90, 120, and 180 min post-injection. After 180 min, all mice were returned to their home cages with ad libitum access to water and food. After sufficient recovery (~3–4 d), an insulin tolerance test (ITT) was performed after a 5 h fast in a similar manner as the GTT, with an i.p. injection of insulin (0.75 units/kg). Blood samples were collected from the tail in individual cages at 15, 30, 60, 90, and 120 min post-injection.
Brain and Body Dissections
After ~1 week recovery from ITT, females were dosed at 0900h and decapitated after sedation with ketamine (100 µl of 100 mg/ml, i.p.) at 1000h. Food was removed at the last dosing. Trunk blood was collected in a K+ EDTA collection tube. Plasma was prepared for peptide analysis by adding a protease inhibitor, 4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF, 1 mg/mL) to each collection tube. Samples were maintained on ice until centrifugation at 3,000 rpm for 10 min at 4°C. Plasma was stored at –80°C until analysis. Leptin and interleukin-6 (Il-6) were determined by multiplex assay (#MMHMAG-44K; EMD Millipore, Billerica, MA, USA). Total plasma E2 levels were measured using Mouse/Rat Estradiol ELISA kit (ES180S-100; Calbiotech, Spring Valley, CA, USA).
The brain was immediately extracted from the skull and rinsed in ice-cold Sorensen’s buffer for 30 sec., then cut using a brain matrix (Ted Pella, Redding, CA, USA) into one mm thick coronal rostral and caudal blocks corresponding to Plates 42 to 47 and Plates 48 to 53, respectively, from The Mouse Brain in Stereotaxic Coordinates (Paxinos & Franklin 2008, 3rd Edition)29. Blocks of the basal hypothalamus (BH) were transferred to RNAlater (Life Technologies) and stored overnight at 4°C. The rostral and caudal parts of the arcuate nucleus were dissected from slices using a dissecting microscope. Dissected tissue was stored at –80°C. Total RNA was extracted from the combined nucleus (rostral and caudal arcuate) using Ambion RNAqueous-Micro Kits (Life Technologies) per the manufacturer’s protocol. Total RNA was also DNase I-treated, using the extraction kits, at 37°C for 30 min. to minimize any genomic DNA contamination. RNA quantity and quality were determined using a NanoDrop ND-2000 spectrophotometer (ThermoFisher, Waltham, MA, USA) and an Agilent 2100 Bioanalyzer and RNA Nano Chips (Agilent Technologies, Santa Clara, CA, USA). Only samples with RNA Integrity Number > 8 were used in the analysis of gene expression.
Quantitative Real-Time PCR
Complementary DNA (cDNA) was synthesized from 200 ng of total RNA using Superscript III reverse transcriptase (Life Technologies), 4 μl 5x Buffer, 25 mM MgCl2, 10 mM dNTP (Clontech Laboratories, Mountain View, CA, USA), 100 ng random hexamer primers (Promega, Madison, WI, USA), 40 U/μl Rnasin (Promega) and 100 mM DTT in DEPC-treated water (Bioexpress, Kaysville, UT, USA) in total volume of 20 μl. Reverse transcription was conducted using the following protocol: 5 min at 25°C, 60 min at 50°C, 15 min at 70°C. The cDNA was diluted to 1:20 with nuclease-free water (Bioexpress) for a final cDNA concentration 0.5 ng/μl and stored at –20°C. Negative control (no reverse transcriptase) was processed simultaneously with the experimental samples.
All primers were designed to span exon-exon junctions and synthesized by Life Technologies, using Clone Manager 5 software (Sci Ed Software, Cary, NC, USA). See Table 2 for a listing of all the primer sets used for quantitative real-time PCR (qPCR). For qPCR, 4 μl cDNA template was amplified using either PowerSYBR Green master mix (Life Technologies) or Sso Advanced SYBR Green (BioRad, Hercules, CA, USA) on CFX-Connect Real-time PCR instrument (BioRad). Standard curves for each primer pair were prepared using serial dilutions of BH cDNA in triplicate to determine the efficiency (E=10(−1/m) – 1, m=slope) of each primer pair. All efficiencies expressed as percent efficiency were approximately equal (one doubling per cycle, 90–110%). The relative mRNA expression was calculated using the ΔΔCT method utilizing a calibrator of diluted (1:20) cDNA from BH of an untreated male. The amplification protocol for all the genes was as follows: initial denaturing – 95°C for 10 min (PowerSYBR) or 3 min (SsoAdvanced) followed by 40 cycles of amplification at 94°C for 10 sec (denaturing), 60°C for 45 sec (annealing), and completed with a dissociation step for melting point analysis with 60 cycles of 95°C for 10 sec, 65 °C to 95°C (in increments of 0.5°C) for 5 sec and 95°C for 5 sec. The geometric mean of the reference genes Actb, Hprt, and Gapdh was used to calculate δCq values. Quantification values were generated only from samples showing a single product at the expected melting point. All gene expression data were expressed as an n-fold difference relative to the calibrator.
Table 2.
Primer sequences
| Gene Name |
Forward Primer | Reverse Primer | Accession # |
|---|---|---|---|
| Actb | GCCCTGAGGCTCTTTTCCA | TAGTTTCATGGATGCCACAGGA | NM_007393.3 |
| Agrp | CTCCACTGAAGGGCATCAGAA | ATCTAGCACCTCCGCCAAA | NM_007427.2 |
| Cart | GCTCAAGAGTAAACGCATTCC | GTCCCTTCACAAGCACTTCAA | NM_013732 |
| Cx3cl1 | ACGAAATGCGAAATCATGTGC | CTGTGTCGTCTCCAGGACAA | NM_009142.3 |
| Hprt | GCTTGCTGGTGAAAAGGACCTC TCGAAG |
CCCTGAAGTACTCATTATAGTCA AGGGCAT |
NM_013556 |
| Icam1 | GTGATGCTCAGGTATCCATCCA | CACAGTTCTCAAAGCACAGCG | NM_010493.2 |
| Ikk | CCATATCCTGGCTGTCACCT | GGCACCTTGGATGACCTAGA | NM_01159774.1 |
| Il-1 | GCAACTGTTCCTGAACTCAACT | ATCTTTTGGGGTCCGTCAACT | NM_008361.3 |
| Il-6 | GAGATCGACTCTCTGTTCGAGG | GCCCGTTGAAGAAGTCCTG | NM_010479.2 |
| Il-15 | ACATCCATCTCGTGCTACTTGT | GCCTCTGTTTTAGGGAGACCT | NM_008357.2 |
| Insr | GTGTTCGGAACCTGATGAC | GTGATACCAGAGCATAGGAG | NM_010568 |
| Lepr | AGAATGACGCAGGGCTGTAT | TCCTTGTGCCCAGGAACAAT | NM_146146.2 |
| Npy | ACTGACCCTCGCTCTATCTC | TCTCAGGGCTGGATCTCTTG | NM_023456 |
| Pai1 | TTCAGCCCTTGCTTGCCTC | ACACTTTTACTCCGAAGTCGGT | NM_008871.2 |
| Pomc | GGAAGATGCCGAGATTCTGC | TCCGTTGCCAGGAAACAC | NM_008895 |
| Socs3 | TTCACGGCTGCCAACATCT | GCTAGTCCCGAAGCGAAATCT | NM_007707.3 |
| Stat3 | TTCCTGGCACCTTGGATTG | CGAAGGTTGTGCTGATAGAG | NM_213659.2 |
| Tlr4 | ATGGCATGGCTTACACCACC | GAGGCCAATTTTGTCTCCACA | NM_021297.2 |
| Traf6 | ATCTCTGAGGATCATCAAGTAC ATTGT |
TGTGTGTATTAACCTGGCACTTC TG |
NM_009424.2 |
Statistical Analysis
All data were expressed as mean ± SEM, and analyzed by a multi-factorial (steroid and diet) ANOVA followed by a post-hoc Bonferroni test using Graphpad Prism software (GraphPad Software, Inc., La Jolla, CA 92037 USA). Body weight, GTT, and ITT data were analyzed using repeated-measures, two-way ANOVA with a post-hoc Bonferroni test. All gene expression data were analyzed using two-way ANOVA with a post-hoc Bonferroni test within each genotype because expression was normalized to the CON-oil samples of each genotype. In all experiments, effects were considered significant at α ≤ 0.05.
Results
E2 reduces weight gain from LA and SFA
We fed one of three HFDs with a range of LA concentrations (1%, 15%, 22.5%), or a low-fat control diet (CON) to ovx female mice with and without E2 treatment for 8 weeks and measured weight gain. By the second week, weight gain for 1% and 22.5% oil became greater than CON; weight gain for 15% oil became greater than CON by week 3 (Figure 1A-C). Weight gain from week 2 onwards was also greater for oil vs. E2 treated mice within all three HFDs. At weeks 6–8, weight gain of 1% and 22.5% E2 became greater than CON. The final body weights of oil treated 1%, 15%, and 22.5% were greater than CON (Figure 1D). Diet (F (3, 88) = 14.64; p < 0.0001), steroid (F (1, 88) = 39.79; p < 0.0001), and their interaction (F (3, 88) = 2.845; p < 0.05) affected body weights. Within diets, E2 treatment reduced body weights in 15% and 22.5%, but not 1% or CON.
Figure 1: Body weight gain.
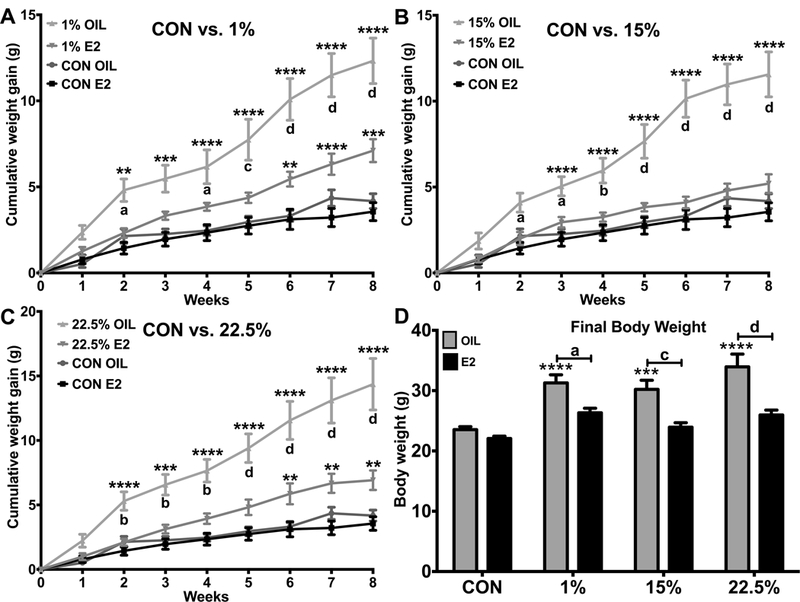
A. Weekly weight gain of CON vs. 1% fed oil and E2 treated mice. Two-way ANOVA: interaction time*treatment (txt): F (24, 352) = 16.11; p < 0.0001. B. Weekly weight gain of CON vs. 15% fed oil and E2 treated mice. Two-way ANOVA: interaction time*txt: F (24, 352) = 17.23; p < 0.0001. C. Weekly weight gain of CON vs. 22.5% fed oil and E2 treated mice. Two-way ANOVA: interaction time*txt: F (24, 352) = 16; p < 0.0001. D. Cumulative weight gain over 8 weeks. Two-way ANOVA: interaction: diet*steroid: F (3, 88) = 2.845; p < 0.05. Data are represented as means ± SEM. Sample size for each group was 12. Data were analyzed by two-way ANOVA (repeated measures or standard) with post-hoc Bonferroni multiple pairwise comparisons test. Letters denote comparisons within diet between steroid treatments; asterisks denote comparisons within steroid treatment between HFD and CON. Comparisons within steroid, between HFDs are denoted with asterisks over capped lines. (*& a = p<0.5; ** & b = p<0.01; *** & c = p<0.001; **** & d = p<0.0001).
HFD effects on intake and body composition opposed by E2
We determined weekly energy intake (Figure 2A) by multiplying food intake in grams by the caloric density of diets. Steroid (F (1, 56) = 11.9; p < 0.01) and diet (F (3, 56) = 11.19; p < 0.0001) affected food intake, but there was only a trend towards an effect of E2 within any diet. E2-treated 15% (p<0.001) and 22.5% (p<0.0001) consumed more food than E2-treated CON; all oil-treated HFD groups consumed more food than oil-treated CON. Feeding efficiency (Figure 2B) was determined by dividing weekly weight gained in grams by food consumed that week in kcals. The effects of E2 (F (1, 39) = 11.07; p < 0.01) resulted in a greater FE in 22.5% oil than their E2-treated counterparts (p<0.0001). The effects of diet on FE (F (3, 39) = 5.079; p < 0.01) were seen in all HFD oils being more efficient than E2-treated.
Figure 2: Feeding and body composition.
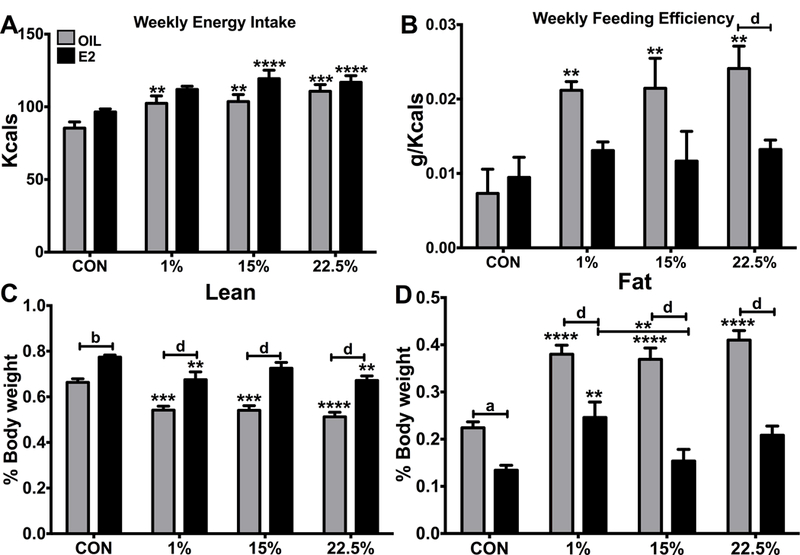
A. Weekly average food intake. Two-way ANOVA: diet: F (3, 56) = 11.19; p < 0.0001, steroid: F (1, 56) = 11.9; p < 0.01. B. Weekly average feeding efficiency. Two-way ANOVA: diet: F (5, 39) = 5.079; p < 0.01, steroid: F (1, 39) = 11.07; p < 0.01. C. Lean mass as a percent of body weight. Two-way ANOVA: diet: F (3, 87) = 14.21; p < 0.0001, steroid: F (1, 87) = 95.56; p < 0.0001. D. Fat mass as a percent of body weight. Two-way ANOVA: diet: F (3, 87) = 17.17; p < 0.0001, steroid: F (1, 87) = 112.6; p < 0.0001. Data are represented as means ± SEM. Sample size for each group was 12. Data were analyzed by two-way ANOVA with post-hoc Bonferroni multiple pairwise comparison tests. Letters denote comparisons within diet between steroid treatments; asterisks denote comparisons within steroid treatment between HFD and CON. Comparisons within steroid, between HFDs are denoted with asterisks over capped lines. (*& a = p<0.5; ** & b = p<0.01; *** & c = p<0.001; **** & d = p<0.0001).
Lean mass (Figure 2C) and fat mass (Figure 2D), determined by MRI and reported as percentage of body weight, were, respectively, affected by diet (F (3, 87) = 14.21; p < 0.0001, F (3, 87) = 17.17; p < 0.0001) and by steroid (F (1, 87) = 95.56; p < 0.0001, F (1, 87) = 112.6; p < 0.0001). Fat mass (F (3, 87) = 3.786; p <0.05) but not lean mass (F (3, 87) = 1.117; p = 0.3468) showed significant variability from their interaction. Lean mass was reduced in 1% and 22.5% both oil and E2 compared to CON, but only 15% oil had a reduction, in 15% E2 was more protective. Fat mass was higher in all oil treated HFDs than CON, but also higher in 1% E2 compared to CON. In 15% and 22.5% E2 was again protective. Fat mass in E2-treated 1% was also higher than E2-treated 15%; showing that E2 is more protective from the dystrophic effects of HFD for a FA profile balanced between LA and SFA.
LA slows glucose disposal
Fasting BG was affected by steroid (F (1, 86) = 9.146; p < 0.01), but due to variability from diets did not produce any differences; CON oil vs. 22.5% oil were nearly so (p = 0.0672). Differences in glucose metabolism were, however, uncovered during GTT and ITT. GTT and ITT were performed after 8 weeks of feeding CON, 1%, 15% and 22.5% diets to determine the effects on glucose metabolism in oil- and E2-treated female mice. At several time points of the GTT, for both oil and E2 treated animals, 22.5% had higher BG than control and other HFDs (Figure 3A-B). Time (F (6, 264) = 152.9; p < 0.0001) and the interaction of time and diet (F (18, 264) = 4.502; p < 0.0001) were significant sources of variation within oil treated animals, whereas time (F (6, 240) = 90.46; p < 0.0001), diet (F (3, 40) = 4.582; p < 0.01), and their interaction (F (18, 240) = 1.986; p < 0.05) were sources for E2 treated animals. At 90 min post-injection 22.5% oil had higher BG than 1% oil, and at 120 minutes 22.5% oil had higher BG than 1% oil and CON oil. All oil treated HFDs had higher BG than CON at 180 minutes. E2 accelerated glucose disposal in all diet except 22.5%, which had higher BG than CON at 30 through 120 min post-injection. At 120 min, 22.5% BG was also higher than 1%, and 15% BG was higher than CON. The area under the curve (AUC) analysis of GTT BG measurements was higher for oil compared to E2 groups within each diet, although the difference between oil and E2 groups was smaller in the high LA HFD. Between diets, 22.5% E2 and oil, and 15% E2, had elevated AUC (Figure 3E). Diet and E2 (F (3, 84) = 8.143; p < 0.0001, F (1, 84) = 63.05; p < 0.0001), but not their interaction, were significant sources of variation in GTT AUC data. The trends and differences in GTT data suggest a dose-response of LA for glucose intolerance.
Figure 3: Glucose metabolism.
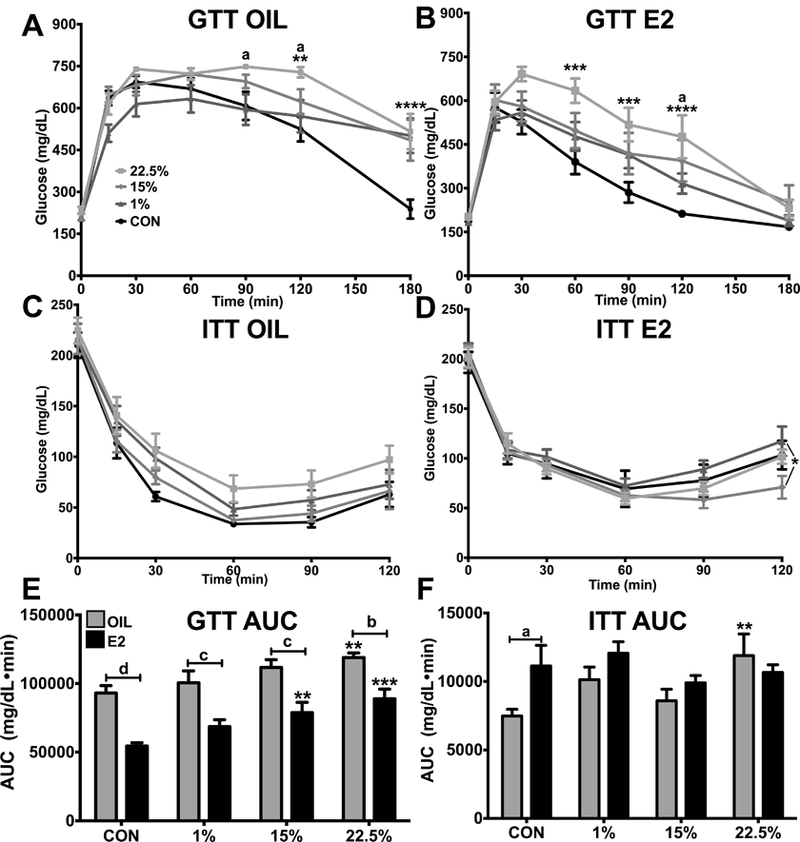
A. 180 min glucose tolerance test between oil-treated females. Two-way ANOVA: interaction time*diet: F (18, 264) = 4.02; p < 0.0001. B. 180 min glucose tolerance test between E2-treated females. Two-way ANOVA: interaction time*diet: F (18, 240) = 1.986; p < 0.05. C. 120 min insulin tolerance test between oil-treated females. Two-way ANOVA: time: F (5, 210) = 207.2; p < 0.0001. D. 120 min insulin tolerance test between E2-treated females. Two-way ANOVA: time: F (5, 195) = 124.9; p < 0.0001. E. Area under the curve of blood glucose during glucose tolerance test. Two-way ANOVA: diet: F (3, 88) = 9.86; p < 0.0001, steroid: F (1, 88) = 64.69; p < 0.0001. F. Area under the curve of blood glucose during insulin tolerance test. Two-way ANOVA: steroid: F (1, 82) = 3.687; p = 0.0583. Data are represented as means ± SEM. Sample size for each group was 12. Data were analyzed by two-way ANOVA with post-hoc Bonferroni multiple pairwise comparison tests. Letters denote comparisons within diet between steroid treatments; asterisks denote comparisons within steroid treatment between HFD and CON. Comparisons within steroid, between HFDs are denoted with asterisks over capped lines. (*& a = p<0.5; ** & b = p<0.01; *** & c = p<0.001; **** & d = p<0.0001).
BG suppression by insulin injection was similar across diets (Figure 3C-D). For both oil and E2 treated mice, only time (F (5, 210) = 207.2; p < 0.0001, F (5, 195) = 124.9; p < 0.0001) significantly affected inter-group variability. Within oil treated mice, diet trended towards being significant (F (3, 42) = 2.325; p = 0.0886) and 22.5% trended towards higher BG in the later time points. At 120 min post-injection, 1% E2 had a greater recovery and was closer to its initial BG, being higher than 15% E2 and CON E2, but not 22.5% E2. The higher trending BG over the course of ITT in 22.5% oil-treated females resulted in a higher AUC (Figure 3F). The glucose metabolism impairment of LA, as a response to insulin challenge, was less than during glucose challenge.
CO2 production, but not O2 consumption, reduced by HFD
O2 consumption was not altered by diet (Figure 4A). The day/night cycle (F (1, 170) = 33.89; p < 0.0001) and steroid (F (7, 170) = 17.65; p < 0.0001) both affected variability; during the night E2 treated mice had higher O2 consumption than oil treated within all diets, whereas only 15% showed a higher O2 consumption from E2 during the day. Although trending higher in all diets, E2 treatment induced more CO2 production only in 15% at night (Figure 4B). Day/night (F (1, 170) = 22.37; p < 0.0001) and diet/E2 (F (7, 170) = 22.37; p < 0.0001) were both significant sources of variability. CO2 production was suppressed by all HFDs during both the day and night, except for 15% E2 during the day, which was the same as CON.
Figure 4: Calorimetry.
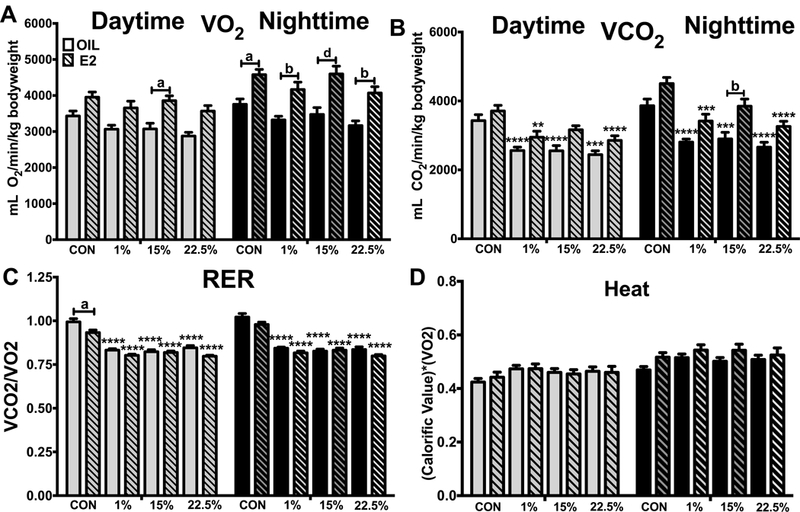
A. Volume of oxygen consumption during the night and day. Two-way ANOVA: txt: F (7, 170) = 17.65; p < 0.0001, time: F (1, 170) = 33.89; p < 0.0001. B. Volume of carbon dioxide production during the night and day. Two-way ANOVA: txt: F (7, 170) = 22.33; p < 0.0001, time: F (1, 170) = 32.28; p < 0.0001. C. Ratio of carbon dioxide production to volume of oxygen consumption during the night and day. Two-way ANOVA: txt: F (7, 170) = 78.14; p < 0.0001, time: F (1, 170) = 4.764; p < 0.05. D. Heat production during the night and day. Two-way ANOVA: txt: F (7, 170) = 2.279; p < 0.05, time: F (1, 170) = 44.59; p < 0.0001. Data are represented as means ± SEM. Sample size for each group was 12. Data were analyzed by two-way ANOVA with post-hoc Bonferroni multiple pairwise comparison tests. Letters denote comparisons within diet between steroid treatments; asterisks denote comparisons within steroid treatment between HFD and CON. Comparisons within steroid, between HFDs are denoted with asterisks over capped lines. (*& a = p<0.5; ** & b = p<0.01; *** & c = p<0.001; **** & d = p<0.0001).
The respiratory exchange ratio (RER), the ratio of CO2 produced to O2 consumed, followed the CO2 pattern of being lower in all HFD fed mice regardless of the day/night cycle or steroid treatment (Figure 4C). Daytime RER was lower within CON diet for E2 treated animals due to a higher trend of daytime O2 consumption compared to CO2 production. Time (F 1, 170) = 4.764; p < 0.05) and treatment (F (7, 170) = 78.14; p < 0.0001) caused variability. Heat production, a product of O2 consumption and the estimated caloric value of the diet, was not different between diets or time of day (Figure 4D). Although both treatment (F (7, 170) = 44.59; p < 0.0001) and time (F (7, 170) = 2.279; p < 0.05) affected variability, their interaction did not (F (7, 170) = 0.5128; p = 0.8241), producing little difference between groups.
Spontaneous activity as measured by movement in the X and Z-axes during calorimetry was not affected by diet or steroid (Figure 5A-C). The day/night cycle was the sole source of variation in X total (F (1, 170) = 76.99; p < 0.0001), X ambulatory (F (1, 170) = 45.57; p < 0.0001), and Z total (F (1, 170) = 77.3; p < 0.0001) counts; all mice, regardless of diet or E2, had increased nighttime activity.
Figure 5: Spontaneous activity.
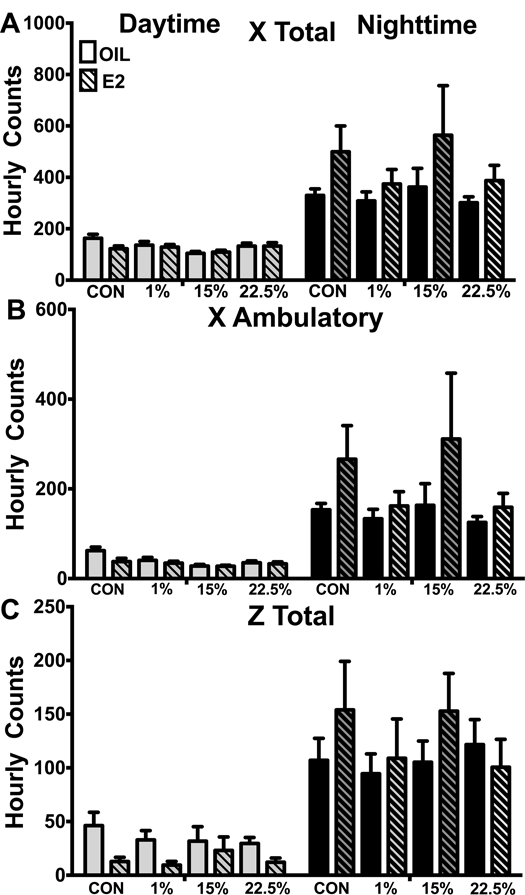
A. Total beam breaks in the x-axis during the night and day. Two-way ANOVA: time: F (1, 170) = 76.99; p < 0.0001. B. Novel beam breaks in the x-axis during the night and day. Two-way ANOVA: time: F (1, 170) = 47.57; p < 0.0001. C. Total beam breaks in the z-axis during the night and day. Two-way ANOVA: time: F (1, 170) = 77.3; p < 0.0001. Data are represented as means ± SEM. Sample size for each group was 12. Data were analyzed by two-way ANOVA with post-hoc Bonferroni multiple pairwise comparison tests. Letters denote comparisons within diet between steroid treatments; asterisks denote comparisons within steroid treatment between HFD and CON. Comparisons within steroid, between HFDs are denoted with asterisks over capped lines. (*& a = p<0.5; ** & b = p<0.01; *** & c = p<0.001; **** & d = p<0.0001).
SFA suppresses leptin and IL6 compared to LA in E2 deficient mice
Serum E2 was higher in all E2 treated mice compared to oil treatment (Figure 6A). Diet did not affect circulating E2 (F (3, 72) = 0.5621; p = 0.6418). All HFD mice, regardless of E2 treatment, had higher circulating leptin than CON, which is consistent with their higher fat mass (Figure 6B). Diet was the primary driver of variation (F (3, 56) = 79.74; p < 0.0001). 1% oil, but not 1% E2, had lower circulating leptin than their 15% and 22.5% counterparts, despite having similar fat masses. Circulating IL6 levels followed a similar pattern as leptin (Figure 6C). Nearly all HFD groups had higher IL6 than CON (Figure 6B). Diet (F (3, 48) = 110.5; p < 0.0001), E2 (F (1, 48) = 8.547; p < 0.01), and their interaction (F (3, 48) = 12.57; p < 0.0001) were all significant variables. 1% oil had lower IL6 than 15% and 22.5% oil, being suppressed all the way to the CON level. IL6 was also suppressed in 1% E2 compared to 15% and 22.5% E2, but not to the same degree as in oil treatment.
Figure 6: Plasma E2 and peptides.
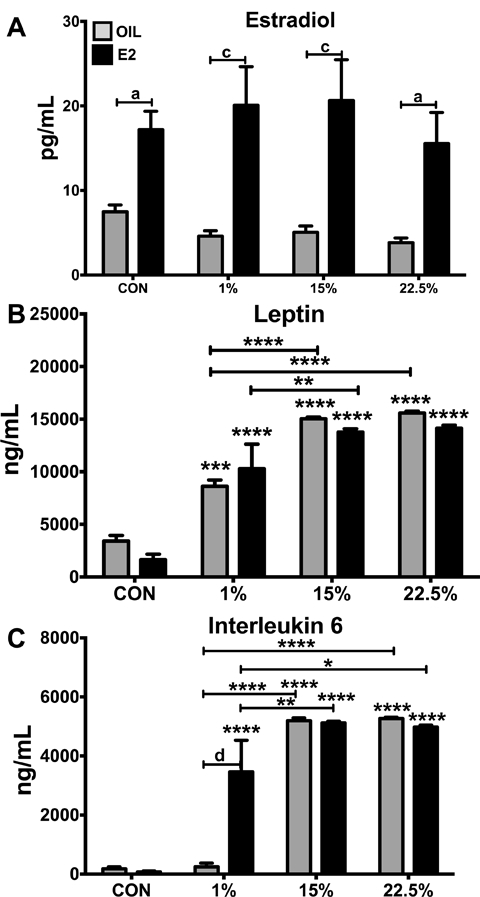
A. Plasma E2 (pg/mL). Two-way ANOVA: steroid: F (1, 72) = 48.03; p < 0.0001. B. Plasma leptin (pg/mL). Two-way ANOVA: diet: F (3, 56) = 79.74; p < 0.0001. C. Plasma IL-6 (pg/mL). Two-way ANOVA: interaction diet*steroid: F (3, 48) = 12.57; p < 0.0001. Data are represented as means ± SEM. Sample size for each group was 12. Data were analyzed by two-way ANOVA with post-hoc Bonferroni multiple pairwise comparison tests. Letters denote comparisons within diet between steroid treatments; asterisks denote comparisons within steroid treatment between HFD and CON. Comparisons within steroid, between HFDs are denoted with asterisks over capped lines. (*& a = p<0.5; ** & b = p<0.01; *** & c = p<0.001; **** & d = p<0.0001).
Arcuate neuropeptide and inflammatory gene expression
The expression of the anorexigenic neuropeptides Pomc and Cart was not affected by diet (Figure 7A-B). Pomc expression was suppressed by E2 in CON and 22.5% (F (1, 64) = 17.62; p < 0.0001. No treatment differences altered Npy expression (Figure 7C). Diet influenced Agrp expression (F (3, 63) = 2.93; p < 0.05), which was suppressed by 22.5% in E2 treated mice (Figure 8D); a trend towards lower expression is apparent in the HFD-fed groups.
Figure 7: Expression of arcuate nucleus neuropeptides.
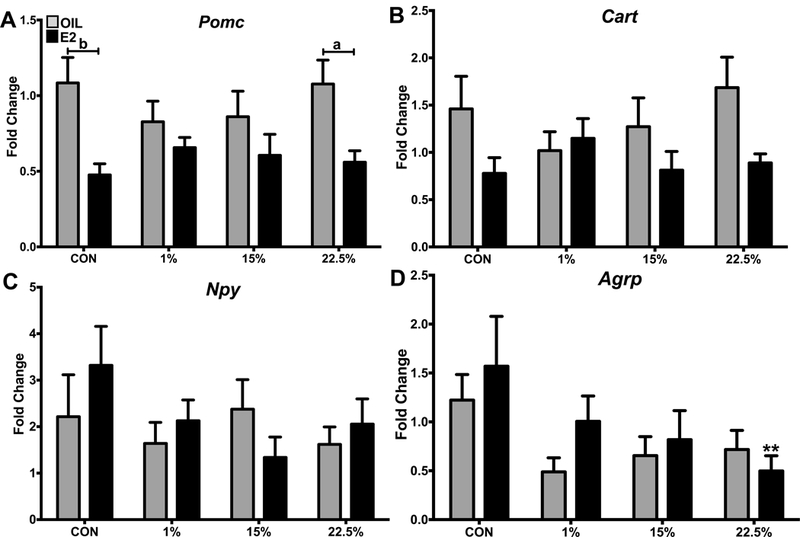
A. Expression of Pomc relative to reference genes. Two-way ANOVA: steroid: F (1, 64) = 17.62; p < 0.0001. B. Expression of Cart relative to reference genes. Two-way ANOVA: steroid: F (1, 64) = 6.689; p < 0.05. C. Expression of Npy relative to reference genes. No significant effect of diet or steroid. D. Expression of Agrp relative to reference genes. Two-way ANOVA: diet: F (3, 63) = 2.93; p < 0.05. Data are represented as means ± SEM. Sample size for each group was 12. Data were analyzed by two-way ANOVA (repeated measures or standard) with post-hoc Bonferroni multiple pairwise comparisons test. Letters denote comparisons within diet between steroid treatments; asterisks denote comparisons within steroid treatment between HFD and CON. Comparisons within steroid, between HFDs are denoted with asterisks over capped lines. (*& a = p<0.5; ** & b = p<0.01; *** & c = p<0.001; **** & d = p<0.0001).
Figure 8: Expression of arcuate nucleus inflammatory genes.
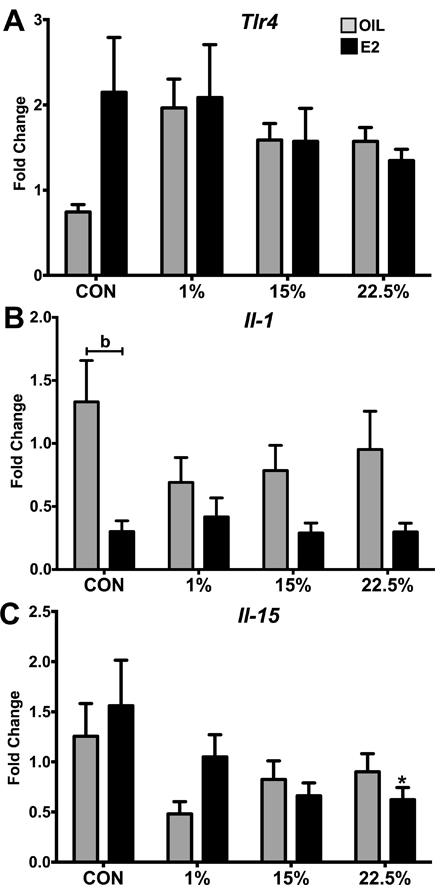
A. Expression of Tlr4 relative to reference genes. No significant effect of diet or steroid. B. Expression of Il-1 relative to reference genes. Two-way ANOVA: steroid: F (1, 61) = 17.6; p < 0.0001. C. Expression of Il-15 relative to reference genes. Two-way ANOVA: diet: F (3, 63) = 3.219; p < 0.05. Data are represented as mean ± SEM. Sample size for each group was 12. Data were analyzed by two-way ANOVA with post-hoc Bonferroni multiple pairwise comparison tests. Letters denote comparisons within diet between steroid treatments; asterisks denote comparisons within steroid treatment between HFD and CON. Comparisons within steroid, between HFDs are denoted with asterisks over capped lines. (*& a = p<0.5; ** & b = p<0.01; *** & c = p<0.001; **** & d = p<0.0001).
Tlr4 expression trended lower in the CON oil group compared to CON E2 (p = 0.0631) but high variability prevented this suppression from reaching significance (F (3, 63) = 0.9228; p = 0.4351) or E2 (F (1, 63) = 1.37; p = 0.2462) (Figure 8A). IL1 expression was suppressed by E2 (F (1, 61) = 17.6; p < 0.0001) as seen in the difference between CON-fed oil and E2, but not within other diets (Figure 8B). IL15 was mostly affected by diet (F (3, 63) = 3.219; p = 0.05), being suppressed by HFDs and also by E2 within 22.5% HFD (Figure 8C). Although circulating leptin was higher in HFD fed animals, and highest from LA, arcuate nucleus expression of its receptor Lepr was not different between groups (data not shown). The expression of Insr, Stat3, Socs3, Irak1, Traf6, and Ikk were not different between groups (data not shown).
Discussion
Here we showed that E2 suppressed weight gain from both SFA and LA HFD. Glucose metabolism, however, was affected in an FA-dependent manner. During both GTT and ITT, 22.5% diet impaired glucose disposal in E2-deficient (oil-treated) females, while E2 treatment failed to rescue high-LA consuming females to levels commensurate with the other groups. There was also a slight potentiation of leptin secretion, and a more substantial potentiation of IL-6 secretion, by LA, especially when comparing E2 deficient females. These peptides did not cause a difference in weight gain, but may be involved in glucose metabolism differences.
Previously, E2 has been shown to decrease food intake, acting through estrogen receptor-α (ERα) and cholecystokinin29, and to reduce post-ovx weight gain30. In our study, the HFD fed, E2 treated mice did not eat less food than those treated with oil. This may be due to the route of administration; the anorectic effects of E2 are more pronounced in direct hindbrain compared to oral administration29. Hypothalamic E2 administration inhibits food intake, but this effect is acute rather than chronic31. Although no exogenous administration of E2 can perfectly mimic ovarian secretion, oral delivery is more similar to it than is delivery through central cannulation.
Furthermore, pair feeding of ovx to intact mice resulted in 25% greater weight gain in ovx mice32, a similar gain as reported herein, showing that hyperphagia is not necessary for E2 deficiency-induced weight gain. The anorectic effects of E2 may be of an acute, secondary nature relative to chronic metabolic effects. The effects of E2 on energy and glucose homeostasis may involve both ERE-dependent and ERE-independent signaling through ERα. Recently, we demonstrated that E2 protects against the effects of diet-induce obesity through ERE-independent ERα signaling by reducing adiposity and increasing glucose clearance33. These signaling mechanisms are no doubt activated in our current study. Within E2 treated animals, 1% and 22.5% began gaining weight faster than 15% and CON at the later stages of the experiment. This was not sufficient to result in a difference in cumulative weight gain at 8 weeks, but may have if feeding continued. In a 24 week HFD feeding experiment, weight gain was ongoing in female, intact mice consuming Research Diets D1249234, which is similar to the 15% diet in our study.
We have previously shown that in male mice LA disrupts glucose metabolism more than SFA23. In that study, all HFD mice had impaired glucose disposal from glucose challenge, but only 22.5% were impaired from insulin challenge. In the present study, glucose disposal during glucose challenge, and not insulin challenge, was impaired by LA. LA is an independent variable for hyperglycemia22, likely through a mechanism related to the Randle Effect35. CPT1 expression is higher in liver cells of rats fed n-6 PUFA compared to SFA36. Inhibition of CPT1 by methyl palmoxirate treatment reduces rats’ ability to oxidize corn oil but not medium chain saturated triglycerides37. The short and medium chain SFAs in coconut oil are also protective of damage from alcohol38 and alloxan39, conferring protection to beta cells from potential lipotoxicity.
Another difference between SFA and PUFA is greater PPAR activation by PUFA40. PPAR signaling is adaptive, shunting FAs to desired uses as concentrations increase, but the type of FA activating these receptors affects the response. LA and other n-6 PUFA induce adipogenesis through PPAR activation, whereas n-3 PUFA do not41. The resulting inflammation from this adipogenic environment, as well as the downstream eicosanoids of n-6 PUFA42, could have a negative effect on blood glucose. The estrogen receptor system acts on, and is acted on, PPAR gamma43, a potential reason for E2 opposition to the glucose metabolism impairing effects of HFD.
Similarly, activation of ERα increases the rate of fat oxidation and decreases tissue accumulation of diacylgycerol44. The clearance of FAs from circulation and tissues by E2 may be the primary, although indirect, mechanism of improving glucose metabolism. By relieving cells of FAs inhibitory to glucose oxidation, the oxidation rate, and consequently the rate of glucose drawn from blood, is increased. Because of this estrogen mediated boosting of fat clearance, FA profile might be a more important health concern for men and post-menopausal women than for cycling women.
O2 consumption trended lower in HFD oil groups than CON, and this trend was stronger during the night. During the night, E2 caused an increase in O2 consumption, especially in HFD fed mice. A limitation of using whole animal O2 consumption as a proxy for metabolic rate is the inability to differentiate between O2 used in cellular respiration and O2 used in non-respiratory oxidation. Higher tissue and circulating PUFA can take up O2 to produce peroxyls and peroxides45. It is likely that the trend towards lower O2 consumption in HFD-fed oil-treated mice would be a significant difference if non-respiratory O2 use could be removed. An equation, similar to those developed by Bauer46 for estimation of tissue n-3 and n-6 PUFA content, which estimates FA peroxidation from factors such as PUFA intake, metal catalyst concentration, and O2/CO2 ratio, would be a useful addition to indirect calorimetrics. An open container of highly oxidizable material would register as “respiring” according to the current method of measuring VO2.
The presence of higher circulating and tissue FAs in HFD-fed mice is supported by their reduced CO2 production. A reduction in CO2 production, particularly in the presence of a relatively stable O2 consumption, indicates an increased ratio of FAs as energy substrate. Respiratory quotient (CO2/O2, has also been used as an independent predictor of body mass changes47. There is evidence from the basic chemistry of the Bohr effect on hemoglobin48, as well as the hypercapnic activation of vitamin K2 dependent enzymes49, that CO2 supports the metabolic rate. Boulder and other high altitude cities50 have substantially lower rates of obesity and associated metabolic disease. Since a low-fat, glucose-based metabolism has the same effect on capnia, or blood CO2 level, as altitude, this seems like a more obvious place to start in explaining low obesity rates common in high altitude populations than their idiosyncratic diets and cultures.
Circulating leptin was correlated with fat mass. Since leptin is mostly produced by white adipose tissue51, concentration is determined at the level of secretion by body fatness. In this study, we show that although all HFD mice have higher serum leptin than CON, 1% oil was an intermediate between CON and the higher LA HFD oil mice. The lower leptin of 1% oil compared to 15% and 22.5% did not cause a difference in weight gain or arcuate leptin receptor mRNA. An even greater difference was observed in circulating IL-6, where 1% oil was similar to CON. Another study measuring E2’s effects on cytokines showed an increase in IL-6 from E2 while feeding an HFD made from corn oil and lard52, both good sources of LA.
This is consistent with our results, where the oil-treated, 1% LA HFD-fed mice had low levels of IL-6 compared to the elevated IL-6 levels in E2-treated, HFD-fed mice. Serum leptin and IL-6 reduction in E2 and LA deficient mice may come from their molecular similarity. Leptin an IL-6 are both cytokines with receptors from the same family, and are induced similarly by agents such as LPS, other cytokines, and E253. Higher circulating cytokines likely play a part in the glucose intolerance seen in 22.5%. Elevated peripheral IL-6 is commonly seen in insulin resistance54, and blocks the suppression of hepatic gluconeogenesis by insulin55. This difference in circulating IL-6, like the difference in leptin, did not influence arcuate gene expression. Arcuate cytokine mRNA, including Il-6, was not different between groups. The moderate suppression by 22.5% of arcuate Pomc and Agrp did not correlate food intake, which was unaffected by FA.
It is reasonable to conclude that peripheral hormone and inflammatory activity is more reflective of dietary influence, such as type and concentration of FAs, than is central. The hypothalamus is not protected by a complete blood brain barrier, but is still less accessible to circulating molecules than is the periphery. It may be that a greater change in hypothalamic gene expression may require a longer dietary intervention. It has been shown previously that approximately 5 weeks post-ovx, expression of energy balance related genes in the arcuate nucleus is similar to that of intact females, despite an enduring effect on body weight56. The normalization of neuropeptide levels at a higher plateau of obesity is reminiscent of the set point observations on leptin during weight loss, where the reduction in leptin from adipose loss is perceived as a danger and results in an attempt to defend the body weight by preventing further weight loss.
There is a significant challenge in experimentally capturing both obesity and pre-obesity events causative of weight gain. Inflammation measured in obese animals can be interpreted as a symptom or a cause of the obesity. Early onset inflammation, before an animal becomes obese, relies on the assumption of future obesity and consistent production. IL-6 can stimulate the pancreas to release insulin and control glycemia57, but over a different course of time can blunt insulin’s ability to suppress hepatic glucose production55. In this study, we chose to measure hypothalamic gene expression and serum protein at time of sacrifice, when oil treated HFD mice were obese, and found that circulating IL-6 and leptin were related to glucose intolerance but not to obesity or HI. Future experiments may include pre-obesity HI measurements to address this challenge.
Overall, our results show that E2 confers greater resistance to female mice from HFD induced obesity from different types of FAs, but does not protect against impaired glucose disposal from LA. These data suggest that young, cycling women who present with markers of pre-diabetes could potentially slow or halt its progression, which may follow reduced ovarian steroid production during menopause, by consuming a reduced-LA diet. For post-menopausal women, E2 treatment may combat obesity, but additional benefit may be had from a low LA diet for preserving proper glucose metabolism. Our finding that E2 increased circulating IL-6 in HFD-fed mice, especially higher LA HFD, is evidence against E2 as a risk-free obesity treatment. Estrogen replacement in post-menopausal women has long been controversial because of the increased risk of some cancers. Endometrial cancer risk is greatly increased by estrogen replacement, and further in obesity, but is reduced by progesterone58. Indeed, progesterone in combination with E2 is required to induce higher leptin secretion in women59, and may play a role in the resistance to obesity related disease in cycling women. Reducing dietary fat is a safe strategy to prevent obesity in women regardless of menopausal status, and targeting LA for reduction may specifically benefit glucose metabolism.
Acknowledgments
Funding: This research was supported by funds from USDA-NIFA NJ06107 and from National Institutes of Health R00DK083457, R00DK083457-S1, and P30ES005022.
Acknowledgements
The authors thank Dr. Sara Campbell for the use of the EMD Millipore MAGPIX® Multiplex® System and Dr. Judith Storch for the use of the Comprehensive Lab Animal Monitoring System and EchoMRI Body Composition Analyzer.
Footnotes
Declaration of Interest: The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- 1.Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA 2014. February 26;311(8):806–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moore TR. Adolescent and adult obesity in women: a tidal wave just beginning. Clin Obstet Gynecol 2004;47(4):884–889-981. [DOI] [PubMed] [Google Scholar]
- 3.Hodson L, Banerjee R, Rial B, Arlt W, Adiels M, Boren J, et al. Menopausal Status and Abdominal Obesity Are Significant Determinants of Hepatic Lipid Metabolism in Women. J Am Heart Assoc 2015;4(10):e002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patil SR. Prevalence of carotid artery calcification in postmenopausal women and its correlation with atherogenic risk factors. J Nat Sci Biol Med. India: Medknow Publications & Media Pvt Ltd; 2015. August;6(Suppl 1):S1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gallego MPO, López PB, Angel M, Armero T, Alemán JA, Albero JS, et al. Metabolic syndrome and its components in Spanish postmenopausal women. Nutr Hosp 2015;32(2):656–66. [DOI] [PubMed] [Google Scholar]
- 6.Johnson ZP, Lowe J, Michopoulos V, Moore CJ, Wilson ME, Toufexis D. Oestradiol differentially influences feeding behaviour depending on diet composition in female rhesus monkeys. J Neuroendocrinol 2013;25(8):729–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paolisso G, Rizzo MR, Mazziotti G, Rotondi M, Tagliamonte MR, Varricchio G, et al. Lack of association between changes in plasma leptin concentration and in food intake during the menstrual cycle. Eur J Clin Invest 1999;29(6):490–5. [DOI] [PubMed] [Google Scholar]
- 8.Buffenstein R, Poppitt SD, McDevitt RM, Prentice AM. Food intake and the menstrual cycle: A retrospective analysis, with implications for appetite research. Physiol Behav 1995;58(6):1067–77. [DOI] [PubMed] [Google Scholar]
- 9.Dye L, Blundell JE. Menstrual cycle and appetite control: implications for weight regulation. Hum Reprod 1997;12(6):1142–51. [DOI] [PubMed] [Google Scholar]
- 10.Sullivan EL, Shearin J, Koegler FH, Cameron JL. Selective estrogen receptor modulator promotes weight loss in ovariectomized female rhesus monkeys (Macaca mulatta) by decreasing food intake and increasing activity. AJP Endocrinol Metab 2012;302(7):E759–67. [DOI] [PubMed] [Google Scholar]
- 11.Lobo RA, Davis SR, De Villiers TJ, Gompei A, Henderson VW, Hodis HN, et al. Prevention of diseases after menopause. Climacteric 2014;17(5):540–56. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Vol. 372, Nature 1994. p. 425–32. [DOI] [PubMed] [Google Scholar]
- 13.Thaler JP, Choi SJ, Schwartz MW, Wisse BE. Hypothalamic inflammation and energy homeostasis: resolving the paradox. Front Neuroendocrinol. Elsevier Inc; 2010. January;31(1):79–84. [DOI] [PubMed] [Google Scholar]
- 14.Cai D, Liu T. Hypothalamic inflammation: a double-edged sword to nutritional diseases. Ann N Y Acad Sci 2011. December;1243:E1–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wisse BE, Schwartz MW. Does Hypothalamic Inflammation Cause Obesity? Cell Metab. Elsevier Inc; 2009. October;10(4):241–2. [DOI] [PubMed] [Google Scholar]
- 16.Velloso LA, Araújo EP, de Souza CT. Diet-induced inflammation of the hypothalamus in obesity. Neuroimmunomodulation 2008. January;15(3):189–93. [DOI] [PubMed] [Google Scholar]
- 17.Roepke TA. Oestrogen Modulates Hypothalamic Control of Energy Homeostasis Through Multiple Mechanisms. J Nueroendocrinology 2009;21(2):141–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Willett WC. Dietary fat and obesity: an unconvincing relation. Am J Clin Nutr 1998. December;68(6):1149–50. [DOI] [PubMed] [Google Scholar]
- 19.Heini AF, Weinsier RL. Divergent Trends in Obesity and Fat Intake Patterns: The American Paradox. Am J Med 1997;102:259–64. [DOI] [PubMed] [Google Scholar]
- 20.Taubes G The Soft Science of Dietary Fat. Science (80- ) 2001;291(March):1–16. [DOI] [PubMed] [Google Scholar]
- 21.Blasbalg TL, Hibbeln JR, Ramsden CE, Majchrzak SF, Rawlings RR. Changes in consumption of omega-3 and omega-6 fatty acids in the United States during the 20th century. Am J Clin Nutr 2011;93(5):950–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ikemoto S, Takahashi M, Tsunoda N, Maruyama K, Itakura H, Ezaki O. High-fat diet-induced hyperglycemia and obesity in mice: Differential effects of dietary oils. Metabolism 1996;45(12):1539–46. [DOI] [PubMed] [Google Scholar]
- 23.Mamounis KJ, Yasrebi A, Roepke TA. Linoleic acid causes greater weight gain than saturated fat without hypothalamic inflammation in the male mouse. J Nutr Biochem. Elsevier B.V; 2016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diniz YS, Cicogna AC, Padovani CR, Santana LS, Faine L a., Novelli ELB. Diets Rich in Saturated and Polyunsaturated Fatty Acids: Metabolic Shifting and Cardiac Health. Nutrition 2004. February;20(2):230–4. [DOI] [PubMed] [Google Scholar]
- 25.Alvheim AR, Malde MK, Osei-Hyiaman D, Lin YH, Pawlosky RJ, Madsen L, et al. Dietary Linoleic Acid Elevates Endogenous 2-AG and Anandamide and Induces Obesity. Obesity 2012. October;20(10):1984–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alvheim AR, Torstensen BE, Lin YH, Lillefosse HH, Lock EJ, Madsen L, et al. Dietary linoleic acid elevates the endocannabinoids 2-AG and anandamide and promotes weight gain in mice fed a low fat diet. Lipids 2014;49(1):59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akoum S El, Lamontagne V, Cloutier I, Tanguay J. Nature of fatty acids in high fat diets differentially delineates obesity-linked metabolic syndrome components in male and female C57BL / 6J mice. Diabetol Metab Syndr. BioMed Central Ltd; 2011;3(34):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates, Compact, Third Edition: The coronal plates and diagrams 3rd ed. Academic Press; 2008. 256 p. [Google Scholar]
- 29.Thammacharoen S, Lutz TA, Geary N, Asarian L. Hindbrain administration of estradiol inhibits feeding and activates estrogen receptor-α-expressing cells in the nucleus tractus solitarius of ovariectomized rats. Endocrinology 2008;149(4):1609–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mamounis KJ, Yang JA, Yasrebi A, Roepke TA. Estrogen response element-independent signaling partially restores post-ovariectomy body weight gain but is not sufficient for 17β-estradiol’s control of energy homeostasis. Steroids 2014;81:88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Santollo J, Torregrossa A- M, Eckel LA. Estradiol acts in the medial preoptic area; arcuate nucleus, and dorsal raphe neurons to reduce food intake in ovariectomized rats. Horm Behav 2011;60(1):86–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rogers NH, Ii JWP, Strissel KJ, Obin MS, Greenberg AS. Reduced Energy Expenditure and Increased Inflammation Are Early Events in the Development of Ovariectomy-Induced Obesity 2009;150(May):2161–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yasrebi A, Rivera JA, Krumm EA, Yang JA, Roepke TA. Activation of estrogen response element-independent ER-alpha signaling protects female mice from diet-induced obesity. Endocrinology 2017;158(2):319–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chakraborty TR, Donthireddy L, Adhikary D, Chakraborty S. Long-Term High Fat Diet Has a Profound Effect on Body Weight, Hormone Levels, and Estrous Cycle in Mice. Med Sci Monit 2016;22:1601–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963;281(7285):785–9. [DOI] [PubMed] [Google Scholar]
- 36.Power GW, Yaqoob P, Harvey DJJ, Newsholme EAA, Calder PCC. The effect of dietary lipid manipulation on hepatic mitochondrial phospholipid fatty acid composition and carnitine palmitoyltransferase I activity. Biochem Mol Biol Int 1994;34(4):671–84. [PubMed] [Google Scholar]
- 37.Friedman MI, Ramirez I, Bowden CR, Tordoff MG. Fuel partitioning and food intake : role for mitochondrial fatty acid transport. Am J Physiol Regul Integr Comp Physiol 1990;258(1):R216–21. [DOI] [PubMed] [Google Scholar]
- 38.Lieber CS, Lefèvre A, Spritz N, Feinman L, DeCarli LM. Difference in hepatic metabolism of long- and medium-chain fatty acids: the role of fatty acid chain length in the production of the alcoholic fatty liver. J Clin Invest 1967;46(9):1451–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodriguez RR, Cattaneo P, Houssay BA, Uno B. Acides gras non satures et diabete alloxanique. Biol Comptes Rendus 1952;8(11–12):1099–101. [PubMed] [Google Scholar]
- 40.Georgiadi A, Kersten S. Mechanisms of Gene Regulation by Fatty Acids. Adv Nutr 2012;3(2):127–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Madsen L, Koefoed R, Kristiansen K. Regulation of adipocyte differentiation and function by polyunsaturated fatty acids 2005;1740:266–86. [DOI] [PubMed] [Google Scholar]
- 42.Ricciotti E, Fitzgerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol 2011;31(5):986–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sugii S, Evans RM. Epigenetic codes of PPARy in metabolic disease. FEBS Lett 2011;585(13):2121–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Camporez JPG, Jornayvaz FR, Lee H- Y, Kanda S, Guigni B a, Kahn M, et al. Cellular mechanism by which estradiol protects female ovariectomized mice from high-fat diet-induced hepatic and muscle insulin resistance. Endocrinology 2013. March;154(3):1021–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yin H, Xu L, Porter NA. Free Radical Lipid Peroxidation: Mechanisms and Analysis. Chem Rev 2011;111(10):5944–72. [DOI] [PubMed] [Google Scholar]
- 46.Bauer JE, Waldron MK, Spencer AL, Hannah SS. Predictive equations for the quantitation of polyunsaturated fats in dog plasma and neutrophils from dietary fatty acid profiles. J Nutr 2002;132(6 Suppl 2): 1642S–5S. [DOI] [PubMed] [Google Scholar]
- 47.Longo K a, Charoenthongtrakul S, Giuliana DJ, Govek EK, McDonagh T, Distefano PS, et al. The 24-hour respiratory quotient predicts energy intake and changes in body mass. Am J Physiol Regul Integr Comp Physiol 2010;298(3):R747–54. [DOI] [PubMed] [Google Scholar]
- 48.Tyuma I The Bohr Effect and the Haldane Effect in Human Hemoglobin. Jpn J Physiol 1984;34(2):205–16. [DOI] [PubMed] [Google Scholar]
- 49.Rishavy MA, Hallgren KW, Berkner KL. The vitamin K-dependent carboxylase generates gamma-carboxylated glutamates by using CO2 to facilitate glutamate deprotonation in a concerted mechanism that drives catalysis. J Biol Chem 2011;286(52):44821–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sherry B, Blanck HM, Galuska DA, Pan L, Dietz WH, Balluz L. Vital Signs: State-Specific Obesity Prevalence Among Adults — United States, 2009. Morb Mortal Wkly Rep 2010;59(30):951–5. [PubMed] [Google Scholar]
- 51.Wang P, Mariman E, Renes J, Keijer J. The secretory function of adipocytes in the physiology of white adipose tissue. J Cell Physiol 2008. July;216:3–13. [DOI] [PubMed] [Google Scholar]
- 52.Riant E, Waget A, Cogo H, Arnal JF, Burcelin R, Gourdy P. Estrogens protect against high-fat diet-induced insulin resistance and glucose intolerance in mice. Endocrinology 2009;150(5):2109–17. [DOI] [PubMed] [Google Scholar]
- 53.Lord GM. Leptin as a Proinflammatory Cytokine. Obes Kidney Contrib to Nephrol 2006;151:151–64. [DOI] [PubMed] [Google Scholar]
- 54.Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. 2005;11(2):183–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim HJ, Higashimori T, Park SY, Choi H, Dong JY, Kim YJ, et al. Differential effects of interleukin-6 and-10 on skeletal muscle and liver insulin action in vivo. Diabetes 2004;53(4):1060–7. [DOI] [PubMed] [Google Scholar]
- 56.Clegg DJ, Brown LM, Zigman JM, Kemp CJ, Strader AD, Benoit SC, et al. Estradiol-dependent decrease in the orexigenic potency of ghrelin in female rats. Diabetes 2007;56(4):1051–8. [DOI] [PubMed] [Google Scholar]
- 57.Suzuki T, Imai J, Yamada T, Ishigaki Y, Kaneko K, Uno K, et al. Interleukin-6 enhances glucose-stimulated insulin secretion from pancreatic β-cells: Potential involvement of the PLC-IP3-dependent pathway. Diabetes 2011;60(2):537–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carlson MJ, Thiel KW, Yang S, Leslie KK. Catch it before it kills: progesterone, obesity, and the prevention of endometrial cancer. Discov Med 2012;14(76):215–22. [PMC free article] [PubMed] [Google Scholar]
- 59.Messinis IE, Papageorgiou I, Milingos S, Asprodini E, Kollios G, Seferiadis K. Oestradiol plus progesterone treatment increases serum leptin concentrations in normal women. Hum Reprod 2001;16(9):1827–32. [DOI] [PubMed] [Google Scholar]