

Summary
The electron transport chain (ETC) is an important participant in cellular energy conversion, but its biogenesis presents the cell with numerous challenges. In order to address these complexities, the cell utilizes ETC assembly factors, which include the LYR protein family. Each member of this family interacts with the mitochondrial acyl carrier protein (ACP), the scaffold protein upon which the mitochondrial fatty acid synthesis pathway (mtFAS) builds fatty acyl chains from acetyl-CoA. We demonstrate that the acylated form of ACP is an acetyl-CoA-dependent allosteric activator of the LYR protein family to stimulate ETC biogenesis. By tuning ETC assembly to the abundance of acetyl-CoA, which is the major fuel of the TCA cycle and ETC, this system could provide an elegant mechanism for coordinating the assembly of ETC complexes with one another and with substrate availability.
ETOC BLURB
We demonstrate that mitochondrial fatty acid synthesis (mtFAS), through the generation of an acylated form of the acyl carrier protein (acyl-ACP). Acyl-ACP interacts with members of the LYR protein family, which coordinately activate mitochondrial respiration, primarily through promoting electron transport chain assembly.
Introduction
Mitochondrial acetyl-CoA, which is the common catabolic product of carbohydrates, fats and many amino acids, is the primary substrate that fuels oxidative phosphorylation. It is oxidized by the TCA cycle to provide reduced NADH and FADH2 that feed the electron transport chain (ETC). The ETC complexes, comprising subunits encoded by both the nuclear and mitochondrial genomes, couple the passing of electrons through redox-active cofactors to the establishment of an electrochemical gradient across the inner mitochondrial membrane. In the end, ATP synthase (complex V) exploits this membrane potential to generate ATP. While this system provides an efficient source of usable cellular energy, the biogenesis of these complexes presents the cell with numerous challenges. First, complexes I-V are not simple enzymes, but, rather, highly intricate multimeric assemblies comprised of 44, 4, 11, 20, and 19 subunits, respectively. As such, the cell must coordinate the synthesis and interaction of dozens of individual subunits, translated from two distinct genomes, in order to form mature and active complexes. Second, individual subunits are often hydrophobic and prone to aggregation during the assembly process. Finally, these subunits often contain redox-active cofactors that can facilitate deleterious reactions when not secluded in an assembled complex. In order to cope with these challenges, the cell utilizes dozens of complex-specific assembly factors to manage ETC complex assembly (Ghezzi and Zeviani, 2012).
Eukaryotes have a collection of late-stage ETC assembly factors known as the LYR protein family, which is characterized by a Leu-Tyr-Arg (LYR) motif. In the yeast S. cerevisiae, this family is composed of five proteins—Sdh6, Sdh7, Mzm1, Isd11, and Fmc1. Sdh6 and Sdh7, Mzm1, and Fmc1, support the assembly of complex II, complex III, and complex V, respectively, whereas Isd11 is an essential component of the iron-sulfur cluster biogenesis machinery (Adam et al., 2006; Angerer et al., 2014; Atkinson et al., 2010; Atkinson et al., 2011; Cui et al., 2012; Ghezzi et al., 2009; Lefebvre-Legendre et al., 2001; Na et al., 2014; Sánchez et al., 2013; Wiedemann et al., 2006). LYR proteins support individual complex assembly through direct physical interactions with so-called LYR target proteins (Figure S1A). For example, the complex III-specific LYR protein Mzm1 specifically binds the Rip1 subunit and supports its integration into complex III as the final step in the assembly process (Figure S1A) (Atkinson et al., 2011). By regulating the integration of the final subunit(s) to a pre-formed membrane-bound sub-assembly, LYR proteins facilitate the activation of their respective complexes. In mammals, this family has expanded considerably to include, in addition to direct orthologues of Sdh6, Sdh7, Mzm1, and Isd11, two stable accessory subunits of complex I (Angerer et al., 2014; Sackmann et al., 1991), a regulator of mitochondrial ribosome biogenesis (Brown et al., 2017), and several other LYR proteins with unknown functions. Fmc1, whose function remains poorly understood, has no obvious orthologue in mammalian cells.
Recent studies have demonstrated that several LYR proteins independently interact with the mitochondrial acyl carrier protein (ACP), an essential component of the mitochondrial fatty acid synthesis pathway (mtFAS) (Angerer, 2015; Floyd et al., 2016; Hiltunen et al., 2010; Hiltunen et al., 2009; Huttlin et al., 2015). This prokaryote-like pathway, which has been maintained in eukaryotic mitochondria, is structurally and functionally distinct from the FASN-dependent cytosolic fatty acid synthesis pathway. In order to catalyze the initiation and elongation of nascent acyl chains, mtFAS employs a number of enzymes that interact transiently with ACP, which functions as a soluble scaffold for fatty acid synthesis. ACP utilizes a 4′-phosphopantetheine prosthetic group (4′-PP), which is covalently attached to an invariant Ser residue and provides the terminal thiol upon which fatty acids are synthesized (Majerus et al., 1965; Stuible et al., 1998). Classically, the primary role of mtFAS and ACP was thought to be the synthesis of octanoate, an eight-carbon fatty acid, which is subsequently converted to lipoic acid, a cofactor required for a number of mitochondrial enzymes including pyruvate dehydrogenase (PDH), α-ketoglutarate dehydrogenase (α-KGDH) and glycine cleavage proteins (Brody et al., 1997). In addition to lipoic acid synthesis, mtFAS was also shown to be required for mitochondrial biogenesis as mtFASΔ cells exhibit profound defects in mitochondrial translation and in the abundance of mitochondrial proteins (Kursu et al., 2013). Curiously, in vitro biochemical studies indicate that mtFAS can synthesize ACP-bound acyl chains significantly longer than eight carbons; however, no functional importance has yet to be attributed to these extended acyl chains (Zhang et al., 2005).
The ACP-LYR associations were initially observed in the context of large-scale proteomics-based interaction studies (Angerer, 2015; Floyd et al., 2016; Huttlin et al., 2015; Van Vranken et al., 2016). These observations were subsequently corroborated by structural studies, which demonstrate the importance of the Leu-Tyr-Arg motif in ACP-LYR binding and highlight the unique nature of the ACP-LYR interaction (Boniecki et al., 2017; Brown et al., 2017; Cory et al., 2017; Fiedorczuk et al., 2016; Zhu et al., 2016). Indeed, these structures illustrate an acylated form of ACP with its 4′-PP-bound acyl chain inserted into the hydrophobic core of its LYR binding partner (Figure 1A). These observations raise many questions regarding the evolutionary purpose of this unique post-translational modification. Owing to its intimate connection with the LYR protein family, we hypothesized that ACP acylation might play a functional role in ETC complex assembly. Herein, we demonstrate that mtFAS-mediated acylation of ACP represents an acetyl-CoA-dependent post-translational modification capable of allosterically activating the LYR protein family to support ETC assembly.
Figure 1. LYR proteins require ACP to execute their functions as complex-specific ETC assembly factors.
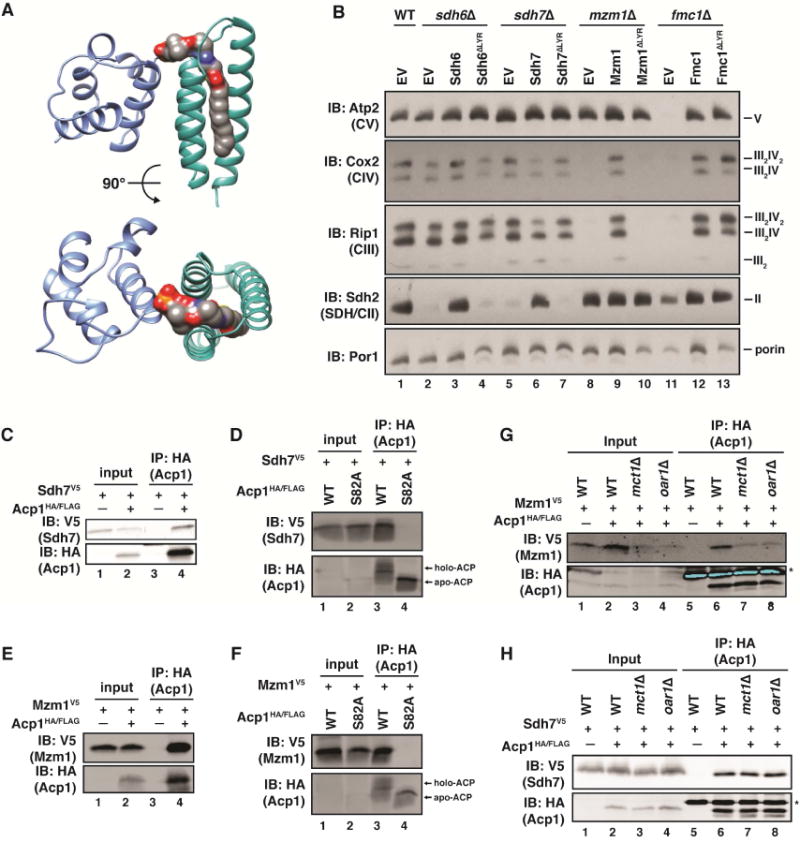
A. A structural model depicting the interaction between ACP (blue) and ISD11 (LYR; green). The 4′-PP and a bound acyl chain are colored by element. PDB ID: 5USR (Cory et al., 2017). B. Mitochondrial lysates generated from the indicated strains harboring the indicated plasmids were separated by BN-PAGE and immunoblotted with the indicated antibodies (EV – empty vector). C-F. Acp1HA/FLAG or Acp1S82A-HA/FLAG were immunoprecipitated from cells expressing Sdh7V5 (C,D) or Mzm1V5 (E,F). The inputs and eluates were immunoblotted with the indicated antibodies. G,H. Acp1HA/FLAG was immunprecipitated from WT, mct1Δ, and oar1Δ cells expressing Mzm1V5 (G) or Sdh7V5 (H). The inputs and eluates were immunoblotted with the indicated antibodies. Inputs represent 5% of the total lysate.
Results
LYR proteins require the Leu-Tyr-Arg motif to support ETC complex assembly
In order to begin to understand the function of ACP in LYR-mediated ETC complex assembly we selected the yeast S. cerevisiae as our primary model and sought to investigate the role of ACP in supporting the function of each LYR protein, independently. Consistent with previous reports, deletion of individual genes encoding LYR proteins results in the specific destabilization of individual complexes when assessed by blue native (BN)-PAGE (Figure 1B), as well as respiratory growth defects (Figure S1B-E) (Adam et al., 2006; Angerer et al., 2014; Atkinson et al., 2010; Atkinson et al., 2011; Cui et al., 2012; Ghezzi et al., 2009; Lefebvre-Legendre et al., 2001; Na et al., 2014; Sánchez et al., 2013; Wiedemann et al., 2006). Appropriately, re-expression of each LYR protein in the context of their respective deletion strains can restore both respiration-dependent growth and complex assembly (Figure 1B and S1B-E). In order to independently prevent the interaction of each LYR family member with ACP, mutant proteins were generated in which the Leu-Tyr-Arg motif was substituted with three Ala residues (ΔLYR), which structural studies suggest should abrogate the ACP-LYR interaction (Boniecki et al., 2017; Brown et al., 2017; Cory et al., 2017; Fiedorczuk et al., 2016; Zhu et al., 2016). Likely reflecting a defect in ACP binding, these mutant proteins are less stable than their WT counterparts and therefore they needed to be overexpressed in order to accumulate to a similar (or even greater) extent as the corresponding WT protein (Figure S1F). Even when overexpressed, Sdh6ΔLYR, Sdh7ΔLYR, and Mzm1ΔLYR failed to restore complex assembly and, therefore, were unable to complement the respiratory growth defects associated with the respective mutant strains (Figure 1B and S1B-D). Fmc1ΔLYR, on the other hand, was able to rescue both growth and ETC assembly in fmc1Δ cells (Figure 1B and S1E). While the source of this discrepancy remains unclear, Fmc1 frequently behaves atypically when compared to other members of the yeast LYR protein family and may have evolved ACP-independent activity. Taken together these data demonstrate that the LYR proteins require the Leu-Tyr-Arg motif, which mediates the ACP interaction, to function as complex-specific assembly factors.
Formation of ACP-LYR complexes depends on 4′-PP but not ACP acylation
High-resolution structures of ACP-LYR complexes show that the 4′-PP-bound acyl chain of ACP penetrates into the helical core of the LYR proteins (Figure 1A), suggesting this acyl chain may be required for the ACP-LYR interaction (Boniecki et al., 2017; Brown et al., 2017; Cory et al., 2017; Fiedorczuk et al., 2016; Van Vranken et al., 2016; Zhu et al., 2016). To test this, the ACP-LYR interactions were first confirmed by immunoprecipitation of Acp1HA/FLAG, the yeast ACP, from mitochondria expressing Sdh6V5, Sdh7V5, Mzm1V5, Fmc1V5 and Isd11V5 fusion proteins (Figure 1C-H and S1G-O). Next, we wanted to test the dependence of this interaction on the presence of the ACP-bound 4′-PP. In order to address this question an Acp1HA/FLAG mutant was generated in which Ser82, the residue to which 4′-PP is conjugated, was substituted with an Ala (Acp1S82A-HA/FLAG). Ser82 and 4′-PP were critical for the majority of ACP-LYR interactions, as the Acp1S82A mutant protein, which migrates faster than its WT counterpart, was incapable of interacting with Sdh6, Sdh7, Mzm1, and Isd11 (Figure 1D,F and S1H,J). Interestingly, Fmc1 maintained interaction with Acp1S82A, further suggesting that Fmc1 is atypical among the LYR protein family members (Figure S1L).
Having demonstrated the importance of the 4′-PP in the formation of ACP-LYR complexes, we next investigated the importance of ACP acylation in these interactions. Acp1HA/FLAG and the LYRV5 fusion proteins were expressed in strains in which MCT1 or OAR1 had been deleted (mtFASΔ). These genes encode the malonyl-CoA:ACP transferase (MCT1) and the 3-oxoacyl-ACP reductase (OAR1) and are both required for mtFAS activity (Figure S2A) (Hiltunen et al., 2009). As such, these mutant strains are unable to synthesize ACP-bound acyl chains. Interestingly, the Sdh6 LYR protein was destabilized in mtFASΔ cells (Figure S1M), which precluded any evaluation of the importance of ACP acylation in the Acp1-Sdh6 interaction. Mzm1 was partially destabilized, but the pool of Mzm1 that persisted remained competent for binding Acp1 in the mct1Δ and oar1Δ strains (Figure 1G). Sdh7, Isd11, and Fmc1 all accumulate normally in cells lacking Mct1 and Oar1 and bind the non-acylated form of Acp1 equivalently to the acylated form (Figure 1H and S1N,O). Thus, ACP acylation is not required for the formation of ACP-LYR complexes.
Yeast mitochondria accumulate an acylated form of ACP
Although ACP acylation is not required for LYR binding, the intimate connections between the acyl chain and LYR proteins suggest that an acylated form of ACP might still be important for LYR protein function. To begin to address this question, we first wanted to define the array of ACP species that accumulate in mitochondria in vivo. Because ACP acylation is dependent on a thioester bond between the 4′-PP and the acyl chain (Figure 2A), these species are both chemically and biologically labile. Furthermore, thiol-blocking reagents like N-ethylmaleimide (NEM) can stabilize modified ACP intermediates during sample preparation (Post-Beittenmiller et al., 1991; Shintani and Ohlrogge, 1994). Taking these factors into consideration, we developed an assay that maintains acylation and enables visualization of the various ACP species that accumulate.
Figure 2. Yeast mitochondria accumulate an acylated form of ACP.
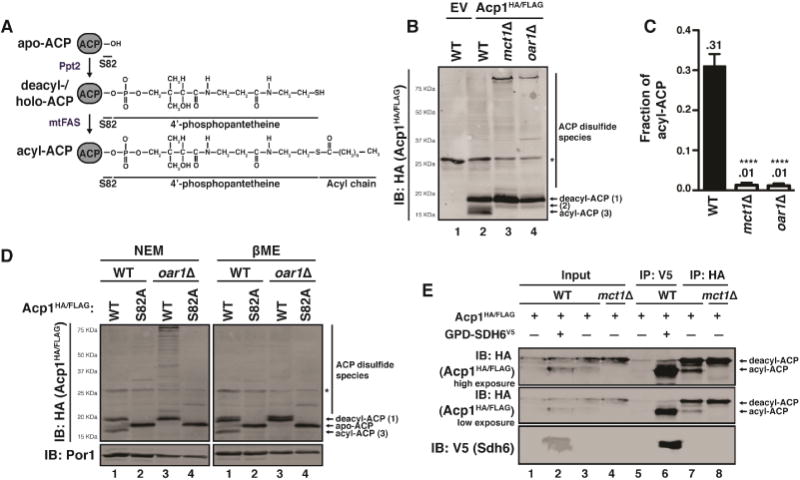
A. Depiction of various ACP species. B. Mitochondrial lysates expressing Acp1HA/FLAG were separated by SDS-PAGE in the presence of 10mM NEM and immunoblotted for HA. * indicates non-specific bands. C. The fraction of acylated Acp1HA/FLAG from (B) was quantified using densitometry. N≥3. Error bars represent standard deviation. ****p<0.0001. D. WT or oar1Δ mitochondrial lysates expressing Acp1HA/FLAG or Acp1S82A-HA/FLAG were separated by SDS-PAGE in the presence of 10mM NEM or 1% βME and immunoblotted for HA. E. Acp1HA/FLAG or Sdh6V5 (expressed from the GPD1 promoter) were immunoprecipitated from mitochondrial lysates expressing the indicated proteins, separated by SDS-PAGE in the presence of 10 mM NEM, and immunoblotted for HA (Acp1HA/FLAG) and V5 (Sdh6V5). Inputs represent 5% of the total lysate.
Mitochondrial lysates from yeast cells expressing Acp1HA/FLAG were resolved by SDS-PAGE to visualize specific Acp1-containing species by immunoblot (Figure 2A,B). In WT cells, Acp1HA/FLAG resolved into three clearly defined bands—identified as 1, 2, and 3— that migrated with apparent molecular weights between 15 and 20 KDa (Figure 2B). In order to facilitate the identification of any potential acyl-ACP species, the mct1Δ and oar1Δ strains were again employed. These genes are required for mtFAS-dependent acylation of ACP and therefore any bands that are dependent on these genes are likely to represent acylated ACP species. Band 1 (~55% of total Acp1HA/FLAG), accumulates significantly in mct1Δ and oar1Δ cells (mtFASΔ; ~90% of total Acp1HA/FLAG) (Figure 2B,C and S2B). Because the presence of this band is not sensitive to mtFAS deficiency, and in fact, accumulates in this context, this band likely represents holo- or deacyl-ACP, which has the 4′-PP cofactor but no acylation (Figure 2A,B). Band 2 (~15% of total Acp1HA/FLAG), migrates similarly to an Acp1S82A mutant and, therefore, could represent apo-ACP lacking the 4′-PP (Figure 2A,B,D and S2B). Importantly, this designation is not definitive and it remains possible that this band represents an alternative ACP species of unknown modification. Finally, Band 3 (~30% of total Acp1HA/FLAG) readily accumulates in WT cells but is undetectable in mtFASΔ lysates (Figure 2B,C and S2B). This band also fails to accumulate in cells expressing Acp1S82A-HA/FLAG, which have no 4′-PP (Figure 2D). Because the accumulation of Band 3 is dependent on mtFAS activity as well as on 4′-PP, we conclude that it is an acylated form of ACP, which we will define as acyl-ACP (Figure 2A). The faster electrophoretic mobility of ACP upon acylation is somewhat surprising but consistent with previous studies of ACP acylation in plants (Post-Beittenmiller et al., 1991).
A number of other ACP-containing species, which migrate slower than deacyl-ACP, accumulate specifically in mtFASΔ cells (Figure 2B,D). These bands fail to accumulate in mtFASΔ cells expressing Acp1S82A and are also sensitive to treatment with reducing agents like β-mercaptoethanol (βME; Figure 2D). We, therefore, conclude that these bands likely represent spurious ACP disulfide-linked species. Because the mature form of Acp1 contains no Cys residues, the only free thiol capable of participating in disulfide bonds is the terminal thiol of the 4′-PP prosthetic group. Since these higher molecular weight spurious disulfide species could only form when this thiol is unmodified, we interpret these disulfide species as marks of ACP deacylation but do not believe they are of any biological importance.
LYR proteins interact preferentially with acyl-ACP
We next wanted to determine if the observed acyl-ACP species represents the acylated ACP species observed in high-resolution ACP-LYR structures (Boniecki et al., 2017; Brown et al., 2017; Cory et al., 2017; Fiedorczuk et al., 2016; Zhu et al., 2016). To that end, LYR proteins Sdh6V5 and Mzm1V5 were immunoprecipitated from cells expressing Acp1HA/FLAG. The resulting eluates were then assessed by immunoblot to determine which ACP species interact with these LYR proteins. Consistent with the ACP-LYR structures, Sdh6 and Mzm1 both exhibited a clear preference for binding to acyl-ACP over deacyl-ACP (Figure 2E and S2C; compare lanes 6 and 7). Despite this preference for acyl-ACP, Sdh6V5 and Mzm1V5 both appear to interact weakly with deacyl-ACP (Figure 2E and S2C). This corroborates the prior conclusion that ACP acylation is not required for ACP-LYR interactions. Taken together, these data suggest that while LYR proteins can bind deacyl-ACP, they exhibit a greater affinity for acyl-ACP. These data raise the possibility that acylation of ACP could serve as a post-translational regulator of the LYR proteins to allosterically activate the ETC assembly network.
ACP acylation, but not mtFAS, is required for ETC biogenesis
To begin to test this hypothesis, we sought to determine if ACP acylation is necessary and sufficient to support ETC biogenesis. Deletion of genes encoding mtFAS enzymes results in pleiotropic mitochondrial defects, including attenuated mitochondrial translation, depletion of lipoic acid, and defects in ETC subunit accumulation (Kursu et al., 2013; Schonauer et al., 2008). These defects result in cells that are unable to grow on carbon sources like glycerol, which require mitochondrial respiration for proliferation (Figure S3A). Remarkably, respiration-dependent growth can be restored in mtFASΔ cells simply by ectopic mitochondria-targeted expression of a peroxisomal medium-chain fatty acyl-CoA synthetase known as FAA2 (mito-FAA2; Figure 3A) (Harington et al., 1994; Kastaniotis et al., 2004; Kursu et al., 2013). Stemming from the pleiotropy of defects described earlier, mtFASΔ cells exhibited an almost complete loss of all ETC complexes (Figure 3B and S3C). As with respiratory growth, expression of mito-FAA2 also led to a significant, but incomplete, restoration of ETC complex abundance in mtFASΔ cells (Figure 3B). Therefore, ectopically targeting a single acyl-CoA synthetase to mitochondria is sufficient to render the role of mtFAS in ETC biogenesis and respiratory growth obsolete.
Figure 3. ACP acylation is required for ETC biogenesis.
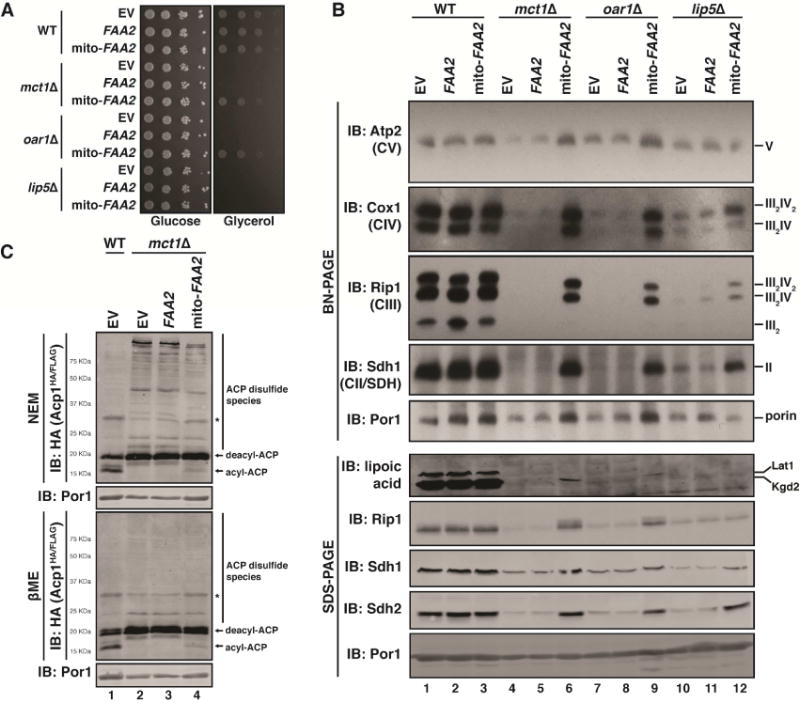
A. Ten-fold serial dilutions of the indicated strains harboring the indicated plasmids were plated on synthetic medium with either glucose or glycerol and grown for 2-3 days. B. Mitochondrial lysates generated from the indicated strains harboring the indicated plasmids were separated by BN-PAGE (upper panel) or SDS-PAGE (lower panel) and immunoblotted with the indicated antibodies. C. Mitochondrial lysates expressing Acp1HA/FLAG were separated by SDS-PAGE in the presence of 10mM NEM or 1% βME and immunoblotted for HA. * indicates non-specific bands.
In addition to the synthesis of stably ACP-bound acyl chains, mtFAS is also required for the synthesis of lipoic acid (Brody et al., 1997). In order to test the importance of lipoic acid synthesis in ETC complex assembly, mitochondrial lysates from lip5Δ cells were resolved by BN-PAGE. LIP5 is required for the conversion of ACP-bound octanoate to lipoic acid but is not directly involved in fatty acid synthesis (Figure S2A). Consistent with previous studies, lip5Δ cells contain detectable levels of both complex II and complex III and IV-containing super-complexes (Figure 3B) (Kursu et al., 2013). Therefore, defects in lipoic acid synthesis alone cannot explain the full range of defects observed in mtFASΔ cells. Furthermore, expression of mito-FAA2 in lip5Δ cells resulted in a complete restoration of complex II and a partial restoration of complex III and IV without rescuing lipoic acid abundance (Figure 3B). These data suggest that the necessity of mtFAS for ETC complex assembly is independent of its role in lipoic acid synthesis.
Having demonstrated that re-localization of Faa2 to mitochondria is capable of restoring ETC biogenesis in mtFASΔ cells, we next wanted to determine the mechanism responsible for this effect. First, we wanted to know if the ability of mito-FAA2 to rescue respiration-dependent growth in mtFASΔ cells was dependent on an ACP-bound 4′-PP. Therefore, mito-FAA2 was expressed in cells lacking PPT2, which encodes the matrix-localized 4′-PP transferase required for the covalent attachment of 4′-PP to ACP (Stuible et al., 1998). Cells lacking Ppt2 are mtFAS-deficient and therefore unable to assemble the ETC or to grow on respiratory carbon sources like glycerol (Figure S3A,D). Unlike other mtFASΔ mutant strains, however, expression of mito-FAA2 failed to restore respiratory growth in ppt2Δ cells (Figure 3A and S3D). Therefore, cells expressing mito-FAA2 require acylation-competent ACP to restore respiration when lacking mtFAS. This genetic result led to the hypothesis that mito-Faa2 may restore ETC biogenesis in mtFASΔ cells by generating acyl-CoA species in the mitochondrial matrix that are then used as substrates for mtFAS-independent acylation of ACP. Under normal conditions, peroxisomal Faa2 catalyzes, through the ligation of coenzyme A (CoA; Figure S3B), the activation of nine to twelve carbon acyl chains, which is the approximate length of the ACP-bound acyl chains present in high-resolution structures of ACP-LYR complexes (Johnson et al., 1994; Knoll et al., 1994). As expected, mtFASΔ cells expressing either an empty vector or WT peroxisomal FAA2 had no detectable acyl-Acp1HA/FLAG (Figure 3C). This, however, was partially restored by expression of mito-FAA2 in mtFASΔ, but not ppt2Δ cells, which lack acylation-competent ACP (Figure 3C and S3E). This modest restoration of acylation is corroborated by a marked decrease in the abundance of the high molecular weight disulfide species (Figure 3C). Taken together, these data demonstrate that mito-FAA2 is capable of supporting ACP acylation independent of mtFAS. Furthermore, acyl-ACP, but not mtFAS or lipoic acid, is required for ETC biogenesis.
ACP acylation and ETC biogenesis are sensitive to limitation of mitochondrial acetyl-CoA
In addition to being the primary substrate of oxidative phosphorylation and the point at which major catabolic pathways converge, acetyl-CoA is also the source of carbon for mtFAS-dependent acylation of ACP (Figure S2A) (Hoja et al., 2004). As such, it has been hypothesized that ACP acylation may serve as a functional link between acetyl-CoA abundance and ETC assembly—capable of connecting substrate availability with oxidative capacity (Kursu et al., 2013). In order to begin to test this hypothesis, we sought to determine if ACP acylation is sensitive to perturbations in the levels of mitochondrial acetyl-CoA. As such, we limited the availability of mitochondrial metabolic substrates using two genetic models of mitochondrial acetyl-CoA restriction—mpc1Δ and lip5Δ (described earlier) (Figure S4A). Mpc1 is a requisite subunit of the mitochondrial pyruvate carrier and deletion of this gene impairs mitochondrial pyruvate import, thereby limiting pyruvate dehydrogenase (PDH)-dependent generation of acetyl-CoA (Figure S4A) (Bricker et al., 2012). This defect in acetyl-CoA accumulation is corroborated by decreased protein acetylation (Figure S4B). Importantly, this mutation decreases but does not eliminate mitochondrial acetyl-CoA because of a well-characterized but inefficient PDH bypass in yeast (Kursu et al., 2013; Orlandi et al., 2014; Wei et al., 2009). To determine if ETC biogenesis is sensitive to a diminution of mitochondrial acetyl-CoA, mitochondrial lysates isolated from WT and mpc1Δ cells were resolved by BN-PAGE and SDS-PAGE. Cells lacking Mpc1 (or Lip5; shown in Figure 3B) have clear defects in the steady state abundance of complex II and complex III and IV-containing super-complexes, while complex V was comparable to WT (Figure 4A). Interestingly, despite a profound depletion of assembled complex II and III, the steady-state abundance of the individual subunits of these two complexes was only subtly affected (Figure 4A). This suggests that the defects in complex II and III are likely at the level of subunit assembly rather than expression.
Figure 4. ACP acylation is sensitive to perturbation in mitochondrial acetyl-CoA.
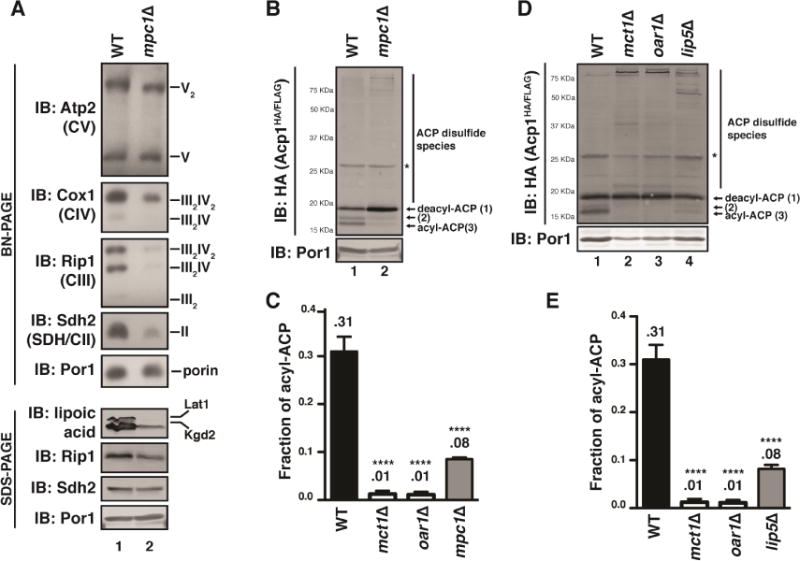
A. Mitochondrial lysates of the indicated strains were separated by BN-PAGE (upper panel) or SDS-PAGE (lower panel) and immunoblotted with the indicated antibodies. B,D. Mitochondrial lysates generated from mpc1Δ (B) or lip5Δ (D) strains expressing Acp1HA/FLAG were separated by SDS-PAGE in the presence of 10mM NEM and immunoblotted for HA. * indicates non-specific bands. C,E. The fraction of acylated Acp1HA/FLAG from B and D was quantified using densitometry for mpc1Δ (C) and lip5Δ (E) cells. N≥3. Error bars represent standard deviation. ****p<0.0001.
Consistent with the defects in ETC biogenesis, there was a significant and specific depletion of acyl-ACP in mpc1Δ cells (~8% of total Acp1HA/FLAG; Figure 4B,C and S4C,D). Furthermore, this is coincident with an accumulation of both deacyl-ACP and ACP-containing disulfide species (Figure 4B and S4C,D). This phenotype is reminiscent of lip5Δ cells, which also have defects in PDH-dependent synthesis of acetyl-CoA (due to a loss of lipoic acid) and ETC assembly (Figure 3B). Importantly, these cells also have a reduction in the pool of acyl-ACP similar to the mpc1Δ strain (~8% of total Acp1HA/FLAG; Figure 4D,E and Figure S4E). Therefore, the defects in ETC biogenesis observed in the lip5Δ strain likely stem from a decrease in acyl-ACP and not from a loss of lipoic acid per se. Taken together, these data demonstrate that both ACP acylation and ETC biogenesis are sensitive to chronic perturbations in mitochondrial acetyl-CoA availability.
Acetyl-CoA-independent re-acylation of ACP in mpc1Δ cells is sufficient to restore ETC complex assembly
To determine if the defect in ETC assembly observed in acetyl-CoA-limited conditions was the direct result of ACP deacylation, mito-FAA2 was expressed in mpc1Δ cells. Although still respiration-competent, mpc1Δ cells have a strong growth defect in respiratory carbon sources, which is consistent with their defects in ETC biogenesis (Figure 5A and S5A). Importantly, expression of mito-FAA2 in mpc1Δ cells resulted in a partial rescue of this growth defect (Figure 5A) and also restored the steady state abundance of complex II and complex III and IV-containing super-complexes to WT levels (Figure 5B). Consistent with this increase in ETC complex abundance, mito-FAA2 also restored the levels of acyl-ACP to that of WT cells (Figure 5C). Therefore, mtFAS-independent acylation of ACP is sufficient to restore ETC biogenesis in acetyl-CoA-limited conditions. Taken together, these data demonstrate that limitation of mitochondrial acetyl-CoA prevents ETC assembly and activation by restricting ACP acylation. Thus, ACP acylation functions as an acetyl-CoA-dependent post-translation modification required for ETC assembly.
Figure 5. Expression of mito-FAA2 restores ACP acylation, OXPHOS assembly and growth in mpc1Δ cells.
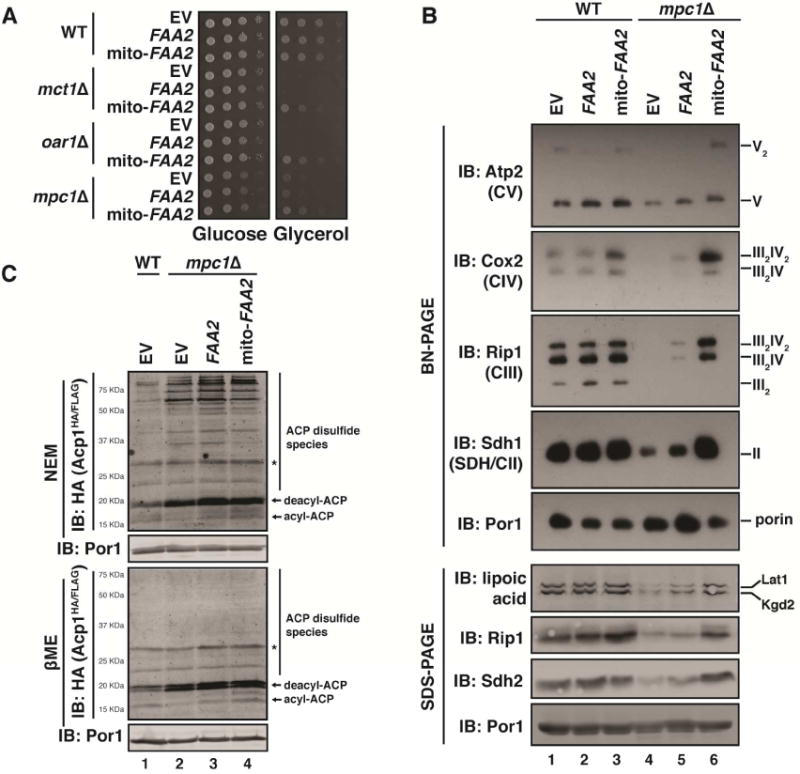
A. Ten-fold serial dilutions of the indicated strains harboring the indicated plasmids were plated on synthetic medium with either glucose or glycerol and grown for 2-3 days. B. Mitochondrial lysates generated from the indicated strains harboring the indicated plasmids were separated by BN-PAGE (upper panel) or SDS-PAGE (lower panel) and immunoblotted with the indicated antibodies. C. Mitochondrial lysates expressing Acp1HA/FLAG were separated by SDS-PAGE in the presence of 10mM NEM or 1% βME and immunoblotted for HA. * indicates non-specific bands.
mct1Δ and mpc1Δ cells have unique proteomic profiles
The pleiotropic phenotype observed in cells deficient in mtFAS likely results from extreme and chronic ACP deacylation (Figure 2B,C), resulting in a failure to translate the mitochondrial genome, a loss of lipoic acid, and an inhibition of the LYR network of ETC assembly factors. This is in stark contrast to the phenotype of mpc1Δ cells, which results from acetyl-CoA limitation and a partial depletion of acyl-ACP (Figure 4E,G). As such, mtFAS deficiency likely represents a non-physiological mitochondrial insult that masks direct effects in ETC biogenesis. The decreased abundance of acyl-ACP observed in mpc1Δ cells (and lip5Δ cells), on the other hand, might more accurately model a physiologically relevant situation in which acetyl-CoA and acyl-ACP become limited.
We compared the consequences of MCT1 and MPC1 deletion on the cellular proteome using quantitative tandem mass spectrometry after isobaric labeling (Figure 6A). In total, we quantified the steady state abundance of over 4700 proteins and found notable changes throughout the cell. Reproducibility was high across the three biological replicates from each group and all samples segregated according to their genotype (Figure 6B). Compared to WT cells, the mct1Δ strain exhibited a significant remodeling of the cellular proteome with 245 and 95 proteins significantly depleted and accumulated, respectively (Figure 6C, S6A and Table S1). The mpc1Δ strain, on the other hand, showed more modest effects with just 25 proteins being depleted and 80 increased (Figure 6D, S6B and Table S2).
Figure 6. Assessing the phenotypic differences between mct1Δ and mpc1Δ cells.
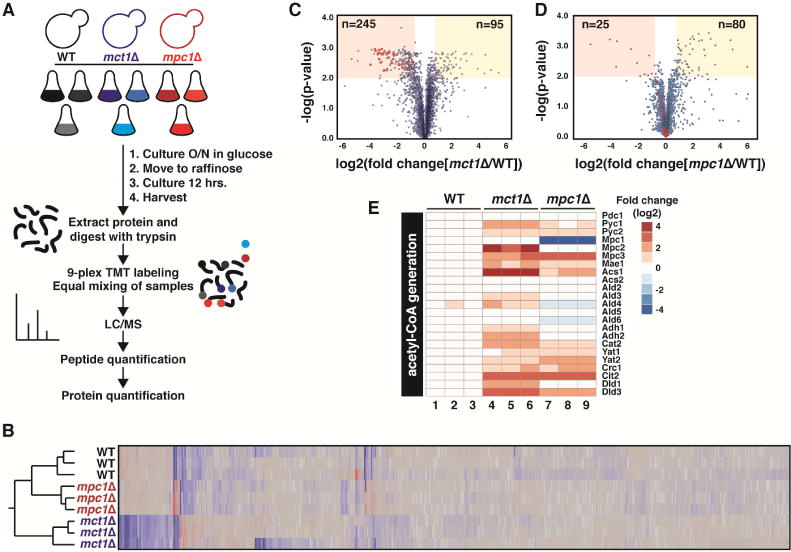
A. Proteomics experimental overview. B. Heat map and dendrogram plot demonstrating that all biological replicates cluster as expected. C,D. Volcano plots highlighting significantly (p≤0.01) up-(fold change ≥ 1.25) and down-(fold change ≤ 0.75) regulated proteins comparing WT and mct1Δ (C) and WT and mpc1Δ (D). Proteins involved in oxidative phosphorylation are emphasized in red. E. Heat map showing the fold change (log2) of the indicated proteins involved in acetyl-CoA generation.
mpc1Δ cells have a proteomic response consistent with mitochondrial acetyl-CoA depletion
Despite considerable differences in their respective proteomic profiles, mct1Δ and mpc1Δ strains both exhibited a significant increase in the expression of genes involved in mitochondrial acetyl-CoA generation (Figure 6E). This is consistent with a depletion of mitochondrial acetyl-CoA in both strains. Presumably, the cell senses metabolic substrate starvation in mitochondria and responds by upregulating compensatory pathways in a failed attempt to fully restore metabolic homeostasis. This is consistent with a defect in lipoic acid synthesis in mct1Δ cells and supports the notion that mitochondria in mpc1Δ cells are indeed limited, but not fully depleted, for mitochondrial acetyl-CoA.
Mitochondrial acetyl-CoA-limited cells have no defect in mitochondrial translation
These comparative proteomics data demonstrated that cells devoid of mtFAS failed to accumulate any of the subunits of the ETC complexes, consistent with a profound defect in mitochondrial translation (Figure 7A and S6C). This is corroborated by a profound depletion of protein subunits of the mitochondrial ribosome and RNase P (Figure 7B) as well as mitochondrial genome-encoded ETC subunits (Figure S7A). Acetyl-CoA-limited (mpc1Δ) cells, on the other hand, have a much milder phenotype that is consistent with normal mitochondrial translation. ETC subunits are largely unaffected (Figure 7A), as are the protein subunits of the mitochondrial ribosome, RNase P, and mitochondrial genome-encoded proteins (Figure 7B and S7A). Taken together, these data demonstrate that chronic and complete deacylation of ACP results in profound defects in mitochondrial translation, while limitation of mitochondrial acetyl-CoA has no effect on this process.
Figure 7. Acetyl-CoA limitation prevents LYR-mediated ETC assembly and activation.
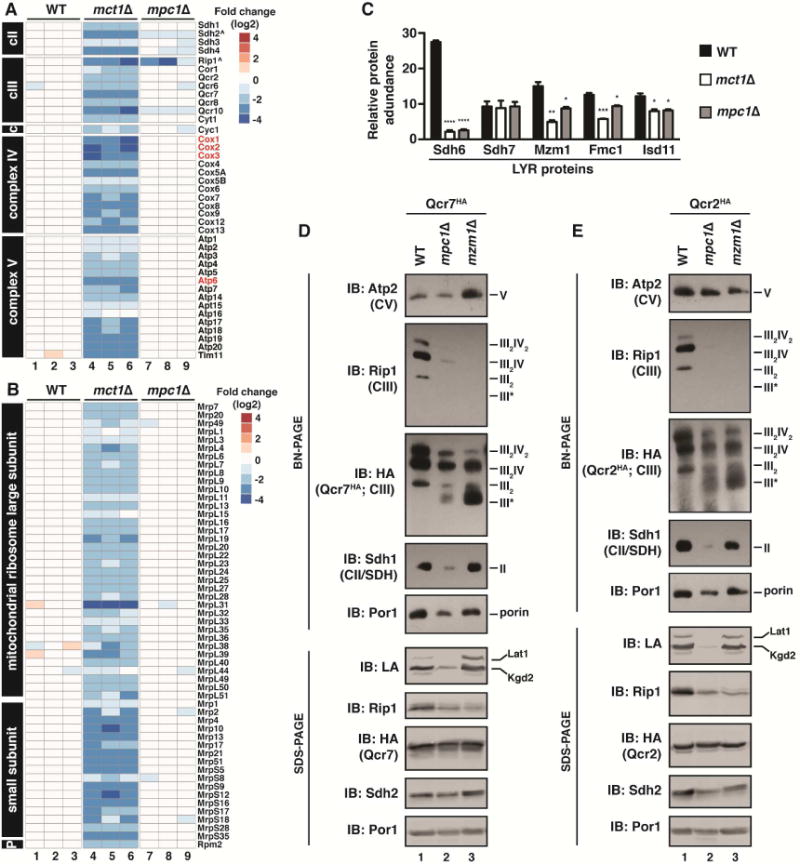
A,B. Heat map showing the fold change (log2) of the indicated proteins involved in mitochondrial respiration (A) and mitochondrial protein synthesis (B) in mct1Δ and mpc1Δ cells compared to WT. ^LYR target proteins. Proteins in red are encoded by the mitochondrial genome. C. Relative abundance of the indicated proteins in WT, mct1Δ, and mpc1Δ cells (N=3). Error bars represent the standard deviation. *p<0.05, **p<0.005, ***p<0.0005, ****p<0.00005. D,E. Mitochondrial lysates isolated from cells expressing either Qcr7HA (D) or Qcr2HA (E) were separated by BN-PAGE (upper panel) or SDS-PAGE (lower panel) and immunoblotted with the indicated antibodies.
Mitochondrial acetyl-CoA limited cells fail to support LYR-mediated ETC biogenesis
Having demonstrated that mpc1Δ cells have normal mitochondrial translation, we hypothesized that the observed decrease in ETC complex assembly might result from a failure to activate the LYR network of ETC assembly factors. Although the majority of ETC complex subunits accumulate normally in mpc1Δ cells, there were two notable subunits that were consistently and significantly depleted—Sdh2 and Rip1—the direct LYR target proteins of Sdh6/Sdh7 and Mzm1, respectively (Figure 7A) (Atkinson et al., 2011; Na et al., 2014). Furthermore, the LYR proteins themselves, which were depleted in the mct1Δ strain, were also significantly depleted in mpc1Δ cells (Figure 7C). Consistent with what we had observed in mct1Δ cells, Acp1 can still bind LYR proteins Sdh7 and Mzm1 in mpc1Δ cells (Figure S7B,C). This suggests that the partial depletion of mitochondrial acetyl-CoA conferred by loss of MPC1 results in impaired activation of the LYR network without obvious defects in ACP-LYR interactions. Taken together, these data suggest the interesting possibility that, in acyl-ACP-limiting conditions, the reduced pool of acyl-ACP prioritizes the activation of mitochondrial translation but initiates a metabolic checkpoint that prevents ETC assembly by inactivating the LYR network.
We next compared the defects in complex III assembly, subunit accumulation, and LYR abundance in mpc1Δ cells with those that lack the complex III-specific LYR, Mzm1. Both mpc1Δ and mzm1Δ cells exhibited decreased abundance of Rip1 as well as fully assembled complex III (as assessed by Rip1 immunoblot); however, the phenotype was more pronounced in cells lacking Mzm1 (Figure 7D,E). Importantly, mpc1Δ cells retain some Mzm1 and a small pool of acyl-ACP, which together can likely support modest complex III biogenesis (Figure 7C and 4D). Deletion of MZM1 specifically abrogates the integration of the LYR target Rip1 and its binding partner Qcr10 into complex III (Atkinson et al., 2011). By BN-PAGE, mzm1Δ cells exhibited a faster migrating complex III specific sub-assembly (as assessed by Qcr7HA or Qcr2HA immunoblot) that contained every complex III subunit except Rip1 and Qcr10 (Atkinson et al., 2011). Consistent with a specific defect in Mzm1-mediated complex III assembly, mpc1Δ cells also accumulate a similar sub-assembly that lacks Rip1 and Qcr10 (Figure 7D,E and S7D,E). Thus, complex III assembly stalls at the point in which the LYR target protein (Rip1) and its binding partner Qcr10 are integrated. This explains why Rip1 and Qcr10 are destabilized in mpc1Δ cells, while all other subunits accumulate normally (Figure S7F). Taken together these data suggest that, in mitochondrial acetyl-CoA-limited conditions, ACP deacylation prevents the complete assembly and activation of complexes II and III through impaired incorporation of LYR target proteins, like Rip1 and Sdh2 (Figure S7G).
Discussion
Previous studies have described a role for yeast mtFAS in mitochondrial biogenesis, as mutants exhibit a deficiency in mitochondrial translation and loss of PDH activity. These effects were attributed to primary defects in RNase P activity and lipoic acid synthesis, respectively (Kursu et al., 2013; Schonauer et al., 2008). More recently, large-scale proteome-wide interaction studies and high-resolution structural data have demonstrated that ACP interacts with members of the LYR protein family (Angerer, 2015; Boniecki et al., 2017; Brown et al., 2017; Cory et al., 2017; Fiedorczuk et al., 2016; Floyd et al., 2016; Huttlin et al., 2015; Zhu et al., 2016). Because the LYR proteins have important roles in ETC complex biogenesis, we hypothesized that ACP might also play an important role in this process. Motivated by this gap in understanding, we sought to define the role of ACP and mtFAS in directly supporting ETC complex biogenesis. The data presented herein illuminate an unexpected role of mtFAS in stimulating direct ETC assembly through the demonstration that the acylated form of ACP is an acetyl-CoA-dependent allosteric activator of this process. Acyl-ACP directly interacts with each member of the LYR protein family and, in an acyl chain-dependent manner, stimulates their ETC complex assembly function. As such, we propose that mtFAS could provide a functional link between the abundance of mitochondrial acetyl-CoA and the biogenesis of the complexes needed for its metabolism.
The maintenance of the prokaryotic-type fatty acid synthesis pathway, mtFAS, in eukaryotic mitochondria represents a long-standing evolutionary mystery (Hiltunen et al., 2010). Indeed, this pathway is required for the synthesis of octanoate, the eight-carbon precursor of the critical mitochondrial cofactor, lipoic acid (Brody et al., 1997). Because eukaryotic cells are unable to scavenge lipoic acid, which is required for PDH and α-KGDH activity, mitochondrial metabolism depends on this pathway (Feng et al., 2009). This seems an inadequate rationale for the pan-eukaryotic conservation of this multi-protein system, however, because simple re-targeting of the Faa2 acyl-CoA synthetase is sufficient to restore respiratory growth (Figure 3). Moreover, mtFAS is capable of synthesizing ACP-bound acyl chains significantly longer than eight carbons (Zhang et al., 2005). An early clue towards understanding the role of these extended acyl chains came in the form of high-resolution structures of ACP-LYR complexes. These models suggest the importance of the extended acyl chains by demonstrating that ACP embeds a 4′-PP-conjugated acyl chain into the core of the hydrophobic three-helix bundle of an LYR protein (Figure 1A) (Boniecki et al., 2017; Brown et al., 2017; Cory et al., 2017; Fiedorczuk et al., 2016; Zhu et al., 2016). This observation raised a number of questions regarding the functional importance of this unique interaction. In nutrient-replete conditions WT cells readily accumulate an acylated form of ACP, defined here as acyl-ACP (~30% of total Acp1HA/FLAG; Figure 2). It is this fraction that is likely dedicated to LYR-dependent functions like ETC assembly and FeS cluster biogenesis, while the remaining fraction is available for LYR-independent functions, like lipoic acid synthesis. Focusing on the LYR-dependent function in ETC assembly, we propose that acyl-ACP functions as an allosteric activator of the LYR network. The following pieces of data support this hypothesis. First, acyl-ACP interacts with each member of the LYR protein family (Figure 1) (Boniecki et al., 2017; Brown et al., 2017; Cory et al., 2017; Fiedorczuk et al., 2016; Zhu et al., 2016). Second, LYR proteins that have been mutated to block interaction with acyl-ACP lose their function as ETC assembly factors (Figure 1). Third, ACP acylation is not a formal requirement for LYR binding (Figure 1 and 2). Fourth, ACP acylation is required for ETC assembly (Figure 3). Finally, LYR proteins appear to have a higher affinity for acyl-ACP than for deacyl-ACP (Figure 2). Taken together, these data, combined with previously published structures of ACP-LYR complexes, support a model in which ACP acylation functions as an allosteric activator of the LYR protein network. In this way, ACP acylation can be sensed by LYR proteins to trigger ETC activation through the integration of LYR target proteins into their respective complexes.
Each member of the LYR family plays a very specific role in support of cellular respiration including cofactor synthesis, activation of mitochondrial translation, and biogenesis of the respiratory chain (Adam et al., 2006; Angerer et al., 2014; Atkinson et al., 2010; Atkinson et al., 2011; Cui et al., 2012; Ghezzi et al., 2009; Lefebvre-Legendre et al., 2001; Na et al., 2014; Sánchez et al., 2013; Wiedemann et al., 2006). Importantly, it is very unlikely to be a coincidence that LYR proteins are responsible for the final step in complex assembly and activation. In the context of complex III, for example, Mzm1 is required for the addition of the final two subunits thereby converting an inactive sub-complex into a fully active complex III (Atkinson et al., 2011). This concept holds for other LYR-dependent complexes including complexes I and II. Therefore, activation of mitochondrial respiration is dependent on the LYR protein family and we demonstrate herein that LYR protein activity depends on ACP acylation. We found that mutation of the eponymous Leu-Tyr-Arg motif, which is critical for mediating the ACP-LYR interaction, prevents LYR assembly activity (Figure 1). Furthermore, in strains in which ACP acylation is ablated or severely diminished, LYR proteins fail to execute their specific functions in support of ETC biogenesis (Figure 3 and 4). Because each LYR protein requires acyl-ACP as a single common cue, we propose that that this family of assembly factors has evolved in order to coordinate ETC assembly, cofactor synthesis, and subunit translation that are all required for respiration.
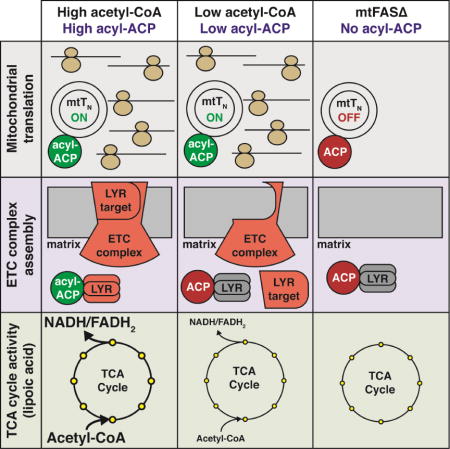
Acyl-ACP allosterically activates the LYR network of complex assembly factors by embedding a 4′-PP-bound acyl chain into the hydrophobic core of LYR proteins. But why use an acyl chain? Why couple the activation of a catabolic process, in this case cellular respiration, with an anabolic process, in this case mtFAS-dependent ACP acylation? We propose that the answer centers on mitochondrial acetyl-CoA. In addition to being the primary substrate of oxidative phosphorylation, acetyl-CoA is also the primary source of carbon for the synthesis of ACP-bound acyl chains. Therefore, ACP acylation could serve as a functional link between mitochondrial acetyl-CoA abundance and ETC biogenesis. Importantly, ACP acylation is sensitive to specific dampening of mitochondrial acetyl-CoA synthesis. As such, substrate limitation and PDH inactivation, as modeled by MPC1 and LIP5 deletion, respectively, each result in a greater than three-fold depletion of the acyl-ACP pool (Figure 4). This decrease in acyl-ACP results in a primary defect in complex II and III assembly, which can be rescued by re-acylation of ACP in these acetyl-CoA-limited conditions (Figure 4 and 5). Interestingly, in mitochondrial acetyl-CoA-limited cells (mpc1Δ), we observed a highly specific defect in LYR-mediated complex assembly (Figure 7), with the LYR target proteins Rip1 and Sdh2 being the only ETC complex subunits that are significantly and reproducibly depleted, presumably due to impaired complex integration (Figure 7). Furthermore, the LYR proteins themselves are significantly depleted in these cells. Therefore, we conclude that limitation of mitochondrial acetyl-CoA causes depletion of acyl-ACP, thereby initiating a metabolic checkpoint to prevent complex activation by restricting incorporation of LYR target proteins into their respective complexes.
mtFAS-dependent acylation of ACP is required for numerous critical mitochondrial processes including mitochondrial translation, ETC biogenesis, and lipoic acid synthesis (Hiltunen et al., 2010; Kursu et al., 2013; Schonauer et al., 2008). As such, complete mtFAS deficiency results in a pleiotropic phenotype that leads to a near complete loss of all respiratory chain complex subunits. While mtFAS loss of function studies highlight the importance of this pathway in maintaining mitochondrial physiology, these mutations likely represent a non-physiological mitochondrial defect. Furthermore, they mask the role of mtFAS and ACP in directly supporting ETC complex assembly. Therefore, we focused our studies on mtFAS-dependent processes in the context of mitochondrial acetyl-CoA limitation. As expected, these cells exhibited a more specific phenotype than mtFASΔ cells, with no apparent defect in mitochondrial translation (Figure 6 and 7). Instead, the extent of the defects appears to be lipoic acid synthesis, which correlates with a lack of substrate, and LYR-dependent ETC biogenesis (Figure 4). Therefore, the limited amount of acyl-ACP in mpc1Δ cells is sufficient to support normal mitochondrial translation but insufficient for LYR activation (Figure 7), suggesting the existence of multiple thresholds of ACP acylation. Based on the strains utilized in this investigation, it is possible to establish three tiers of ACP acylation state—no acylation (mtFASΔ), low acylation (mpc1Δ), and high acylation (WT). In the complete absence of acylation (mtFASΔ), mitochondrial translation is absent leading to the observed extreme phenotype. Somewhere between no acylation and the low acylation observed in the mpc1Δ cells is a threshold at which point mitochondrial translation is activated. In mpc1Δ cells (low acylation), translation is normal but LYR-dependent ETC biogenesis is attenuated. This second threshold, therefore, appears to initiate complex assembly. In other words, we propose that increasing or decreasing levels of mitochondrial acetyl-CoA would lead to a corresponding change in the levels of acyl-ACP. This modulation in acyl-ACP is sensed by the LYR network, which tunes complex assembly as appropriate. This model would depend on differential affinities of individual LYR proteins for acyl-ACP. As such, when acyl-ACP becomes limiting, LYR proteins with a greater affinity for acyl-ACP will be preferentially activated while those LYR proteins with a weaker affinity for acyl-ACP will remain inactive.
Overall, we present a model in which acylation of ACP functions as an acetyl-CoA-dependent post-translational modification that stimulates ETC biogenesis. It executes this function through direct physical interaction with, and allosteric activation of, the LYR protein family. In addition to ETC biogenesis, acetyl-CoA, mtFAS, and ACP are also required for the activity of the TCA cycle enzymes PDH and α-KGDH (through the synthesis of lipoic acid) and activation of mitochondrial translation. As such, this pathway could provide the cell an elegant mechanism whereby the primary substrate of mitochondrial metabolism can signal the assembly and activation of the two main systems of mitochondrial catabolism (Figure S7H). While this study highlights the mechanism by which acetyl-CoA, mtFAS, and ACP regulate ETC biogenesis, it will be important moving forward to understand the role of this system in the maintenance of mitochondrial metabolic homeostasis. Importantly, all the genes studied in this investigation are highly conserved throughout eukaryotic evolution. Therefore, our data will be highly informative when considering the physiological implication of this system in mammals.
Star Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| HA epitope | BioLegend | MMS-101P-500 |
| V5 epitope | Abcam | ab9116 |
| FLAG epitope | Sigma Aldrich | F7425 |
| Sdh1 | Dr. Dennis Winge | N/A |
| Sdh2 | Dr. Dennis Winge | N/A |
| Rip1 | Dr. Dennis Winge | N/A |
| MTCO1 | Abcam | ab14705 |
| Atp2 | Dr. Dennis Winge | N/A |
| Por1 | Abcam | ab110326 |
| LA | Abcam | ab58724 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| β-mercaptoethanol | Sigma Aldrich | M6250 (SHBC3203V) |
| N-ethylmaleimide | Sigma Aldrich | E3876 (SLBV2285) |
| Digitonin special-grade (water-soluble) | Gold Biotechnology | D-180 (3936.112917A) |
| Lyticase from Anthrobacter luteus | Sigma Aldrich | L4025 (SLB4644) |
| Protease inhibitor cocktail (for use with yeast) | Sigma Aldrich | P8215 (026M4064V) |
| NativePAGE 20X Running Buffer | Invitrogen | BN2001 (1874446) |
| NativePAGE 20X Cathode Buffer Additive | Invitrogen | BN2002 (1877160) |
| Monoclonal anti-HA agarose | Sigma Aldrich | A2095 (026M4810V) |
| Anti-V5 agarose | Sigma Aldrich | A7345 (075M4767V) |
| LysC | Wako | 125-05061 |
| Trypsin | Thermo Fisher | 9057 |
| TMT10plex Isobaric Label Reagent Set plus TMT11-131C Label Reagent | Thermo Fisher | A34808 |
| Critical Commercial Assays | ||
| Peirce BCA Protein Assay Kit | Thermo Scientific | 23225 (RL242698) |
| Peirce Quantitative Colorimetric Peptide Assay | Thermo Scientific | 23275 |
| Experimental Models: Yeast strains | ||
| acp1::HygMX+ACP1HA/FLAG | This study | yJR2698 |
| ppt2::NatMX | This study | yJR3049 |
| mct1::NatMX | This study | yJR2885 |
| oar1::NatMX | This study | yJR3205 |
| cem1::NatMX | This study | yJR3130 |
| htd2::NatMX | This study | yJR3214 |
| lip5::NatMX | This study | yJR3209 |
| lip2::NatMX | This study | yJR3071 |
| mpc1::HygMX | This study | yJR3149 |
| sdh6::NatMX | This study | yJR2701 |
| sdh7::KanMX | This study | yJR2703 |
| mzm1::KanMX | This study | yJR2814 |
| fmc1::HygMX | This study | yJR3284 |
| WT (BY4741) | This study | yJR2884 |
| Recombinant DNA | ||
| Plasmid: ACP1HA/FLAG | This study | pJR13982 |
| Plasmid: ACP1S82A-HA/FLAG | This study | pJR13985 |
| Plasmid: SDH6V5 | This study | pJR13986 |
| Plasmid: SDH7V5 | This study | pJR13987 |
| Plasmid: MZM1V5 | This study | pJR13988 |
| Plasmid: FMC1V5 | This study | pJR13990 |
| Plasmid: ISD11V5 | This study | pJR13989 |
| Plasmid: PGPD-SDH6V5 | This study | pJR13777 |
| Plasmid: PGPD-MZM1V5 | This study | pJR13779 |
| Plasmid: PGPD-SDH6ΔLYR-V5 | This study | pJR13792 |
| Plasmid: PGPD-SDH7ΔLYR-V5 | This study | pJR13793 |
| Plasmid: PGPD-MZM1ΔLYR-V5 | This study | pJR13794 |
| Plasmid: PGPD-FMC1ΔLYR-V5 | This study | pJR13795 |
| Plasmid: PADH-FAA2 | This study | jrp14001 |
| Plasmid: PADH-mito-FAA2 | This study | jrp14002 |
| Plasmid: QCR7HA | This study | jrp14003 |
| Plasmid: QCR2HA | This study | jrp14004 |
| Software | ||
| Prism | GraphPad Software | Version 6 |
| FIJI | ImageJ | Version 1.0 |
| Other | ||
| NativePAGE 3-12% Bis-Tris Gel (15 well) | Invitrogen | BN2012BX10 (17092660) |
| NativePAGE 4-16% Bis-Tris Gel (15 well) | Invitrogen | BN2112BX10 (17022860) |
| Deposited data | ||
| doi:10.17632/b6bgtp9trh.1 (Mendeley) | ||
Contact for Reagent and Resource Sharing
Please contact Jared Rutter (rutter@biochem.utah.edu).
Experimental Model and Subject Details
Yeast Strains
Saccharomyces cerevisiae BY4741 (MATa, his3 leu2 met15 ura3), was used as the wild-type strains where indicated. Each mutant was generated in diploid cells using a standard PCR-based homologous recombination method. The genotypes of all strains used in this study are listed in Key Resources Table. Yeast transformation was performed by the standard TE/LiAc method and transformed cells were recovered and grown in synthetic complete glucose (SD) medium lacking the appropriate amino acid(s) for selection purposes. Medium used in this study is synthetic minimal medium supplemented with 2% glucose or 2% raffinose and appropriate amino acids.
Method Details
Yeast Growth Assays
Growth assays were performed using synthetic minimal media supplemented with the appropriate amino acids and indicated carbon source. For plate-based growth assays, overnight cultures were back-diluted to equivalent ODs and spotted as 10-fold serial dilutions. Cells were allowed to grow for 2-3 days at 30°C.
Isolation of yeast mitochondria (non-acylation conditions)
Cell pellet was washed once with ddH2O and incubated in TD buffer (100 mM Tris-SO4, pH 9.4 and 100 mM DTT) for 15 min at 30°C. Cell pel lets were wahed once with SP buffer (1.2 M Sorbitol and 20 mM potassium phosphate, pH 7.4). Spheroplasts were obtained by incubating cells in SP buffer supplemented with 0.3 mg/mL lyticase for 1 hour at 30°C to remove the cell wall. Spheroplasts were gently washed once and homogenized in ice-cold SH buffer (0.6 M sorbitol, 20 mM HEPES-KOH, pH 7.4, yPIC) using a dounce homogenizer applied with 20 strokes. Crude mitochondria were then isolated by differential centrifugation. As such, whole cell lysates were spun at 3,000xg for 5 mins. The resulting supernatant was then spun again at 10,000Xg for 10 mins to isolate crude mitochondria. Mitochondrial protein content was quantified using a Peirce BCA Protein Assay Kit (Thermo Scientific).
Isolation of yeast mitochondria for ACP acylation studies
Cell pellet was washed once with ddH2O and incubated in TD buffer (100 mM Tris-SO4, pH 9.4 and 100 mM DTT) for 5 min at 30°C. Cell pell ets were washed twice with SP buffer (1.2 M Sorbitol and 20 mM potassium phosphate, pH 7.4). Spheroplasts were obtained by incubating cells in SP buffer supplemented with 0.3 mg/mL lyticase for 30 mins. at 30°C to remove the cell wall. Spheroplasts were gently washed once and homogenized in ice-cold SH buffer+NEM (0.6 M sorbitol, 20 mM HEPES-KOH, pH 7.4, yPIC, 10mM NEM) using a dounce homogenizer applied with 20 strokes. Crude mitochondria were then isolated by differential centrifugation. As such, whole cell lysates were spun at 3,000xg for 5 mins. The resulting supernatant was then spun again at 10,000Xg for 10 mins to isolate crude mitochondria. Mitochondrial protein content was quantified using a Peirce BCA Protein Assay Kit (Thermo Scientific). Mitochondria was solubilized in 2X Laemelli Buffer supplemented with 10mM NEM or 1% βME to 1 mg/mL and incubated for 10 mins. At 65°C. 30μg of sample was loaded immediately following heating.
Immunoprecipitation
Crude mitochondria were isolated and resuspended to a concentration of 5 mg/mL in XWA buffer. Mitochondria was solubilized in 0.7% digitonin for 30 mins. Followed by centrifugation at 20,000 × g for 20 mins. Cleared mitochondrial lysates were incubated with anti-HA antibody-conjugated or anti-V5 antibody-conjugated agarose (Sigma) for 1-2 hrs. at 4°C. The agarose was washed 3-5 times and eluted in Laemmli buffer (65°C, 10 mins). Elutions were resolved by SDS-PAGE and assessed by immunoblot. In order to reliably maintain ACP acylation during immunoprecipitation 10mM N-ethylmaleimide was added to IP buffers.
Steady-state protein analysis
Yeast mitochondria were solubilized in Laemmli buffer. Samples were resolved by SDS-PAGE and assessed by immunoblot. In order to maintain ACP acylation during electrophoresis, 10 mM NEM was added to the Laemmli buffer.
Blue native polyacrylamide gel electrophoresis (BN-PAGE)
BN-PAGE was performed as described previously (Wittig et al., 2006). All buffers were purchased from Invitrogen. Mitochondria were resuspended in 1X lysis buffer (Invitrogen) supplemented with yPIC (Sigma) and solubilized with 1% digitonin for 15 mins on ice. Cleared mitochondrial lysates were prepared by centrifugation at 20,000×g for 15-20 mins. Lysates were resolved on a 3%-12% or 4%-16% gradient native gel (Invitrogen).
Proteomics
Cell lysis and protein digestion, and peptide labeling
In order to fully use the sample multiplexing ability, an 11-plex experiment was designed containing the following proteome-wide comparisons: wt (n=3), mct1Δ (n=3), mpc1Δ (n=30), and wt_extra (samples for an unrelated experiment) (n=2). Cell pellets from each condition were resuspended at 4°C in buffer containing 8M urea, 50mM EPPS pH 8.5, 50mM NaCl, and protease inhibitors. Chilled zirconium oxide beads were added to cell slurries. Cells were lysed using a MiniBeadbeater (Biospec products, Bartlesville, OK) at 4°C in 2mL screw cap tubes for 5 cycles of 30 seconds each, with 1 min pauses between cycles to avoid overheating. After centrifugation, clarified lysates were transferred to new tubes. Bicinchoninic acid (BCA) protein assay (Thermo Fischer Scientific) was performed to determine protein concentration. Proteins were then subjected to disulfide reduction with 5mM tris (2 carboxyethyl) phosphine (TCEP), (room temperature, 30 min) and alkylation with 10 mM iodoacetamide (room temperature, 30 min in the dark). 15 mM dithiotreitol was used to quench excess iodoacetamide (room temperature, 15 min in the dark). Proteins (200ug) were then chloroform/methanol precipitated and washed with methanol prior to air drying. Samples were resuspended in 8 M urea, 50 mM EPPS, pH 8.5, and then diluted to <1M urea with 50mM EPPS, pH 8.5. Proteins were digested for 16 hours with LysC (1:100 enzyme:protein ratio) at room temperature, followed by trypsin (1:100 enzyme:protein ratio) for 6 hours at 37°C. Peptides were quantified using Pierce Quantitative Colorimetric Peptide Assay. TMT-11 reagents (0.8mg) were dissolved in 40uL anhydrous acetonitrile, and 7uL was used to label 70ug peptides in 30% (v/v) acetonitrile. Labeling proceeded for 1 hour at room temperature, until reaction was quenched using 7uL 5% hydroxylamine. TMT-labeled peptides were then pooled, vacuum centrifuged to dryness, and cleaned using 50mg Sep-Pak (Waters).
Offline basic pH reversed-phase (BPRP) fractionation
The pooled TMT-labeled peptide sample was fractionated using BPRP HPLC. We used an Agilent 1260 Infinity pump equipped with a degasser and a single wavelength detector (set at 220 nm). Peptides were subjected to a 50 min linear gradient from 8% to 40% acetonitrile in 10 mM ammonium bicarbonate pH 8 at a flow rate of 0.6 mL/min over an Agilent 300Extend C18 column (3.5 m particles, 4.6 mm ID and 250 mm in length). We collected a total of 96 fractions, then consolidated those into 24 and vacuum centrifuged to dryness. Twelve of the 24 fractions were resuspended in a 5% acetonitrile, 1% formic acid solution. Fractions were desalted via StageTip, dried via vacuum centrifugation, and reconstituted in 5% acetonitrile, 5% formic acid for LC-MS/MS processing.
Liquid chromatography and tandem mass spectrometry
Mass spectrometry data were collected using an Orbitrap Fusion Lumos mass spectrometer (Thermo Fischer Scientific) equipped with a Proxeon EASY-nLC 1000 liquid chromatography (LC) system (Thermo Fisher Scientific). Peptides were separated on a 100μm inner diameter microcapillary column packed with ~35 cm of Accucore C18 resin (2.6μm, 150 Å, Thermo Fisher Scientific). Approximately 2ug peptides were separated using a 2.5 h gradient of acidic acetonitrile. We used the multinotch MS3-based TMT method (McAlister et al., 2014). The scan sequence began with a MS1 spectrum (Orbitrap analysis; resolution 120,000; mass range 400−1400 Th). MS2 analysis followed collision-induced dissociation (CID, CE = 35) with a maximum ion injection time of 150 ms and an isolation window of 0.5 Da. The 10 most abundant MS1 ions of charge states 2-6 were selected for MS2/MS3 analysis. To obtain quantitative information, MS3 precursors were fragmented by high-energy collision-induced dissociation (HCD, CE = 55) and analyzed in the Orbitrap (resolution was 50,000 at 200 Th) with a maximum ion injection time of 150 ms and a charge state-dependent variable isolation window of 0.7 to 1.2 Da (Paulo et al., 2016).
Data Analysis
MS2 mass spectra were processed using a SEQUEST-based software pipeline (Huttlin et al., 2010; McAlister et al., 2012; McAlister et al., 2014). Spectra were converted to mzXML using a modified version of ReAdW.exe. Database searching used the yeast proteome downloaded from Uniprot (UniProt-Consortium, 2015) in both forward and reverse directions, along with common contaminating protein sequences. Searches were performed using a peptide mass tolerance of 20 ppm, and a fragment ion tolerance of 0.9 Da. These wide-mass-tolerance windows were chosen to maximize sensitivity in conjunction with SEQUEST searches and linear discriminant analysis (Beausoleil et al., 2006; Huttlin et al., 2010). TMT tags on lysine residues and peptide N termini (+229.163 Da) and carbamidomethylation of cysteine residues (+57.021 Da) were set as static modifications, while oxidation of methionine residues (+15.995 Da) was set as a variable modification.
Peptide-spectrum matches (PSMs) were adjusted to a 1% false discovery rate (FDR) (Elias and Gygi, 2007). Linear discriminant analysis was used to filter PSMs, as described previously (Huttlin et al., 2010), while considering the following parameters: XCorr, ΔCn, missed cleavages, adjusted PPM, peptide length, fraction of ions matched, charge state, and precursor mass accuracy. PSMs were identified, quantified, and collapsed to a 1% peptide false discovery rate (FDR) and then collapsed further to a final protein-level FDR of 1%. PSMs were quantified from MS3 scans; those with poor quality, MS3 spectra with total TMT reporter signal-to-noise ratio that is <200, or no MS3 spectra were excluded from quantitation. Protein quantitation was performed by summing the signal-to-noise values for all peptides for a given protein. Each TMT channel was summed across all quantified proteins and normalized to enforce equal protein loading. Each protein’s quantitative measurement was then scaled to 100. 9 channels were used (wt vs mct1Δ vs mpc1Δ).
Quantification and Statistical Analysis
The signal-to-noise ratios for each of the six channels were scaled to represent the percent of total signal in the six channels. Student’s t-tests were performed in Excel (2-tailed). P-values were adjusted using the Benjamini-Hochberg approach for the 4,716 proteins that were tested. Significance was determined based on an adjusted P-value of <0.01. Heat maps and dendrograms in Figure 5 were generated using JMP. Heat maps in Figure 5 and 6 were generated using R. The Volcano plot was generated in Excel. PRISM software was used to graph all quantitative data and perform statistical analyses. P values for pairwise comparisons were determined using a Student’s t test. Error bars represent the standard deviation.
Data and Software Availability
Source data available at: doi:10.17632/b6bgtp9trh.1.
Supplementary Material
Highlights (3-4; 85 characters each).
Acyl-ACP is an allosteric activator of LYR motif-containing ETC assembly factors.
ACP acylation is sensitive to perturbations in mitochondrial acetyl-CoA synthesis.
ACP and mtFAS coordinate the activation of mitochondrial respiration.
Acknowledgments
We would like to thank Marjorie Riches Gunn for her support of this work. SMN was supported by a UMDF postdoctoral fellowship. This work was supported partially by grants from the NIH (GM115174 and GM115129 to JR and GM97645 to SPG) and the Nora Eccles Treadwell Foundation (to JR). JR is an Investigator of the Howard Hughes Medical Institute and this work was supported by HHMI.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Conceptualization, J.G.V. and J.R.; Methodology, J.G.V., K.J.C., S.P.G., D.R.W., and J.R.; Investigation, J.G.V., S.M.N., K.J.C., M.J., Y.O., S.P.G., D.R.W., and J.R.; Formal analysis, J.G.V., S.M.N., K.J.C., M.J., Y.O., J.A.B., J.P.G., S.P.G., D.R.W., and J.R.; Writing – Original draft, J.G.V.; Writing – Review and Editing, J.G.V., S.M.N., K.J.C., S.P.G., D.R.W., and J.R.; Funding acquisition, D.R.W., S.P.G. and J.R.
Declaration of interests
Nothing to declare.
References
- Adam AC, Bornhövd C, Prokisch H, Neupert W, Hell K. The Nfs1 interacting protein Isd11 has an essential role in Fe/S cluster biogenesis in mitochondria. EMBO J. 2006;25:174–183. doi: 10.1038/sj.emboj.7600905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angerer H. Eukaryotic LYR Proteins Interact with Mitochondrial Protein Complexes. Biology. 2015;4:133–150. doi: 10.3390/biology4010133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angerer H, Radermacher M, Mańkowska M, Steger M, Zwicker K, Heide H, Wittig I, Brandt U, Zickermann V. The LYR protein subunit NB4M/NDUFA6 of mitochondrial complex I anchors an acyl carrier protein and is essential for catalytic activity. Proc Natl Acad Sci USA. 2014;111:5207–5212. doi: 10.1073/pnas.1322438111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson A, Khalimonchuk O, Smith P, Sabic H, Eide D, Winge DR. Mzm1 Influences a Labile Pool of Mitochondrial Zinc Important for Respiratory Function. J Biol Chem. 2010;285:19450–19459. doi: 10.1074/jbc.M110.109793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson A, Smith P, Fox JL, Cui TZ, Khalimonchuk O, Winge DR. The LYR Protein Mzm1 Functions in the Insertion of the Rieske Fe/S Protein in Yeast Mitochondria. Mol Cell Biol. 2011;31:3988–3996. doi: 10.1128/MCB.05673-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotech. 2006;24:1285–1292. doi: 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
- Boniecki MT, Freibert SA, Mühlenhoff U, Lill R, Cygler M. Structure and functional dynamics of the mitochondrial Fe/S cluster synthesis complex. Nat Comm. 2017;8:1287. doi: 10.1038/s41467-017-01497-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricker DK, Taylor EB, Schell JC, Orsak T, Boutron A, Chen YC, Cox JE, Cardon CM, Van Vranken JG, Dephoure N, Redin C, Boudina S, Gygi SP, Brivet M, Thummel CS, Rutter J. A Mitochondrial Pyruvate Carrier Required for Pyruvate Uptake in Yeast, Drosophila, and Humans. Science. 2012;337:96. doi: 10.1126/science.1218099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody S, Oh C, Hoja U. Mitochondrial acyl carrier protein is involved in lipoic acid synthesis in Saccharomyces cerevisiae. FEBS Lett. 1997;408:217–220. doi: 10.1016/s0014-5793(97)00428-6. [DOI] [PubMed] [Google Scholar]
- Brown A, Rathore S, Kimanius D, Aibara S, Bai X-c, Rorbach J, Amunts A, Ramakrishnan V. Structures of the human mitochondrial ribosome in native states of assembly. Nat Struct Biol. 2017;24:866. doi: 10.1038/nsmb.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cory SA, Van Vranken JG, Brignole EJ, Patra S, Winge DR, Drennan CL, Rutter J, Barondeau DP. Structure of human Fe–S assembly subcomplex reveals unexpected cysteine desulfurase architecture and acyl-ACP–ISD11 interactions. Proc Natl Acad Sci USA. 2017;114:E5325–E5334. doi: 10.1073/pnas.1702849114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui TZ, Smith PM, Fox JL, Khalimonchuk O, Winge DR. Late-Stage Maturation of the Rieske Fe/S Protein: Mzm1 Stabilizes Rip1 but Does Not Facilitate Its Translocation by the AAA ATPase Bcs1. Mol Cell Biol. 2012;32:4400–4409. doi: 10.1128/MCB.00441-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Meth. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- Feng D, Witkowski A, Smith S. Down-regulation of Mitochondrial Acyl Carrier Protein in Mammalian Cells Compromises Protein Lipoylation and Respiratory Complex I and Results in Cell Death. J Biol Chem. 2009;284:11436–11445. doi: 10.1074/jbc.M806991200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedorczuk K, Letts JA, Degliesposti G, Kaszuba K, Skehel M, Sazanov LA. Atomic structure of the entire mammalian mitochondrial complex I. Nature. 2016;538:406–410. doi: 10.1038/nature19794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd Brendan J, Wilkerson Emily M, Veling Mike T, Minogue Catie E, Xia C, Beebe Emily T, Wrobel Russell L, Cho H, Kremer Laura S, Alston Charlotte L, Gromek Katarzyna A, Dolan Brendan K, Ulbrich A, Stefely Jonathan A, Bohl Sarah L, Werner Kelly M, Jochem A, Westphall Michael S, Rensvold Jarred W, Taylor Robert W, Prokisch H, Kim J-JaP, Coon Joshua J, Pagliarini David J. Mitochondrial Protein Interaction Mapping Identifies Regulators of Respiratory Chain Function. Mol Cell. 2016;63:621–632. doi: 10.1016/j.molcel.2016.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezzi D, Goffrini P, Uziel G, Horvath R, Klopstock T, Lochmuller H, D’Adamo P, Gasparini P, Strom TM, Prokisch H, Invernizzi F, Ferrero I, Zeviani M. SDHAF1, encoding a LYR complex-II specific assembly factor, is mutated in SDH-defective infantile leukoencephalopathy. Nat Genet. 2009;41:654–656. doi: 10.1038/ng.378. [DOI] [PubMed] [Google Scholar]
- Ghezzi D, Zeviani M. Assembly Factors of Human Mitochondrial Respiratory Chain Complexes: Physiology and Pathophysiology. Adv Exp Med Biol. 2012;748:65–106. doi: 10.1007/978-1-4614-3573-0_4. [DOI] [PubMed] [Google Scholar]
- Harington A, Schwarz E, Slonimski PP, Herbert CJ. Subcellular relocalization of a long-chain fatty acid CoA ligase by a suppressor mutation alleviates a respiration deficiency in Saccharomyces cerevisiae. EMBO J. 1994;13:5531–5538. doi: 10.1002/j.1460-2075.1994.tb06890.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiltunen JK, Autio KJ, Schonauer MS, Kursu VAS, Dieckmann CL, Kastaniotis AJ. Mitochondrial fatty acid synthesis and respiration. Bioenergetics. 2010;1797:1195–1202. doi: 10.1016/j.bbabio.2010.03.006. [DOI] [PubMed] [Google Scholar]
- Hiltunen JK, Schonauer MS, Autio KJ, Mittelmeier TM, Kastaniotis AJ, Dieckmann CL. Mitochondrial Fatty Acid Synthesis Type II: More than Just Fatty Acids. J Biol Chem. 2009;284:9011–9015. doi: 10.1074/jbc.R800068200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoja U, Marthol S, Hofmann J, Stegner S, Schulz R, Meier S, Greiner E, Schweizer E. HFA1 Encoding an Organelle-specific Acetyl-CoA Carboxylase Controls Mitochondrial Fatty Acid Synthesis in Saccharomyces cerevisiae. J Biol Chem. 2004;279:21779–21786. doi: 10.1074/jbc.M401071200. [DOI] [PubMed] [Google Scholar]
- Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villén J, Haas W, Sowa ME, Gygi SP. A Tissue-Specific Atlas of Mouse Protein Phosphorylation and Expression. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin Edward L, Ting L, Bruckner Raphael J, Gebreab F, Gygi Melanie P, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, Dong R, Guarani V, Vaites Laura P, Ordureau A, Rad R, Erickson Brian K, Wühr M, Chick J, Zhai B, Kolippakkam D, Mintseris J, Obar Robert A, Harris T, Artavanis-Tsakonas S, Sowa Mathew E, De Camilli P, Paulo Joao A, Harper JW, Gygi Steven P. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell. 2015;162:425–440. doi: 10.1016/j.cell.2015.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DR, Knoll LJ, Levin DE, Gordon JI. Saccharomyces cerevisiae contains four fatty acid activation (FAA) genes: an assessment of their role in regulating protein N-myristoylation and cellular lipid metabolism. J Cell Biol. 1994;127:751–762. doi: 10.1083/jcb.127.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastaniotis AJ, Autio KJ, Sormunen RT, Hiltunen JK. Htd2p/Yhr067p is a yeast 3-hydroxyacyl-ACP dehydratase essential for mitochondrial function and morphology. Mol Microbiol. 2004;53:1407–1421. doi: 10.1111/j.1365-2958.2004.04191.x. [DOI] [PubMed] [Google Scholar]
- Knoll LJ, Johnson DR, Gordon JI. Biochemical studies of three Saccharomyces cerevisiae acyl-CoA synthetases, Faa1p, Faa2p, and Faa3p. Biol Chem. 1994;269:16348–16356. [PubMed] [Google Scholar]
- Kursu VAS, Pietikäinen LP, Fontanesi F, Aaltonen MJ, Suomi F, Raghavan Nair R, Schonauer MS, Dieckmann CL, Barrientos A, Hiltunen JK, Kastaniotis AJ. Defects in mitochondrial fatty acid synthesis result in failure of multiple aspects of mitochondrial biogenesis in Saccharomyces cerevisiae. Mol Microbiol. 2013;90:824–840. doi: 10.1111/mmi.12402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre-Legendre L, Vaillier J, Benabdelhak H, Velours J, Slonimski PP, di Rago JP. Identification of a Nuclear Gene (FMC1) Required for the Assembly/Stability of Yeast Mitochondrial F1-ATPase in Heat Stress Conditions. J Biol Chem. 2001;276:6789–6796. doi: 10.1074/jbc.M009557200. [DOI] [PubMed] [Google Scholar]
- Majerus PW, Alberts AW, Vagelos PR. Acyl carrier protein, IV. The identification of 4′-phosphopantetheine as the prosthetic group of the acyl carrier protein. Proc Natl Acad Sci USA. 1965;53:410–417. doi: 10.1073/pnas.53.2.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, Huttlin EL, Haas W, Ting L, Jedrychowski MP, Rogers JC, Kuhn K, Pike I, Grothe RA, Blethrow JD, Gygi SP. Increasing the Multiplexing Capacity of TMTs Using Reporter Ion Isotopologues with Isobaric Masses. Anal Chem. 2012;84:7469–7478. doi: 10.1021/ac301572t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, Nusinow DP, Jedrychowski MP, Wühr M, Huttlin EL, Erickson BK, Rad R, Haas W, Gygi SP. MultiNotch MS3 Enables Accurate, Sensitive, and Multiplexed Detection of Differential Expression across Cancer Cell Line Proteomes. Anal Chem. 2014;86:7150–7158. doi: 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na U, Yu W, Cox JE, Bricker DK, Brockmann K, Rutter J, Thummel CS, Winge DR. The LYR Factors SDHAF1 and SDHAF3 Mediate Maturation of the Iron-Sulfur Subunit of Succinate Dehydrogenase. Cell Metab. 2014;20:253–266. doi: 10.1016/j.cmet.2014.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlandi I, Coppola DP, Vai M. Rewiring yeast acetate metabolism through MPC1 loss of function leads to mitochondrial damage and decreases chronological lifespan. Microbial Cell. 2014;1:393–405. doi: 10.15698/mic2014.12.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulo JA, O’Connell JD, Everley RA, O’Brien J, Gygi MA, Gygi SP. Quantitative mass spectrometry-based multiplexing compares the abundance of 5000 S. cerevisiae proteins across 10 carbon sources. J Proteomics. 2016;148:85–93. doi: 10.1016/j.jprot.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post-Beittenmiller D, Jaworski JG, Ohlrogge JB. In vivo pools of free and acylated acyl carrier proteins in spinach. Evidence for sites of regulation of fatty acid biosynthesis. J Biol Chem. 1991;266:1858–1865. [PubMed] [Google Scholar]
- Sackmann U, Zensen R, Rohlen D, Jahnke U, Weiss H. The acyl-carrier protein in Neurospora crassa mitochondria is a subunit of NADH: ubiquinone reductase (complex I) Eur J Biochem. 1991;200:463–469. doi: 10.1111/j.1432-1033.1991.tb16205.x. [DOI] [PubMed] [Google Scholar]
- Sánchez E, Lobo T, Fox JL, Zeviani M, Winge DR, Fernández-Vizarra E. LYRM7/MZM1L is a UQCRFS1 chaperone involved in the last steps of mitochondrial Complex III assembly in human cells. Biochem Biophys ACTA. 2013;1827:285–293. doi: 10.1016/j.bbabio.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonauer MS, Kastaniotis AJ, Hiltunen JK, Dieckmann CL. Intersection of RNA Processing and the Type II Fatty Acid Synthesis Pathway in Yeast Mitochondria. Mol Cell Biol. 2008;28:6646–6657. doi: 10.1128/MCB.01162-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani DK, Ohlrogge JB. The characterization of a mitochondrial acyl carrier protein isoform isolated from Arabidopsis thaliana. Plant Physiol. 1994;104:1221–1229. doi: 10.1104/pp.104.4.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuible HP, Meier S, Wagner C, Hannappel E, Schweizer E. A Novel Phosphopantetheine: Protein Transferase Activating Yeast Mitochondrial Acyl Carrier Protein. J Biol Chem. 1998;273:22334–22339. doi: 10.1074/jbc.273.35.22334. [DOI] [PubMed] [Google Scholar]
- UniProt-Consortium. UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–D212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vranken JG, Jeong MY, Wei P, Chen YC, Gygi SP, Winge DR, Rutter J. The mitochondrial acyl carrier protein (ACP) coordinates mitochondrial fatty acid synthesis with iron sulfur cluster biogenesis. eLife. 2016;5:e17828. doi: 10.7554/eLife.17828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Lin M, Oliver DJ, Schnable PS. The roles of aldehyde dehydrogenases (ALDHs) in the PDH bypass of Arabidopsis. BMC Biochem. 2009;10:7–7. doi: 10.1186/1471-2091-10-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann N, Urzica E, Guiard B, Müller H, Lohaus C, Meyer HE, Ryan MT, Meisinger C, Mühlenhoff U, Lill R, Pfanner N. Essential role of Isd11 in mitochondrial iron–sulfur cluster synthesis on Isu scaffold proteins. EMBO J. 2006;25:184–195. doi: 10.1038/sj.emboj.7600906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittig I, Braun HP, Schagger H. Blue native PAGE. Nat Prot. 2006;1:418–428. doi: 10.1038/nprot.2006.62. [DOI] [PubMed] [Google Scholar]
- Zhang L, Joshi AK, Hofmann J, Schweizer E, Smith S. Cloning, Expression, and Characterization of the Human Mitochondrial β-Ketoacyl Synthase. J Biol Chem. 2005;280:12422–12429. doi: 10.1074/jbc.M413686200. [DOI] [PubMed] [Google Scholar]
- Zhu J, Vinothkumar KR, Hirst J. Structure of mammalian respiratory complex I. Nature. 2016;536:354. doi: 10.1038/nature19095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.