

Abstract
Background
Analysis of muscle biopsies allowed to characterize the pathophysiological changes of Duchenne and Becker muscular dystrophies (D/BMD) leading to the clinical phenotype. Muscle tissue is often investigated during interventional dose finding studies to show in situ proof of concept and pharmacodynamics effect of the tested drug. Less invasive readouts are needed to objectively monitor patients' health status, muscle quality, and response to treatment. The identification of serum biomarkers correlating with clinical function and able to anticipate functional scales is particularly needed for personalized patient management and to support drug development programs.
Methods
A large‐scale proteomic approach was used to identify serum biomarkers describing pathophysiological changes (e.g. loss of muscle mass), association with clinical function, prediction of disease milestones, association with in vivo 31P magnetic resonance spectroscopy data and dystrophin levels in muscles. Cross‐sectional comparisons were performed to compare DMD patients, BMD patients, and healthy controls. A group of DMD patients was followed up for a median of 4.4 years to allow monitoring of individual disease trajectories based on yearly visits.
Results
Cross‐sectional comparison enabled to identify 10 proteins discriminating between healthy controls, DMD and BMD patients. Several proteins (285) were able to separate DMD from healthy, while 121 proteins differentiated between BMD and DMD; only 13 proteins separated BMD and healthy individuals. The concentration of specific proteins in serum was significantly associated with patients' performance (e.g. BMP6 serum levels and elbow flexion) or dystrophin levels (e.g. TIMP2) in BMD patients. Analysis of longitudinal trajectories allowed to identify 427 proteins affected over time indicating loss of muscle mass, replacement of muscle by adipose tissue, and cardiac involvement. Over‐representation analysis of longitudinal data allowed to highlight proteins that could be used as pharmacodynamic biomarkers for drugs currently in clinical development.
Conclusions
Serum proteomic analysis allowed to not only discriminate among DMD, BMD, and healthy subjects, but it enabled to detect significant associations with clinical function, dystrophin levels, and disease progression.
Keywords: Biomarker, Duchenne muscular dystrophy, Becker muscular dystrophy, Longitudinal analysis, Proteomics, Sarcopenia
Introduction
Duchenne muscular dystrophy (DMD) is a severe muscle‐wasting disease caused by protein truncating mutations in the dystrophin encoding DMD gene.1, 2, 3, 4 The identification of biomarkers for DMD is a priority to the field to identify prognostic factors and to provide objective readouts to reliably evaluate patients' response to drugs in clinical trials.5 Several muscle specific molecular entities have been detected in biofluids such as serum, plasma, and urine showing that it is possible to obtain direct muscle‐related information without an invasive muscle biopsy.6, 7, 8, 9, 10, 11, 12, 13, 14, 15 Muscle‐derived proteins such as muscle creatine kinase (CK) are known to be highly elevated in patients' blood in the early phases of the disease and to decrease over time as muscle mass is lost.15 In several interventional clinical trials, CK levels dropped, with a range of possible plausible explanations such as reduced muscle damage after treatment, but also reduced exercise due to hospitalization or even a decrease in muscle mass. Indeed, other factors that make interpretation of CK levels difficult are day‐to‐day variability, seasonal variability, and exercise‐dependent fluctuations. All these represent obstacles for the use of CK as pharmacodynamic biomarker.
Other muscle‐related proteins such as myosin light chain 3 (MYL3) show profiles comparable to CK and as such probably also make poor pharmacodynamic biomarkers.8, 11 Only a few proteins were reported to be stably elevated in DMD such as MMP‐96 and LEP.16 While these may be attractive biomarkers, little is known about whether serum concentration vary after therapeutic intervention in animal studies or clinical trials.
So far, studies in the literature have focused on the identification of biomarkers able to discriminate between healthy and DMD using different technologies, describing how the identified proteins belong to pathways known to be associated with lack of dystrophin or with the secondary pathology. While these studies provide complementary information about the disease status, longitudinal studies allowing to model individual disease trajectories are lacking. This is particularly important given that clinical differences are observed in patients carrying the same or comparable causative mutation. Published studies often used age as a proxy for disease severity,8 due to the unavailability of longitudinal samples that would enable modelling the intra‐individual variation and that represent a better simulation of evolving pathologic processes. There is only one recent report showing how a restricted number of proteins and steroid hormones can monitor safety and efficacy of glucocorticoids treatment in a longitudinal setup.17
In this study, we assessed protein concentrations in serum samples of DMD patients and healthy controls to identify relevant biological molecules able to discriminate between healthy and disease. We then compared the identified signature with patients affected by the milder allelic form of the disease called Becker muscular dystrophy (BMD). We further examined BMD patients to identify associations between serum proteomic data and clinical data, in vivo 31P phosphorous magnetic resonance spectroscopy (31P MRS) data, muscle derived gene expression data, and dystrophin levels in muscle. Finally, we modelled longitudinal proteomic profiles of a cohort of DMD patients followed up for several years, providing estimates of proteomic changes over time and prediction of disease milestones.
Materials and methods
Patients participants
In the cross‐sectional comparison, 15 DMD patients and 9 healthy age matched controls were included. The median age was 9 years for both groups, while the mean was 9 years for DMD and 10 years for the control group. Metadata are provided in Table S1. A total of 62 BMD patients were studied of whom 33 were followed up at the Leiden University Medical Center (LUMC) and 29 at the John Walton Muscular Dystrophy Centre of the University of Newcastle. Ten healthy adult controls were included. The median age was 39 (age range 19–66) for the Leiden BMDs, 35 (age range 19–67) for the Newcastle BMDs, and 46 (age range 24–64) for the healthy controls. Muscle strength was measured for a subset of BMD patients by using a quantitative muscle assessment (QMA) system (www.qmasystem.com) to assess the maximal voluntary isometric contraction.18 Measurements of the following muscle groups were obtained: handgrip, elbow flexion and extension, and knee and hip flexion and extension. Measurements were performed as described by Hogrel et al.19 For each muscle, the highest value of three consecutive measurements was used as the maximum strength (in kg). If a measurement differed more than 10% from previous measurements, it was discarded, and an extra measurement was performed. Handgrip strength was assessed on the dominant hand. Participants were sitting in an upright position with their arm parallel to their trunk and were encouraged to squeeze the dynamometer of the QMA system as hard as possible. Elbow flexion was also assessed in a supine position with the forearm in a neutral position and the elbow at the side flexed at 90° and the strap attached around the wrist. Hip flexion and extension were assessed in supine position with the hip and knee flexed at 90° and the strap attached proximal to the knee. For knee flexion and knee extension, patients were tested in sitting position, with hip and knee flexed at 90° flexion with the strap attached around their ankle. The highest value of three consecutive measurements was used as the maximum strength. If a measurement differed more than 10% from previous measurements, it was discarded, and an extra measurement was performed. Dystrophin was quantified for 25 patients. 31P MRS data were available for 23 BMD patients. Twenty‐one muscle biopsies were available to perform gene expression analysis.
Fourteen DMD patients were enrolled in a longitudinal study, five of whom were also included in the cross‐sectional study. The median age was 8.6 years (range 5.4–14.4 years of age). Eleven of them were followed‐up at the LUMC, while three were followed‐up at the John Walton Muscular Dystrophy Research Centre in Newcastle. The median follow‐up time was 4.4 years (range 0.4–5.3 years). A total of 56 samples were analysed with an average of four samples per patients. All patients' information is reported in Table S2.
The study has been approved by the Institutional Review Board of the involved clinical centers. Informed consent forms were obtained for all participants. The investigation was conducted according to the declaration of Helsinki.
Serum preparation
Venous blood was allowed to clot for 30 min in red capped tubes followed by centrifugation for 10 min at 2350 g. Serum was carefully removed, aliquoted, and conserved at −80 °C pending use.
Proteomics
The commercially available SOMAscan® multiplex assay consists of individual affinity molecules called SOMAmer® (slow off‐rate modified DNA aptamer) reagents, each with very high affinity to their protein targets.20, 21 In brief, a biological sample in each well of a 96‐well plate is incubated with a mixture of the SOMAmer reagents. After bead‐based immobilization and washing steps, target‐bound SOMAmer reagents were purified and quantified on a custom Agilent hybridization array. For the cross‐sectional analysis, a mix of 1305 somamers was used (performed by the Neurochemistry Lab at VU University Medical Center Amsterdam, The Netherlands), while for the longitudinal study a mix of 4006 somamers was used (performed at Somalogic Inc., Boulder, USA).
Analysis of muscle tissue
Gene expression analysis was performed in muscle biopsies obtained for a subset of BMD patients. Healthy control muscle derived total RNA was purchased from Ambion. To obtain gene expression data, total RNA was purified using Tripure and resuspended in RNase‐free water. cDNA synthesis was performed using the Transcriptor Reverse Transcriptase (cat. n. 03531287001, Roche), random hexamers (cat. n. SO142, ThermoFisher Scientific), RNAsin ribonuclease inhibitor (cat. n. N2515, Promega), and dNTPs (cat. n. 10297018, ThermoFisher Scientific) in a total volume of 20 μL according to the manufacturer's instructions. A selection of genes was performed based on genes known to be differentially expressed in DMD based on available human and mouse data.22 Primer pairs were designed spanning at least one splice junction, and RT‐qPCR was used to quantify gene expression using SYBR green as intercalating dye. Melting curve analysis was performed to filter out primers leading to non‐specific amplification products. Data analysis was performed using the LinReg software to correct for PCR efficiency.23 The fold change over healthy skeletal muscle derived total RNA (cat. n. AM7982, Ambion) was used for downstream analysis. Dystrophin quantification was performed by western blot as previously described.24
31P phosphorous magnetic resonance spectroscopy
In vivo 31P MRS datasets were obtained from the right lower leg using a 7T MR scanner (Philips, Achieva, Best, The Netherlands) for 23 BMD patients as described before.25, 26 Metabolite ratios and tissue pH were calculated for muscles individually and then averaged for all muscles. The following ratios were used: inorganic phosphate over ATP (Pi/ATP) Pi over phosphocreatine (Pi/PCr), phosphodiesters over ATP (PDE/ATP), and PCr over ATP (PCr/ATP).
Statistics and pathway analysis
Analysis of proteomic cross‐sectional data was performed after hybridization control normalization, median signal normalization, and log10 transformation. A linear model with FDR multiple testing correction was used to compare protein levels in DMD patients and controls. The analysis of BMD patients focused on those proteins found to be differentially expressed in DMD. A linear model with FDR multiple testing correction was used to compare BMD patients and healthy controls after correcting for cohort effects. An adjusted P‐value of less than 0.05 was considered significant. Linear models were also used to determine the association of protein concentration in serum with functional scores, 31P MRS, muscle gene expression, and dystrophin amounts in the LUMC cohort after correcting for patients age. Analysis of longitudinal protein profiles was performed after normalization and log10 transformation. To model the longitudinal proteins and identify which show a statistically significant change over time, we used a linear mixed effects model. In the fixed effects part, we have assumed linear evolutions in time, and in the random effects part we have assumed random intercepts and random slopes. For some proteins where convergence problems were observed, we have considered a simpler random effects structure, i.e. a single random intercepts term. We tested if the mean change in time of each protein is statistically significant using the likelihood ratio test. All analyses have been performed in R. Pathway analysis of both cross‐sectional and longitudinal proteomic data was performed using the online tool GeneTrail2 (version 1.5).27 The over‐representation analysis was applied using as reference gene‐set all proteins detected by the SOMAscan assay depleted by the significant ones. Wikipathways was the sole category tested for this analysis.
Results
Cross‐sectional analysis of DMD, BMD, and healthy controls
Proteomic analysis of serum samples by SOMAscan enabled the quantification of 1305 proteins in 15 DMD patients and nine healthy controls. A total of 285 aptamers (directed to a total of 276 unique targets) detected significant changes between DMD patients and healthy controls (Figure 1A and Table S3); protein concentration was reduced in DMD patients for 120 proteins, while 165 proteins were found to be elevated in DMD patients. Overlap analysis with two previously published datasets comparing DMD and healthy individuals using the same technology8 revealed that 34 proteins were able to discriminate between DMD patients and healthy controls in all cohorts (Figure 1B). All 34 proteins except GAPDH showed concordant directional change towards either a consistent increase or decrease in patients compared with healthy controls in the two studies (Table 1), suggesting that 33 proteins are bona fide diagnostic biomarkers for DMD. Two of the identified proteins (APOE and C3) were quantified with three independent aptamers, and another four (F9, FGA‐FGB‐FGG, LYN, and PLG) were detected with two different aptamers. The directional change was consistent for all proteins recognized by multiple SOMAmers indicating a first layer of internal validation for these entities. Examples of down‐regulated and up‐regulated proteins are provided in Figure 1C. Hierarchical clustering of the differentially expressed proteins identified in healthy and DMD individuals followed by correlation analysis showed that many of the differentially expressed proteins are co‐expressed in healthy individuals and that co‐expression is lost in the DMD context (Figure 1D‐E). Pathway analysis identified 44 pathways affected in DMD patients compared with healthy controls encompassing insulin signalling, energy‐related pathways such as glycolysis and gluconeogenesis, Wnt signalling, TGFβ pathway as well as pathways affecting cardiac tissue (e.g. calcium regulation of cardiac cells, microRNAs in cardiomyocyte hypertrophy, and cardiac hypertrophic response) (Table S4).
Figure 1.
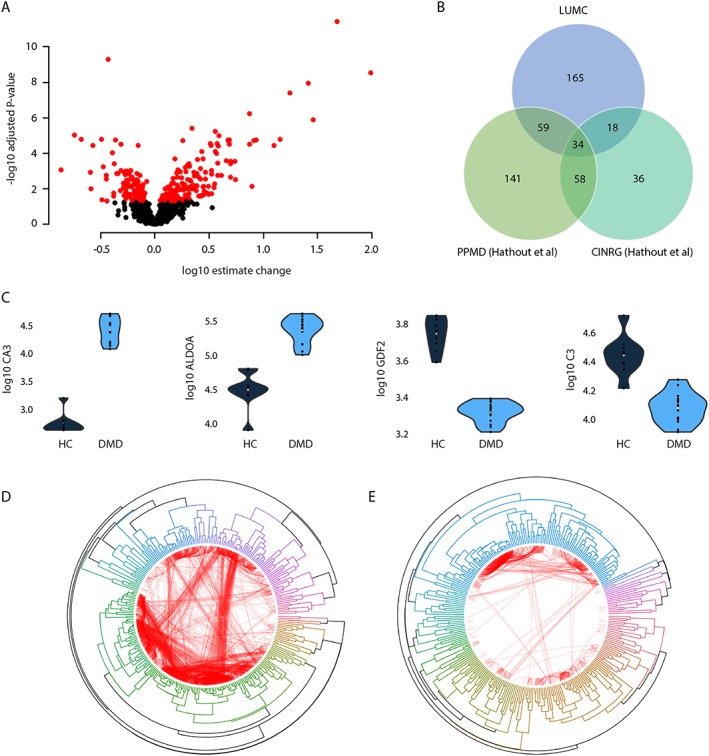
Cross‐sectional analysis of DMD patients. (A) Volcano plot showing the estimated change in DMD patients compared with healthy controls (x‐axis) and the −log10 of the adjusted P‐values. Black circles represent proteins that are not differentially represented in patients compared to healthy controls, while red circles represent the 285 proteins surviving multiple testing correction. (B) Overlap with known proteins able to discriminate between DMD and healthy controls identified using SOMAmers. (C) Violin plots showing four examples of proteins differentially represented between DMD patients and healthy controls. Carbonic anhydrase 3 (CA3) and fructose‐bisphosphate aldolase A (ALDOA) were elevated in patients over controls, while growth differentiation factor 2 (GDF2) and complement component 3 (C3) levels were reduced in DMD patients compared with healthy controls (adjusted P < 0.01 for all). (D–E) Circular plots showing correlation‐based hierarchical clustering of the 285 differentially expressed proteins in healthy individuals (D) and DMD patients (E). Connections shown in red represent correlations between protein levels in each group. Only correlations above 0.8 are shown. The thickness of the lines is proportional to the correlation strength.
Table 1.
List showing the overlap of 32 differentially represented proteins between cases and controls in our cohort and in the previously published cohorts by Hathout and collaborators8 using the Somalogic platform
| Gene ID | Uniprot ID | This dataset | PPMD Hathout et al. | CINRG Hathout et al. |
|---|---|---|---|---|
| CDH5 | P33151 | Down | Down | Down |
| TNFRSF17 | Q02223 | Down | Down | Down |
| CD109 | Q6YHK3 | Down | Down | Down |
| NTRK3 | Q16288 | Down | Down | Down |
| FCER2 | P06734 | Down | Down | Down |
| ACAN | P16112 | Down | Down | Down |
| CADM1 | Q9BY67 | Down | Down | Down |
| ROBO2 | Q9HCK4 | Down | Down | Down |
| RET | P07949 | Down | Down | Down |
| FAP | Q12884 | Down | Down | Down |
| CD86 | P42081 | Down | Down | Down |
| BCAM | P50895 | Down | Down | Down |
| NTRK2 | Q16620 | Down | Down | Down |
| CD55 | P08174 | Down | Down | Down |
| PSPN | O60542 | Up | Up | Up |
| ANP32B | Q92688 | Up | Up | Up |
| TNNI3 | P19429 | Up | Up | Up |
| GPI | P06744 | Up | Up | Up |
| PLA2G2A | P14555 | Up | Up | Up |
| MAPK12 | P53778 | Up | Up | Up |
| FGA FGB FGG | P02671 P02675 P02679 | Up | Up | Up |
| LDHB | P07195 | Up | Up | Up |
| CAMK2A | Q9UQM7 | Up | Up | Up |
| TPT1 | P13693 | Up | Up | Up |
| MB | P02144 | Up | Up | Up |
| FABP3 | P05413 | Up | Up | Up |
| CAMK2D | Q13557 | Up | Up | Up |
| TKT | P29401 | Up | Up | Up |
| CA3 | P07451 | Up | Up | Up |
| CKM | P06732 | Up | Up | Up |
| MDH1 | P40925 | Up | Up | Up |
| GAPDH | P04406 | Up | Up | Down |
| TNNI2 | P48788 | Up | Up | Up |
| GPT | P24298 | Up | Up | Up |
To understand which proteins could be affected by dystrophin presence or dystrophin reintroduction in DMD patients, we considered the 285 proteins affected in DMD patients and studied whether their concentration was affected in two independent cohorts of BMD patients; 13 of these SOMAmer targets were able to discriminate between BMD patients and healthy individuals (Figure 2A). Four proteins were reduced in BMD patients compared with healthy controls, while nine proteins were found to be elevated. The identified proteins were involved in muscle function and contraction. All markers except 2 (PGD and MDK) showed changes in the same direction as for DMD patients, but the amplitude of the change was somewhat lower in BMD patients compared with DMD (e.g. CA3 and MAPK12) (Table 2). The comparison of DMD and BMD individuals highlighted 121 SOMAmer targets as being differentially represented, with 33 proteins being elevated and 88 being reduced in BMD compared with DMD patients (Figure 2B). Overlap analysis showed that 10 proteins were able to discriminate between the three groups (Table 2).
Figure 2.
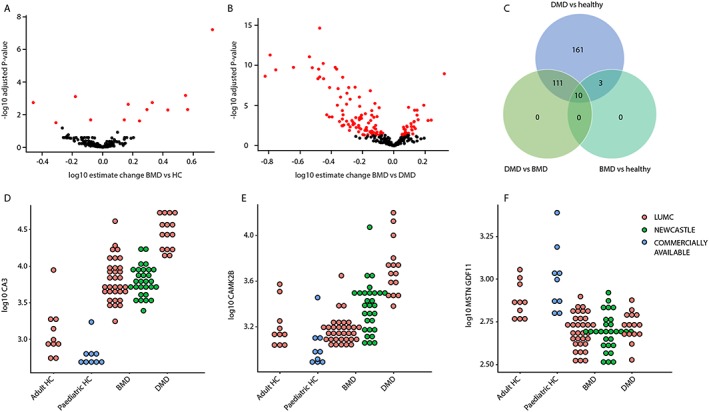
Cross‐sectional analysis of BMD patients. (A) Volcano plot showing the estimated change in BMD patients compared with healthy controls (x‐axis) and the −log10 of the adjusted P‐values. Black circles represent proteins that are not differentially represented in BMD patients compared to healthy controls, while red circles represent the 13 proteins surviving multiple testing correction. (B) Volcano plot showing the estimated change in BMD patients compared with DMD patients. Red circles represent the 121 proteins that were significant after multiple testing correction. (C) Venn diagram showing the overlap between proteins differentially expressed in the DMD, BMD, and healthy. (D–F) Dot plots showing three examples of proteins able to discriminate among DMD, BMD, and healthy (D), or discriminating between DMD and healthy but not between BMD and healthy (E), or able to discriminate between dystrophic and non‐dystrophic but unable to separate DMD and BMD (F).
Table 2.
List of proteins differentially represented in sera of BMD patients compared with healthy controls. The table also shows whether the directional change is in line with the observations in DMD patients
| Uniprot ID | Gene name | Fold change | P‐value | Adjusted P‐value | Same direction as DMD | Different in DMD vs. BMD |
|---|---|---|---|---|---|---|
| P07451 | CA3 | 5.33 | 2.11E − 10 | 6.03E − 08 | Yes | Yes |
| P48788 | TNNI2 | 3.53 | 4.70E − 06 | 6.70E − 04 | Yes | Yes |
| O95390/O14793 | GDF11 MSTN | 0.66 | 8.04E − 06 | 7.64E − 04 | Yes | No |
| P52209 | PGD | 0.35 | 2.68E − 05 | 1.84E − 03 | No | Yes |
| Q00872 | MYBPC1 | 2.13 | 3.22E − 05 | 1.84E − 03 | Yes | Yes |
| P53778 | MAPK12 | 1.48 | 4.81E − 05 | 2.28E − 03 | Yes | Yes |
| P02144 | MB | 1.97 | 1.40E − 04 | 4.98E − 03 | Yes | Yes |
| P12277 P06732 | CKB CKM | 3.65 | 1.40E − 04 | 4.98E − 03 | Yes | Yes |
| P06732 | CKM | 2.70 | 1.63E − 04 | 5.16E − 03 | Yes | Yes |
| P24298 | GPT | 1.39 | 7.45E − 04 | 2.03E − 02 | Yes | Yes |
| P41217 | CD200 | 0.84 | 7.84E − 04 | 2.03E − 02 | Yes | No |
| P04075 | ALDOA | 1.76 | 9.95E − 04 | 2.36E − 02 | Yes | Yes |
| P21741 | MDK | 0.49 | 1.39E − 03 | 3.04E − 02 | No | No |
For the Dutch cohort of BMD patients, 31P MRS data, upper and lower limb functional data, ambulation status, dystrophin levels in muscle, and targeted gene expression data were obtained, allowing to investigate whether associations exist with circulating proteins levels. No significant associations were found between serum protein levels, 31P MRS, and muscle derived gene expression data after multiple testing correction. Serum concentration of four proteins encoded by genes IGF2R, CDNF, RGMA, and BMP6 (corresponding to Uniprot IDs P11717, Q49AH0, Q96B86, and P22004) were associated with upper limb function as measured by elbow flection (Figure 3A‐C). TIMP2 and SERPIND1 (Uniprot IDs P16035 and P05546) were significantly associated with dystrophin levels in muscle quantified by western blot (Figure 3D).
Figure 3.
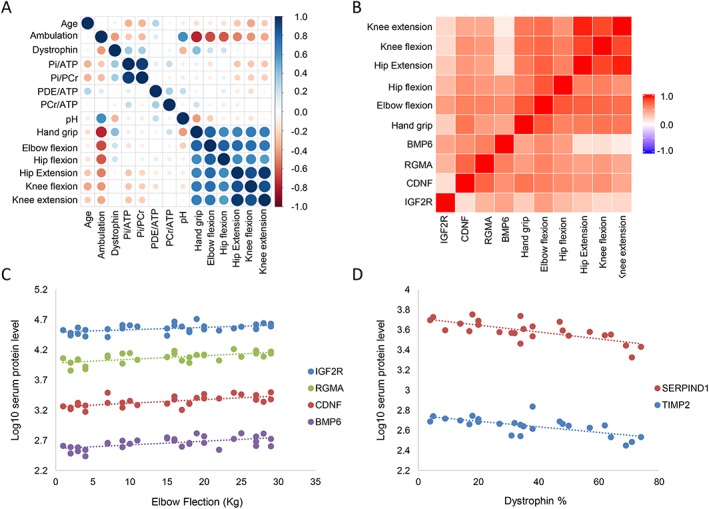
Association of serum proteomic signature with 31P MRS data, functional data, and dystrophin levels. (A) Correlation plot showing the high degree of (Pearson) correlation between the different functional scores and moderate correlation among 31P MRS data with the exception of the Pi/PCr and Pi/ATP ratios. This plot was used to identify potential correlating covariates to exclude during modelling. (B) Heat map showing the Pearson correlation of IGF2R, CDNF, RGMA, and BMP6 with upper and lower limb functional data. (C) Scatter plot showing the linear association between elbow flection and the concentration of these four proteins in serum. (D) Scatter plot showing the association between TIMP2 and SERPIND1 with dystrophin protein levels in muscle.
Longitudinal analysis of Duchenne muscular dystrophy patients
To study whether disease progression is reflected in the biomarker peripheral signature over time, we studied a longitudinal cohort composed of 14 patients followed up for an average of 4 years (Table S2). A total of 4006 proteins were quantified in 56 samples. The vast majority were unchanged (3579 constituting 89% of all quantified proteins), while 427 (427 unique Uniprot IDs) changed over time: 83 (2%) increased, while 344 (9%) decreased over time (Figure 4A). The estimated change per year in log scale per protein was calculated and is presented in Figure 4B. Proteins down‐regulated over time were enriched for muscle specific proteins or proteins involved in muscle function, while proteins up‐regulated over time were mostly proteins involved in fat formation or response to oxidative stress. The proteins with the largest estimated increase over time were leptin (gene name LEP, estimated mean log increase of 0.289 per year), nicotinamide phosphoribosyltransferase (gene name NAMPT), and carnosine dipeptidase 1 (gene name CNDP1); the proteins with largest estimated decrease over time were phospholipase A2 group IIA (gene name PLA2G2A), myosin binding protein C slow type (gene name MYBPC1), and creatine kinase M‐type (gene name CKM). The estimated changes per year for each individual protein are listed in Table S5. Among the 427 proteins affected by time, 66 were differentially represented between cases and controls in the cross‐sectional proteomic study, underlining that most of the proteins changing over time and possibly tracking disease progression are not differentially represented between patients and controls (Figure 4C). Among these 66, only six (SNRPF, N6AMT1, ERP29, CNTFR, KEAP1, and C5) increased over time in DMD patients; four of these (SNRPF, N6AMT1, ERP29, and KEAP1) were elevated compared with healthy controls. The remaining proteins differentially represented between cases and controls did not change over time constituting a stable set of proteins able to discriminate between healthy and disease status.
Figure 4.
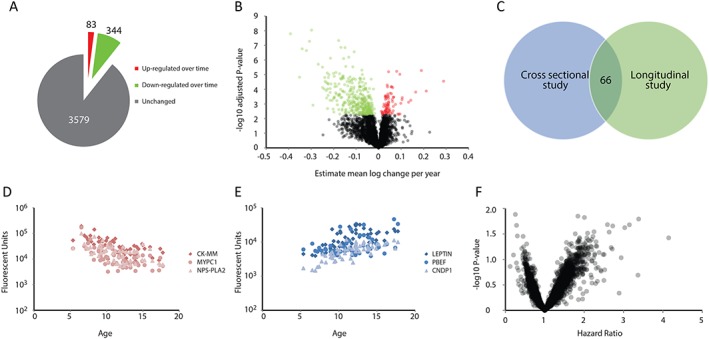
Longitudinal proteomic analysis of DMD patients. (A) Pie chart showing the number of proteins unaffected, increasing, and decreasing over time in DMD patients followed‐up longitudinally. (B) Volcano plot showing the estimated mean log change per year (x axis) for each protein and the −log10 of the P‐value (y axis). Black circles represent proteins stable during disease progression, green and red circles represent proteins with decreasing and increasing concentration over time, respectively. (C) Venn diagram showing the overlap of proteins identified in the cross‐sectional and longitudinal proteomic studies. (D–E) Scatter plots showing exemplar proteins decreasing (D) and increasing (F) over time in DMD patients. Age is plotted on the x axis, while protein concentration is plotted on the y axis. (F) Volcano plot showing the hazard ratios (x axis) and −log10 of the P‐value of the survival analysis modelling the time to loss of ambulation. No protein is significant after multiple testing correction.
Given that all patients were able to walk when the first sample was obtained and wheelchair dependent when the last sample was collected, we investigated whether proteins were associated with an increased risk of wheelchair dependency. None of the 4006 proteins showed a significant association after multiple testing correction; however, 34 proteins had unadjusted P‐value below 0.05, 18 of which with hazard ratio above 2, and four of them with hazard ratio below 0.5 (Figure 4F and Table S6). The most extreme positive hazard ratio was recorded for CPB1 meaning that a unit increase (in a log2 transformed scale) in CPB1 was associated with a 414% increased risk of wheelchair dependency. Over‐representation analysis of the proteins affected over time enabled the identification of 27 pathways affected in DMD patients during disease progression (23 and 4 represented, respectively, by the proteins decreasing and increasing over time) (Table S4). The pathways represented by the proteins reduced over time were typical DMD pathways (e.g. striated muscle contraction, calcium regulation, energy production pathways, and focal adhesion), while adipogenesis, differentiation of white and brown adipocytes, transcriptional regulation of white adipocyte differentiation, and the NRF2 pathway were enriched in subset of proteins increasing over time.
Discussion
DMD is a lethal disease caused by the absence of dystrophin, leading to chronic muscle damage followed up by fibro‐fatty substitution of muscle mass.28, 29 Downstream effects of lack of dystrophin have been characterized by histological, gene expression, and proteomic analyses of muscles biopsies obtained from affected patients (and animal models) enabling the identification of morphological alterations and pathological pathways behind the clinical presentation.30, 31, 32, 33 The examination of muscle biopsies is still often performed during interventional clinical trials to study the effect of the drug (e.g. dystrophin restoration by antisense oligonucleotides or read‐through compounds or levels of fibrosis by anti‐fibrotic compounds). Obtaining muscle biopsies is however an invasive procedure that both patients and investigators would ideally avoid if similar information could be obtained by studying blood derived samples.
In this work, we first studied a large number of proteins in serum to identify blood biomarkers reflecting the pathophysiological changes occurring in striated muscles of patients affected by DMD compared with healthy controls. Our observations show that several of the known pathological pathways known to affect DMD muscles can be monitored peripherally. We confirm many of the already known proteins found to be differentially represented between cases and controls and provides a list of 33 bona fide diagnostic biomarkers able to discriminate between healthy and diseased. We expanded the list with 165 new proteins that were not identified in previous studies. Interestingly, many pathways (e.g. focal adhesion and striated muscle contraction) that have been identified by gene expression studies of muscle biopsies31, 34 were detected and quantified by the analysis of serum samples, providing evidence that muscle biopsies could be avoided when the aim is to monitor these particular pathways.
We then compared the obtained signature with two independent cohorts of BMD patients. A strong normalization towards healthy controls was observed in BMD; only 13 proteins were found to be affected compared with healthy controls, while 121 proteins were differentially represented in BMD compared with DMD. Interestingly, 10 proteins were able to discriminate between the three groups, indicating that a shift towards the levels observed in BMD patients could be observed following dystrophin restoration in DMD patients.
The availability of in vivo 31P MRS data as well as functional data, gene expression data, and dystrophin levels in muscle for the Dutch cohort of BMD patients enabled us to study whether associations exist between these outcomes and serum protein levels. We identified four proteins associated with upper limb function and two proteins associated with dystrophin levels as assessed by Western blot. Three of the four proteins correlating with elbow flection were related to TGF‐β: IGF2R is involved in the control of extracellular levels TGF‐β (even though not specifically for muscle),35 RGMA functions as a bone morphogenetic protein (BMP) co‐receptor enhancing BMP signalling,36 and BMP6 was discovered to attenuate TGF‐β signalling in Dupuytren derived fibroblasts.37 The last protein correlating with elbow flexion was CDNF, a neurotrophic factor acting on dopamine neurons, which has been linked to improved motor function in Parkinson's animal models. The concentration of TIMP2 and SERPIND1 were negatively associated with dystrophin percentage in muscle. TIMP2 has been linked to muscle fibrosis in DMD,38 and the negative correlation with dystrophin might provide information on the capacity of muscle tissue to synthesize dystrophin. It should be mentioned, however, that dystrophin levels were determined only for single muscles and that they may vary between muscle groups. SERPIND1 is heparin cofactor 2, and it was recently found to be elevated in DMD patients' sera8; the negative correlation between dystrophin levels and SERPIND1 could be due to the fact that coagulation abnormalities could lead to microcirculation insufficiency and muscle ischemia further reducing muscle fibre viability and their capacity to produce dystrophin.39
To further explore whether peripheral proteomic profiling could be used to model disease progression, we studied the concentration of 4006 proteins in DMD patients followed up over time for a median of 4.4 years. Importantly, our study is the first to quantify and analyse a large number of proteins in a longitudinal setting, providing a detailed overview of the pathological features that can be monitored peripherally as disease progresses. In total, 427 proteins were significantly affected by disease progression. The estimated changes of each protein per year were computed. Most of the proteins which decreased over time (344) were related to muscle degeneration, heart condition, focal adhesion, and energy production; the 83 proteins which increased over time were mostly connected to fat formation and oxidative stress. The downtrend of muscle related proteins has been widely described in DMD patients,7, 8, 9, 11, 12, 13, 15 and it has been related to disease caused muscle damage/breakdown as well as muscle weakness15, sarcopenia40 and cachexia.41It is not clear to which extent the observed changes are caused by the disease itself or to the inability of the patients to use the affected muscles. There are only a few papers reporting the association of blood derived adipose markers such as leptin with DMD.16, 17, 42 The increase in leptin levels has been associated with fat mass42 and treatment with corticosteroids17 in DMD patients; an increase in leptin has however been reported in steroid naïve DMD patients16 together with other markers of metabolic syndrome such as hypertriglyceridemia, hyperinsulinemia and insulin resistance. While the fat derived signature present in blood is certainly confounded by the treatment with corticosteroids in our study, we did not observe changes characteristic of the metabolic syndrome such as an increase of glucose levels (mean 5.5 standard deviation 1.2 mmol/L of non‐fasting glucose) and glycated haemoglobin (mean 37.7 standard deviation 1.9 mmol/mol Hb). Because biomarker profiling shows some signs of pre‐symptomatic metabolic syndrome, we argue that the substitution of muscle mass by adipose tissue (hallmark of the disease) contributes to the fat derived signature observed in patients' serum with markers such as AGT, CNTFR, LEP, NAMPT, and SOCS3. Prospective studies including both MRI, histological analysis, and serum biomarkers should aim to clarify whether a direct and linear relationship exists between muscle fat fraction and peripheral markers of adipose tissue.
Modelling of longitudinal profiles enabled us to identify proteins that could be used as pharmacodynamic and safety biomarkers in dose finding studies for different types of drugs. Several drugs aim to correct different secondary effects of lack of dystrophin leading to the need of different biomarkers in order to be able to monitor the drug effects. As an example, effects of drugs aiming to increase muscle mass (e.g. IGF‐1 overexpression43, 44) could be monitored by studying AKT protein concentration in serum, which was decreasing over time in the studied population; a second example could be a drug aiming to stabilize calcium homeostasis in muscle fibres and in cardiomyocytes (e.g. Rycalls), the effect of which may be monitored by e.g. gene ATP1B2 45 and Ca2+/calmodulin‐dependent protein kinases (CAMK2A, CAMK2B, and CAMK2D 46) all linked to calcium homeostasis in muscle cells and captured in our assays. Not all therapeutic approaches may however find a direct pharmacodynamic biomarker to be evaluated in serum as dystrophin restoring drugs are likely to still require muscle biopsies to prove correct dystrophin expression and localization in dose optimization trials; however dystrophin restoration should result in an overall normalization of the signal towards healthy controls that could be monitored peripherally and hopefully avoid the need for muscle biopsies in the larger phase III studies.
Our study has some limitations such as the long intervals between samples, the overall longitudinal study duration poorly reflecting the duration of clinical trials, the lack of controls for food intake and corticosteroid use. Another limitation of our study is represented by the impossibility to test whether associations exist between serum biomarkers levels and patients walking performance (e.g. 6‐minute walk test data), cardiac and pulmonary data as well as daily activity data as these data were not available or there were too many missing data points. Despite these limitations, our study is the first study modelling a large number of proteins longitudinally in DMD patients, proving evidence that in‐depth information over the pathophysiologic pathways can be monitored over time using non‐invasive methodologies. We were able to identify significant changes in protein concentrations over time in longitudinal samples despite the low patient numbers thanks to the relatively large effect sizes observed in the DMD population and low intra‐assay variation of SOMAscan data. We believe that this study provides a list of candidate biomarkers to be studied in more detail prospectively in natural history studies, which may help investigators design clinical trials and evaluate patients' progression and response to therapy in dose findings studies.
Conflict of interest
None.
Supporting information
Table S1. Characteristics of the DMD patients involved in the cross‐sectional proteomic study.
Table S2. Characteristics of the DMD patients involved in the longitudinal proteomic study.
Table S3. List of the 285 proteins differentially represented between DMD patients and healthy controls.
Table S4. List of the significant pathways identified by over‐representation analysis in the cross‐sectional proteomic comparison between DMD and healthy individuals and in the longitudinal study.
Table S5. List of proteins affected over time in the longitudinal proteomic comparison. Estimated changes per year and adjusted P‐values are provided.
Table S6. List of proteins identified to be associated with an increased risk of wheelchair dependency. The association was not significant after multiple testing correction.
Acknowledgements
We are grateful to Biomarin for providing us with serum samples of paediatric healthy controls. We acknowledge the support by the European Community's Seventh Framework Programme (FP7/2007–2013) under grant agreements 305121 ‘Integrated European Project on Omics Research of Rare Neuromuscular and Neurodegenerative Diseases (NEUROMICS)’, 305444 ‘RD‐Connect’ and 601573 SCOPE‐DMD. P.S. and A.A.R. would like to acknowledge the Association Française contre les Myopathies (grant n. 17724), ZonMw (grant n. 113302001) and Parent Project Muscular Dystrophy (PPMD) for supporting this work. F.M. is supported by the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London. The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia, and Muscle.47
Spitali, P. , Hettne, K. , Tsonaka, R. , Charrout, M. , van den Bergen, J. , Koeks, Z. , Kan, H. E. , Hooijmans, M. T. , Roos, A. , Straub, V. , Muntoni, F. , Al‐Khalili‐Szigyarto, C. , Koel‐Simmelink, M. J. A. , Teunissen, C. E. , Lochmüller, H. , Niks, E. H. , and Aartsma‐Rus, A. (2018) Tracking disease progression non‐invasively in Duchenne and Becker muscular dystrophies. Journal of Cachexia, Sarcopenia and Muscle, 9: 715–726. 10.1002/jcsm.12304.
References
- 1. Manzur AY, Muntoni F. Diagnosis and new treatments in muscular dystrophies. J Neurol Neurosurg Psychiatry 2009;80:706–714. [DOI] [PubMed] [Google Scholar]
- 2. Ellis JA, Vroom E, Muntoni F. 195th ENMC International Workshop: newborn screening for Duchenne muscular dystrophy 14–16th December, 2012, Naarden, The Netherlands. Neuromuscul Disord 2013;23:682–689. [DOI] [PubMed] [Google Scholar]
- 3. Prins KW, Humston JL, Mehta A, Tate V, Ralston E, Ervasti JM. Dystrophin is a microtubule‐associated protein. J Cell Biol 2009;186:363–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Way M, Pope B, Cross RA, Kendrick‐Jones J, Weeds AG. Expression of the N‐terminal domain of dystrophin in E. coli and demonstration of binding to F‐actin. FEBS Lett 1992;301:243–245. [DOI] [PubMed] [Google Scholar]
- 5. Ferlini A, Flanigan KM, Lochmuller H, Muntoni F, ‘t Hoen PAC, McNally E. 204th ENMC International Workshop on Biomarkers in Duchenne Muscular Dystrophy 24‐26 January 2014, Naarden, The Netherlands. Neuromuscul Disord 2015;25:184–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nadarajah VD, van Putten M, Chaouch A, Garrood P, Straub V, Lochmüller H, et al. Serum matrix metalloproteinase‐9 (MMP‐9) as a biomarker for monitoring disease progression in Duchenne muscular dystrophy (DMD). Neuromuscul Disord 2011;21:569–578. [DOI] [PubMed] [Google Scholar]
- 7. Hathout Y, Marathi RL, Rayavarapu S, Zhang A, Brown KJ, Seol H, et al. Discovery of serum protein biomarkers in the mdx mouse model and cross‐species comparison to Duchenne muscular dystrophy patients. Hum Mol Genet 2014;23:6458–6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hathout Y, Brody E, Clemens PR, Cripe L, DeLisle RK, Furlong P, et al. Large‐scale serum protein biomarker discovery in Duchenne muscular dystrophy. Proc Natl Acad Sci 2015;112:7153–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rouillon J, Poupiot J, Zocevic A, Richard I, Svinartchouk F. Serum proteomic profiling reveals fragments of MYOM3 as potential biomarkers for monitoring the outcome of therapeutic interventions in muscular dystrophies. Hum Mol Genet 2015;24:1–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jeanson‐Leh L, Lameth J, Krimi S, Buisset J, Amor F, Le Guiner C, et al. Serum Profiling Identifies Novel Muscle miRNA and Cardiomyopathy‐Related miRNA Biomarkers in Golden Retriever Muscular Dystrophy Dogs and Duchenne Muscular Dystrophy Patients. Am J Pathol 2014;184:2885–2898. [DOI] [PubMed] [Google Scholar]
- 11. Ayoglu B, Chaouch A, Lochmüller H, Politano L, Bertini E, Spitali P, et al. Affinity proteomics within rare diseases: a BIO‐NMD study for blood biomarkers of muscular dystrophies. EMBO Mol Med 2014;6:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oonk S, Spitali P, Hiller M, Switzar L, Dalebout H, Calissano M, et al. Comparative mass spectrometric and immunoassay‐based proteome analysis in serum of Duchenne muscular dystrophy patients. Proteomics Clin Appl 2015;10:290–299. [DOI] [PubMed] [Google Scholar]
- 13. Cynthia Martin F, Hiller M, Spitali P, Oonk S, Dalebout H, Palmblad M, et al. Fibronectin is a serum biomarker for Duchenne muscular dystrophy. Proteomics ‐ Clin Appl 2014;8:269–278. [DOI] [PubMed] [Google Scholar]
- 14. Coenen‐Stass AML, McClorey G, Manzano R, Betts CA, Blain A, Saleh AF, et al. Identification of novel, therapy‐responsive protein biomarkers in a mouse model of Duchenne muscular dystrophy by aptamer‐based serum proteomics. Sci Rep 2015;5:17014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Burch PM, Pogoryelova O, Goldstein R, Bennett D, Guglieri M, Straub V, et al. Muscle‐derived proteins as serum biomarkers for monitoring disease progression in three forms of muscular dystrophy. J Neuromuscul Dis 2015;2:241–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rodríguez‐Cruz M, Cruz‐Guzmán OR, Escobar RE, López‐Alarcón M. Leptin and metabolic syndrome in patients with Duchenne/Becker muscular dystrophy. Acta Neurol Scand 2016;133:253–260. [DOI] [PubMed] [Google Scholar]
- 17. Hathout Y, Conklin LS, Seol H, Gordish‐dressman H, Brown KJ, Morgenroth LP, et al. Serum pharmacodynamic biomarkers for chronic corticosteroid treatment of children. Sci Rep 2016;6:31727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van den Bergen JC, Wokke BH, Janson AA, van Duinen SG, Hulsker MA, Ginjaar HB, et al. Dystrophin levels and clinical severity in Becker muscular dystrophy patients. J Neurol Neurosurg Psychiatry 2014;85:747–753. [DOI] [PubMed] [Google Scholar]
- 19. Hogrel JY, Payan CA, Ollivier G, Tanant V, Attarian S, Couillandre A, et al. Development of a French isometric strength normative database for adults using quantitative muscle testing. Arch Phys Med Rehabil 2007;88:1289–1297. [DOI] [PubMed] [Google Scholar]
- 20. Rohloff JC, Gelinas AD, Jarvis TC, Ochsner UA, Schneider DJ, Gold L, et al. Nucleic acid ligands with protein‐like side chains: modified aptamers and their use as diagnostic and therapeutic agents. Mol Ther ‐ Nucleic Acids 2014;e201:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gold L, Ayers D, Bertino J, Bock C, Bock A, Brody EN, et al. Aptamer‐based multiplexed proteomic technology for biomarker discovery. PLoS One 2010;5:e15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van Putten M, Kumar D, Hulsker M, Hoogaars WMH, Plomp JJ, van Opstal A, et al. Comparison of skeletal muscle pathology and motor function of dystrophin and utrophin deficient mouse strains. Neuromuscul Disord 2012;22:406–417. [DOI] [PubMed] [Google Scholar]
- 23. Ruijter JM, Ramakers C, Hoogaars WMH, Karlen Y, Bakker O, van den Hoff MJB, et al. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res 2009;37:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Spitali P, Van Den Bergen JC, Verhaart IEC, Wokke B, Janson AAM, Van Den Eijnde R, et al. DMD transcript imbalance determines dystrophin levels. FASEB J 2013;27:4909–4916. [DOI] [PubMed] [Google Scholar]
- 25. Hooijmans MT, Doorenweerd N, Baligand C, Verschuuren JJGM, Ronen I, Niks EH, et al. Spatially localized phosphorous metabolism of skeletal muscle in Duchenne muscular dystrophy patients: 24‐month follow‐up. PLoS One 2017;12:e0182086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hooijmans MT, Niks EH, Burakiewicz J, Verschuuren JJGM, Webb AG, Kan HE. Elevated phosphodiester and T2 levels can be measured in the absence of fat infiltration in Duchenne muscular dystrophy patients. NMR Biomed 2017;30:e3667. [DOI] [PubMed] [Google Scholar]
- 27. Stöckel D, Kehl T, Trampert P, Schneider L, Backes C, Ludwig N, et al. Multi‐omics enrichment analysis using the GeneTrail2 web service. Bioinformatics 2016;32:1502–1508. [DOI] [PubMed] [Google Scholar]
- 28. Janssen B, Voet N, Geurts A, van Engelen B, Heerschap A. Quantitative MRI reveals decelerated fatty infiltration in muscles of active FSHD patients. Neurology 2016;86:1700–1707. [DOI] [PubMed] [Google Scholar]
- 29. Duijnisveld BJ, Henseler JF, Reijnierse M, Fiocco M, Kan HE, Nelissen RGHH. Quantitative Dixon MRI sequences to relate muscle atrophy and fatty degeneration with range of motion and muscle force in brachial plexus injury. Magn Reson Imaging 2017;36:98–104. [DOI] [PubMed] [Google Scholar]
- 30. Kinali M, Arechavala‐Gomeza V, Cirak S, Glover A, Guglieri M, Feng L, et al. Muscle histology vs MRI in Duchenne muscular dystrophy. Neurology 2011;76:346–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Haslett JN, Sanoudou D, Kho AT, Han M, Bennett RR, Kohane IS, et al. Gene expression profiling of Duchenne muscular dystrophy skeletal muscle. Neurogenetics 2003;4:163–171. [DOI] [PubMed] [Google Scholar]
- 32. Doran P, Wilton SD, Fletcher S, Ohlendieck K. Proteomic profiling of antisense‐induced exon skipping reveals reversal of pathobiochemical abnormalities in dystrophic mdx diaphragm. Proteomics 2009;9:671–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pescatori M, Broccolini A, Minetti C, Bertini E, Bruno C, D'amico A, et al. Gene expression profiling in the early phases of DMD: a constant molecular signature characterizes DMD muscle from early postnatal life throughout disease progression. FASEB J 2007;21:1210–1226. [DOI] [PubMed] [Google Scholar]
- 34. Haslett JN, Sanoudou D, Kho AT, Bennett RR, Greenberg SA, Kohane IS, et al. Gene expression comparison of biopsies from Duchenne muscular dystrophy (DMD) and normal skeletal muscle. Proc Natl Acad Sci U S A 2002;99:15000–15005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wylie AA, Pulford DJ, McVie‐Wylie AJ, Waterland RA, Evans HK, Chen Y‐T, et al. Tissue‐specific inactivation of murine M6P/IGF2R. Am J Pathol 2003;162:321–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xia Y, Yu PB, Sidis Y, Beppu H, Bloch KD, Schneyer AL, et al. Repulsive guidance molecule RGMa alters utilization of bone morphogenetic protein (BMP) type II receptors by BMP2 and BMP4. J Biol Chem 2007;282:18129–18140. [DOI] [PubMed] [Google Scholar]
- 37. Krause C, Kloen P, ten Dijke P. Elevated transforming growth factor β and mitogen‐activated protein kinase pathways mediate fibrotic traits of Dupuytren's disease fibroblasts. Fibrogenesis Tissue Repair 2011;4:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mann CJ, Perdiguero E, Kharraz Y, Aguilar S, Pessina P, Serrano AL, et al. Aberrant repair and fibrosis development in skeletal muscle. Skelet Muscle 2011;1:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Saito T. Coagulation and fibrinolysis abnormalities in patients with muscular dystrophy. In: Fibrinolysis and Thrombolysis. InTech; 2014.
- 40. Curcio F, Ferro G, Basile C, Liguori I, Parrella P, Pirozzi F, et al. Biomarkers in sarcopenia: a multifactorial approach. Exp Gerontol 2016;85:1–8. [DOI] [PubMed] [Google Scholar]
- 41. Stephens NA, Skipworth RJE, Gallagher IJ, Greig CA, Guttridge DC, Ross JA, et al. Evaluating potential biomarkers of cachexia and survival in skeletal muscle of upper gastrointestinal cancer patients. J Cachexia Sarcopenia Muscle 2015;6:53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Söderpalm A‐C, Magnusson P, Åhlander A‐C, Karlsson J, Kroksmark A‐K, Tulinius M, et al. Low bone mineral density and decreased bone turnover in Duchenne muscular dystrophy. Neuromuscul Disord 2007;17:919–928. [DOI] [PubMed] [Google Scholar]
- 43. Kang JK, Malerba A, Popplewell L, Foster K, Dickson G. Antisense‐induced myostatin exon skipping leads to muscle hypertrophy in mice following octa‐guanidine morpholino oligomer treatment. Mol Ther 2011;19:159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ridgley JA, Pinniger GJ, Hamer PW, Grounds MD. The physiological effects of IGF‐1 (class 1:Ea transgene) over‐expression on exercise‐induced damage and adaptation in dystrophic muscles of mdx mice. Pflügers Arch ‐ Eur J Physiol 2009;457:1121–1132. [DOI] [PubMed] [Google Scholar]
- 45. Gritz SM, Radcliffe RA. Genetic effects of ATP1A2 in familial hemiplegic migraine type II and animal models. Hum Genomics 2013;7:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Tissue‐based map of the human proteome. Science 2015;347, 1260419–1260419. [DOI] [PubMed] [Google Scholar]
- 47. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Characteristics of the DMD patients involved in the cross‐sectional proteomic study.
Table S2. Characteristics of the DMD patients involved in the longitudinal proteomic study.
Table S3. List of the 285 proteins differentially represented between DMD patients and healthy controls.
Table S4. List of the significant pathways identified by over‐representation analysis in the cross‐sectional proteomic comparison between DMD and healthy individuals and in the longitudinal study.
Table S5. List of proteins affected over time in the longitudinal proteomic comparison. Estimated changes per year and adjusted P‐values are provided.
Table S6. List of proteins identified to be associated with an increased risk of wheelchair dependency. The association was not significant after multiple testing correction.