

Abstract
Background
Skeletal muscle is characterized by an efficient regeneration potential that is often impaired during myopathies. Understanding the molecular players involved in muscle homeostasis and regeneration could help to find new therapies against muscle degenerative disorders. Previous studies revealed that the Ser/Thr kinase p21 protein‐activated kinase 1 (Pak1) was specifically down‐regulated in the atrophying gastrocnemius of Yoshida hepatoma‐bearing rats. In this study, we evaluated the role of group I Paks during cancer‐related atrophy and muscle regeneration.
Methods
We examined Pak1 expression levels in the mouse Tibialis Anterior muscles during cancer cachexia induced by grafting colon adenocarcinoma C26 cells and in vitro by dexamethasone treatment. We investigated whether the overexpression of Pak1 counteracts muscle wasting in C26‐bearing mice and in vitro also during interleukin‐6 (IL6)‐induced or dexamethasone‐induced C2C12 atrophy. Moreover, we analysed the involvement of group I Paks on myogenic differentiation in vivo and in vitro using the group I chemical inhibitor IPA‐3.
Results
We found that Pak1 expression levels are reduced during cancer‐induced cachexia in the Tibialis Anterior muscles of colon adenocarcinoma C26‐bearing mice and in vitro during dexamethasone‐induced myotube atrophy. Electroporation of muscles of C26‐bearing mice with plasmids directing the synthesis of PAK1 preserves fiber size in cachectic muscles by restraining the expression of atrogin‐1 and MuRF1 and possibly by inducing myogenin expression. Consistently, the overexpression of PAK1 reduces the dexamethasone‐induced expression of MuRF1 in myotubes and increases the phospho‐FOXO3/FOXO3 ratio. Interestingly, the ectopic expression of PAK1 counteracts atrophy in vitro by restraining the IL6‐Stat3 signalling pathway measured in luciferase‐based assays and by reducing rates of protein degradation in atrophying myotubes exposed to IL6. On the other hand, we observed that the inhibition of group I Paks has no effect on myotube atrophy in vitro and is associated with impaired muscle regeneration in vivo and in vitro. In fact, we found that mice treated with the group I inhibitor IPA‐3 display a delayed recovery from cardiotoxin‐induced muscle injury. This is consistent with in vitro experiments showing that IPA‐3 impairs myogenin expression and myotube formation in vessel‐associated myogenic progenitors, C2C12 myoblasts, and satellite cells. Finally, we observed that IPA‐3 reduces p38α/β phosphorylation that is required to proceed through various stages of satellite cells differentiation: activation, asymmetric division, and ultimately myotube formation.
Conclusions
Our data provide novel evidence that is consistent with group I Paks playing a central role in the regulation of muscle homeostasis, atrophy and myogenesis.
Keywords: Paks, Muscle, Regeneration, Cachexia, Atrophy
Introduction
Skeletal muscle is one of the most abundant tissues in the human body and it is involved in the generation of voluntary movements and in the control of body metabolism. Overstretching, trauma and excessive training are responsible for acute muscle damage, which is counterbalanced by considerable regenerative potential to preserve muscle function over time.1 After injury, the inflammatory response promotes muscle stem cells activation and differentiation to eventually repair damaged muscle tissue by generating new myofibers.2 Impairment of this regeneration process is responsible for deterioration of quality of life, precedes death, and occurs not only in sarcopenia (i.e. age‐associated muscle wasting) but also in several chronic conditions such as Duchenne Muscular Dystrophy and cancer cachexia.3
Cancer cachexia is a multifactorial syndrome characterized by massive muscle wasting, which cannot be reversed by increased nutrition. Cachexia appears in nearly 50% of patients with advanced stage of cancer and leads to progressive functional impairment, decreased quality of life with respiratory complications, and death can occur following the loss of ~75% of skeletal muscle mass.4, 5, 6 We have recently found that three genes encoding for proteins of the C‐X‐C motif chemokine receptor 4 pathway are down‐regulated in the atrophying gastrocnemius of Yoshida hepatoma‐bearing rats: stromal cell‐derived factor 1, adenylate cyclase 7, and p21 protein‐activated kinase 1 (Pak1).7, 8
Paks are Ser/Thr kinases acting as downstream effectors of the p21 small GTPases Rac1 and Cdc42. The six members of Pak family are divided into two groups depending on their structure and regulation. Paks 1, 2, and 3 belong to group I, while Paks 4, 5, and 6 belong to group II.9 The interaction between the small GTPases and the Paks relies on their GTPase binding domain also called Cdc42‐Rac interactive binding domain.
At the cellular level, Paks functions range from the modulation of cytoskeleton dynamics to the control of cell survival and cell cycle progression as well as transcriptional regulation via the MAPK cascade.10 In muscles, Pak1 is part of a ternary complex with Dishevelled and Muscle‐Specific Kinase that is important for the Acetylcholine Receptor (AChR) clustering during the formation of the neuromuscular junction.11
Cdc42, the GTPase upstream of Pak1, has been shown to be important for p38 activation, a MAPK involved in the activation of satellite cells, the regulation of their asymmetric division and myoblast‐to‐myotube transition.12, 13, 14, 15, 16, 17 During myoblast differentiation, the activation of p38 depends on cell‐to‐cell contact and TNFα stimulation.18, 19 Upon cell‐to‐cell contact and cadherin ligation, the intracellular region of Cell adhesion molecule‐related/down‐regulated by oncogenes (Cdo), a multifunctional cell surface protein, mediates the assembly of a scaffold structure formed by C‐Jun‐amino‐terminal kinase‐interacting protein 4 (JLP) and Bcl2/adenovirus E1B 19 kDa protein‐interacting protein 2 that in turn recruits p38 and the GTPase Cdc42, respectively, in close proximity of the plasma membrane. The switch from Cdc42‐GDP to Cdc42‐GTP results in the activation of p38 and the ensuing signalling cascade regulating myoblast differentiation.20, 21, 22, 23, 24 Once activated, p38α/β (hereafter p38) participates in many key steps of the myoblast differentiation process25 by phosphorylating and activating several myogenic transcription factors, including Mef2a, Mef2c, and Mef2d13, 15, 26 or E47 that heterodimerizes with MyoD to enhance its transcriptional activity.27 Moreover, p38 is important to up‐regulate myogenin and down‐regulate Pax7 expression, a hallmark of satellite cell differentiation,28, 29, 30 as well as to trigger cell‐cycle exit.14, 31
Nonetheless, how the signal is transmitted from Cdc42 to p38 or to other kinases is unknown. Very recently, group I Paks have been implicated in the N‐cadherin/Cdo/Cdc42 signalling pathway and in the control of myogenesis.32 However, this conclusion was reached by using mice that were knocked out for the Pak1 or Pak2 genes, making difficult to discriminate the roles of Paks in muscle during development and in the adulthood. We avoided this by electroporating plasmids expressing PAK1 in muscles of adult post‐puberal mice or by treating them with IPA‐3, a group I Paks inhibitor,33 to transiently modulate the activity of Paks.
Here, we show that Pak1 levels are down‐regulated in two models of muscle wasting: (i) cancer‐related cachexia of colon adenocarcinoma‐bearing mice (C26) in vivo and (ii) dexamethasone‐induced atrophy in vitro. Ectopic expression of Pak1 counteracts muscle atrophy in in vivo and in vitro models. Interestingly, we found that IPA‐3 administered in vivo impairs regeneration of injured muscles, thus confirming the role of group I Paks in this process. Moreover, IPA‐3 treatment in vitro affects myogenin expression and myotube formation of vessel‐associated myogenic progenitors (mesoangioblasts, Mabs), C2C12, and satellite cells, ultimately reducing p38 phosphorylation. Overall, our findings support a role for group I Paks in muscle differentiation and tissue homeostasis.
Materials and methods
Cell cultures
The Mabs cell line was kindly provided by Giulio Cossu's laboratory.34 C2C12 mouse myoblast cell line was purchased from ATCC (American Type Culture Collection, Bethesda, MD, USA) company (CRL‐1772). Mabs or C2C12 were seeded on Falcon dishes at 37°C with 5% CO2 in growth medium (GM), Dulbecco modified Eagle medium (DMEM), supplemented with 10% heat‐inactivated fetal bovine serum (FBS), 100 U/ml penicillin, 100 mg/ml streptomycin, 1 mM sodium pyruvate, and 10 mM HEPES. For some of the experiments shown in Figures 1 and 2, C2C12 were differentiated into myotubes by growing them in DMEM supplemented with 2% Horse Serum at 37°C with 8% CO2.
Figure 1.

Pak1 expression is reduced in TA muscles of cachectic C26‐bearing mice and its ectopic expression preserves myofiber area of cachectic mice by reducing the expression of atrogin‐1 and MuRF1 and possibly by inducing myogenin. (A) Representative western blot revealing total Pak1 in crude protein extracts from TA of colon adenocarcinoma‐bearing mice (C26) compared with controls (PBS). Vinculin is used as a loading control. Twenty microgram of lysates of C2C12 myoblasts previously transfected for 24 h with GFP‐PAK1 expressing plasmids have been used as controls as well as non‐transfected cells. (B) The bar graph illustrates the densitometric quantification of Pak1/vinculin signal ratio for experiments as represented in (A) (n = 4–6, unpaired t ‐test, **P ≤ 0.01) GUSB has been used as housekeeping gene. (C) The mRNA levels of Pak1 in TA from C26‐bearing mice were determined by quantitative polymerase chain reaction (n = 6–10, unpaired t‐test, *P = 0.02). (D) Representative images of cross‐sections of TA from C26‐bearing mice or PBS‐injected ones, previously electroporated in vivo with DsRed2‐PAK1, are shown. Scale bar: 100 μm. (E) Frequency histograms showing the distribution of cross‐sectional areas of muscle fibers of TA either from PBS‐injected mice or C26‐injected ones for 14 days and transfected with empty vector or DsRed2‐PAK1. (F) The mean cross‐sectional area is shown for the four conditions described earlier (n = 219 fibers from five electroporated TA from PBS‐injected mice; n = 339 fibers from seven electroporated TA from C26‐injected mice, unpaired t‐test, ****P ≤ 0.001). Atrogin‐1 (G) and MuRF1 (H) expressions (reported in AU) inversely correlate with transfected human GFP‐PAK1 in muscle from C26‐bearing mice. On the x and y axes, the relative amount of expression of the genes indicated is reported. These numbers have been obtained by comparing the CT of the samples with that of standards of which serial dilutions have been run and probed for the same genes in the same plate. Pearson's test is shown for correlation analysis, n = 6–7. (I) The mRNA levels of myogenin in TA from C26‐bearing mice electroporated with plasmids for GFP or GFP‐PAK1 were determined by quantitative polymerase chain reaction (n = 8–9, unpaired t‐test, *P ≤ 0.05) TBP has been used as housekeeping gene. SEM is indicated in all figures. PAK1, p21 protein‐activated kinase 1; TA, tibialis anterior; AU, arbitrary units; GUSB, β‐glucuronidase; TBP, Tata‐Binding Protein.
Figure 2.
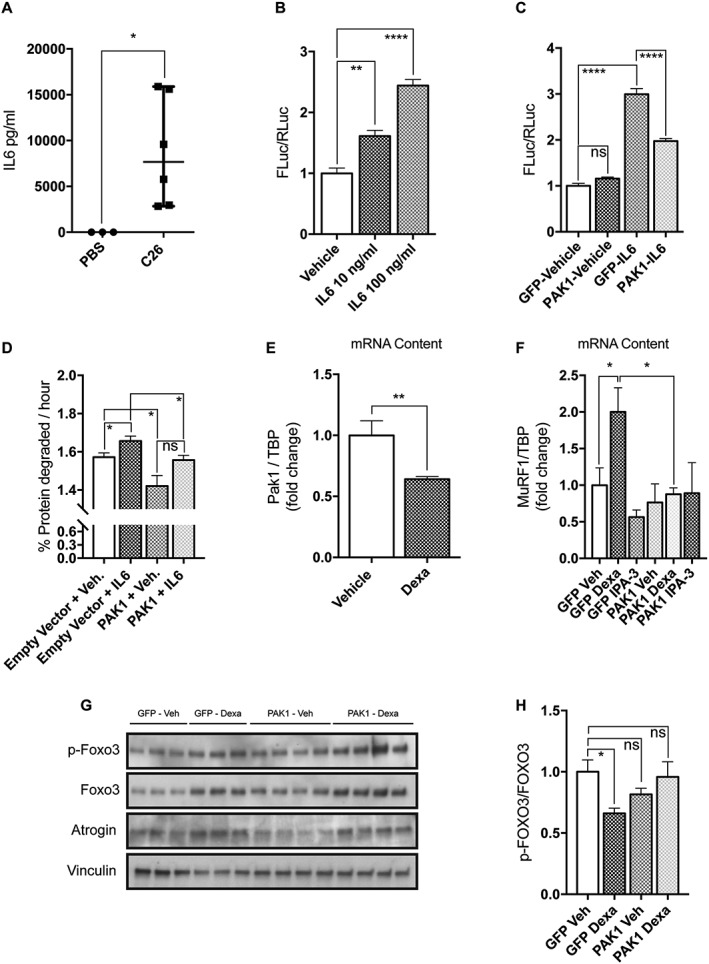
PAK1 overexpression exerts anti‐atrophic effects in two models of in vitro C2C12 atrophy. (A) The circulating levels of IL6 in the plasma of C26‐bearing mice is drastically increased with respect to PBS‐injected mice (n = 3–6, Mann Whitney's test, *P = 0.02). (B) Upon 5 h treatment with 10 or 100 ng/mL murine IL6, C2C12 myoblasts transiently expressing Stat3 4X‐FLuc reporter plasmids induce FLuc (n = 3, one‐way anova followed by Dunnett's test, **P ≤ 0.01, ****P ≤ 0.0001). (C) IL6‐induced Stat3‐FLuc signal is reduced in myoblasts expressing GFP‐PAK1 with respect to those expressing GFP. Myoblasts treated for 5 h with IL6 (100 ng/mL) were previously transfected with Stat3 4X‐FLuc plasmid, Renilla luciferase plasmids, and GFP or PAK1‐expressing plasmids. The results of three independent experiments are shown. Mean is reported (n = 14, unpaired t‐test, ***P = 0.0002). (D) Rates of long‐lived protein degradation were measured in myotubes transfected on the third day of differentiation with plasmids for pDSRed2‐PAK1 or empty vector and differentiated for one more day, when they were exposed for 24 h to 10 ng/mL IL6 (n = 4, unpaired t‐test, *P ≤ 0.05). (E) The mRNA content of Pak1 is reduced in atrophying myotubes exposed for 24 h to 10 μM dexamethasone (n = 6, Mann Whitney's test, **P = 0.002, ****P ≤ 0.0001). (F) Myotubes transfected on the third day of differentiation with plasmids for GFP‐PAK1 or only GFP were exposed for 24 h on the fourth day of differentiation to vehicle or 1 μM dexamethasone or 10 μM IPA‐3. In these conditions, the mRNA levels of MuRF1 were determined by quantitative polymerase chain reaction (n = 4, unpaired t‐test, *P ≤ 0.05). (G) Myoblasts were transfected, as indicated earlier, for 24 h, total protein was then extracted. Immunoblot analysis reveals the protein content of p‐FOXO3, FOXO3, atrogin‐1, and vinculin that is used as loading control. (H) Quantification of the ratio between p‐FOXO3 over total FOXO3 is shown (n = 6–8, unpaired t‐test, *P ≤ 0.05). SEM is indicated in all figures. IL6, interleukin‐6; FLuc, firefly luciferase; PAK1, p21 protein‐activated kinase 1; TBP, Tata‐Binding Protein.
Satellite cells isolation and culture
Hind limb muscles were isolated from C57BL/6 mice. Muscles were then subjected to mechanical dissociation followed by enzymatic digestion for 60 min at 37°C. The enzymatic mix was composed of 2 μg/mL collagenase A (Roche 10103586001), 2,4 U/mL dispase II (Roche 04942078001), and 0,01 mg/mL DNase I (Roche 04716728001) in D‐PBS with Calcium and Magnesium. Enzymatic digestion was stopped by addition of Hank's Balanced Salt Solution (Thermo Fisher Scientific 14025050) and cell suspension was filtered through a 100, 70, 40 μm cell strainer. Red Blood Cells were removed using RBC Lysis Buffer (Santa Cruz sc‐296258) and cell suspension was filtered through 30‐μm cell strainer. Cells were sorted using the MACS separation technology. The antibodies used were CD45 (Miltenyi 130‐052‐301), CD31 (Miltenyi 130‐097‐418), and a7‐integrin (Miltenyi 130‐104‐261). SCs were selected as CD45−/CD31−/a7‐integrin+ cells. The average yield of SCs from hind limbs of a wild type C57BL/6 mouse was 2 * 105 cells.
Prior to starting any treatment, freshly isolated CD45−/CD31−/a7‐integrin+ cells were seeded on matrigel‐coated dishes at 37°C with 5% CO2 in satellite cell growth medium (sGM), DMEM supplemented with 20% heat‐inactivated FBS, 10% horse serum, and 2% chicken embryo extract, 100 U/ml penicillin, 100 mg/ml streptomycin, 1 mM sodium pyruvate, and 10 mM HEPES and cultured for at least 48 h before treatment.
Plasmids and drugs
The GFP or human GFP‐PAK1 expressing plasmids were kind gifts of Prof. Parsons (Kings College London, UK) and described in Parsons et al.35 PDsRed2‐humanPAK1 was kindly donated by Prof. Jonathan Chernoff (Fox‐Chase Cancer Center, Philadelphia, Pennsylvania, United States) and described in Joseph et al.,32 while the empty vector was purchased from Clontech Laboratories Inc. 4XStat3‐Firefly Luciferase (FLuc) plasmids were kindly donated by Prof. Besser (Max‐Delbrück‐Center, Berlin, Germany). PRL‐TK plasmids were purchased from Promega. C2C12 cells were treated with 1 or 10 μM dexamethasone (Sigma‐Aldrich D2915), as indicated in the legend, or mouse interleukin‐6 (IL‐6) at 10 and 100 ng/mL (SPACE Import Export Srl, 406‐ML‐025) dissolved in water or water for controls. Cells were treated with 10 μM IPA‐3 (Sigma‐Aldrich I2285) dissolved in dimethyl sulfoxide (DMSO) or DMSO for controls. The medium, supplemented with IPA‐3 or DMSO, was refreshed every day or every 2 days depending on the experiment. For experiments shown in Figure 2, except for Figure 2B and 2C, treatments lasted 24 h and started 24 h after cell transfection with PAK1‐expressing plasmids.
Luciferase‐based assays
C2C12 myoblasts were transfected with GFP or GFP‐PAK1‐expressing plasmids in combination with 4XStat3‐FLuc and PRL‐TK plasmids using Lipofectamine 2000 (Life Technologies Europe BV), according to manufacturer's instructions and the day after they were treated with vehicle or IL6 for 5 h. Samples were processed using Dual‐Luciferase Reporter Assay System (Promega Italia, Srl) and F‐Luc signal quantitated using a luminometer (LumatLB9507, Berthord, Wild‐bad, Germany).
Protein degradation and synthesis in C2C12 myotubes
C2C12 myotubes were transfected on the third day of differentiation with Empty vector (pDSRed2) or pDSRed2‐PAK1 using Lipofectamine 3000 (Life Technologies Europe BV), according to manufacturer's instructions. The day after, they were incubated with L‐[Ring‐3,5‐3H]‐tyrosine (2 μCi/mL; Perkin‐Elmer) (pulse medium) for 24 h to label long‐lived proteins and then washed three times with chase medium (containing 2 mM unlabelled tyrosine), before collecting aliquots from the medium at specific times. Both pulse and chase media contained the treatment to test (vehicle, IL6, IPA‐3, dexamethasone, or combination of them). Such aliquots were then combined with TCA (10% final concentration) to pellet overall proteins.36 After collecting the last aliquot, cells were resuspended in 2 M NaOH and dissolved in scintillation fluid (Ultima Gold, Perkin‐Elmer) and counted with the Liquid Scintillation Analyzer Tri‐Carb 2800TR (Perkin‐Elmer).
Cell differentiation measurements
The percentage of myogenin positive cells (Myog +) was calculated as the ratio between the myogenin expressing nuclei and the total number of nuclei for each field.
Fusion index (F ind) was determined as the percentage of nuclei included in Myosin Heavy Chain (MyHC)‐expressing myotubes (containing at least three nuclei) vs. the total number of nuclei for each field.37
The percent variance for F ind and for Myog + are defined as
Myotube diameter was evaluated taking three short‐axis measurements at ¼, ½, and ¾ along the length of a given myotube and averaged. More than 30 myotubes per condition were measured and data replicated in at least three independent experiments.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde (PFA) for 10 min at room temperature (RT) and permeabilized in 0,1% Triton X‐100 for 5 min. Cells were then blocked with PBS, 10% FBS and 0,1% Triton X‐100, for 1 h at RT. Incubation with the primary antibody was performed for 1 h at RT, then cells were washed three times and incubated with the secondary antibody for 30 min at RT. The antibodies used were the following: mouse anti‐myogenin (1:250, eBioscience 14‐5643), mouse anti‐MHC (1:2 MF20, DSHB), anti‐mouse secondary antibody Alexa Fluor 555 conjugated (1:100, Life technologies A‐21425), and anti‐mouse secondary antibody Alexa Fluor 488 conjugated (1:100, Life technologies A‐11001). The samples were washed three times and nuclei were counterstained with Hoechst 33342 (Thermo Fisher Scientific, #3570) (1 mg/mL, 5 min at RT). Images were acquired with LEICA fluorescent microscope (DMI6000B).
Cryosections were fixed with 4% of PFA for 10 min, washed three times, and permeabilized with PBS 0,3% Triton X‐100 (30 min at RT). Sections were then saturated with PBS containing 0,1% Triton X‐100 and 10% goat serum (30 min at RT) and incubated with the primary antibody (1 h at RT or O/N at 4°C), washed three times, and incubated with the secondary antibody 30 min at RT. Next, the samples were washed three times and incubated with DAPI (1:500, Molecular Probes D1306) 10 min at RT, washed again and mounted. Primary antibody: rabbit anti‐laminin (1:300, Sigma‐Aldrich L9393), secondary antibody anti‐rabbit Alexa Fluor 488 conjugated (1:250, Life Technologies A‐11008). Images were acquired with Nikon ECLIPSE TE2000‐E fluorescent microscope. The total corrected cellular fluorescence (TCCF) was evaluated using ImageJ software (National Institutes of Health) as integrated density—(area of selected cell * mean fluorescence of background readings).38
EdU‐labelling
Forty‐eight hours upon isolation, SCs were incubated for 24 h with 10 μM EdU (Thermo Fisher #C10337) at 37°C and 5% CO2 in sGM. Cells were then fixed with 4% PFA for 15 min. EdU‐labelled cells were detected using the Click‐iT EdU Imaging Kit according to the manufacturer's instructions. For each experimental group, 125 microscopic fields were automatically analysed using Cell Profiler.
Cross‐sectional area analysis
Cryosections stained with anti‐laminin antibody were analysed using an ImageJ macro, which automatically detects single fibers within the laminin fluorescent signal. Wrongly detected fibers were manually corrected before starting the cross‐sectional area (CSA) measurement. For each sample, the area of more than 2000 single fibers was measured. Using ImageJ, CSAs of transfected and untransfected fibers from the same muscle were measured and compared for a total of 220–340 fibers for each condition and 5–7 electroporated muscles were analysed per condition.
Histological analysis
Evaluation of centrally nucleated fibers (CNFs) fraction has been performed by haematoxylin and eosin staining. Four sections for each sample (n = 2) were randomly selected and pictures of the entire section were acquired with Zeiss Lab A1 AX10 microscope. The resulting images were automatically combined using Photoshop in order to reconstitute the entire section. The images obtained were manually analysed with the ImageJ software to evaluate percentage of the CNFs.
Immunoblotting
After the removal of culture medium, cells were washed in plate with PBS and homogenized in lysis buffer (Millipore cell signalling lysis buffer #43‐040 or RIPA) supplemented with protease inhibitor cocktail 200X (Sigma), inhibitor phosphatase cocktail I and II 100X (Sigma). Samples were then incubated in ice for 30 min with the lysis buffer and cell debris was separated by centrifugation at 14 000 rpm for 30 min at 4°C. Muscle lysates were prepared as indicated in Martinelli et al.7 Protein concentrations were determined by Bradford colorimetric assay (Bio‐Rad) or BCA assay (ThermoFisher). Total protein extracts (15 or 20 μg) were then separated by SDS‐PAGE. Gels were transferred to membranes, saturated with blocking solution (5% milk or BSA and 0,1% Tween‐20 in PBS), and incubated with primary antibodies overnight at 4°C. The antibodies used were as follows: rabbit anti‐Pak1 (1:1000, Cell Signalling 2602), rabbit anti‐Pak1/2/3 (1:1000, Cell Signalling 2604), rabbit anti‐phospho‐PAK1 (Ser144)/PAK2 (Ser141) (1:1000, Cell Signalling 2606), rabbit anti‐phospho‐PAK1 (Thr423)/PAK2 (Thr402) (1:1000, Cell Signalling 2601), rabbit anti‐FoxO3a (1:1000, Cell Signalling 2497), rabbit anti‐phospho‐FoxO3a (Ser253) (1:1000, Cell Signalling 9466), mouse anti‐atrogin1 (1:1000, Abcam ab168372), rabbit anti‐phospho p38 (1:1000, Cell Signalling 9211), rabbit anti‐p38 (1:1000, Cell Signalling 9212), mouse anti‐myogenin (1:500, e‐Bioscience 14‐5643), rabbit anti‐tubulin antibody (1:500, Santa Cruz sc‐9104), rabbit anti‐actin (1:1000, Sigma A2066), mouse anti‐vinculin (1:1000, Abcam ab18058), and mouse anti‐vinculin (1:5000, Sigma, V9264). Following the incubations with primary antibodies, membranes were then washed three times with the washing solution (0,1% Tween‐20 in PBS) and incubated with anti‐mouse or anti‐rabbit secondary antibodies conjugated with horseradish peroxidase (1:2500, Jackson ImmunoResearch) or conjugated with alkaline phosphatases (1:7500, Promega) for 1 h at RT. The blots were further washed three times and visualized with an enhanced chemiluminescent immunoblotting detection system or Tropix CDP‐Star (Life Technologies). Densitometric analysis was performed using ImageJ software. Phosphorylated and total proteins were normalized with tubulin or actin or vinculin. Finally, the ratio between phosphorylated and total protein was indicated.
RNA isolation, reverse transcription, and quantitative real‐time polymerase chain reaction
Total RNA was isolated from the muscles with QIAzol Lysis Reagent (Qiagen, Venlo, Netherlands). RNA concentration, purity, and integrity were measured in a spectrophotometer (NANODROP 1000, Thermo SCIENTIFIC, Waltham, MA, USA). Analysis of mRNA/μg in muscle and in myotubes was performed using TaqMan reverse transcription reagents (Life Tech) or the fluorescent intercalating DNA SYBR Green (Qiagen). β‐Glucuronidase or Tata‐Binding Protein were used as housekeeping genes.
Animal procedures
For adult mouse skeletal muscle electroporation, TA muscles of 8‐week‐old male BALB/c mice were in vivo transfected as in Piccirillo and Goldberg36 with the previously described plasmids, while 1 day later mice were injected with 1 * 106 of C26 cells in 200 μL subcutaneously or with equal volume of PBS. Muscles were dissected 14 days after muscle in vivo transfection and analysed by an individual unaware of the electroporation conditions. Experiments on animals were conducted according to the rules of good animal experimentation I.A.C.U.C. n °432 of 12 March 2006 and under ethical approval released on 11/12/2012 from Italian Ministry of Health, protocol #20/01‐D.
Four 3‐month‐old C57BL/6 mice were treated with one intraperitoneal (IP) injection of 4 mg/kg body weight (BW) of IPA‐3 in PBS or PBS alone [2 days before and immediately following cardiotoxin (CTX) treatment]. For the CTX muscle‐crush injury, mice were anesthetized with an intramuscular injection of physiologic saline (10 ml/kg) containing ketamine (5 mg/mL) and xylazine (1 mg/mL) and then 20 μl of 10 μM CTX isolated from Naja pallida (Latoxan L81‐02) were intramuscularly administered into the tibialis anterior (TA) muscle. Mice were further treated with IP injection of IPA‐3 or PBS 3 and 7 days after CTX injection. PBS and IPA‐3 treated mice were sacrificed 14 days after CTX injury, the TA was collected and snap frozen in OCT for cryosectioning with a Leica cryostat. Experiments on animals were conducted according to the rules of good animal experimentation I.A.C.U.C. n °432 of 12 March 2006 and under ethical approval released on 16/09/2011 from Italian Ministry of Health, protocol #163/2011‐B.
Cytokines
Interleukin‐6 levels in murine plasma were analysed by the Luminex® xMAP technology, exploiting fluorescent‐coded magnetic beads coated with specific antibodies. The tests were run using kits and protocols from Merck Millipore. The sensitivity of the assay was 2,2 pg/mL.
Statistical analysis
Statistical analysis was performed by unpaired Student's t‐test or Mann Whitney's test or One‐way anova followed by Dunnett's test (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001). Results are presented as the means ± SEM. All the experiments were repeated at least twice. P‐values ≤ 0.05 were considered significant. For statistical analyses, GraphPad Prism software (GraphPad, version 7.0) was used.
Results
The p21 protein‐activated kinase 1 expression is reduced in tibialis anterior muscles of cachectic mice and its ectopic expression preserves myofiber cross‐sectional area from C26‐induced cachexia
Because we recently reported that in microarray analysis Pak1 expression was specifically down‐regulated only in the skeletal muscle of Yoshida hepatoma‐bearing rats and not in muscles of rodents undergoing other kinds of muscle wasting (i.e. fasting, uremia, diabetes, disuse),8 we asked whether the Pak1 protein level was also reduced in cachectic muscles of mice. To this end, we induced cachexia in BALB/c mice by injecting colon adenocarcinoma C26 cells that is a highly characterized model of cancer cachexia.39
Mice were injected in their right flank with 1 × 106 C26 cells subcutaneously and food intake and BW were monitored daily as in Martinelli et al.7 In accordance with institutional guidelines, animals were euthanized when at least four out of five signs of distress (loss of mobility, kyphosis, ruffled fur, low body temperature, tremor) were present or when more than 20% of BW was lost in 72 h. We found that the expression of Pak1 is reduced to one fourth at the protein level (Figure 1A and 1B) and to half at the mRNA level (Figure 1C) in TA of C26‐bearing animals, confirming Pak1 down‐regulation in a second model of cancer cachexia. Further, we checked PAK1 expression in TA from immunodeficient mice bearing a human renal carcinoma RXF393 that also results in cachexia40 and confirmed a trend toward reduced levels of mRNAs encoding for PAK1 (Figure S1).
To understand if Pak1 down‐regulation has a functional relevance for the progression of C26‐induced muscle wasting, we electroporated plasmids directing the synthesis of PAK1 and the corresponding empty vector in the TA of mice that were further injected with C26 cells the day after electroporation. Fourteen days later, muscles were dissected and analysed for fiber CSA and the expression of atrogenes, such as atrogin‐1 and MuRF1, 8 was evaluated by quantitative polymerase chain reaction (Q‐PCR). While muscle overexpression of PAK1 has no appreciable effect on fiber CSA of control mice (Figure 1D–F), it preserves fibers from atrophy in C26‐bearing mice, as shown by 20% increase of mean CSA (Figure 1F). We could discriminate exogenous from endogenous Pak1, because the human PAK1 mRNA expressed by the transfected plasmid could be detected by Q‐PCR using specific probes. When we compared the expression levels of exogenous PAK1 electroporated in TA of C26‐carrying mice with atrogin‐1 or MuRF1, we found significant inverse correlations, indicating that the highest PAK1 expression associates with the lowest levels of the ubiquitin ligase transcripts (Figure 1G and 1H). In contrast, the expression of myogenin is significantly induced in muscles from C26‐bearing mice electroporated with the PAK1 plasmid (Figure 1I), while PAX7 and MyoD genes were unaffected by PAK1 overexpression (Figure S2A–C). No change in the expression of atrogin‐1, MuRF1, and myogenin was also observed in PAK1‐overexpressing muscles from PBS‐injected mice (Figure S3).
The p21 protein‐activated kinase 1 overexpression counteracts C2C12 atrophy induced by either interleukin‐6 or dexamethasone
In the attempt to understand if reduced amount of Pak1 may affect the muscle wasting process, we performed further gain‐of‐function experiments in vitro. First, we measured the plasma level of IL6 that is known to be one of the major inflammatory mediators of muscle wasting in cancer‐bearing animals as in cancer patients.41, 42, 43 As expected, we found a significant increase in circulating IL6 only in C26‐bearing mice (Figure 2A). We next treated myoblasts that were previously transfected with a plasmid expressing FLuc under the control of four Stat3 binding sites with two concentrations of IL6, including the one measured in vivo (i.e. 10 ng/mL). A dose‐dependent increase in FLuc‐based signal was found upon IL6 treatment (Figure 2B). Interestingly, PAK1 overexpressed for 48 h restrains the IL6‐induced FLuc signal, indicating that increased Pak1 can counteract the IL6 signalling leading to C2C12 atrophy in vitro (Figure 2C). Indeed, transfected PAK1 is activated as indicated by immunoblot analysis, showing that exogenous GFP‐PAK1 is phosphorylated in threonine 432 (Figure S4). To better understand the mechanism behind the anti‐atrophic effect exerted by PAK1, we measured rates of long‐lived protein degradation in myotubes transfected with plasmids overexpressing PAK1 for 24 h or with the corresponding empty vector. Interestingly, we found that PAK1 expression negatively affects protein degradation rates and restrains IL6‐induced proteolysis (Figure 2D). Indeed, we also found reduced protein degradation rates in cells expressing PAK1 in the absence of IL6 treatment (Figure 2D). This result partially contrasts with the observation that atrogin‐1 and MuRF1 mRNA levels are not affected by PAK1 overexpression in mature myotubes or in murine muscles (Figures 2F and S3). However, we observed a correlation between PAK1 expression and a trend to reduction in the protein content of endogenous p97/VCP (P = 0.06) (Figure S5), a member of the AAA‐ATPase superfamily that we have previously shown to be involved in muscle protein degradation.36
Further, we monitored Pak1 expression levels in an in vitro model of myotube atrophy, which recapitulates some of the features of cancer cachexia.44, 45, 46 Fully mature myotubes were exposed for 24 h with doses of dexamethasone known to reduce myotube diameters and induce the expression of the atrogenes.8, 44 Pak1 expression, measured by Q‐PCR, decreases after cells are treated with 10 μM dexamethasone (Figure 2E), as observed in cachectic muscles in in vivo models (Figure 1C).
Notably, differently from dexamethasone treatment, the inhibition of group I Paks with the specific compound IPA‐3 does not reduce protein synthesis, nor it exacerbates the dexamethasone‐dependent effect on protein synthesis (Figure S6A) or protein degradation (Figure S6B). We conclude that inhibiting the enzymatic activity of Pak1 is not sufficient to recapitulate myotube atrophy at least in vitro. Most interestingly, when we measured mRNA levels of MuRF1 in dexamethasone‐exposed myotubes, previously transfected with plasmids encoding for PAK1 or GFP, we found that exogenous PAK1 is able to restrain dexamethasone‐induced MuRF1 expression, while GFP does not (Figure 2F). Consistently, the ratio between phosphoFOXO3 and FOXO3 is maintained by PAK1 overexpression as in normal not atrophying cells and it does not decrease as in dexamethasone‐exposed myoblasts (Figure 2G and 2H). PAK1 overexpression is also able to restrain partially atrogin‐1 protein induction in dexamethasone‐treated myotubes (Figures 2G and S7).
Overall, these data suggest that activating Pak1 can have beneficial effects on C2C12 cells and muscles undergoing cancer‐induced wasting.
The p21 protein‐activated kinase 1 inhibition delays skeletal muscle regeneration following cardiotoxin injury in vivo
Because IL6 plays a role not only in muscle wasting but also in regeneration following muscle injury47 and because we observed increased myogenin expression in C26‐bearing mice electroporated with a PAK1 expressing plasmid, we wondered whether the inhibition of group I Paks by the in vivo administration of its inhibitor IPA‐3 had any effect on muscle regeneration. Because muscle regeneration is hardly activated in physiological conditions, we treated hind limb muscles of C57BL/6 mice with CTX injections48 to induce muscle injury and to activate the muscle repair machinery. To make sure that the homodimer inactive forms of group I Paks were inhibited at the moment of muscle injury, mice were pre‐treated with IP administration of IPA‐3 (4 mg/kg) or PBS. Mice were further treated with IP injection of IPA‐3 or PBS the same day of CTX injury as well as 3 and 7 days later (Figure 3A).
Figure 3.
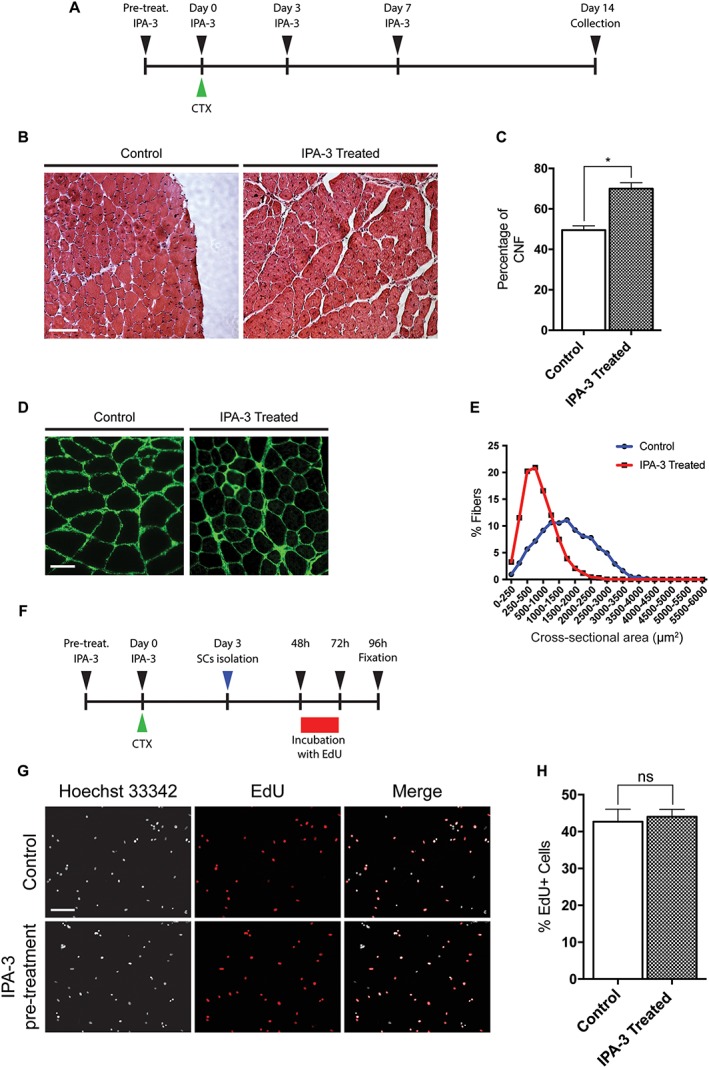
IPA‐3 in vivo treatment delays muscle regeneration. (A) Graphical representation of the IPA‐3 treatments prior and following CTX treatment. (B) Representative images of haematoxylin and eosin histological staining on mouse TA sections 14 days after CTX injection. Scale bar: 100 μm. (C) The bar graph shows the percentage of CNF 14 days after CTX injury in the TA of control and IPA‐3 treated mice (n = 2, unpaired t‐test, *P ≤ 0.05). (D) Immunofluorescence images showing laminin expression in mouse TA sections 14 days after CTX injection. Scale bar: 50 μm. (E) Graphical representation of CSA distribution in TA of IPA‐3 treated and control mice 14 days after CTX injury. The values represent the mean of two muscle samples (n = 2). (F) Schematic representation of the EdU labelling of SC following in vivo IPA‐3 pre‐treatment and CTX injury. (G) Immunofluorescence microphotographs of SCs 24 h upon EdU pulse (green). Scale bar: 100 μm. (H) Bar plot showing the percentage of EdU positive SCs for the experiment reported in G (n = 3, unpaired t‐test). SEM is indicated in all figures. CTX, cardiotoxin; CNF, centrally nucleated fiber; TA, tibialis anterior.
Hind limb muscles were collected 14 days following injury. Histological analysis of TA of mice treated with IPA‐3 reveals altered regeneration process when compared with untreated controls. Haematoxylin and eosin stained TA muscles isolated from IPA‐3‐treated mice display a higher number of CNFs compared with controls (Figure 3B and 3C). Consistently, the CSA of treated muscles is smaller than controls (Figure 3D and 3E). To test whether IPA‐3 pre‐treatment impaired satellite cells proliferation, we isolated SC 3 days after CTX injury from control or IPA‐3 pre‐treated mice. Cells were cultured with sGM (20% FBS, 10% HS, and 2% chicken embryo extract) for 48 h and then incubated for 24 h with EdU (ethynyl‐2′‐deoxyuridine), a thymidine analogue, which incorporates into dividing cells during the S‐phase. As shown in Figure 3F–H, SC isolated from IPA‐3 pre‐treated mice did not show any difference in EdU incorporation in vitro when compared with SC isolated from control mice. Taken together, these results suggest that IPA‐3 treatment slows down muscle regeneration following CTX injury without affecting the proliferation rate of SC, implicating the activity of the specific group I Paks in satellite differentiation in vivo.
The IPA‐3 treatment affects myogenin expression during mesoangioblasts and C2C12 differentiation
Because muscle regeneration in vivo is mainly driven by myoblast proliferation and differentiation, we analysed the effect of IPA‐3 treatment on myoblast differentiation in vitro. To first confirm that IPA‐3 treatment was impairing Pak1 activation, we treated C2C12 in GM with different concentrations of IPA‐3. Among the three members of group I Paks, only Pak1 and Pak2 are expressed in skeletal muscle and among them, Pak1 is the most abundant in both C2C12 and Mabs.49, 50, 51 To test whether IPA‐3 affects Pak1 activity, we plated C2C12 (250 cells/mm2) in GM and after 24 h, cells were treated with different concentrations of IPA‐3. As shown in Figure 4A, phosphorylation (p‐Pak1) on S14452 is reduced by IPA‐3 treatment. Inhibition of activation at IPA‐3 concentrations of 20 and 30 μM is stronger than at 10 μM. However, these concentrations were toxic over long period of culture (data not shown). Thus, in the experiments reported in the succeeding text, we used 10 μM IPA‐3. To analyse the effect of IPA‐3 treatment during myogenic differentiation in vitro, we tested two myogenic cell lines: C2C12 and a line of Mabs.34 Cells were plated at low confluence (250 cells/mm2) and cultured for 9 days in GM. One day after plating the GM was supplemented with IPA‐3 or DMSO as control. The medium was refreshed every 2 days to minimize cell stress during differentiation. As shown in Figure 4B–D, we observed a significant difference in the percentage of myogenin+ cells when IPA‐3‐treated cells were compared with controls in both Mabs and C2C12. Moreover, as shown in Figure 4E and 4F, the percentage of myogenin+ cells was lower in the treated cells than in the control throughout the entire length of the experiment (9 days). We further evaluated myogenin cell content measuring the total corrected fluorescence intensity of differentiating Mabs treated with or without 10 μM of IPA‐3 5 days post plating. As shown in Figure S8, there is a significant difference between control and treated cells within each replicate, suggesting that the reduced percentage of myogenin expressing cells upon IPA‐3 treatment is also accompanied by lower levels of myogenin expression at the single cell level. This result suggests that upon group I Paks inhibition, the expression of myogenin is significantly impaired in differentiating myoblasts.
Figure 4.
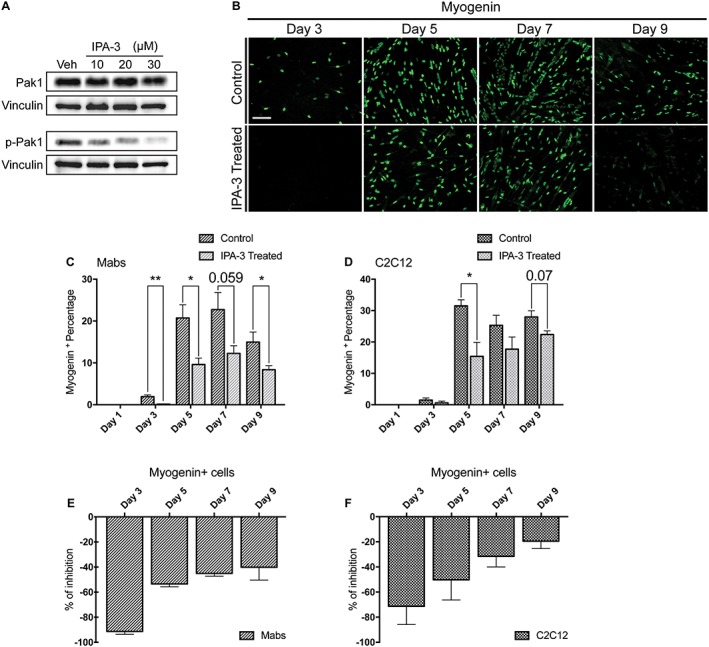
IPA‐3 treatment reduces myogenin expression throughout Mabs and C2C12 myogenic differentiation process. (A) Immunoblot analysis revealing p‐Pak1 (S144) and total Pak1 in C2C12 protein extracts following 2 h treatment with various concentrations of the inhibitor IPA‐3 or vehicle (dimethyl sulfoxide). Pak1 and p‐Pak1 signals have been developed on separate filters and vinculin serves as a loading control. (B) Immunofluorescence microphotographs of Mabs labelled with an antibody against myogenin (green). (C, D) Bar plot representing the percentage of myogenin positive cells for Mabs and C2C12, respectively. (E) The graph illustrates the percentage difference of myogenin positive cells during Mabs myogenic differentiation program. (F) Graph showing the different percentage of myogenin positive cells during C2C12 differentiation. The values represented are means of at least three independent experiments + SEM. Statistical significance has been evaluated using the unpaired t‐test, *P ≤ 0.05, **P ≤ 0.01. Scale bar: 100 μm. PAK1, p21 protein‐activated kinase 1; Mabs, mesoangioblasts.
The p21 protein‐activated kinase 1 inhibition affects myogenic terminal differentiation
Myogenesis is a finely orchestrated process requiring the sequential activation of signalling proteins and transcription factors.53 We asked whether the observed decrease in myogenin expression caused by the inhibition of Pak1 activity could have consequences on the outcome of the differentiation process. To test this possibility, Mabs and C2C12 were plated at low density and cultured for 9 days to induce differentiation following cell contact after growth in GM. One day after plating, the medium was supplemented with IPA‐3 or DMSO and the differentiation efficiency was assessed upon immunostaining with an antibody against the MyHC, a late muscle‐specific differentiation marker. MyHC expression was significantly higher at Day 5 post plating in control cells when compared with the treated ones (Figure 5A). Later on, MyHC‐expressing myotubes appeared also in the IPA‐3‐treated samples albeit at a significantly lower efficiency. The remarkable differentiation inhibition was accompanied by a significant decrease in the fusion index (Figure 5B–E). At Day 9, the difference in fusion index was less striking, but myotubes formed by IPA‐3‐treated cells were thinner than controls (Figure 5F). So, the in vitro inhibition of group I Paks in myogenic cell lines is accompanied by a drastic reduction of the myogenic potential of Mabs and C2C12, possibly as a consequence of the inefficient activation of the p38 MAP kinase.
Figure 5.
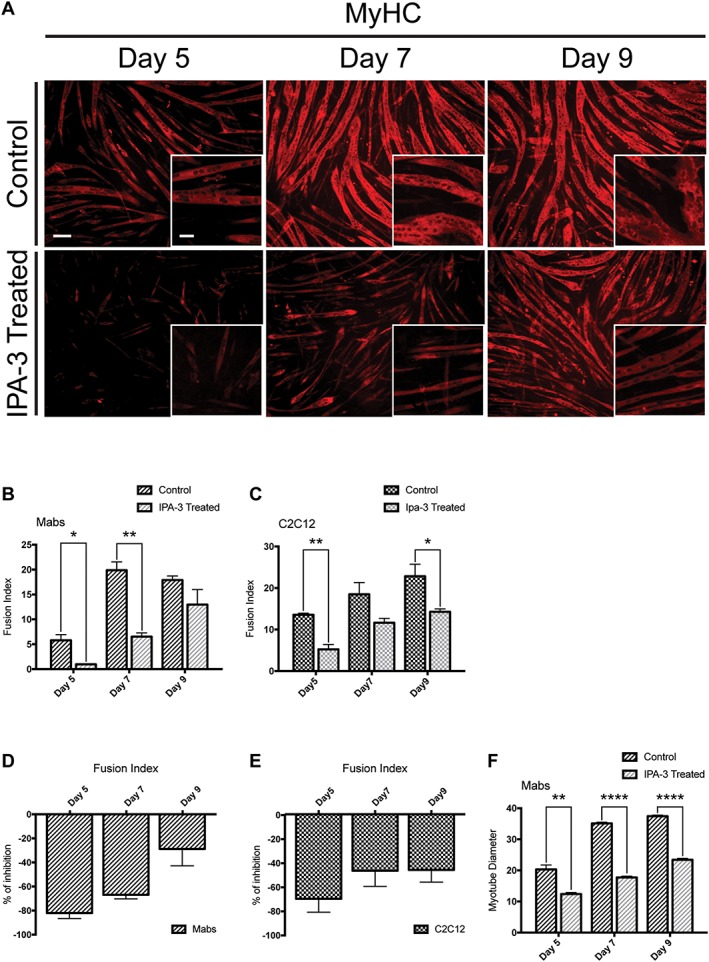
IPA‐3 treatment affects MyHC expression and myotube formation of differentiating myogenic cells. (A) Immunofluorescence images, showing MyHC expression (red) in IPA‐3 treated differentiating Mabs. The insets display a higher magnification (×20). (B, C) Average fusion index for Mabs and C2C12 following IPA‐3 treatment. (D, E) Bar plots representing the percentage of inhibition for the fusion index between IPA‐3 treated and control Mabs or C2C12. (F) Bar plot of Mabs‐derived myotube diameter (μm). The plotted values are means of three independent experiments + SEM. Statistical significance has been evaluated using the unpaired t‐test, *P ≤ 0.05, **P ≤ 0.01, ****P ≤ 0.0001. Scale bar for ×20 magnification: 50 μm. Scale bar for ×10 magnification: 100 μm. MyHC, myosin heavy chain; Mabs, mesoangioblasts.
The IPA‐3 treatment reduces p38 phosphorylation during mesoangioblasts and C2C12 differentiation
Pak1 is a downstream effector of the small G protein Cdc42, which mediates the activation of p38 during myoblast differentiation following cell‐to‐cell contact.21, 22, 23, 24, 54 Given the role of p38 in myoblast differentiation, we asked whether the observed negative effect of Pak1 inhibition on Mabs and C2C12 myogenesis correlated with an impaired activation of p38. To this end, we tested the effect of IPA‐3 on p38 phosphorylation during differentiation of mouse Mabs and C2C12. Mabs and C2C12 were cultured (350 cells/mm2) in GM for 5 days in order to promote cell‐to‐cell contact and myogenic differentiation. Starting from Day 1 post‐plating, the medium was supplemented with IPA‐3 in treated cells or DMSO in controls. Because the stability in solution of dissolved IPA‐3 is not known, the medium was refreshed every 24 h.
In both Mabs and C2C12, Pak1 inhibition resulted in decreased p38 phosphorylation (p‐p38) that was accompanied by lower and delayed myogenin expression (Figure 6A–F), in agreement with p38 involvement in myogenin transcription.30 The reduction of p38 phosphorylation in IPA‐3 is observed already at Day 2 for C2C12 treated cells, while it occurs during Days 3 and 4 in differentiating Mabs. Furthermore, during Mabs differentiation, the expression of myogenin is significantly decreased in treated cells from Day 3.
Figure 6.
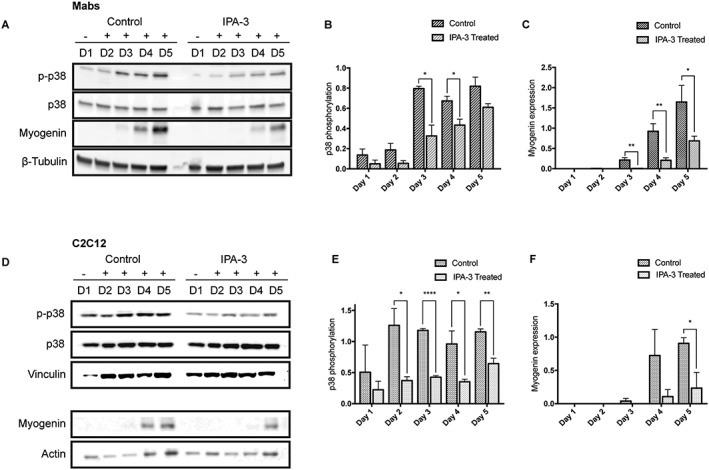
IPA‐3 treatment reduces p38 phosphorylation during Mabs and C2C12 differentiation. Representative western blot revealing total p38, p‐p38, and myogenin expression in crude protein extracts from IPA‐3 treated or control Mabs (A) and C2C12 (D) during 5 days of myogenic differentiation. β‐Tubulin, vinculin, or actin are used as loading controls. The bar graphs illustrate the ratio between p‐p38 and total p38 signals during Mabs (B) or C2C12 (E) differentiation, as determined by the densitometric quantitation of western blots such as the one in panel A and D. The bar graphs represent the densitometric quantitation of myogenin expression for Mabs (C) and C2C12 (F). The values are mean of at least three independent experiments + SEM. Statistical significance was evaluated by the unpaired t‐test, *P ≤ 0.05, **P ≤ 0.01, ****P ≤ 0.0001. Mabs, mesoangioblasts.
These results imply group I Paks in the signalling cascade triggering p38 phosphorylation in myogenic cells. The reduction in p38 activation upon IPA‐3 treatment impairs and delays the myogenic differentiation process, as revealed by reduced myogenin expression.
The p21 protein‐activated kinase 1 inhibition delays cell‐cycle exit without affecting cell proliferation rate
Because Mabs and C2C12 differentiation is strictly dependent on cell‐to‐cell contact, we asked if the reduced p38 phosphorylation and the impaired differentiation upon Pak1 inhibition resulted from an altered proliferation rate in treated cells. A decreased proliferation would delay attainment of confluence and activation of p38. In order to evaluate this possibility, Mabs were cultured for 4 days after plating at low density (250 cells/mm2). IPA‐3 or DMSO was added 24 h after plating and the medium was refreshed every 24 h. Cell number was assessed by counting the nuclei stained with Hoechst 33342 (Figure 7A). In the initial 2 days of treatment, we observed no difference in cell number in control and treated‐cells, suggesting that IPA‐3 does not impair proliferation. However, control cells stopped proliferating at Day 3 post‐plating and they did not increase significantly in number at Day 4, while IPA‐3‐treated cells kept on dividing (Figure 7B). This result suggests a delayed exit from the cell cycle for IPA‐3‐treated cells, consistently, with the reduced activity of p38 and its role in the regulation of cell‐cycle exit during skeletal muscle differentiation.31
Figure 7.
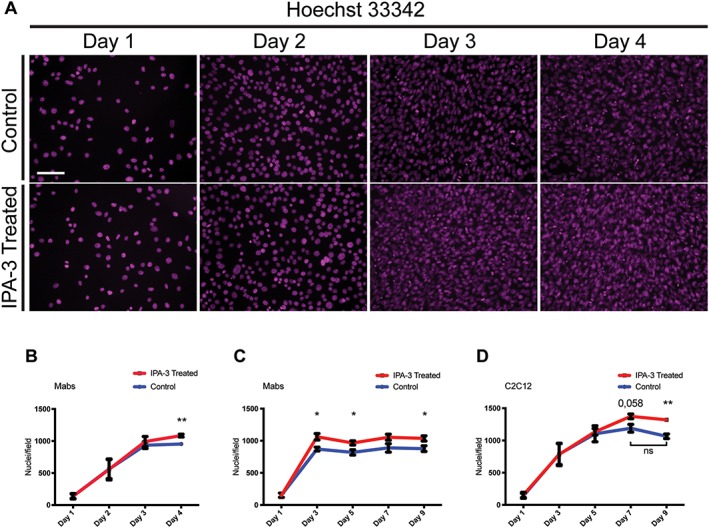
IPA‐3 treatment delays Mabs cell cycle exit without affecting proliferation rate. (A) The panels show Mabs nuclei stained with Hoechst 33342 (magenta) at four time points after plating in growth medium supplemented with DMSO (control) or with IPA‐3. (B) The graph illustrates the number of Mabs nuclei/field during the first 4 days of differentiation. (C, D) The graphs show the number of nuclei/field during 9 days of differentiation for Mabs and C2C12, respectively. Field area is approx. 2,84 * 105 μm2. The values are mean of at least three independent experiments + SEM. Statistical significance has been evaluated using the unpaired t‐test, *P ≤ 0.05, **P ≤ 0.01. Scale bar: 100 μm. Mabs, mesoangioblasts.
To further investigate the effect of Pak1 inhibition on cell proliferation during a longer period of time, Mabs and C2C12 cells were plated at low density and their number was assessed in the following 9 days of culture. IPA‐3 or DMSO were added 24 h after plating and the medium was refreshed every 48 h.
C2C12 and Mabs showed a slightly different response. Mabs treated with IPA‐3 reached a higher cell number compared with control cells during the first 3 days, maintaining this higher density throughout the 9 days of culture (Figure 7C). On the other hand, C2C12 cells showed a slower proliferation compared with Mabs and the number of treated‐cells is similar to the number of control cells until Day 5. However, we observed that while control cells stop their growth at Day 5 post‐plating, IPA‐3 treated cells continue to proliferate reaching a significantly higher number at Day 9 (Figure 7D). This result suggests that the delayed exit from the cell cycle observed for Mabs following group I Paks inhibition occurs also in C2C12 cells.
Group I p21 protein‐activated kinase inhibition affects satellite cell differentiation
Satellite cells are quiescent in physiological conditions and are activated following muscle damage. Once activated, SC re‐enter cell cycle, proliferate, and differentiate contributing to muscle regeneration. During SC activation, p38 acts as a molecular switch and its phosphorylation is detected within 30 min following muscle dissection.16 Because we observed impaired muscle regeneration in vivo following IPA‐3 administration and Pak1 down‐regulation in cachectic muscles that are defective for regeneration, we asked whether IPA‐3 treatment impacted on the differentiation process of satellite cells in vitro.
We isolated SC from hind limb muscles of C57BL/6 mice and plated as ~100 cells/mm2 (3000 cells/well on matrigel‐coated 96‐well plates) in sGM. Two days after plating, cells were treated with IPA‐3 or DMSO as control in sGM and were fixed 3, 4, and 6 days after treatment. To reduce cell stress, the medium was refreshed every 2 days. As expected, the decrease in Pax7 expression is accompanied by an increase in the fraction of cells expressing MyoD. No significant difference between control and IPA‐3 treated cells was observed at various timepoints during this process (Figure S9). In addition, both control and treated cells express myogenin already at Day 3. However, the percentage of cells that are positive for myogenin staining is significantly higher in controls than in treated cells (Figure 8A and 8C). At Days 4 and 6, the percentage of myogenin‐expressing cells remains higher in control cells.
Figure 8.
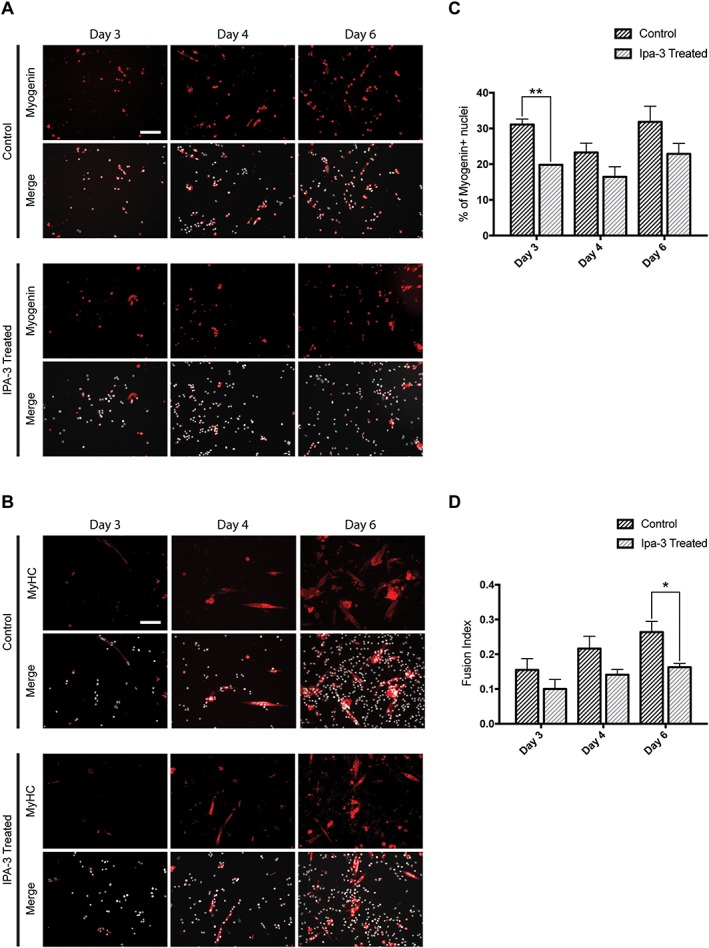
IPA‐3 treatment affects satellite cells differentiation. (A, B) Immunofluorescence showing myogenin (red) and myosin heavy chain (MyHC; red) expression during SC differentiation. Nuclei are represented in grey. (C) Bar plot of the percentage of myogenin positive cells. (D) The bar graph represents fusion index during SC differentiation. The values are mean of three independent experiments + SEM. Statistical significance has been evaluated using the unpaired t‐test, *P ≤ 0.05, **P ≤ 0.01. Scale bar: 100 μm.
The effect of IPA‐3 treatment on SC terminal differentiation was assessed by staining with anti‐MyHC. MyHC expression is higher in control cells than in treated cells (Figure 8B and 8D). These results suggest that IPA‐3 treatment affects the terminal differentiation of satellite cells in vitro.
Discussion
Skeletal muscle homeostasis ensures the maintenance of fiber size through the balance of catabolic and anabolic processes together with regular fiber turnover, involving sporadic fusion of activated muscle stem cells.2 Moreover, the maintenance of a functional muscle architecture following damage is guaranteed by efficient regeneration, which involves a complex cross‐talk between immune cells and several muscle‐resident stem cells. Skeletal muscle function and structure are compromised in muscle myopathies, such as DMD and cancer cachexia, negatively impacting on quality of life.
Pak1 is a Ser/Thr kinase that is involved in cytoskeleton remodelling, cell survival, cell cycle progression, and transcriptional regulation via the MAPK cascade.10 This protein is expressed in skeletal muscle and has been implicated in the regulation of myogenin and AChR expression following denervation of mature fibers.55, 56
Moreover, in skeletal muscle, Pak1 has been associated with the regulation of glucose uptake through insulin‐induced Glut4 translocation56, 57, 58 and in contraction modulation by phosphorylating Myosin Light Chain Kinase.59, 60 In addition, we have recently shown that Pak1 is down‐regulated in atrophying gastrocnemius of Yoshida hepatoma‐bearing rats and others have reported it to be up‐regulated in soleus muscles following Axokine administration.7, 8, 61 All this evidence points to a central role of Pak1 in processes related to muscle physiology. However, we still miss a clear picture of the molecular mechanisms underlying these pleiotropic effects.
Here, we report that the levels of Pak1 protein and mRNA are decreased in the cachectic TA of C26‐bearing mice (Figure 1A–C) and tended to be less in cachectic muscles of RXF393‐carrying mice (Figure S1). Moreover, CSA analysis of TA from C26‐bearing mice previously electroporated with DsRed2‐PAK1 or DsRed2 expressing plasmids suggested that overexpressed Pak1 preserves fiber area during cancer‐induced muscle wasting (Figure 1D–F). Skeletal muscle atrophy is an established adverse effect caused by treatment with glucocorticoids.62 Consistently, we also observed reduced mRNA levels of Pak1 in atrophying myotubes following dexamethasone exposure in vitro (Figure 2E). Dexamethasone treatment causes myotube atrophy, as seen by reduced protein synthesis (Figure S6A), while at the same time induces a down‐regulation of Pak1 expression and MuRF1 induction (Figure 2E and 2F). Interestingly, PAK1 overexpression is able to restrain dexamethasone‐induced MuRF1 expression (Figure 2F) and maintains FOXO3 inactive in the cytosol, as seen by increased ratio of phospho‐FOXO3/over total (Figure 2G and 2H). However, treatment with the group I Paks inhibitor IPA‐3 is not sufficient to induce atrophy, which may suggest a new activity‐independent function for Pak1 or the requirements of additional players to induce atrophy (Figures 2F, S6A, and S6AB).
We confirmed that in C26‐bearing mice, the circulating levels of IL6 in the plasma are drastically increased compared with control mice and that a reporter plasmid expressing the FLuc under the control of a Stat3‐responsive promoter is activated in C2C12 cells upon exposure to either 10 or 100 ng/mL IL6 (Figure 2A and 2B).
It has been shown that Stat3‐based signalling is enhanced in skeletal muscle of C26‐bearing mice and it is both necessary and sufficient to induce muscle wasting during cancer.40, 63 After overexpressing PAK1 in myoblasts, IL6‐induced Stat3‐FLuc signal is significantly reduced compared with controls (Figure 2C) and IL6‐induced proteolysis is restrained (Figure 2D).
Taken together, these results support Pak1 as beneficial to counteract either the IL6/Stat3‐based signalling leading to atrophy in vitro or the decreased fiber areas of muscles undergoing cancer cachexia in vivo. The Pak1 substrates that mediate these anti‐atrophic effects remain to be established. However, based on our results (Figures 2F–H and S7), FOXO3 comes out as an interesting candidate worth further testing.
Because muscle regeneration is impaired in cancer cachexia,3 we also asked if Pak1 plays a role in skeletal muscle regeneration after CTX insult. We observed that the muscle fibers of IPA‐3‐treated mice display a reduced regeneration after CTX injury, as shown by the persistence of a higher fraction of CNFs and a smaller CSA compared with controls (Figure 3A–E). SC are directly responsible for fibers repair in vivo, and upon muscle damage, they exit from quiescence and rapidly proliferate for 5 days post‐injury when they start to differentiate and fuse to damaged fibers.2, 64 We asked whether the slower regeneration rate observed in muscles of IPA‐3 treated mice was caused by an impairment of SC proliferation following muscle injury. However, we observed that IPA‐3 pre‐treatment does not affect SC proliferation upon CTX injury (Figure 3F–H). Collectively, these data suggest that Pak1 is required for muscle regeneration in vivo. This conclusion is consistent with what was very recently reported by Joseph et al. using mice knocked out for the Pak genes.32
Myogenesis in vivo is driven by the activation of satellite cells, which in turn differentiate into proliferating myoblasts committed toward terminal differentiation. To assess if the delayed muscle regeneration that is observed following IPA‐3 treatment was related to an impairment of myoblast differentiation, we analysed myogenin expression during Mabs and C2C12 differentiation in vitro. Myogenin is a member of a family of four related basic helix–loop–helix transcription factors: MyoD, Myf5, myogenin, and Mrf465 and their coordinated expression is necessary for cell commitment and completion of myogenesis.26, 53, 66 The higher myogenin expression in control cells throughout the differentiation process compared with the treated ones suggests that the expression of myogenin is impaired following IPA‐3 treatment (Figure 4).28, 30 Consistently, the beneficial effects of PAK1 in protecting muscle fiber size of adult mice undergoing cancer cachexia may be mediated by PAK1‐induced myogenin expression (Figure 1I), but further experiments are needed to explore this in depth. Myogenesis is achieved through the sequential expression in a temporally orchestrated fashion of specific subsets of genes, allowing the proper completion of the process.53 Thus, we asked whether the reduction of myogenin expression following chemical inhibition of group I Paks would result in the impairment of terminal myogenic differentiation. To this end, we analysed the expression of the late myogenic marker MyHC. Both Mabs and C2C12 show delayed MyHC expression in treated cells compared with controls. Furthermore, myoblasts treated with IPA‐3 during differentiation were characterized by a lower fusion index and diameter, indicating that the entire myogenic program is compromised following group I Paks inhibition (Figure 5A–F).
The p38 MAP kinase plays many regulatory roles at several steps during the regeneration process of adult muscle. Both TNFα and cell–cell contact can activate p38 during myoblast differentiation.20, 21, 22, 23, 24 When myoblasts reach a high density and make contacts, the surface protein Cdo recruits the JLP scaffold protein that in turn promotes the co‐localization of p38 with other proteins, such as Cdc42 that causes its activation and myogenic differentiation. In this scenario, however, the molecular link between the activation of the Cdc42 switch and the phosphorylation of T180 and Y182 in the activation loop of p38 is not clear.
However, this cascade should not be considered as exclusive for p38 activation because Cdo−/− myoblasts, despite evident defects in completing differentiation, are still able to activate p38 and express the myogenic marker MyHC, albeit less efficiently.15, 21, 22 It is noteworthy that the exogenous expression of the Rho‐specific GEF Geft induces myogenesis of C2C12 cells via activation of RhoA, Rac1, and Cdc42 and its downstream effectors including Pak1.54 Pak1 acts downstream of Cdc42 and its involvement in the activation of p38, following IL‐1 stimulation or after cell‐to‐cell contact disruption in the epithelial‐to‐mesenchymal transition, has been reported.67, 68 Thus, we speculated that group I Paks could also be involved in linking Cdc42 and p38 activation during myogenesis as it has also been recently reported by Joseph and coworkers.32
Our results demonstrate that group I Paks inhibition correlates with a decreased p38 phosphorylation during Mabs and C2C12 differentiation in vitro (Figure 6), consistent with group I Paks playing a role in the myogenic cascade activating p38. The observed down‐regulation of p38 activation in the treated cells is not complete, probably due to incomplete group I Paks inhibition or to redundant p38 activation mechanisms, such as the one induced by TNFα.18, 19 We ruled out the possibility that IPA‐3 treatment affects p38 activation by slowing down cell proliferation and by delaying cell contact. Results in Figure 7 rather support the conclusion that the delay in myogenic differentiation is a consequence of the negative modulation of p38 activation following group I Paks inhibition. In fact, IPA‐3 does not affect cell proliferation, but treated Mabs show a delayed exit from the cell cycle, consistent with the involvement of p38 activation in cell cycle exit.14, 31 The activation of p38 and cell cycle exit are both necessary to induce myogenic differentiation.
Muscle regeneration in vivo is mainly driven by SC, quiescent muscle stem cells located between the muscle fibers and the basal lamina. We observed that group I Paks inhibition affects satellite cells differentiation as revealed by the decrease in myogenin expression and fusion index for treated cells compared with controls (Figure 8A–D). However, Pak1 is poorly expressed in quiescent satellite cells, while its expression increases more than five folds after 4 days of culture.69 Because p38 phosphorylation occurs within 30 min upon SC isolation, Pak1 is probably not involved in this process and the effect of IPA‐3 treatment are probably related to other functions of Pak1, such as cytoskeleton remodelling or the regulation of Glut4 translocation or to Pak2 inhibition.
Taken together, our data add novel evidence to Pak1 playing a central role in the regulation of muscle homeostasis atrophy and myogenesis. In addition to the already described functions in the modulation of cytoskeleton remodelling, glucose uptake, myogenin, and AChR expression following denervation of mature fibers, we report here that Pak1 is down‐regulated in muscle during cancer cachexia or in dexamethasone‐induced atrophy. Its ectopic expression, on the other hand, counteracts muscle wasting associated with cancer cachexia or IL6 or dexamethasone treatment in vitro. Moreover, group I Paks positively modulates muscle regeneration, probably by acting between Cdc42 and the MAPK cascade, leading to p38 phosphorylation during myoblast differentiation. Chemical inhibition of group I Paks resulted in diminished p38 activation following cell‐to‐cell contact, delayed exit from the cell cycle, and a reduced efficiency in myogenic differentiation in vitro accompanied by impaired regeneration in vivo.
More than 35 proteins whose activities are modulated via direct phosphorylation by Pak1 have been reported.70 Which of these substrates play a functional role in the phosphorylation network that modulates the mechanisms underlying its pleiotropic functions in the muscle remains to be established.
Conflict of interest
A.C.P., A.D.R.C., M.C., G.B.M., C.F., G.D., L.C., C.G., R.P., and G.C. declare that they have no conflict of interest.
Supporting information
Fig S1 The mRNA levels of Pak1 tends to be decreased in TA from RXF393‐bearing mice. PAK1 expression was determined by quantitative PCR (n = 5). Gusb was used as housekeeping gene. SEM is reported. Unpaired T test, ns, p = 0.1528. Mice were sacrificed 18 days after tumor inoculation. Consider 72 as reference for RXF393 in vivo inoculation.
Fig. S2 PAK1 overexpression in Tibialis Anterior of C26‐bearing mice does not alter the expression of PAX7 or MyoD. Human PAK1‐GFP or GFP expressing plasmids were electroporated into the Tibialis Anterior of mice that had been injected with C26 tumor cells the day after electroporation. The expression of PAX7 (A), MyoD (B), exogenous (human) PAK1 (C) and a housekeeping gene (TBP) were measured by Q‐PCR on muscle dissected 14 days after electroporation (n = 6–9, unpaired T‐Test, ***p = 0.0004). SEM is reported.
Fig S3 The expression of atrogin‐1, MuRF1 and myogenin is not affected by PAK1 overexpression in muscles of PBS‐injected mice. The mRNA levels of atrogin‐1 (A), MuRF1 (B) and myogenin (C) in TA from PBS‐injected mice electroporated for 14 days with empty vector or PAK1 were determined by quantitative PCR (n = 7). IPO8 or TBP were used as housekeeping genes. Unpaired T test, ns.
Fig. S4 Phosphorylation of ectopically expressed PAK1 occurs in transfected myoblasts. Immunoblot analysis revealing p‐PAK1 (T423) and total PAK1 in C2C12 protein extracts. Cells extracts were analyzed 24 h after transfection of plasmids expressing GFP‐human PAK1 or a corresponding empty vector. PAK1 and p‐PAK1 signals have been developed on separate membranes. Vinculin served as a loading control and has been blotted on both membranes.
Fig S5 The endogenous p97/VCP content tends to be decreased in C2C12 by PAK1 overexpression. (A) Myoblasts were transfected with empty vector or Pak1 expressing vectors for 24 h and differentiated into myotubes, total protein was then extracted. Immunoblot analysis reveals the protein content of p97 and vinculin, that is used as loading control. (B) The bar graph illustrates the densitometric quantification of p97/vinculin signal ratio for experiments as represented in A (n = 6, 5) Unpaired T test, p = 0,06.
Fig. S6 Pak1 inhibition by IPA‐3 does not affect protein synthesis or protein degradation of normal or dexamethasone‐treated atrophying myotubes. Overall protein synthesis measured as total CPM (counts per minute) per well (A) and percentage of protein degraded per hour (B) of 4‐days differentiated myotubes treated for 30 h with vehicle (DMSO), 1μM dexamethasone, 10μM IPA‐3 or both, are plotted (n = 4, Unpaired T‐Test, *p < 0.05, **p < 0.01, ****p < 0.0001). Total protein synthesis was calculated by incorporation of L‐[Ring‐3,5‐3H]‐tyrosine into cell proteins, 73 following the procedure described in Piccirillo and Goldberg.36
Fig. S7 PAK1‐overexpressing myoblasts express lower levels of atrogin‐1 protein after dexamethasone treatment. Densitometric analysis of the blot from the main manuscript (Figure 2G) shows quantification of the protein content of atrogin‐1 in myoblasts transfected with plasmid for GFP or PAK1 and treated for 24 h with 1μM dexamethasone (Dexa) or vehicle. Vinculin was used as loading control (n = 6–8, Unpaired T‐Test, *p < 0.05, ***p < 0.001).
Fig. S8 IPA‐3 treated Mabs display a lower myogenin expression compared to control. Protein content for each cell was evaluated as total corrected fluorescence intensity for myogenin positive nuclei. The graph shows the difference of myogenin expression between control and IPA‐3 treated cells for each biological replicate. Values are represented as box plots with 2.5–97.5% confidence intervals. The total number of cells counted for each group is 100. Statistical significance has been evaluated using the Unpaired t‐test, *p ≤ 0.05, ***p = 0.001.
Fig. S9 IPA‐3 treatment does not affect Satellite cells expression of Pax7 and MyoD. Immunofluorescence showing Pax7 (green) (A, B) and MyoD (red) (D, E) expression during SC differentiation for control and IPA‐3 treated cells. Nuclei are represented in grey. The plot shows the percentage of Pax7 positive cells (C) or MyoD positive cells (F) for the experiments reported in A, B and D, E. The values are mean of three independent experiments + SEM. Statistical significance has been evaluated using the Unpaired t‐test. Scale bar: 100 μm.
Acknowledgements
We are grateful to the Italian Association for Cancer Research (AIRC grant 14135 to G.C. and AIRC Start‐UP 11423 to R.P.), to the European Research Council (grant N 322749 to G.C.), and the Union under the Marie Curie International Reintegration Grant (PIRG08‐GA‐2010‐277008 to R.P.). The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia, and Muscle.71 All authors declare that the submitted work has not been published before (neither in English nor in any other language) and that the work is not under consideration for publication elsewhere.
Cerquone Perpetuini, A. , Re Cecconi, A. D. , Chiappa, M. , Martinelli, G. B. , Fuoco, C. , Desiderio, G. , Castagnoli, L. , Gargioli, C. , Piccirillo, R. , and Cesareni, G. (2018) Group I Paks support muscle regeneration and counteract cancer‐associated muscle atrophy. Journal of Cachexia, Sarcopenia and Muscle, 9: 727–746. 10.1002/jcsm.12303.
Contributor Information
Cesare Gargioli, Email: cesare.gargioli@uniroma2.it.
Rosanna Piccirillo, Email: rosanna.piccirillo@marionegri.it.
Gianni Cesareni, Email: cesareni@uniroma2.it.
References
- 1. Piccirillo R, Demontis F, Goldberg AL, Perrimon N. Mechanisms of muscle growth and atrophy in mammals and Drosophila. Dev Dyn 2014;243:201–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yin H, Price F, Rudnicki MA. Satellite cells and the muscle stem cell niche. Physiol Rev 2013;93:23–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. He WA, Berardi E, Cardillo VM, Acharyya S, Aulino P, Thomas‐Ahner J, et al. NF‐κB‐mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J. Clin. Invest. American Society for Clin Investig 2013;123:4821–4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wigmore SJ, Plester CE, Ross JA, Fearon KC. Contribution of anorexia and hypermetabolism to weight loss in anicteric patients with pancreatic cancer. Br J Surg 1997;84:196–197. [PubMed] [Google Scholar]
- 5. Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol Elsevier 2011;12:489–495. [DOI] [PubMed] [Google Scholar]
- 6. Assi M, Rébillard A. The Janus‐faced role of antioxidants in cancer cachexia: new insights on the established concepts. Oxid Med Cell Longev Hindawi Publishing Corporation 2016;2016:9579868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Martinelli GB, Olivari D, Re Cecconi AD, Talamini L, Ottoboni L, Lecker SH, et al. Activation of the SDF1/CXCR4 pathway retards muscle atrophy during cancer cachexia. Oncogene. Nature Publishing Group 2016;35:6212–6222. [DOI] [PubMed] [Google Scholar]
- 8. Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, et al. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB JFederation of American Societies for Experimental Biology 2004;18:39–51. [DOI] [PubMed] [Google Scholar]
- 9. Jaffer ZM, Chernoff J. p21‐Activated kinases: three more join the Pak. Int J Biochem Cell Biol 2002;34:713–717. [DOI] [PubMed] [Google Scholar]
- 10. Bokoch GM. Biology of the p21‐activated kinases. Annu Rev Biochem Annual Reviews 4139 El Camino Way, PO Box 10139, Palo Alto, CA 94303–0139, USA; 2003;72:743–781. [DOI] [PubMed] [Google Scholar]
- 11. Luo ZG, Wang Q, Zhou JZ, Wang J, Luo Z, Liu M, et al. Regulation of AChR clustering by Dishevelled interacting with MuSK and PAK1. Neuron 2002;35:489–505. [DOI] [PubMed] [Google Scholar]
- 12. Cuenda A. Stress‐activated protein kinase‐2/p38 and a rapamycin‐sensitive pathway are required for C2C12 myogenesis. J Biol Chem 1999;274:4341–4346. [DOI] [PubMed] [Google Scholar]
- 13. Zetser A, Gredinger E, Bengal E. p38 mitogen‐activated protein kinase pathway promotes skeletal muscle differentiation. Participation of the Mef2c transcription factor. Journal of Biological Chemistry American Society for Biochemistry and Mol Biol 1999;274:5193–5200. [DOI] [PubMed] [Google Scholar]
- 14. Puri PL, Wu Z, Zhang P, Wood LD, Bhakta KS, Han J, et al. Induction of terminal differentiation by constitutive activation of p38 MAP kinase in human rhabdomyosarcoma cells. Genes Dev 2000;14:574–584. [PMC free article] [PubMed] [Google Scholar]
- 15. Wu Z, Woodring PJ, Bhakta KS, Tamura K, Wen F, Feramisco JR, et al. p38 and extracellular signal‐regulated kinases regulate the myogenic program at multiple steps. Mol Cell Biol 2000;20:3951–3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jones NC, Tyner KJ, Nibarger L, Stanley HM, Cornelison DDW, Fedorov YV, et al. The p38α/β MAPK functions as a molecular switch to activate the quiescent satellite cell. J Cell Biol 2005;169:105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Troy A, Cadwallader AB, Fedorov Y, Tyner K, Tanaka KK, Olwin BB. Coordination of satellite cell activation and self‐renewal by Par‐complex‐dependent asymmetric activation of p38α/β MAPK. Cell Stem Cell 2012;11:541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen S‐E, Jin B, Li Y‐P. TNF‐alpha regulates myogenesis and muscle regeneration by activating p38 MAPK. AJP: Cell Physiology 2007;292:C1660–C1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhan M, Jin B, Chen S‐E, Reecy JM, Li Y‐P. TACE release of TNF‐α mediates mechanotransduction‐induced activation of p38 MAPK and myogenesis. Journal of Cell Science The Company of Biologists Ltd 2007;120:692–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cole F, Zhang W, Geyra A, Kang J‐S, Krauss RS. Positive regulation of myogenic bHLH factors and skeletal muscle development by the cell surface receptor CDO. Developmental Cell Elsevier 2004;7:843–854. [DOI] [PubMed] [Google Scholar]
- 21. Takaesu G, Kang JS, Bae GU, Yi MJ, Lee CM, Reddy EP, et al. Activation of p38/MAPK in myogenesis via binding of the scaffold protein JLP to the cell surface protein Cdo. J Cell Biol 2006;175:383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kang JS, Bae GU, Yi MJ, Yang YJ, Oh JE, Takaesu G, et al. A Cdo‐Bnip‐2‐Cdc42 signaling pathway regulates p38/MAPK activity and myogenic differentiation. J Cell Biol 2008;182:497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bae GU, Kim BG, Lee HJ, Oh JE, Lee SJ, Zhang W, et al. Cdo binds Abl to promote p38/mitogen‐activated protein kinase activity and myogenic differentiation. Mol Cell Biol 2009;29:4130–4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lu M, Krauss RS. N‐cadherin ligation, but not Sonic hedgehog binding, initiates Cdo‐dependent p38/MAPK signaling in skeletal myoblasts. Proc Natl Acad Sci 2010;107:4212–4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Knight JD, Tian R, Lee RE, Wang F, Beauvais A, Zou H, et al. A novel whole‐cell lysate kinase assay identifies substrates of the p38 MAPK in differentiating myoblasts. Skelet Muscle 2012;2:5–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Penn BH, Bergstrom DA, Dilworth FJ, Bengal E, Tapscott SJ. A MyoD‐generated feed‐forward circuit temporally patterns gene expression during skeletal muscle differentiation. Genes Dev 2004;18:2348–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lluís F, Ballestar E, Suelves M, Esteller M, Muñoz‐Cánoves P. E47 phosphorylation by p38 MAPK promotes MyoD/E47 association and muscle‐specific gene transcription. EMBO J 2005;24:974–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Simone C, Forcales SV, Hill DA, Imbalzano AN, Latella L, Puri PL. p38 pathway targets SWI‐SNF chromatin‐remodeling complex to muscle‐specific loci. Nat Genet 2004;36:738–743. [DOI] [PubMed] [Google Scholar]
- 29. Palacios D, Mozzetta C, Consalvi S, Caretti G, Saccone V, Proserpio V, et al. TNF/p38α/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell 2010;7:455–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Forcales SV, Albini S, Giordani L, Malecova B, Cignolo L, Chernov A, et al. Signal‐dependent incorporation of MyoD‐BAF60c into Brg1‐based SWI/SNF chromatin‐remodelling complex. EMBO J EMBO Press 2012;31:316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Briata P, Forcales SV, Ponassi M, Corte G, Chen C‐Y, Karin M, et al. p38‐Dependent phosphorylation of the mRNA decay‐promoting factor KSRP controls the stability of select myogenic transcripts. Mol Cell 2005;20:891–903. [DOI] [PubMed] [Google Scholar]
- 32. Joseph GA, Lu M, Radu M, Lee JK, Burden SJ, Chernoff J, et al. Group I Paks promote skeletal myoblast differentiation in vivo and in vitro. American Society for Microbiology. Molecul Cellul Biol 2016; MCB.00222–16–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Deacon SW, Beeser A, Fukui JA, Rennefahrt UEE, Myers C, Chernoff J, et al. An isoform‐selective, small‐molecule inhibitor targets the autoregulatory mechanism of p21‐activated kinase. Chem Biol 2008;15:322–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Minasi MG, Riminucci M, De Angelis L, Borello U, Berarducci B, Innocenzi A, et al. The meso‐angioblast: a multipotent, self‐renewing cell that originates from the dorsal aorta and differentiates into most mesodermal tissues. Development 2002;129:2773–2783. [DOI] [PubMed] [Google Scholar]
- 35. Parsons M, Monypenny J, Ameer‐Beg SM, Millard TH, Machesky LM, Peter M, et al. Spatially distinct binding of Cdc42 to PAK1 and N‐WASP in breast carcinoma cells. Molecular and Cellular Biology. American Society for Microbiology 2005;25:1680–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Piccirillo R, Goldberg AL. The p97/VCP ATPase is critical in muscle atrophy and the accelerated degradation of muscle proteins. EMBO J 2012;31:3334–3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yoshida S, Fujisawa‐Sehara A, Taki T, Arai K, Nabeshima Y. Lysophosphatidic acid and bFGF control different modes in proliferating myoblasts. J Cell Biol. The Rockefeller University Press 1996;132:181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McCloy RA, Rogers S, Caldon CE, Lorca T, Castro A, Burgess A. Partial inhibition of Cdk1 in G 2phase overrides the SAC and decouples mitotic events. Cell Cycle 2014;13:1400–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bonetto A, Rupert JE, Barreto R, Zimmers TA. The colon‐26 carcinoma tumor‐bearing mouse as a model for the study of cancer cachexia. JoVE 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pretto F, Ghilardi C, Moschetta M, Bassi A, Rovida A, Scarlato V, et al. Sunitinib prevents cachexia and prolongs survival of mice bearing renal cancer by restraining STAT3 and MuRF‐1 activation in muscle. Oncotarget 2015;6:3043–3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bonetto A, Aydogdu T, Kunzevitzky N, Guttridge DC, Khuri S, Koniaris LG, et al. STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PLoS ONE Public Libr Sci 2011;e22538:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Scott HR, McMillan DC, Crilly A, McArdle CS, Milroy R. The relationship between weight loss and interleukin 6 in non‐small‐cell lung cancer. Br J Cancer Nature Publishing Group 1996;73:1560–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fearon KC, McMillan DC, Preston T, Winstanley FP, Cruickshank AM, Shenkin A. Elevated circulating interleukin‐6 is associated with an acute‐phase response but reduced fixed hepatic protein synthesis in patients with cancer. Ann Surg Lippincott, Williams, and Wilkins 1991;213:26–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, et al. The IGF‐1/PI3K/Akt pathway prevents expression of muscle atrophy‐induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell 2004;14:395–403. [DOI] [PubMed] [Google Scholar]
- 45. Braun TP, Grossberg AJ, Krasnow SM, Levasseur PR, Szumowski M, Zhu XX, et al. Cancer‐ and endotoxin‐induced cachexia require intact glucocorticoid signaling in skeletal muscle. FASEB J Federation of American Societies for Experimental Biology 2013;27:3572–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Clarke BA, Drujan D, Willis MS, Murphy LO, Corpina RA, Burova E, et al. The E3 ligase MuRF1 degrades myosin heavy chain protein in dexamethasone‐treated skeletal muscle. Cell Metabolism. Elsevier 2007;6:376–385. [DOI] [PubMed] [Google Scholar]
- 47. Fujita R, Kawano F, Ohira T, Nakai N, Shibaguchi T, Nishimoto N, et al. Anti‐interleukin‐6 receptor antibody (MR16‐1) promotes muscle regeneration via modulation of gene expressions in infiltrated macrophages. Biochim Biophys Acta 2014;1840:3170–3180. [DOI] [PubMed] [Google Scholar]
- 48. Lin Shiau SY, Huang MC, Lee CY. Mechanism of action of cobra cardiotoxin in the skeletal muscle. J Pharmacol Exp Ther 1976;196:758–770. [PubMed] [Google Scholar]
- 49. Manser E, Leung T, Salihuddin H, Zhao ZS, Lim L. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature 1994;367:40–46. [DOI] [PubMed] [Google Scholar]
- 50. Manser E, Chong C, Zhao ZS, Leung T, Michael G, Hall C, et al. Molecular cloning of a new member of the p21‐Cdc42/Rac‐activated kinase (PAK) family. J Biol Chem 1995;270:25070–25078. [DOI] [PubMed] [Google Scholar]
- 51. Teo M, Manser E, Lim L. Identification and molecular cloning of a p21cdc42/rac1‐activated serine/threonine kinase that is rapidly activated by thrombin in platelets. J Biol Chem 1995;270:26690–26697. [DOI] [PubMed] [Google Scholar]
- 52. Chong C, Tan L, Lim L, Manser E. The mechanism of PAK activation autophosphorylation events in both regulatory and kinase domains control activity. J Biolog Chem. American Society for Biochemistry and Mol Biol 2001;276:17347–17353. [DOI] [PubMed] [Google Scholar]
- 53. Bergstrom DA, Penn BH, Strand A, Perry RLS, Rudnicki MA, Tapscott SJ. Promoter‐specific regulation of MyoD binding and signal transduction cooperate to pattern gene expression. Mol Cell 2002;9:587–600. [DOI] [PubMed] [Google Scholar]
- 54. Bryan BA, Mitchell DC, Zhao L, Ma W, Stafford LJ, Teng BB, et al. Modulation of muscle regeneration, myogenesis, and adipogenesis by the Rho family guanine nucleotide exchange factor GEFT. Mol Cell Biol 2005;25:11089–11101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Barnes CJ, Vadlamudi RK, Mishra SK, Jacobson RH, Li F, Kumar R. Functional inactivation of a transcriptional corepressor by a signaling kinase. Nat Struct Biol 2003;10:622–628. [DOI] [PubMed] [Google Scholar]
- 56. Thomas J‐L, Moncollin V, Ravel‐Chapuis A, Valente C, Corda D, Méjat A, et al. PAK1 and CtBP1 regulate the coupling of neuronal activity to muscle chromatin and gene expression. Molecul Cellul Biol. American Society for Microbiology 2015;35:4110–4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wang Z, Oh E, Clapp DW, Chernoff J, Thurmond DC. Inhibition or ablation of p21‐activated kinase (PAK1) disrupts glucose homeostatic mechanisms in vivo. J Biol Chem 2011;286:41359–41367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sylow L, Jensen TE, Kleinert M, Højlund K, Kiens B, Wojtaszewski J, et al. Rac1 signaling is required for insulin‐stimulated glucose uptake and is dysregulated in insulin‐resistant murine and human skeletal muscle. Diabetes. American Diabetes Association 2013;62:1865–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sanders LC. Inhibition of myosin light chain kinase by p21‐activated kinase. Science 1999;283:2083–2085. [DOI] [PubMed] [Google Scholar]
- 60. Wirth A, Schroeter M, Kock‐Hauser C, Manser E, Chalovich JM, de Lanerolle P, et al. Inhibition of contraction and myosin light chain phosphorylation in guinea‐pig smooth muscle by p21‐activated kinase 1. J Physiol (Lond) Blackwell Publishing Ltd 2003;549:489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tsompanidis A, Vafiadaki E, Blüher S, Kalozoumi G, Mantzoros CS, Sanoudou D. Ciliary neurotrophic factor upregulates follistatin and Pak1, causes overexpression of muscle differentiation related genes and downregulation of established atrophy mediators in skeletal muscle. Metabolism Elsevier BV 2016;1–28. [DOI] [PubMed] [Google Scholar]
- 62. Massaccesi L, Goi G, Tringali C, Barassi A, Venerando B, Papini N. Dexamethasone‐induced skeletal muscle atrophy increases O‐GlcNAcylation in C2C12 cells. J Cell Biochem 2016;117:1833–1842. [DOI] [PubMed] [Google Scholar]
- 63. Bonetto A, Aydogdu T, Jin X, Zhang Z, Zhan R, Puzis L, et al. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL‐6 and in experimental cancer cachexia. Am J Physiol Endocrinol Metab American Physiological Society 2012;303:E410–E421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bentzinger CF, Wang YX, Dumont NA, Rudnicki MA. Cellular dynamics in the muscle satellite cell niche. Nature Publishing Group Nature Publishing Group 2013;14:1062–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yun K, Wold B. Skeletal muscle determination and differentiation: story of a core regulatory network and its context. Curr Opin Cell Biol 1996;8:877–889. [DOI] [PubMed] [Google Scholar]
- 66. Lin Z, Lu M‐H, Schultheiss T, Choi J, Holtzer S, Dilullo C, et al. Sequential appearance of muscle‐specific proteins in myoblasts as a function of time after cell division: evidence for a conserved myoblast differentiation program in skeletal muscle. Cell Motil Cytoskeleton 1994;29:1–19. [DOI] [PubMed] [Google Scholar]
- 67. Zhang S, Han J, Sells MA, Chernoff J, Knaus UG, Ulevitch RJ, et al. Rho family GTPases regulate p38 mitogen‐activated protein kinase through the downstream mediator Pak1. J Biol Chem 1995;270:23934–23936. [DOI] [PubMed] [Google Scholar]
- 68. Sebe A, Masszi A, Zulys M, Yeung T, Speight P, Rotstein OD, et al. Rac, PAK and p38 regulate cell contact‐dependent nuclear translocation of myocardin‐related transcription factor. FEBS Lett 2008;582:291–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fukada S‐I, Uezumi A, Ikemoto M, Masuda S, Segawa M, Tanimura N, et al. Molecular signature of quiescent satellite cells in adult skeletal muscle. Stem Cells 2007;25:2448–2459. [DOI] [PubMed] [Google Scholar]
- 70. Perfetto L, Briganti L, Calderone A, Cerquone Perpetuini A, Iannuccelli M, Langone F, et al. SIGNOR: a database of causal relationships between biological entities. Nucleic Acids Res. Oxford University Press 2016;44:D548–D554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Pretto C, Ghilardi M, Moschetta A, Bassi A, Rovida V, Scarlato, et al. Sunitinib prevents cachexia and prolongs survival of mice bearing renal cancer by restraining STAT3 and MuRF‐1 activation in muscle. Oncotarget 2015;6:3043–3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. White ME, Allen CE, Dayton WR. Effect of sera from fed and fasted pigs on proliferation and protein turnover in cultured myogenic cells. J Anim Sci 1988;66:34–40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1 The mRNA levels of Pak1 tends to be decreased in TA from RXF393‐bearing mice. PAK1 expression was determined by quantitative PCR (n = 5). Gusb was used as housekeeping gene. SEM is reported. Unpaired T test, ns, p = 0.1528. Mice were sacrificed 18 days after tumor inoculation. Consider 72 as reference for RXF393 in vivo inoculation.
Fig. S2 PAK1 overexpression in Tibialis Anterior of C26‐bearing mice does not alter the expression of PAX7 or MyoD. Human PAK1‐GFP or GFP expressing plasmids were electroporated into the Tibialis Anterior of mice that had been injected with C26 tumor cells the day after electroporation. The expression of PAX7 (A), MyoD (B), exogenous (human) PAK1 (C) and a housekeeping gene (TBP) were measured by Q‐PCR on muscle dissected 14 days after electroporation (n = 6–9, unpaired T‐Test, ***p = 0.0004). SEM is reported.
Fig S3 The expression of atrogin‐1, MuRF1 and myogenin is not affected by PAK1 overexpression in muscles of PBS‐injected mice. The mRNA levels of atrogin‐1 (A), MuRF1 (B) and myogenin (C) in TA from PBS‐injected mice electroporated for 14 days with empty vector or PAK1 were determined by quantitative PCR (n = 7). IPO8 or TBP were used as housekeeping genes. Unpaired T test, ns.
Fig. S4 Phosphorylation of ectopically expressed PAK1 occurs in transfected myoblasts. Immunoblot analysis revealing p‐PAK1 (T423) and total PAK1 in C2C12 protein extracts. Cells extracts were analyzed 24 h after transfection of plasmids expressing GFP‐human PAK1 or a corresponding empty vector. PAK1 and p‐PAK1 signals have been developed on separate membranes. Vinculin served as a loading control and has been blotted on both membranes.
Fig S5 The endogenous p97/VCP content tends to be decreased in C2C12 by PAK1 overexpression. (A) Myoblasts were transfected with empty vector or Pak1 expressing vectors for 24 h and differentiated into myotubes, total protein was then extracted. Immunoblot analysis reveals the protein content of p97 and vinculin, that is used as loading control. (B) The bar graph illustrates the densitometric quantification of p97/vinculin signal ratio for experiments as represented in A (n = 6, 5) Unpaired T test, p = 0,06.
Fig. S6 Pak1 inhibition by IPA‐3 does not affect protein synthesis or protein degradation of normal or dexamethasone‐treated atrophying myotubes. Overall protein synthesis measured as total CPM (counts per minute) per well (A) and percentage of protein degraded per hour (B) of 4‐days differentiated myotubes treated for 30 h with vehicle (DMSO), 1μM dexamethasone, 10μM IPA‐3 or both, are plotted (n = 4, Unpaired T‐Test, *p < 0.05, **p < 0.01, ****p < 0.0001). Total protein synthesis was calculated by incorporation of L‐[Ring‐3,5‐3H]‐tyrosine into cell proteins, 73 following the procedure described in Piccirillo and Goldberg.36
Fig. S7 PAK1‐overexpressing myoblasts express lower levels of atrogin‐1 protein after dexamethasone treatment. Densitometric analysis of the blot from the main manuscript (Figure 2G) shows quantification of the protein content of atrogin‐1 in myoblasts transfected with plasmid for GFP or PAK1 and treated for 24 h with 1μM dexamethasone (Dexa) or vehicle. Vinculin was used as loading control (n = 6–8, Unpaired T‐Test, *p < 0.05, ***p < 0.001).
Fig. S8 IPA‐3 treated Mabs display a lower myogenin expression compared to control. Protein content for each cell was evaluated as total corrected fluorescence intensity for myogenin positive nuclei. The graph shows the difference of myogenin expression between control and IPA‐3 treated cells for each biological replicate. Values are represented as box plots with 2.5–97.5% confidence intervals. The total number of cells counted for each group is 100. Statistical significance has been evaluated using the Unpaired t‐test, *p ≤ 0.05, ***p = 0.001.
Fig. S9 IPA‐3 treatment does not affect Satellite cells expression of Pax7 and MyoD. Immunofluorescence showing Pax7 (green) (A, B) and MyoD (red) (D, E) expression during SC differentiation for control and IPA‐3 treated cells. Nuclei are represented in grey. The plot shows the percentage of Pax7 positive cells (C) or MyoD positive cells (F) for the experiments reported in A, B and D, E. The values are mean of three independent experiments + SEM. Statistical significance has been evaluated using the Unpaired t‐test. Scale bar: 100 μm.