

Abstract
Background
Nuclear receptor interaction protein (NRIP) is a calcium/calmodulin (CaM) binding protein. Nuclear receptor interaction protein interacts with CaM to activate calcineurin and CaMKII signalling. The conventional NRIP knockout mice (global knockout) showed muscular abnormality with reduction of muscle oxidative functions and motor function defects.
Methods
To investigate the role of NRIP on neuromuscular system, we generated muscle‐restricted NRIP knockout mice [conditional knockout (cKO)]. The muscle functions (including oxidative muscle markers and muscle strength) and lumbar motor neuron functions [motor neuron number, axon denervation, neuromuscular junction (NMJ)] were tested. The laser‐captured microdissection at NMJ of skeletal muscles and adenovirus gene therapy for rescued effects were performed.
Results
The cKO mice showed muscular abnormality with reduction of muscle oxidative functions and impaired motor performances as global knockout mice. To our surprise, cKO mice also displayed motor neuron degeneration with abnormal architecture of NMJ. Specifically, the cKO mice revealed reduced motor neuron number with small neuronal size in lumbar spinal cord as well as denervating change, small motor endplates, and decreased myonuclei number at NMJ in skeletal muscles. To explore the mechanisms, we screened various muscle‐derived factors and found that myogenin is a potential candidate that myogenin expression was lower in skeletal muscles of cKO mice than wild‐type mice. Because NRIP and myogenin were colocalized around acetylcholine receptors at NMJ, we extracted RNA from synaptic and extrasynaptic regions of muscles using laser capture microdissection and showed that myogenin expression was especially lower at synaptic region in cKO than wild‐type mice. Notably, overexpression of myogenin using intramuscular adenovirus encoding myogenin treatment rescued abnormal NMJ architecture and preserved motor neuron death in cKO mice.
Conclusions
In summary, we demonstrated that deprivation of NRIP decreases myogenin expression at NMJ, possibly leading to abnormal NMJ formation, denervation of acetylcholine receptor, and subsequent loss of spinal motor neuron. Overexpression of myogenin in cKO mice can partially rescue abnormal NMJ architecture and motor neuron death. Therefore, muscular NRIP is a novel trophic factor supporting spinal motor neuron via stabilization of NMJ by myogenin expression.
Keywords: NRIP, Motor neuron, Neuromuscular junction, Myogenin
Introduction
The nuclear receptor interaction protein (NRIP) (also named DCAF6 and IQWD1) consists of 860 amino acids and encodes seven WD40 domains with one IQ motif.1, 2 Nuclear receptor interaction protein interacts with calmodulin (CaM) in the presence of calcium via the IQ domain. Nuclear receptor interaction protein can regulate muscle contraction via CaM to activate calcineurin and CaMKII signalling.3, 4 In limb‐girdle muscular dystrophy, NRIP expression was lower in dystrophic lesions than counterpart normal tissues as noted by mRNA differential display.5 Hence, NRIP is an important factor contributing to normal muscular physiology. However, genetic variations in the NRIP genome location are associated with cardiovascular disease, osteoporosis, and schizophrenia.6, 7, 8 Nuclear receptor interaction protein is also named DCAF6 and belongs to Cul4‐DDB1 associated factors.9, 10 Mutation at R317C of DCAF8 responding to DDB1 binding was identified as the genetic cause of axonal hereditary motor and sensory neuropathy.11 Thus, NRIP might also play a role in neuropsychological diseases.
Motor neuron diseases (MNDs) comprise a group of neurodegenerative disorders characterized by progressive degeneration of motor neurons leading to muscle weakness and atrophy.12, 13 No curative treatment exists for MNDs such as amyotrophic lateral sclerosis (ALS) or spinal and bulbar muscular atrophy (SBMA).14, 15 Recent advances in MND research have shown that the maintenance and survival of motor neuron are determined not only by motor neurons themselves but also by surrounding cells (astrocytes and microglia) and distant tissues (skeletal muscles).15, 16, 17, 18, 19 In a mouse model of ALS, muscle‐restricted expression of mutant superoxide dismutase 1 has toxic effects on spinal motor neurons, with distal axonopathy and the formation of ubiquitinated inclusions.20 Similarly, muscle‐restricted abolishment of transgenic mutant androgen receptor (AR) rescues the abnormal phenotypes of motor neurons and axons in a mouse model of SBMA with mutant AR transgenes.21 These findings provide evidence of the muscle‐related non‐autonomous pathological mechanisms of motor neuron degeneration.
Motor neurons connect to muscle via axonal innervation towards skeletal muscle endplates at the neuromuscular junction (NMJ). Recent study showed that the interaction is bidirectional: motor neurons sustain the integrity of the NMJ and control the muscle contraction, and muscle provides major trophic support for innervating motor neurons in terms of neuron survival, synaptic activity, and axonal function.16, 17, 18, 22, 23 For example, the myogenic transcription factors myogenin and MyoD can inhibit or enhance motor neuron degeneration, respectively.24, 25, 26 Muscle‐derived factors such as neurotrophin‐4 and bone morphogenetic protein (BMP) can support the survival and growth or promote reinnervation of motor neurons.27, 28 Despite the important interaction between motor neurons and skeletal muscles in physiological and pathological conditions, the mechanism of the communication is still not fully understood. Investigating the crosstalk between motor neurons and skeletal muscles can help in understanding the physiology of the neuromuscular unit and provide a novel therapeutic strategy for MNDs.
Recently, we generated global NRIP knockout (KO) mice showing reduced muscle strength, susceptibility to muscle fatigue, and impaired adaptive exercise performance.4 In this study, to examine whether skeletal muscle could retrogradely control the development of motor neurons, we generated muscle‐restricted NRIP KO [conditional KO (cKO)] mice to investigate the role of NRIP in the neuromuscular system. At age 6 weeks, cKO mice showed muscle dystrophy with normal motor neurons, but at age 16 weeks, motor neuron degeneration with abnormal architecture of the NMJ. Nuclear receptor interaction protein KO resulted in the down‐regulation of myogenin at the NMJ, with impaired NMJ architecture, thereby leading to spinal motor neuron degeneration. Gene therapy with an adenovirus encoding myogenin in skeletal muscles rescued the abnormal NMJ phenotypes and prevented motor neuron death. Muscular NRIP may be a novel trophic factor supporting spinal motor neurons by stabilizing the NMJ via myogenin expression.
Methods
Generation of muscle‐restricted nuclear receptor interaction protein knockout mice
The generation of ES cells containing the LoxP‐floxed NRIP allele was previously described.4 The ES cells containing the LoxP‐floxed NRIP allele were injected into blastocytes of C57BL/6J mice and implanted into pseudopregnant foster mothers.29 The chimeric offspring was back‐crossed to C57BL/6J mice (n ≧ 10 generations) to generate the NRIP‐LoxP heterozygous (NRIP flox/+) mice. To obtain muscle‐restricted KO animals, NRIP flox/+ mice were crossed with muscle creatine kinase (MCK)‐Cre heterozygous mice.30 Offspring containing NRIP flox/+ and MCK‐Cre + genotypes was crossed to each other, resulting in six different genotypes: NRIP +/+; MCK‐Cre +, NRIP flox/+; MCK‐Cre +, NRIP flox/flox; MCK‐Cre +, NRIP +/+; MCK‐Cre −, NRIP flox/+; MCK‐Cre −, and NRIP flox/flox; MCK‐Cre −. For subsequent experiments, we used the NRIP flox/flox; MCK‐Cre + mice as the muscle‐restricted NRIP KO mice [named conditional KO (cKO) mice], and control wild‐type (WT) mice were NRIP flox/flox; MCK‐Cre −. Routine genotyping was performed by mouse tail PCR (Qiagen, Valencia, CA, USA). For WT and floxed NRIP genotyping, GU and JD primers were used (Table S1). For Cre recombinase genotyping, four oIMR primers were used (Table S1).
Behavioural analysis
For the rotarod test, mice were trained three times by being placed in a neutral position in the stationary 3‐cm‐diameter cylinder of the rotarod apparatus (Singa RT‐01) with a constant rotating speed of 5 rpm for 30 s. The mice were provided 30 min breaks between the training and test phases. In the test phase, the constant rotating speed was set at 10, 15, and 20 rpm, and the time to falling was recorded when each mouse fell off the rod. For analysis of grip force, before experiments, mice were placed on the electronic balance (Denver Instrument, Bohemia, NY, USA) until they were stable to measure body weight for normalization with grip force strength. The mouse forepaws were placed on the grasping bar of the Grip‐Strength Meter (Somatco, Basile, Italy), and the mouse tail was then lifted and pulled backward until the mouse released the grasping bar. The data were collected from the peak amplifier.
Cresyl violet stain
Lumbar sections (L3–L5) 30 μm thick from the spinal cord were fixed in 4% paraformaldehyde (PFA) and incubated with 1% cresyl violet (Sigma, Louis, MO, USA) containing 2.5% glacial acid for 1 min and rinsed in water to stop the reaction. After dehydration with serial alcohol, sections were mounted with mineral oil and visualized under a Zeiss Axioskop 40 optical microscope. At least 12 sections with distance 120 μm apart from each animal were stained to obtain the average number of α‐motor neurons per ventral horn.
Immunohistochemistry assay
Soleus and gastrocnemius muscles were collected and fixed in 4% PFA. The antigen in paraffin‐embedded sections with thickness 5 μm were retrieved by boiled 0.1 M citrate acid (pH 6.0) and then incubated with indicated antibody at 4°C overnight. After washing with phosphate‐buffered saline (PBS) and horseradish peroxidase (HRP) polymer (BioGenex, Fremont, CA, USA) incubation for 1 h in room temperature, positive signals were detected by HRP polymer reacting with 3,3′‐diaminobenzidine chromogen (BioGenex, Fremont, CA, USA).
Cytochrome c oxidase and succinate dehydrogenase activities
Frozen tissue sections 5 μm thick were incubated with solution containing 80 μM cytochrome c and 1300 U catalase, then mounted with PERMOUNT synthetic organic mounting medium. Cytochrome c oxidase (COX)‐positive fibres were visualized by brown staining. For succinate dehydrogenase (SDH) staining, sections were incubated in a solution containing sodium succinate, phosphate buffer, and nitroblue tetrazoliumand, then mounted with glycerol‐based mounting medium. Succinate dehydrogenase‐positive fibres were visualized by purple staining. Images were acquired under a Zeiss Imager A1 microscope with an AxioCam MRM CCD camera.
Immunofluorescence assay
For calculation of α‐motor neuron number, the mouse vertebrate was separated from the mouse body. Phosphate‐buffered saline was injected into the end of vertebrae by using a butterfly needle to flush out the spinal cord. The lumbar segments (L3–L5) were embedded in optimum cutting temperature (OCT) compound (Tissue‐Tek) and cross‐sectioned into 30 μm thickness by using a cryostat microtome. Sections were stained with the antibodies anti‐neuronal DNA binding protein (NeuN) (Millipore, Temecula , CA, USA 1:500) and anti‐choline acetyltransferase (ChAT) (Millipore, 1:200) overnight at 4°C. After a wash with PBS, fluorescent signals were detected by incubation with Cy3‐conjugated goat anti‐rabbit or 488‐conjugated donkey anti‐goat secondary antibody (Jackson Lab, Sacramento, CA, USA). The images were acquired with a Leica TSC SP5 confocal microscope (Leica Microsystems, Wetzlar, Germany). Every fourth cryostat section was used for immunofluorescence assay (IFA), and the number of motor neurons (20 sections for each animal) was counted.
To calculate the NMJ area, soleus muscles underwent whole‐mount staining. Briefly, soleus muscle was fixed in 2% PFA and teased apart into small muscle bundles by using forceps, then washed with PBS for 30 min. Soleus muscle was incubated with 0.1 M glycine for 1 h and washed three times with PBS containing 0.5% Triton X‐100. Next, soleus was blocked in 2% bovine serum albumin and incubated with the antibodies anti‐neurofilament (Abcam, Cambridge, MA, USA 1:500 dilution) and anti‐synaptophysin (Abcam, 1:200 dilution) overnight at 4°C. After a wash with PBST (PBS containing 0.5% Triton X‐100), the soleus was incubated with a mixture containing the antibodies 488‐conjugated donkey anti‐rabbit secondary and Alexa‐594‐conjugated anti‐bungarotoxin (α‐BTX, Life Technologies, 1:1000), then washed with PBST and mounted in mineral oil (Sigma). Gastrocnemius muscles were fixed with 2% PFA for 2 h at room temperature. Tissues were washed with PBS three times and dehydrated with 30% sucrose and incubated overnight at 4°C. Dehydrated tissues were then embedded in OCT and sectioned to 20 μm thickness. Sections were incubated with anti‐neurofilament and anti‐synaptophysin primary antibodies overnight at 4°C. After PBS washing, slides were incubated with a mixture containing 488‐conjugated donkey anti‐rabbit secondary antibody and Alexa‐594‐conjugated α‐BTX antibody and mounted in DAPI Fluoromount‐G (SouthernBiotech, Birmingham, AL, USA). The fluorescent images from soleus and gastrocnemius muscles were visualized under a Leica TSC SP5 confocal microscope (Leica Microsystems). Fluorescent images from each section were analysed by using ImageJ to measure the size of the NMJ area. An outline was drawn around the endplate of acetylcholine receptor (AChR) cluster area labelled by α‐BTX by using the ImageJ polygon tool; an each NMJ area was calculated by using Prism (GraphPad, La Jolla, CA, USA).
To quantify the denervated endplates, fluorescent images of α‐BTX and synaptophysin were overlapped by using the Leica LAS AF Lite software to visualize the co‐localization of α‐BTX and synaptophysin. The co‐localization of α‐BTX and synaptophysin was defined as innervated endplates; no co‐localization but synaptophysin staining adjacent to α‐BTX was defined as intermediate endplates; no co‐localization or synaptophysin staining adjacent to α‐BTX staining was defined as denervated endplates. The proportion of innervated, intermediate, and denervated endplates was determined as the total number of endplates per section divided by the number of innervated, intermediate, and denervated endplates.
For postsynaptic nuclei calculation, gastrocnemius muscle was embedded in OCT and longitudinally sectioned to 10 μm thickness. Sections were then stained with α‐BTX and SUN2 primary antibody (Abcam, 1:500) for muscle nuclei staining. The postsynaptic nuclei were defined by the co‐localization of α‐BTX and SUN2. SUN2‐positive nuclei without α‐BTX stain were defined as extrasynaptic nuclei.
Laser capture microdissection
Gastrocnemius muscles were embedded in OCT and sectioned to 10 μm thickness. The synaptic region was defined by α‐BTX staining to detect the postsynaptic AChR cluster. The synaptic and extrasynaptic regions were isolated by using the Arcturus Laser Capture Microdissection System (Life Technologies Corp., Grand Island, NY, USA). The isolated synaptic and extrasynaptic sections were collected into a microfuge cap containing lysis buffer (RLT; Qiagen, Hilden, Germany) for total RNA extraction. The gene expression of myogenin was analysed by real‐time quantitative PCR (RT‐qPCR). The myogenin primer sequence is in Table S1.
Western blot analysis
Muscle lysates were extracted by using radioimmunoprecipitation assay lysis buffer (150 mM NaCl, 1% NP‐40, and 50 mM Tris). Proteins were separated on sulfate polyacrylamide gel electrophoresis, then transferred to polyvinylidene difluoride membranes (Millipore), which were blocked in 5% bovine serum albumin at room temperature for 1 h, and then blots were incubated with the primary antibodies anti‐NRIP (Novus, Littleton, CO, USA 1:2000), anti‐ChAT (Abcam, 1;1000), anti‐slow myosin (Abcam, 1:2000), anti‐MyHC (Abcam, 1:1000), anti‐myogenin (SantaCruz, Santa Cruz, CA, USA 1:1000), anti‐Flag (Abcam, 1:2000), and anti‐GAPDH (AbFrontier, Seoul, Korea 1:5000) diluted in blocking buffer at 4°C overnight. After washing blots with 1 × TBST buffer, blots were then incubated with HRP‐conjugated secondary antibody at room temperature for 1 h. After a serial wash with TBST buffer, protein expression was detected by using an ECL western blot detection system (GE Healthcare Life Sciences, Chicago, IL, USA).
Real‐time quantitative PCR
Muscle tissues were explanted and quickly stored in RNAlater solution (Ambion, Waltham, MA, USA). C2C12 cells were scraped off and stored at −80°C. Total RNA was isolated with TRIzol reagent (Invitrogen, Waltham, MA, USA) and treated with DNase (RQ1, Promega, Madison, WI, USA) to remove genomic DNA. One microgram of RNA was reverse transcribed into cDNA for RT‐qPCR. Real‐time quantitative PCR involved use of Power SYBR Green PCR Master Mix (Applied Biosystems, Waltham, MA, USA) or KAPA SYBR FAST qPCR kit Master Mix Universal (Kapa Biosystems, Wilmington, MA, USA) as instructed. The sequences of the primers are in Table S1.
Recombinant adenovirus generation
The myogenin fragment (675 bp) was amplified from the myogenin expression vector (pcDNA3.1‐V5‐myogenin) by PCR reaction; the forward primer (sequence: 5′‐AAAGGTACCATGGAGCTGTAT‐3′) contained the KpnI cutting site, and the reverse primer (sequence: 5′‐TTTAAGCTTTCATTGGGC‐3′) contained the HindIII cutting site. The myogenin was then cloned into the pShuttle‐CMV between the KpnI and HindIII restriction enzyme site. The cloned shuttle vector was co‐transformed with pAdEasy‐1 plasmid into BJ5183 competent cells by electroporation to generate the recombinant plasmid containing the myogenin cDNA (pShuttle‐CMV‐Myogenin). The pShuttle‐CMV‐Myogenin was then isolated by ethanol precipitation and transfected to HEK293A cells for adenovirus encoding myogenin (Ad‐myogenin) production. Adenovirus encoding myogenin viruses were then purified by using an Adeno‐XTM Maxi Purification kit (Clontech, Mountain View, CA, USA) as instructed.
Gene therapy
Mice were anaesthetized by intraperitoneal injection of 2.5% avertin (300 μL), then intramuscular injection of Ad‐shLuc (control) or Ad‐myogenin adenovirus (100 μL, 1 × 109 plaque‐forming units/mL) into bilateral gastrocnemius muscles at age 6 weeks. The mice were monitored daily after recovery until 3 or 10 weeks after gene therapy. The therapeutic effects were measured for motor function, muscle oxidative activity, motor neuron survival, NMJ integration, and axon innervation as previously described.
Statistics
Statistical analysis involved use of Prism 6.0 (GraphPad Software). Data are reported as the mean ± SEM. P‐values were determined by Student's t‐test for comparing two groups or one‐way analysis of variance for more than two groups. P < 0.05 was considered statistically significant.
Study approval
All animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee at the College of Medicine, National Taiwan University. All experimental mice were housed in the animal centre under a 12 h light/dark cycle with free access to food and water.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supporting Information files or on request from the corresponding author.
Results
Muscle‐restricted nuclear receptor interaction protein knockout (conditional knockout) mice reveal muscular dystrophy
We generated cKO mice by crossing mice with two floxed‐NRIP alleles (NRIP flox/flox) to MCK‐Cre mice expressing Cre recombinase under control of a muscle‐specific creatine kinase (MCK) promoter. The promoter activity of MCK is 104‐fold higher in skeletal muscle than in other non‐muscle tissues such as kidney, liver, and spleen.31 The schematic illustration of the genomic structure of the floxed‐NRIP allele is in Figure S1A. A floxed NRIP allele containing a neo cassette with two flanked loxP sequences in intron 1 and a loxP sequence in intron 2 was designed for NRIP deletion. The deletion of NRIP exon 2 caused a frame shift mutation that resulted in premature termination.4 PCR was used to genotype offspring with NRIP flox/flox and MCK‐Cre transgenes (flox/flox; Cre +) to check whether mice possessed the LoxP sequence and Cre recombinase (Figure S1B). The flox/flox; Cre + mice were the cKO mice used in all experiments. Control WT mice were NRIP flox/flox; MCK‐Cre −.
We examined whether NRIP protein was expressed specifically in muscle tissue. In cKO mice, NRIP was not expressed in muscle tissue such as heart and soleus and gastrocnemius muscles but was expressed normally in other tissues including brain, testis, and spinal cord (Figure 1A). Because NRIP can regulate oxidative myofiber expression (such as slow myosin gene expression, mitochondria COX, and SDH) to affect muscle strength, the expression patterns of these proteins were used as markers for oxidative muscle functions.4 cKO mice at age 16 weeks showed decreased protein expression of slow myosin (Figure 1B). The proportion of slow myofibers (Figure 1C), COX‐stained fibres (Figure 1D), and SDH‐stained fibres (Figure 1E) was significantly reduced in cKO mice as compared with the WT. This phenomenon was also observed previously in NRIP global knockout mice.4 As early as age 6 weeks, cKO mice showed a similar reduction of muscle oxidative functions with low slow myosin ( Figure S2A), COX ( Figure S2B), and SDH expression ( Figure S2C) as compared with WT mice. Furthermore, we measured centrally located nuclei, fibrosis, embryonic myosin expression, and infiltration of innate immune cells to see whether NRIP cKO mice showed muscle dystrophy. The histology examination, CD45 molecule, embryonic myosin heavy chain, and fibrosis by Evans blue dye stain in soleus muscles at age 16 weeks showed no significant difference between WT and NRIP cKO ( Figure S3). Taken together, NRIP cKO shows abnormal muscle oxidative functions at a both young and adult age.
Figure 1.
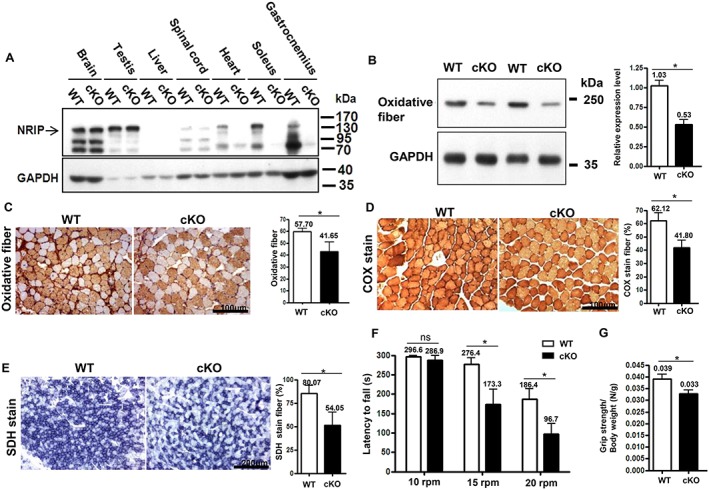
Muscle‐restricted nuclear receptor interaction protein (NRIP) knockout [conditional knockout (cKO)] mice show loss of NRIP in muscle tissues as well as muscle abnormality and impaired motor functions at age 16 weeks. (A) Western blot analysis of NRIP protein in tissues from wild‐type (WT) and cKO mice. GAPDH: internal control. (B) Western blot analysis of protein expression of oxidative myosin fibre (slow myosin). Right: quantification of three independent experiments and values set to 1 for the WT. (C) Representative immunohistochemistry of slow myosin expression. Scale bar, 100 μm. Right: quantification (n = 3). (D) Representative muscle cross sections with cytochrome c oxidase (COX; dark brown) staining. Scale bar, 100 μm. Right: quantification (n = 3). (E) Succinate dehydrogenase (SDH; dark purple) staining. Scale bar, 100 μm. Right: quantification (n = 3). (F) Rotarod test. Both WT and cKO mice were placed on a rod rotating at 10, 15, or 20 rpm, and riding time was measured. n = 7 for each genotype. (G) Grip force analysis. n = 7 for each genotype. WT (NRIP flox/flox; MCK‐Cre − mice). Data are mean ± SEM by Student's t‐test. * P < 0.05. ns, not significant.
Impaired motor function in conditional knockout mice
The body weight was similar between cKO and WT mice at a young and adult age (data not shown). We used rotarod and grip force test to evaluate their motor function performances.4 At age 16 weeks, cKO mice had shorter rotarod maintenance latency under 15 and 20 rpm than WT mice, but the latency under 10 rpm was similar between the two groups (Figure 1F). In addition, grip force was lower for cKO than WT mice (Figure 1G). However, at age 6 weeks, cKO and WT mice did not differ in rotarod and grip force test findings ( Figure S2D and S2E). Collectively, the impaired motor functions occurred at age 16 weeks but not 6 weeks.
Degeneration of lumbar motor neurons in conditional knockout mice
Nuclear receptor interaction protein expression was similar in spinal cords of cKO and WT mice at age 6 and 16 weeks despite the absence of NRIP expression in gastrocnemius muscles of cKO mice (Figure 2A). We used a motor neuron marker, ChAT, to measure motor neuron quantity in spinal cords. Choline acetyltransferase expression was similar between cKO and WT mice at age 6 weeks but was lower in cKO than WT mice at age 16 weeks (Figure 2A). Nissl staining was used for neuron counting in lumbar spinal cords. The spinal neuron was characterized as a pyramidal cell located in the anterior horn with a nucleolus within the nucleus surrounded by large light blue‐stained large cytoplasm (Figure 2B). The number of neurons in the anterior horn was 32.9% lower in cKO than WT mice (P = 0.009) at age 16 weeks but was similar between the groups at age 6 weeks (Figure 2B).
Figure 2.
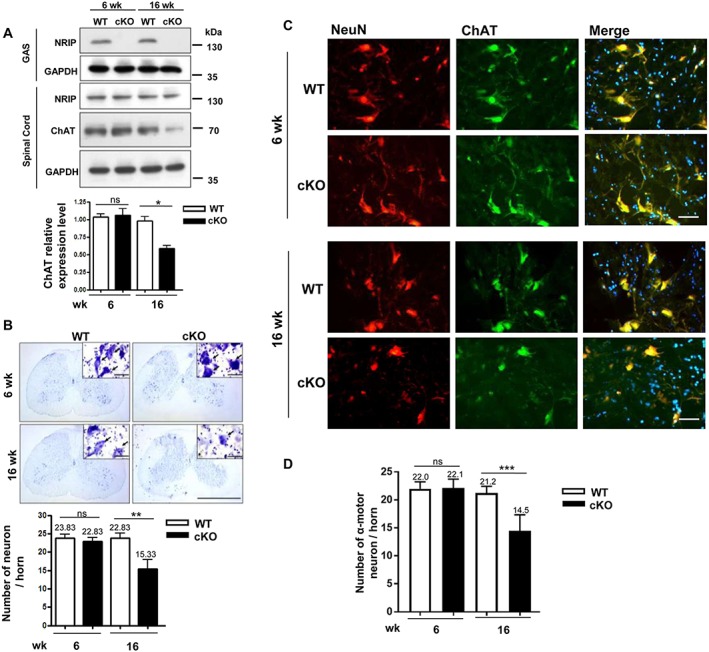
The loss of motor neurons in conditional knockout (cKO) mice. (A) Western blot analysis of the protein expression of a nuclear receptor interaction protein (NRIP) and motor neuron‐specific protein, choline acetyltransferase (ChAT), in gastrocnemius muscle (GAS) and spinal cord of wild‐type (WT) and cKO mice at age 6 and 16 weeks. n = 3 for each group. Lower panel: quantification of ChAT expression (n = 3). (B) Neuronal number measurement in spinal anterior horn by Nissl staining. At least 12 sections of the lumbar spinal cord with distance 120 μm apart were analysed for each animal. Right upper panel: nucleolus located within the nucleus, surrounded by light blue‐stained cytoplasm. n = 6 for each group. Scale bars, 200 μm. Lower panel: quantification of neuronal number per spinal anterior horn. (C) Immunofluorescence assay of neuronal DNA binding protein (NeuN) (red), ChAT (green), and DAPI (blue) expression and co‐expression in lumbar spinal cord by confocal microscopy. Size >500 μm2 was counted as α‐motor neurons. Scale bar: 100 μm. (D) Quantification of motor neuron number per spinal anterior horn shows lower number in cKO than WT mice at age 16 weeks but not 6 weeks. n = 5 for each group. Data are mean ± SEM by one‐way analysis of variance. * P < 0.05, ** P < 0.01, and *** P < 0.001. ns, not significant.
To precisely identify motor neurons, we labelled them with antibodies for NeuN and ChAT on IFA. Anterior horn cells with both NeuN and ChAT immunoactivity and >500 μm2 were considered α‐motor neurons.32 Motor neuron number was lower for cKO than WT mice at age 16 weeks (14.5 vs. 21.2 per anterior horn, P < 0.001) but not 6 weeks (Figure 2C and 2D). In addition, soma area was smaller, and perimeter of spinal motor neurons was shorter for cKO than WT mice at age 16 weeks ( Figure S4). However, motor neuron number (Figure 2D) and soma area and its perimeter were similar between cKO and WT mice at age 6 weeks (data not shown). Thus, muscle‐restricted NRIP KO resulted in motor neuron degeneration with age, as shown by small neuronal size and reduced neuronal survival.
Abnormal architecture of the neuromuscular junction in conditional knockout mice
We analysed the architecture of the NMJ in cKO and WT mice by using α‐BTX (venom of Bungarus multicinctu) to label the AChR of skeletal muscles and synaptophysin and neurofilament antibodies to label presynaptic nerve terminals (Figure 3A). The area of BTX‐labelled AChR represented the NMJ area. In WT mice, the NMJ area in soleus muscle was significantly enlarged from age 6 to 16 weeks (282.9 vs. 434.6 μm2) (Figure 3B). The NMJ area was similar between cKO and WT mice at age 6 weeks but was significantly smaller in cKO than WT mice at 16 weeks (329.0 vs. 434.6 μm2).
Figure 3.
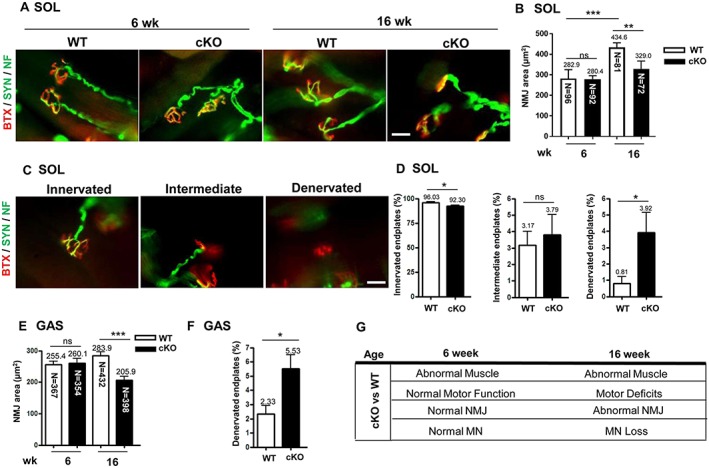
The abnormal architecture of the neuromuscular junction (NMJ) with axonal denervation in conditional knockout (cKO) mice. (A) Fluorescent images of NMJ in soleus muscle (SOL) from mice at age 6 and 16 weeks. Immunofluorescence assay with α‐bungarotoxin (BTX) [red, for acetylcholine receptor (AChR)] and anti‐synaptophysin (Syn) and anti‐neurofilament (NF) antibodies (both green, for nerve terminals) used for analysis of NMJ area and the patterns of axonal innervation. Scale bar: 10 μm. (B) Quantification of NMJ area in soleus muscle in mice at age 6 and 16 weeks. The area of BTX‐labelled AChR represents the NMJ area. n = 5 for 6 week and n = 7 for 16 week mice. The number in each bar indicates the number of examined NMJs. (C) Patterns of axonal innervation in mice at age 16 weeks. Three NMJ innervation patterns are indicated, including innervated (well overlapping of AChR and nerve terminals), intermediate (nerve terminal adjacent to AChR without overlapping), and denervated (no nerve terminal adjacent or overlapped to AChR). n = 3 for each group. Scale bar: 10 μm. (D) Proportion of innervated, intermediate, and denervated endplates in soleus muscle from mice at age 16 weeks. (E) Quantification of NMJ area in gastrocnemius muscle (GAS) from mice at age 16 weeks and (F) proportion of denervated endplates in gastrocnemius muscle. n = 3 for each group. (G) Pathological and motor functional results in cKO mice at age 6 and 16 weeks. Data are mean ± SEM. Panels D and F: by Student's t‐test; panels B and E: one‐way analysis of variance. * P < 0.05, ** P < 0.01, and *** P < 0.001. ns, not significant; WT, wild‐type.
The muscle innervation patterns of motor endplates were classified as ‘innervated’, ‘intermediate’ or ‘denervated’ according to the staining patterns of AChR and nerve terminals24 (Figure 3C). The proportion of innervated motor endplates was lower (92 vs. 96%, P = 0.020) and that of denervated endplates higher (3.9 vs. 0.8%, P = 0.037) in cKO than WT soleus muscles at age 16 weeks (Figure 3D). To further confirm the NMJ abnormality, we examined the NMJ in gastrocnemius muscles. The NMJ area did not differ between cKO and WT gastrocnemius muscles at age 6 weeks but was significantly smaller in cKO than WT gastrocnemius muscles at age 16 weeks (205.9 vs. 283.9 μm2) (Figure 3E), and the proportion of denervated endplates was higher (5.5 vs. 2.3%, P < 0.001) (Figure 3F). In summary, at age 6 weeks, muscle‐restricted NRIP cKO mice showed muscular abnormality without abnormal NMJ architecture, motor neuron number, or motor function (Figure 3G). However, at age 16 weeks, they showed muscular abnormality as well as abnormal NMJ integrity and motor neuron degeneration, along with impaired motor function.
Low myonucleus number at the neuromuscular junction in muscle‐restricted nuclear receptor interaction protein knockout mice
We further investigated the pathogenic mechanisms of abnormal NMJ architecture and motor neuron degeneration in cKO mice. A high nucleus number at the postsynaptic area is essential to maintain postsynaptic components of NMJ.33, 34 We thus evaluated the role of NRIP on nucleus recruitment in NMJ of skeletal muscles. SUN2 is a myonucleus marker that can be used to distinguish the nuclei of myocytes from other cell types, such as fibroblasts, Schwann cells, and blood cells.35 We co‐stained the myonucleus with DAPI and anti‐SUN2 antibody at the postsynaptic region, then quantified myonuclei (Figure 4A). At age 16 weeks, the number of postsynaptic nuclei was lower in cKO than WT mice (2.2 vs. 4.2 per NMJ, P = 0.011) (Figure 4B) but was similar at age 6 weeks. Nuclear receptor interaction protein likely contributes to myonucleus recruitment in the NMJ. Because the NMJ region has high transcriptional activity,34 muscle‐restricted NRIP KO leading to fewer myonuclei at the NMJ might affect gene expression at the NMJ.
Figure 4.
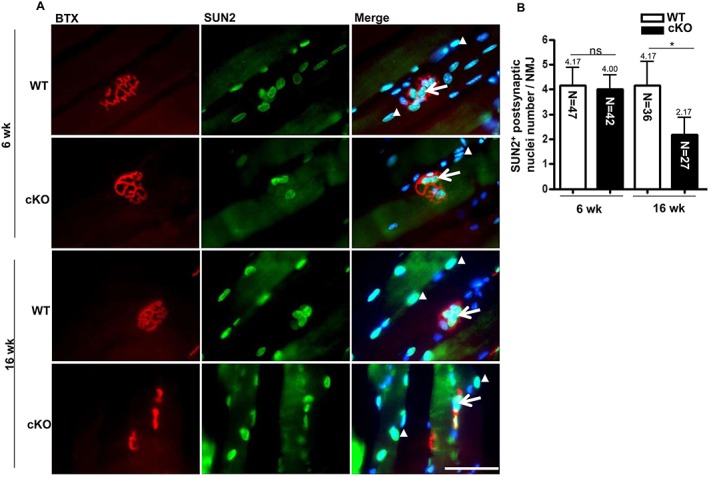
Decrease in postsynaptic nucleus number in conditional knockout (cKO) mice. (A) Staining of postsynaptic and extrasynaptic nucleus in longitudinal sections of muscles from wild‐type (WT) and cKO mice at age 6 and 16 weeks. Acetylcholine receptor at the neuromuscular junction (NMJ) was detected by α‐bungarotoxin (BTX) (red), and myonuclei were detected by anti‐SUN2 antibody (green) with DAPI (blue) for nucleus staining. Arrows indicate postsynaptic nuclei and arrowheads extrasynaptic nuclei. Scale bar, 100 μm. (B) Number of SUN2‐positive nuclei per NMJ. The number in each bar indicates the number of examined NMJs. n = 3 for each group. Data are mean ± SEM by one‐way analysis of variance. * P < 0.05. ns, not significant.
Nuclear receptor interaction protein regulates neuromuscular junction via myogenin expression at postsynaptic sites of muscle
Gain or loss of certain muscle‐related factors may cause retrograde axonal dying back and subsequent motor neuron death,23 which might be the mechanism of motor neuron degeneration under NRIP deficiency in the skeletal muscles of our cKO mice. We screened some published muscle‐derived factors, such as NT4, insulin‐like growth factor‐1, BMP‐4, and myogenin, which are able to protect spinal motor neurons against degeneration ( Figure S5A). Transcriptional analysis showed that myogenin was a potential candidate for NRIP regulation ( Figure S5B), with lower myogenin expression in muscles of cKO than WT mice. Myogenin is a transcriptional factor for myogenesis in muscle.36, 37, 38, 39 Therefore, we hypothesized that NRIP might locate at postsynaptic regions of NMJ to promote myogenin expression and thereby maintain the integrity of NMJ and retrogradely support motor neuron survival.
We examined the relationship between NRIP, myogenin, and AChR in skeletal muscles. Immunofluorescence assay with NRIP antibody and BTX revealed that NRIP was localized around the AChR and predominately located at the NMJ in WT mice at age 16 weeks (Figure 5A). As well, myogenin was co‐localized with NRIP, expressed around the AChR and predominantly concentrated at the NMJ (Figure 5B and 5C). Hence, both NRIP and myogenin proteins were highly concentrated in NMJ regions in normal mice. However, the proportion of myogenin‐immunoactive AChR was significantly lower in cKO than WT mice at age 16 weeks (5.98 vs. 21.38%, P = 0.002) and at age 6 weeks ( Figure S6A and S6B). To further validate the myogenin expression in muscles, we specifically extracted RNA from synaptic and extrasynaptic regions by laser captured microdissection (Figure 5D; upper panel). Myogenin transcript expression was higher in the postsynaptic than extrasynaptic region in both WT and cKO mice at age 16 weeks (Figure 5D; lower panel), with no difference in myogenin expression at the extrasynaptic regions between cKO and WT mice (P = 0.999) but lower myogenin expression at the synaptic regions in cKO than WT mice (P < 0.001). Collectively, NRIP deficiency at the NMJ of skeletal muscle is associated with down‐regulation of myogenin expression at the NMJ.
Figure 5.
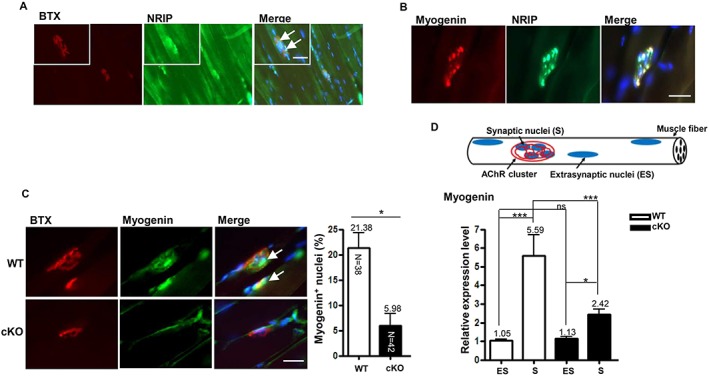
Low myogenin expression at the neuromuscular junction (NMJ) in conditional knockout (cKO) mice at age 16 weeks. (A) Immunofluorescence assay with anti‐nuclear receptor interaction protein (NRIP) antibody (green), bungarotoxin (BTX) (red), and DAPI (blue) of NRIP localized around acetylcholine receptor (AChR) in longitudinal sections of soleus of wild‐type (WT) mice (arrows indicate co‐localization of NRIP and AChR), (B) co‐localization of NRIP (green) and myogenin (red) at NMJ with clustered nuclei (DAPI, blue) in soleus muscle WT mice, and (C) proportion of myogenin (green)‐immunoactive AChR (red) in soleus muscle of cKO and WT mice (arrows indicate co‐localization of myogenin and AChR cluster). Scale bars, 100 μm. The number in each bar indicates the number of examined NMJs. n = 3 for each group. (D) Gastrocnemius muscles of mice at age 16 weeks subjected to laser capture microdissection. RNA was extracted from extrasynaptic and synaptic regions, and the amount of myogenin transcript was analysed by real‐time quantitative PCR. Data were normalized to β‐actin level. n = 7 for each group. Data are mean ± SEM. Panel B: by Student's t‐test; panel D: one‐way analysis of variance. * P < 0.05, ** P < 0.01, and *** P < 0.001.
Intramuscular myogenin gene transfer rescues abnormal phenotypes in conditional knockout mice
To demonstrate that the spinal motor neuron degeneration in NRIP cKO mice directly resulted from down‐regulation of myogenin in skeletal muscles, we injected an Ad‐myogenin into bilateral gastrocnemius muscles of cKO mice at age 6 weeks, followed by pathology analysis at age 9 and 16 weeks (3 and 10 weeks after treatment) (Figure 6A). Nuclear receptor interaction protein cKO mice with intramuscular Ad‐shLuc (control) treatment were a control. Myogenin protein expression in injected muscles was higher in Ad‐myogenin‐treated cKO mice than control‐treated cKO and WT mice 3 weeks after treatment (Figure 6B). As well, the proportion of myocyte nuclei showing immunoactive myogenin signals was higher in Ad‐myogenin‐treated than control muscle at age 9 weeks (Figure 6C). With overexpression of muscular myogenin, the abnormal oxidative muscle function in cKO mice, by counting oxidative muscle fibres and SDH staining of soleus muscles, was significantly improved 3 weeks after treatment (Figure 6D and 6E). In addition, the area of the NMJ was significantly larger in Ad‐myogenin‐treated than control‐treated cKO gastrocnemius muscles at age 9 and 16 weeks (Figure 6F). The proportion of denervating NMJ did not differ between two groups at age 9 weeks but was significantly lower in cKO mice at age 16 weeks with Ad‐myogenin treatment (Figure 6G). With normalization of muscle and NMJ defects in cKO mice after Ad‐myogenin treatment for 10 weeks, the number of spinal motor neurons was higher in the lumbar cord on ChAT and NeuN immunofluorescence staining of cKO mice at age 16 weeks (14.8 vs. 19.3, Figure 6H). To further examine whether there is the autocrine regulatory loop between NRIP and myogenin, we then measured NRIP RNA expression in Ad‐myogenin‐infected gastrocnemius muscles and C2C12 cells by RT‐qPCR. The results showed that myogenin could not control NRIP expression ( Figure S7). Taken together, overexpression of myogenin in skeletal muscles can rescue the abnormal muscle defect, disarranged NMJ architecture, and motor neuron degeneration produced with muscle‐restricted NRIP KO in mice.
Figure 6.
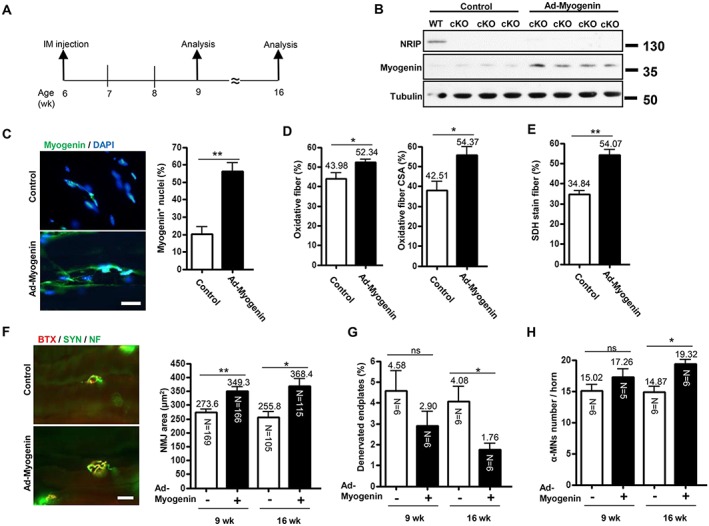
Intramuscular myogenin gene transfer rescues phenotypes of conditional knockout (cKO) mice. (A) Analysis of mice at age 9 and 16 weeks after adenovirus encoding myogenin (Ad‐myogenin) and Ad‐shLuc (control) treatment by intramuscular (i.m.) injection into bilateral gastrocnemius muscle from cKO mice at age 6 weeks. (B) Age 9 weeks: western blot analysis of myogenin and nuclear receptor interaction protein (NRIP) levels in gastrocnemius muscle from cKO mice infected with Ad‐myogenin (n = 4) or control (n = 3), with tubulin as an internal control. (C) Age 9 weeks: immunofluorescence assay of myogenin expression (green) in gastrocnemius muscle from cKO mice with Ad‐myogenin (n = 6) or Ad‐shLuc (n = 6) treatment. Scale bar, 20 μm. (D) Age 9 weeks: quantification of immunohistochemical staining of proportion of oxidative fibres and oxidative fibre cross‐section area (CSA) for slow myosin in soleus muscle from cKO mice with Ad‐myogenin treatment (n = 6) or control (n = 6). (E) Age 9 weeks: proportion of succinate dehydrogenase (SDH)‐stained myofibers in soleus muscle from cKO mice with Ad‐myogenin treatment (n = 6) or control (n = 4). (F) Immunofluorescence assay with α‐bungarotoxin (BTX) (red) and anti‐synaptophysin (Syn) and anti‐neurofilament (NF) antibodies (green) for analysis of neuromuscular junction (NMJ) area and patterns of axonal innervation in gastrocnemius muscle from cKO mice at age 16 weeks. Right: quantification of NMJ area in gastrocnemius muscle from cKO mice at age 9 and 16 weeks. The number in each bar indicates the number of examined NMJs. (G) Proportion of denervated endplates in gastrocnemius muscle from cKO mice. (H) Sections (30 μm) of lumbar spinal cord were stained for choline acetyltransferase and neuronal DNA binding protein for α‐motor neuron labelling. The number of α‐motor neurons per spinal anterior horn was measured. (G and H): the N in each bar indicates the number of mice. Data are mean ± SEM. Panels C–E: by Student's t‐test. Panels F–H: one‐way analysis of variance. * P < 0.05 and ** P < 0.01. ns, not significant.
Discussion
Nuclear receptor interaction protein might participate in various cellular functions, including regulating muscle contraction, promoting cell growth, and inhibiting protein degradation.1, 3, 4, 10 Here, we provide a novel function of NRIP and a novel muscle‐derived neuronal supporting factor indicating that muscular NRIP expression is essential for spinal motor neuron support. We previously generated global NRIP KO mice, which showed loss of muscle strength, susceptibility to muscle fatigue, and impaired adaptive exercise performance.4 In this study, we generated muscle‐restricted NRIP KO mice (cKO) to examine the role of NRIP on the neuromuscular system. The NRIP cKO mice showed muscular abnormality, with impaired motor performance (Figure 1), which could also be detected in global NRIP KO mice. However, muscle‐restricted NRIP KO mice also displayed spinal motor neuron degeneration with abnormal NMJ integrity at adult age (Figure 2 and Figure 3). We found that NRIP and myogenin co‐localized at the NMJ in skeletal muscle, and myogenin expression was reduced at the muscle NMJ in cKO mice. Overexpression of muscular myogenin rescued the abnormal NMJ architecture and prevented motor neuron death in cKO mice. Muscular NRIP could be a novel trophic factor supporting spinal motor neurons by stabilizing the NMJ via myogenin expression.
In our cKO mouse model produced by a Cre/loxP recombination method, the Cre recombinase transgene was driven by a muscle‐specific MCK promoter. Because MCK‐Cre expression starts at the embryonic stage,40 cKO mice would not have NRIP expression in muscular tissues since before birth. With the absence of muscular NRIP expression during early life, muscular abnormality could be noted at age 6 weeks, when there is still no abnormality in NMJ architecture, motor neuron number, or motor functional performance. At age 16 weeks, muscular abnormality as well as abnormal NMJ integrity, motor neuron degeneration, and impaired motor function was detected (Figure 3G). Specifically, adult cKO mice showed reduced motor neuron number and small neuronal size in the lumbar spinal cord as well as denervating change, small motor endplates, and decreased myonuclei number at the NMJ in skeletal muscles. Similarly, in an ALS mouse model, muscle‐restricted G93A‐mutant superoxide dismutase 1 transgenic mice shows skeletal muscle weakness at a young age, with development of motor neuron degeneration at an older age.20 In spinal neurons of SBMA models, knock‐in of muscle‐expressing mutant AR causes early myopathy, followed by the late development of neuronal intranuclear inclusions.41 Therefore, the muscle‐related non‐cell‐autonomous toxicity of MNDs in motor neurons may require a period of time to accumulate before retrograde axonal dying back and degeneration of spinal motor neurons.22, 42, 43, 44
Nuclear receptor interaction protein expression was depleted only in muscle tissues but was normal in spinal cords of cKO mice, which exhibited 32% motor neuron loss at age 16 weeks (Figure 2), so NRIP may regulate certain muscle‐derived factors to protect motor neuron survival. Some important muscle‐derived factors, such as neurotrophin‐4, insulin‐like growth factor‐1, BMP‐4, and myogenin, can protect spinal motor neurons against degeneration.24, 25, 27, 28, 45, 46 By screening these factors, we found myogenin as a candidate for NRIP regulation, with lower myogenin expression in cKO than WT muscles, and further confirmed the low myogenin expression in the NMJ by laser microdissection of RNA from synaptic and extrasynaptic regions of muscles (Figure 5C and 5D). These findings indicate that NRIP deficiency at the NMJ of skeletal muscle is associated with down‐regulated myogenin expression at the NMJ. At age 6 weeks, cKO mice showed reduced myogenin expression at postsynaptic regions of the NMJ ( Figure S6A and S6B) but with normal NMJ size (Figure 3B and 3E). These findings further support the idea of the accumulation of noxious effects (less myogenin) triggering motor neuron degeneration with age.
Myogenin can induce the cluster of AChR to form the NMJ.26, 47 At age 16 weeks, NRIP cKO mice showed disintegrated NMJ, with low myogenin expression at the NMJ, as compared with their age‐matched controls (Figure 3). Hence, NRIP may regulate NMJ via myogenin. Both NRIP and myogenin proteins were highly concentrated in NMJ regions (Figure 5). Myogenin is a muscle‐specific transcription factor that controls myoblast differentiation and AChR gene expression and receptor clustering.48 In a mouse model of ALS, myogenin could support motor neuron survival and maintain muscle innervation.24 Therefore, NRIP likely localizes at postsynaptic regions of NMJ to promote myogenin expression, which can maintain the integrity of NMJ and retrogradely support motor neuron survival.
In addition to maintaining NMJ integrity via myogenin expression, NRIP may also regulate NMJ by acting as a scaffold protein to recruit more nuclei within the NMJ area to affect its formation. In general, normal NMJ contains about three to eight nuclei (synaptic nuclei).35 Here, the deprivation of NRIP in skeletal muscle of cKO mice reduced the myonuclei number at the postsynaptic region in adult but not young mice (Figure 4). Hence, NRIP can recruit more nuclei to form synaptic clusters in postsynaptic regions. Synaptic nuclei transcribe specialized genes to maintain the postsynaptic components of the NMJ.33, 34 The loss of synaptic nuclei might reduce the expression of certain proteins, not only myogenin but also AChR, MuSK and utrophin, and then eliminate the normal functions of the NMJ.48, 49 Currently anchoring proteins such as Syne or desmin can associate with the nuclear envelope for nuclei migration and position.35, 50 Additionally, deprivation of collagen XIII or laminin 2 in skeletal muscles reportedly would destroy postsynaptic AChR clusters resulting in destabilizing presynaptic action via the trans‐synaptic extracellular matrix.51, 52 Further investigation is needed to determine whether and how NRIP anchors nuclei into synapse regions. Nuclear receptor interaction protein‐related NMJ support may involve two mechanisms: (i) induction of myogenin expression at NMJ and (ii) participation in anchoring myonuclei into NMJ areas.
Muscle‐restricted NRIP KO was associated with both down‐regulation of myogenin in skeletal muscles and motor neuron degeneration in the spinal anterior horn with NMJ disarrangement. To investigate the direct relationship between muscular myogenin and spinal motor neuron survival, we overexpressed myogenin in NRIP cKO gastrocnemius muscles by intramuscular Ad‐myogenin injection. Although intramuscular inoculation of certain adenoviral vectors might lead to retrograde gene expression in spinal motor neurons,53, 54 we found myogenin overexpressed in muscles but not the spinal cord after Ad‐myogenin treatment (data not shown). On muscle‐restricted overexpression of myogenin in mice, muscle defects were improved and the NMJ architecture restored. Notably, intramuscular Ad‐myogenin‐treated cKO mice showed less spinal motor neuron degeneration over time (Figure 6). These results indicate that muscular myogenin is able to compensate, at least in part, for the defects in muscles, NMJs, and spinal motor neurons caused by muscle‐restricted NRIP KO. Therefore, myogenin is likely a downstream factor of NRIP in skeletal muscles to support the muscular function and also plays a role on spinal motor neuron survival, probably via maintenance of the normal integrity of the NMJ. The suggestion is consistent with the therapeutic effects of Ad‐myogenin treatment in G93A ALS mice,24 which further supports that myogenin gene transfer in muscle can affect motor neuron survival.
In summary, deprivation of NRIP in skeletal muscles decreases myogenin expression at the NMJ, possibly leading to abnormal NMJ formation, denervation of AChR, and subsequent loss of spinal motor neurons. These findings provide a novel function of NRIP in the neuromuscular system. In addition, muscle‐related non‐autonomous mechanisms play an important role in the pathogenesis of MNDs.21, 41, 55 Considering the trophic effects of NRIP on spinal motor neurons, muscular NRIP–myogenin signalling might be a potential therapeutic target for various MNDs.
Author contributions
S.‐L.C., L.‐K.T., and H.‐H.C. conceived and designed the experiments. H.‐H.C., K.‐Y.L., T.‐C.W., Y.‐C.H., Y.‐H.H., and S.‐W.C. performed the experiments. H.‐H.C., L.‐K.T., P.‐Y.W., Y.‐P.T., and S.‐L.C. analysed the data. S.‐L.C. contributed the reagents, materials, and analysis tools. S.‐L.C., L.‐K.T., and H.‐H.C., contributed to the writing of the manuscript.
Conflict of interest
None declared.
Supporting information
Figure S1. Generation of muscle‐restrictedNRIP knockout (cKO) mice.(A)2 Schematic illustration of the genomic structure of wild‐type (WT) NRIP, loxP‐floxed 3 NRIP, and NRIP‐deleted alleles. The loxP‐floxed allele contains a neo cassette with 4 two flanked loxP sequences in intron 1 and a loxP sequence in intron 2. NRIPexon 2 5 would be deleted by Cre recombinase driven by MCKpromoter.(B) Genotyping by 6 PCR. For floxed NRIP genotyping (upper), GU and JD primers (Supplementary 7 information, TableS1) were used. The band with smaller size (about 680 bp) 8 representsWT NRIP without loxPsequence and the largerone (about 720 bp) 9 NRIP‐containing loxPsequence. NRIPflox/+ represents mice with one WT allele and 10 one floxed allele, NRIPflox/floxmice with two floxed alleles, and NRIP+/+mice with two 11 WT alleles. For genotyping of Cre recombinase (lower), oIMR1085and oIMR6754 12 primers (Supplementary information, Table S1) were used for Cre recombinase loci 13 detection. The oIMR8744 and oIMR8745 primers wereTcrd DNA control primers 14 (The Jackson Laboratory, Supplementary information, Table S1). The band with 15 smaller size (about 200 bp) was used as internal positive control. Mice with16 loxP‐floxed NRIP and Cre‐positive bands were cKO mice. Control wild type (WT) 17 mice were NRIPflox/flox; MCK‐Cre‐.
Figure S2. Muscular abnormalityin cKO mouse soleus muscle at age 6 weeks.(A)20 Immunohistochemistry of the expression of oxidative fibre (slow myosin, n = 3). (B)21 COX (dark brown) expression. n = 3. (C) SDH (dark purple) expression. Scale bar, 100 22 μm. n = 3 for each group. (D) Rotarod maintenance testat age6 weeks (n = 7 for each 23 group). (E) Grip force analysisat age6 weeks (n = 7 for each group). Dataare 24 mean ± SEM by Student t‐test.*P < 0.05
Figure S3. Theassessment of degenerated muscles. (A) HEfor soleus muscle 26 morphology between WT and NRIP cKO at age 16 weeks. There was no 27 difference forcentral located nuclei. Scale bar, 100 μm. (B) CD45 expression for 28 infiltration of innate immune cells. Scale bar, 100 μm. (C) Embryonic myosin 29 expression (eMyHC) for regenerative myofiber marker (n = 3 for each group).30 Scale bar, 100 μm.(D) EBD stain (red) for fibrosis myofiber; Scale bar, 50 μm. All 31 assays: n = 3 for each genotype.
Figure S4.The size and perimeter of spinal motor neurons in in spinal anterior horn of 34 cKO and WT mice at age 16 weeks. Quantification of (A) size of α‐motor neurons 35 (MN) and (B) MN perimeter. n = 3 for each group. Dataare mean ± SEM byStudent t 36 test. *P < 0.05, **P < 0.01.
Figure S5. Screen for muscle‐derived factors that may support motor neuron survival 39 in cKO and WT mice at age 16 weeks. (A) The candidate genes include NT4, IGF‐1, 40 IGF‐2, IGFBP‐4, IGFBP‐6, BMP4, myogenin, and AChRɛ.(B)RT‐qPCR analysis of 41 mRNA expression ingastrocnemius muscles (WT: n = 6, cKO: n = 7). Dataare 42 mean ± SEM byStudent t test.***P < 0.001.
Figure S6. The expression of myogenin at neuromuscular junction in cKO and WT 45 mice at age 6 weeks.(A) Immunofluorescence assay of the proportion of 46 acetylcholine receptors (red) with myogenin immunoactivity (green). Arrow indicates 47 co‐localization of myogenin and AChR cluster. (B) Quantification of (A). The N 48 indicates the examined NMJ number for n = 3 of each genotype. Scale bar, 100 μm. 49 Data are mean ± SEM by Student t test. *P < 0.05. (C) Myogenin expression is activated by NRIP overexpressionin C2C12 cells. Immunofluorescence assay of51 proportion of C2C12 cells withnuclear myogenin (green) expression after treatment 52 with Ad‐NRIP andAd‐GFP (MOI 10) and under differentiation for 2 days. DAPI 53 (blue). Scale bar, 20 μm. Dataare mean ± SEM (n = 3 experiments) byStudent t test. 54 **P < 0.01
Figure S7. The mRNA of NRIP in Ad‐Myogenin infected gastrocnemius and 57 C2C12 cellsby using RT‐qPCR. (A) The RNA extraction from Ad‐Myogenin 58 infected gastrocnemius (n = 3 for each group).(B) The RNA from Ad‐Myogenin 59 infected C2C12 cells (n = 3 for each group).
Acknowledgements
We appreciate Ms Laura Smales for her English editing. Additionally, muscle‐specific NRIP KO mice (cKO) were generated by the Transgenic Mouse Core Facility in National Taiwan University Center for Genomic Medicine. The rotarod and grip force were conducted in the Behavior Core Laboratory of the Neurobiology and Cognitive Science Center; the confocal image analysis at The Imaging Core at the First Core Lab; and the paraffin section preparation in the Animal Center, National Taiwan University. We also thank Dr Shu‐Wha Lin for advice on generating NRIP cKO and Mr Po‐Yuen Wu for technical assistance with the generation of NRIP cKO.
This work was supported by the Ministry of Science and Technology (MOST 104‐2320‐B‐002‐053‐MY3; MOST 105‐2320‐B‐002‐039‐MY3), the National Health Research Institutes (NHRI‐105‐1053ISI), National Taiwan University (10R891903), and the Excellent Translational Medicine Research Projects of National Taiwan University College of Medicine and National Taiwan University Hospital (106R39012). The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle.56
Chen, H.‐H. , Tsai, L.‐K. , Liao, K.‐Y. , Wu, T.‐C. , Huang, Y.‐H. , Huang, Y.‐C. , Chang, S.‐W. , Wang, P.‐Y. , Tsao, Y.‐P. , and Chen, S.‐L. (2018) Muscle‐restricted nuclear receptor interaction protein knockout causes motor neuron degeneration through down‐regulation of myogenin at the neuromuscular junction. Journal of Cachexia, Sarcopenia and Muscle, 9: 771–785. 10.1002/jcsm.12299.
References
- 1. Chen PH, Tsao YP, Wang CC, Chen SL. Nuclear receptor interaction protein, a coactivator of androgen receptors (AR), is regulated by AR and Sp1 to feed forward and activate its own gene expression through AR protein stability. Nucleic Acids Res 2008;36:51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tsai TC, Lee YL, Hsiao WC, Tsao YP, Chen SL. NRIP, a novel nuclear receptor interaction protein, enhances the transcriptional activity of nuclear receptors. J Biol Chem 2005;280:20000–20009. [DOI] [PubMed] [Google Scholar]
- 3. Chang SW, Tsao YP, Lin CY, Chen SL. NRIP, a novel calmodulin binding protein, activates calcineurin to dephosphorylate human papillomavirus E2 protein. J Virol 2011;85:6750–6763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen HH, Chen WP, Yan WL, Huang YC, Chang SW, Fu WM, Su MJ, Yu IS, Tsai TC, Yan YT, Tsao YP, Chen SL. NRIP is newly identified as a Z‐disc protein, activating calmodulin signaling for skeletal muscle contraction and regeneration. J Cell Sci 2015;128:4196–4209. [DOI] [PubMed] [Google Scholar]
- 5. Zhang Y, Ye J, Chen D, Zhao X, Xiao X, Tai S, Yang W, Zhu D. Differential expression profiling between the relative normal and dystrophic muscle tissues from the same LGMD patient. J Transl Med 2006;4:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ehret GB, O'Connor AA, Weder A, Cooper RS, Chakravarti A. Follow‐up of a major linkage peak on chromosome 1 reveals suggestive QTLs associated with essential hypertension: GenNet study. Eur J Hum Genet 2009;17:1650–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cheung CL, Chan BY, Chan V, Ikegawa S, Kou I, Ngai H, Smith D, Luk KD, Huang QY, Mori S, Sham PC, Kung AW. Pre‐B‐cell leukemia homeobox 1 (PBX1) shows functional and possible genetic association with bone mineral density variation. Hum Mol Genet 2009;18:679–687. [DOI] [PubMed] [Google Scholar]
- 8. Shi Y, Li Z, Xu Q, Wang T, Li T, Shen J, Fengyu Z, Jianhua C, Guoquan Z, Weidong J, Baojie L, Yifeng X, Dengtang L, Peng W, Ping Y, Benxiu L, Wensheng S, Chunling W, Shengying Q, Guang H. Common variants on 8p12 and 1q24.2 confer risk of schizophrenia. Nat Genet 2011;43:1224–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chang SW, Su CH, Chen HH, Huang CW, Tsao LP, Tsao YP, Chen SL. DDB2 is a novel AR interacting protein and mediates AR ubiquitination/degradation. Int J Biochem Cell Biol 2012;44:1952–1961. [DOI] [PubMed] [Google Scholar]
- 10. Chen HH, Fan P, Chang SW, Tsao YP, Huang HP, Chen SLNRIP. DCAF6 stabilizes the androgen receptor protein by displacing DDB2 from the CUL4A‐DDB1 E3 ligase complex in prostate cancer. Oncotarget 2017;8:21501–21515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Klein CJ, Wu Y, Vogel P, Goebel HH, Bonnemann C, Zukosky K, Botuyan MV, Duan X, Middha S, Atkinson EJ, Mer G, Dyck PJ. Ubiquitin ligase defect by DCAF8 mutation causes HMSN2 with giant axons. Neurology 2014;82:873–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dion PA, Daoud H, Rouleau GA. Genetics of motor neuron disorders: new insights into pathogenic mechanisms. Nat Rev Genet 2009;10:769–782. [DOI] [PubMed] [Google Scholar]
- 13. Polymenidou M, Cleveland DW. Motor neuron disease: the curious ways of ALS. Nature 2008;454:284–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sun S, Ling SC, Qiu J, Albuquerque CP, Zhou Y, Tokunaga S, Li H, Qiu H, Bui A, Yeo GW, Huang EJ, Eggan K, Zhou H, Fu XD, Lagier‐Tourenne C, Cleveland DW. ALS‐causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1‐snRNP. Nat Commun 2015;6:6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Taylor JP, Brown RH Jr, Cleveland DW. Decoding ALS: from genes to mechanism. Nature 2016;539:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Prior TW, Finanger E. Spinal muscular atrophy. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH , Stephens K, Amemiya A, editors. GeneReviews(R) Seattle (WA) 1993. [PubMed] [Google Scholar]
- 17. Boillee S, Cleveland DW. Gene therapy for ALS delivers. Trends Neurosci 2004;27:235–238. [DOI] [PubMed] [Google Scholar]
- 18. Tsitkanou S, Della Gatta PA, Russell AP. Skeletal muscle satellite cells, mitochondria, and microRNAs: their involvement in the pathogenesis of ALS. Front Physiol 2016;7:403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Haidet‐Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, Song S, Likhite S, Murtha MJ, Foust KD, Rao M, Eagle A, Kammesheidt A, Christensen A, Mendell JR, Burghes AH, Kaspar BK. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol 2011;29:824–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wong M, Martin LJ. Skeletal muscle‐restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum Mol Genet 2010;19:2284–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cortes CJ, Ling SC, Guo LT, Hung G, Tsunemi T, Ly L, Tokunaga S, Lopez E, Sopher BL, Bennett CF, Shelton GD, Cleveland DW, La Spada AR. Muscle expression of mutant androgen receptor accounts for systemic and motor neuron disease phenotypes in spinal and bulbar muscular atrophy. Neuron 2014;82:295–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cary GA, La Spada AR. Androgen receptor function in motor neuron survival and degeneration. Phys Med Rehabil Clin N Am 2008;19:479–494, viii. [DOI] [PubMed] [Google Scholar]
- 23. Kanning KC, Kaplan A, Henderson CE. Motor neuron diversity in development and disease. Annu Rev Neurosci 2010;33:409–440. [DOI] [PubMed] [Google Scholar]
- 24. Park KH, Franciosi S, Leavitt BR. Postnatal muscle modification by myogenic factors modulates neuropathology and survival in an ALS mouse model. Nat Commun 2013;4:2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kablar B, Belliveau AC. Presence of neurotrophic factors in skeletal muscle correlates with survival of spinal cord motor neurons. Dev Dyn 2005;234:659–669. [DOI] [PubMed] [Google Scholar]
- 26. Taniguchi M, Kurahashi H, Noguchi S, Fukudome T, Okinaga T, Tsukahara T, Tajima Y, Ozono K, Nishino I, Nonaka I, Toda T. Aberrant neuromuscular junctions and delayed terminal muscle fiber maturation in alpha‐dystroglycanopathies. Hum Mol Genet 2006;15:1279–1289. [DOI] [PubMed] [Google Scholar]
- 27. Funakoshi H, Belluardo N, Arenas E, Yamamoto Y, Casabona A, Persson H, Ibáñez CF. Muscle‐derived neurotrophin‐4 as an activity‐dependent trophic signal for adult motor neurons. Science 1995;268:1495–1499. [DOI] [PubMed] [Google Scholar]
- 28. Chou HJ, Lai DM, Huang CW, McLennan IS, Wang HD, Wang PY. BMP4 is a peripherally‐derived factor for motor neurons and attenuates glutamate‐induced excitotoxicity in vitro. PLoS One 2013;8:e58441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen CY, Tsai MS, Lin CY, Yu IS, Chen YT, Lin SR, Juan LW, Chen YT, Hsu HM, Lee LJ, Lin SW. Rescue of the genetically engineered Cul4b mutant mouse as a potential model for human X‐linked mental retardation. Hum Mol Genet 2012;21:4270–4285. [DOI] [PubMed] [Google Scholar]
- 30. Bruning JC, Michael MD, Winnay JN, Hayashi T, Horsch D, Accili D, Goodyear LJ, Kahn CR. A muscle‐specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell 1998;2:559–569. [DOI] [PubMed] [Google Scholar]
- 31. Johnson JE, Wold BJ, Hauschka SD. Muscle creatine kinase sequence elements regulating skeletal and cardiac muscle expression in transgenic mice. Mol Cell Biol 1989;9:3393–3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Friesea A, Kaltschmidtb JA, Ladlea DR, Sigrista M, Jessellb TM, Arber S. Gamma and alpha motor neurons distinguished by expression of transcription factor Err3. Proc Natl Acad Sci U S A 2009;106:18588–18593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sanes JR, LJW. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci 2001;2:791–805. [DOI] [PubMed] [Google Scholar]
- 34. Schaeffer L, de Kerchove d'Exaerde A, Changeux JP. Targeting transcription to the neuromuscular synapse. Neuron 2001;31:15–22. [DOI] [PubMed] [Google Scholar]
- 35. Zhang X, Xu R, Zhu B, Yang X, Ding X, Duan S, Xu T, Zhuang Y, Han M. Syne‐1 and Syne‐2 play crucial roles in myonuclear anchorage and motor neuron innervation. Development 2007;134:901–908. [DOI] [PubMed] [Google Scholar]
- 36. Demonbreun AR, Lapidos KA, Heretis K, Levin S, Dale R, Pytel P, Svensson EC, EM MN. Myoferlin regulation by NFAT in muscle injury, regeneration and repair. J Cell Sci 2010;123:2413–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McKinsey TA, Zhang CL, Olson EN. Activation of the myocyte enhancer factor‐2 transcription factor by calcium/calmodulin‐dependent protein kinase‐stimulated binding of 14‐3‐3 to histone deacetylase 5. Proc Natl Acad Sci U S A 2000;97:14400–14405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Armand AS, Bourajjaj M, Martinez‐Martinez S, el Azzouzi H, da Costa Martins PA, Hatzis P, Seidler T, Redondo JM, De Windt LJ. Cooperative synergy between NFAT and MyoD regulates myogenin expression and myogenesis. J Biol Chem 2008;283:29004–29010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Millay DP, Sutherland LB, Bassel‐Duby R, Olson EN. Myomaker is essential for muscle regeneration. Genes Dev 2014;28:1641–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Handschin C, Choi CS, Chin S, Kim S, Kawamori D, Kurpad AJ, Neubauer N, Hu J, Mootha VK, Kim YB, Kulkarni RN, Shulman GI, Spiegelman BM. Abnormal glucose homeostasis in skeletal muscle‐specific PGC‐1alpha knockout mice reveals skeletal muscle‐pancreatic beta cell crosstalk. J Clin Invest 2007;117:3463–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yu Z, Dadgar N, Albertelli M, Gruis K, Jordan C, Robins DM, Lieberman AP. Androgen‐dependent pathology demonstrates myopathic contribution to the Kennedy disease phenotype in a mouse knock‐in model. J Clin Invest 2006;116:2663–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hunter SK, Pereira HM, Keenan KG. The aging neuromuscular system and motor performance. J Appl Physiol (1985) 2016;121:982–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tisdale S, Pellizzoni L. Disease mechanisms and therapeutic approaches in spinal muscular atrophy. J Neurosci 2015;35:8691–8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dauvilliers Y, Siegel JM, Lopez R, Torontali ZA, Peever JH. Cataplexy—clinical aspects, pathophysiology and management strategy. Nat Rev Neurol 2014;10:386–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dobrowolny G, Giacinti C, Pelosi L, Nicoletti C, Winn N, Barberi L, Molinaro M, Rosenthal N, Musarò A. Muscle expression of a local Igf‐1 isoform protects motor neurons in an ALS mouse model. J Cell Biol 2005;168:193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tsai LK, Chen YC, Cheng WC, Ting CH, Dodge JC, Hwu WL, Cheng SH, Passini MA. IGF‐1 delivery to CNS attenuates motor neuron cell death but does not improve motor function in type III SMA mice. Neurobiol Dis 2012;45:272–279. [DOI] [PubMed] [Google Scholar]
- 47. Ferraro E, Molinari F, Berghella L. Molecular control of neuromuscular junction development. J Cachexia Sarcopenia Muscle 2012;3:13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Macpherson PC, Cieslak D, Goldman D. Myogenin‐dependent nAChR clustering in aneural myotubes. Mol Cell Neurosci 2006;31:649–660. [DOI] [PubMed] [Google Scholar]
- 49. Lu PY, Taylor M, Jia HT, Ni JH. Muscle LIM protein promotes expression of the acetylcholine receptor gamma‐subunit gene cooperatively with the myogenin‐E12 complex. Cell Mol Life Sci 2004;61:2386–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kumarapeli AR, Wang X. Genetic modification of the heart: chaperones and the cytoskeleton. J Mol Cell Cardiol 2004;37:1097–1109. [DOI] [PubMed] [Google Scholar]
- 51. Knight D, Tolley LK, Kim DK, Lavidis NA, Noakes PG. Functional analysis of neurotransmission at beta2‐laminin deficient terminals. J Physiol 2003;546:789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Latvanlehto A, Fox MA, Sormunen R, Tu H, Oikarainen T, Koski A, Naumenko N, Shakirzyanova A, Kallio M, Ilves M, Giniatullin R, Sanes JR, Pihlajaniemi T. Muscle‐derived collagen XIII regulates maturation of the skeletal neuromuscular junction. J Neurosci 2010;30:12230–12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Haase G, Pettmann B, Vigne E, Castelnau‐Ptakhine L, Schmalbruch H, Kahn A. Adenovirus‐mediated transfer of the neurotrophin‐3 gene into skeletal muscle of pmn mice: therapeutic effects and mechanisms of action. J Neurol Sci 1998;160:S97–S105. [DOI] [PubMed] [Google Scholar]
- 54. McFall ER, Murray LM, Lunde JA, Jasmin BJ, Kothary R, Parks RJ. A reduction in the human adenovirus virion size through use of a shortened fibre protein does not enhance muscle transduction following systemic or localised delivery in mice. Virology 2014;468–470:444–453. [DOI] [PubMed] [Google Scholar]
- 55. Borselli C, Storrie H, Benesch‐Lee F, Shvartsman D, Cezar C, Lichtman JW, Vandenburgh HH, Mooney DJ. Functional muscle regeneration with combined delivery of angiogenesis and myogenesis factors. Proc Natl Acad Sci U S A 2010;107:3287–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Generation of muscle‐restrictedNRIP knockout (cKO) mice.(A)2 Schematic illustration of the genomic structure of wild‐type (WT) NRIP, loxP‐floxed 3 NRIP, and NRIP‐deleted alleles. The loxP‐floxed allele contains a neo cassette with 4 two flanked loxP sequences in intron 1 and a loxP sequence in intron 2. NRIPexon 2 5 would be deleted by Cre recombinase driven by MCKpromoter.(B) Genotyping by 6 PCR. For floxed NRIP genotyping (upper), GU and JD primers (Supplementary 7 information, TableS1) were used. The band with smaller size (about 680 bp) 8 representsWT NRIP without loxPsequence and the largerone (about 720 bp) 9 NRIP‐containing loxPsequence. NRIPflox/+ represents mice with one WT allele and 10 one floxed allele, NRIPflox/floxmice with two floxed alleles, and NRIP+/+mice with two 11 WT alleles. For genotyping of Cre recombinase (lower), oIMR1085and oIMR6754 12 primers (Supplementary information, Table S1) were used for Cre recombinase loci 13 detection. The oIMR8744 and oIMR8745 primers wereTcrd DNA control primers 14 (The Jackson Laboratory, Supplementary information, Table S1). The band with 15 smaller size (about 200 bp) was used as internal positive control. Mice with16 loxP‐floxed NRIP and Cre‐positive bands were cKO mice. Control wild type (WT) 17 mice were NRIPflox/flox; MCK‐Cre‐.
Figure S2. Muscular abnormalityin cKO mouse soleus muscle at age 6 weeks.(A)20 Immunohistochemistry of the expression of oxidative fibre (slow myosin, n = 3). (B)21 COX (dark brown) expression. n = 3. (C) SDH (dark purple) expression. Scale bar, 100 22 μm. n = 3 for each group. (D) Rotarod maintenance testat age6 weeks (n = 7 for each 23 group). (E) Grip force analysisat age6 weeks (n = 7 for each group). Dataare 24 mean ± SEM by Student t‐test.*P < 0.05
Figure S3. Theassessment of degenerated muscles. (A) HEfor soleus muscle 26 morphology between WT and NRIP cKO at age 16 weeks. There was no 27 difference forcentral located nuclei. Scale bar, 100 μm. (B) CD45 expression for 28 infiltration of innate immune cells. Scale bar, 100 μm. (C) Embryonic myosin 29 expression (eMyHC) for regenerative myofiber marker (n = 3 for each group).30 Scale bar, 100 μm.(D) EBD stain (red) for fibrosis myofiber; Scale bar, 50 μm. All 31 assays: n = 3 for each genotype.
Figure S4.The size and perimeter of spinal motor neurons in in spinal anterior horn of 34 cKO and WT mice at age 16 weeks. Quantification of (A) size of α‐motor neurons 35 (MN) and (B) MN perimeter. n = 3 for each group. Dataare mean ± SEM byStudent t 36 test. *P < 0.05, **P < 0.01.
Figure S5. Screen for muscle‐derived factors that may support motor neuron survival 39 in cKO and WT mice at age 16 weeks. (A) The candidate genes include NT4, IGF‐1, 40 IGF‐2, IGFBP‐4, IGFBP‐6, BMP4, myogenin, and AChRɛ.(B)RT‐qPCR analysis of 41 mRNA expression ingastrocnemius muscles (WT: n = 6, cKO: n = 7). Dataare 42 mean ± SEM byStudent t test.***P < 0.001.
Figure S6. The expression of myogenin at neuromuscular junction in cKO and WT 45 mice at age 6 weeks.(A) Immunofluorescence assay of the proportion of 46 acetylcholine receptors (red) with myogenin immunoactivity (green). Arrow indicates 47 co‐localization of myogenin and AChR cluster. (B) Quantification of (A). The N 48 indicates the examined NMJ number for n = 3 of each genotype. Scale bar, 100 μm. 49 Data are mean ± SEM by Student t test. *P < 0.05. (C) Myogenin expression is activated by NRIP overexpressionin C2C12 cells. Immunofluorescence assay of51 proportion of C2C12 cells withnuclear myogenin (green) expression after treatment 52 with Ad‐NRIP andAd‐GFP (MOI 10) and under differentiation for 2 days. DAPI 53 (blue). Scale bar, 20 μm. Dataare mean ± SEM (n = 3 experiments) byStudent t test. 54 **P < 0.01
Figure S7. The mRNA of NRIP in Ad‐Myogenin infected gastrocnemius and 57 C2C12 cellsby using RT‐qPCR. (A) The RNA extraction from Ad‐Myogenin 58 infected gastrocnemius (n = 3 for each group).(B) The RNA from Ad‐Myogenin 59 infected C2C12 cells (n = 3 for each group).
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the paper and its Supporting Information files or on request from the corresponding author.