

Abstract
Background
Cachexia frequently occurs in women with advanced ovarian cancer (OC), along with enhanced inflammation. Despite being responsible for one third of all cancer deaths, cachexia is generally under‐studied in OC due to a limited number of pre‐clinical animal models. We aimed to address this gap by characterizing the cachectic phenotype in a mouse model of OC.
Methods
Nod SCID gamma mice (n = 6–10) were injected intraperitoneally with 1 × 107 ES‐2 human OC cells to mimic disseminated abdominal disease. Muscle size and strength, as well as bone morphometry, were assessed. Tumour‐derived effects on muscle fibres were investigated in C2C12 myotube cultures. IL‐6 levels were detected in serum and ascites from tumour hosts, as well as in tumour sections.
Results
In about 2 weeks, ES‐2 cells developed abdominal tumours infiltrating omentum, mesentery, and adjacent organs. The ES‐2 tumours caused severe cachexia with marked loss of body weight (–12%, P < 0.01) and ascites accumulation in the peritoneal cavity (4.7 ± 1.5 mL). Skeletal muscles appeared markedly smaller in the tumour‐bearing mice (approximately –35%, P < 0.001). Muscle loss was accompanied by fibre atrophy, consistent with reduced muscle cross‐sectional area (–34%, P < 0.01) and muscle weakness (–50%, P < 0.001). Body composition assessment by dual‐energy X‐ray absorptiometry revealed decreased bone mineral density (–8%, P < 0.01) and bone mineral content (–19%, P < 0.01), also consistent with reduced trabecular bone in both femurs and vertebrae, as suggested by micro‐CT imaging of bone morphometry. In the ES‐2 mouse model, cachexia was also associated with high tumour‐derived IL‐6 levels in plasma and ascites (26.3 and 279.6 pg/mL, respectively) and with elevated phospho‐STAT3 (+274%, P < 0.001), reduced phospho‐AKT (–44%, P < 0.001) and decreased mitochondrial proteins, as well as with increased protein ubiquitination (+42%, P < 0.001) and expression of ubiquitin ligases in the skeletal muscle of tumour hosts. Similarly, ES‐2 conditioned medium directly induced fibre atrophy in C2C12 mouse myotubes (–16%, P < 0.001), consistent with elevated phospho‐STAT3 (+1.4‐fold, P < 0.001) and altered mitochondrial homoeostasis and metabolism, while inhibition of the IL‐6/STAT3 signalling by means of INCB018424 was sufficient to restore the myotubes size.
Conclusions
Our results suggest that the development of ES‐2 OC promotes muscle atrophy in both in vivo and in vitro conditions, accompanied by loss of bone mass, enhanced muscle protein catabolism, abnormal mitochondrial homoeostasis, and elevated IL‐6 levels. Therefore, this represents an appropriate model for the study of OC cachexia. Our model will aid in identifying molecular mediators that could be effectively targeted in order to improve muscle wasting associated with OC.
Keywords: Cancer cachexia, Ovarian cancer, Skeletal muscle, Animal model, ES‐2, IL‐6
Introduction
According to the most recent statistics, 22 240 new cases of ovarian cancer (OC) will be diagnosed by the end of 2018, and 14 070 women will die from the disease in the USA alone, thus representing the most lethal among the gynaecologic malignancies.1 High‐grade serous (HGS)‐OC accounts for 90% of the serous carcinomas and two‐thirds of all OC deaths.2, 3, 4, 5 Early detection of OC is imperative in order to promote better outcomes and to prolong the overall survival. Unfortunately, the symptoms associated with the onset of the disease are often ambiguous, thereby delaying significantly its diagnosis. As a consequence, only 15% of OCs are currently identified at a stage that allows successful treatment. On the contrary, the majority of OC patients is diagnosed when the disease has already spread regionally and/or distally, thus significantly worsening quality of life and resulting into poorer outcomes.6, 7 Current treatment strategies for advanced OCs consist of aggressive surgery aimed at removing the majority of the tumour followed by intraperitoneal chemotherapy with platinum and a taxane, such as taxol. Unfortunately, after an initial positive response, in most cases, the disease recurs due to development of chemoresistance and an aggressive disease course that is often accompanied by the formation of ascites.8, 9, 10 This disease progression is particularly evident in advanced HGS‐OCs and is often associated with hyper‐activation of the pro‐inflammatory IL‐6/STAT3 signalling pathway.2, 3, 11 IL‐6 seems to play a critical role in OC. Indeed, IL‐6 contributes to the occurrence of paraneoplastic thrombocytosis, a condition that promotes tumour growth, while its inhibition indirectly slows down tumour progression.12 High IL‐6 levels in ascites and plasma also associates with shorter survival.12, 13
Evidence from our group suggests that activation of the IL‐6/STAT3 pathway also plays a causative role in the pathogenesis of cancer cachexia, one of the most distressing complications associated with the development of OC.14, 15 Indeed, cachexia usually accompanies the development of ascites and chemoresistance in the most advanced stages of the disease.16 The majority of advanced OC patients develops cachexia, which is a major contributor of morbidity and mortality in these patients. Cachexia is primarily responsible for body and muscle weight loss and correlates with tumour burden, increased pro‐inflammatory cytokine levels, fatigue, and reduced response to chemo‐therapy and radio‐therapy.17, 18, 19 Patients presenting with advanced stage OC often show large tumour and ascites burden that, in turn, results into severe malnourishment because of decreased oral intake and compromised bowel functions. As a consequence, malnutrition and weight loss are frequently diagnosed and together contribute to substantial impairment of anti‐cancer treatments and poorer prognosis.20, 21, 22 Notably, evidence from ours and other groups also suggests that anti‐cancer therapies play a pivotal role in causing the development of muscle loss and muscle weakness.23, 24 Indeed, chemotherapy itself is frequently accompanied by nausea, diarrhoea, anorexia, and fatigue. Cancer patients affected by muscle depletion show increased drug‐related toxicity and reduced survival that are associated with increased levels of pro‐inflammatory cytokines, and these features have been previously implicated in the occurrence of cachexia.14, 25, 26, 27, 28, 29
Despite its significant negative impact on quality of life, no effective treatment is currently available for OC‐related cachexia. Research related to this field remains inadequate, particularly because of the limited availability of optimal pre‐clinical models that are characterized for muscle and bone phenotype or function. Indeed, only one OC xenograft, the mouse bearing the TOV‐21G clear cell carcinoma, has been previously described and partially characterized for the cancer‐associated effects on skeletal muscle.30 While ascites and bloating are primarily used to diagnose OC, these features can mask the underlying sarcopenia and delay the diagnosis of the disease, thus also affecting the overall outcomes.31, 32 Indeed, muscle loss and low skeletal muscle attenuation are often detected in women undergoing primary debulking surgery for the treatment of OCs. Along the same line, recent evidence suggests that baseline sarcopenia represents one of the most accurate prognostic factors for survival in advanced OC and during chemotherapy treatment.16, 33, 34, 35
In the present study, we sought to establish and fully characterize the muscle effects associated with the growth of the ES‐2 human OC in mice. In vivo growth of the ES‐2 cancer cells has been recently reclassified as a suitable model for the study of HGS‐OC, based on cell copy number alterations, TP53 mutations, and low frequency of somatic mutations in protein‐coding regions.4, 5 In our model, in vivo growth of the ES‐2 cancer cells induced profound body wasting, associated with muscle, fat, and trabecular bone depletion, which is also consistent with significant loss of muscle strength. The pro‐inflammatory cytokine IL‐6, along with the activation of the downstream STAT3‐dependent pathway, was associated with the occurrence of muscle wasting in in vivo and in vitro conditions. STAT3 blockade by administration of INCB018424 JAK1/2 pharmacologic inhibitor15, 36 was shown to prevent myofibre atrophy in C2C12 cultures exposed to ES‐2 conditioned medium (CM). Further, similar to what shown in other routinely used experimental models of cancer cachexia, the development of OC resulted in altered activation of regulators of muscle size and markedly exacerbated protein catabolism, thus representing an ideal model for the study of OC‐associated cachexia.
Methods
Cell cultures
Human ES‐2 cells were purchased from ATCC (Manassas, VA, USA; #CRL‐1978) and were cultured in McCoy's medium supplied with 10% foetal bovine serum, 1% glutamine, 1% sodium pyruvate, 1% penicillin/streptomycin, in a 5% CO2, 37°C humidified incubator. Murine C2C12 skeletal myoblasts (ATCC) were grown in high glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% foetal bovine serum, 100 U/mL penicillin, 100 mg/mL streptomycin, 100 mg/mL sodium pyruvate, and 2 mM L‐glutamine and maintained at 37°C in 5% CO2. Myotubes were generated by exposing the myoblasts to DMEM containing 2% horse serum and replacing the medium every other day for 5 days. In order to determine the effects of tumor‐derived mediators on fibre size, myotubes were exposed to 25% or 50% CM, collected from confluent ES‐2 culture plates and subjected to centrifugation to remove cell debris. The JAK1/2 pharmacologic inhibitor INCB018424 (EMD Millipore, Burlington, MA, USA) was dissolved in sterile Dimethyl sulfoxide (DMSO) and administered in the culture medium at 400 nM final concentration for up to 48 h, as previously shown.15
Animals
All animal studies were approved by the Institutional Animal Care and Use Committee at Indiana University School of Medicine and were in compliance with the National Institutes of Health Guidelines for Use and care of Laboratory Animals and with the 1964 Declaration of Helsinki and its later amendments. Female Nod SCID gamma (NSG) (NOD‐scid/IL2Rgnull) immunodeficient mice (In Vivo Therapeutics Core Facility, IU Simon Cancer Center, Indianapolis, IN, USA) were used and housed in a pathogen‐free facility at IU LARC (up to 5 per cage). For the experiments described, animals were randomized into Controls (n = 6) and ES‐2 hosts (n = 10), identified with a code, and the investigators were blinded during allocation, animal handling, and endpoint measurements. 1 × 107 ES‐2 cells were inoculated intraperitoneally (i.p.) in sterile saline.37 The animals were weighed daily. At time of sacrifice, several tissues, including skeletal muscles, were collected, weighed, frozen in liquid nitrogen, and stored at −80°C for further analyses. The tibialis anterior muscle was mounted in OCT and frozen in N2‐cooled isopentane for histology, as previously described.38 The mouse carcasses were fixed for 2 days in 10% neutral buffered formalin and then transferred into 70% ethanol for storage of bone tissues.
Grip strength
The evaluation of the whole body strength in mice was assessed as described in Bonetto et al.39 The absolute grip strength (peak force, expressed in grams) was recorded by means of a grip strength metre (Columbus Instruments, Columbus, OH, USA). Five measurements were completed, and the top three measurements were included in the analysis. In order to avoid habituation, the animals were tested for grip strength up to once per week.
Assessment of muscle cross‐sectional area
For histology and morphometry of skeletal muscle, 8 μm thick cryosections of tibialis anterior muscles taken at the mid‐belly region were stained with haematoxylin and eosin as reported in Bonetto et al.39 All samples were observed under an Axio Observer.Z1 motorized microscope (Zeiss, Oberchoken, Germany), and images were recorded for morphometric examination. For the analysis of the cross‐sectional area, muscle fibres (n = 300–500 per sample) were measured using a Cintiq pen tablet input device (Wacom, Vancouver, WA, USA) and ImageJ 1.43 software,40 as also described in Bonetto et al.39
Assessment of myotube size
Cell layers were fixed in ice‐cold acetone–methanol and incubated with an anti‐Myosin Heavy Chain antibody (MF‐20, 1:200; Developmental Studies Hybridoma Bank, Iowa City, IA, USA) and an AlexaFluor 488‐labelled secondary antibody (Invitrogen, Grand Island, NY, USA). Analysis of myotube size was performed by measuring the average diameter of long, multi‐nucleate fibres (n = 250–350 per condition) avoiding regions of clustered nuclei on a calibrated image using the ImageJ 1.43 software.40
Immunohistochemistry of tumour tissue
For morphological and immunohistochemical analysis of OC tissue, paraffin‐embedded ovarian abdominal tumours 10 μm thick sections were generated. Deparaffinization of the slides was performed through a series of incubations in xylene, 100% ethanol, 95% ethanol, 70% ethanol, and deionized water. Antigen retrieval was performed by treatment in 10 mM citrate buffer solution for 30 min at sub‐boiling temperature. Endogenous peroxidase activity was inhibited by a 10 min incubation in 3% H2O2. Sections were subsequently blocked in Tris‐buffered saline (TBS)‐Tween buffer containing 5% goat serum for 1 h at room temperature. A primary rabbit anti‐IL‐6 antibody (#208113; Abcam, Cambridge, MA, USA) was added (1:50) and incubated overnight in a humidified box at 4°C. Normal rabbit IgG were used as negative controls. Sections were then incubated with the ‘EnVision+System‐HRP‐labelled polymer secondary antibody’ (DAKO, Carpinteria, CA, USA) for 1 h at room temperature. The reaction was then visualized with the DAKO ‘Liquid DAB+substrate chromogen system’ according to the manufacturer's instructions. Slides were counterstained with Harrys Haematoxylin (1:1 in deionized water) for up to 5 min, dehydrated with incubations in 70% ethanol, 95% ethanol, 100% ethanol, and xylene, and mounted with xylene‐based Cytoseal XYL mounting medium (Thermo Fisher Scientific, Suwanee, GA, USA).
MitoTracker staining
The mitochondria polarization was determined using the MitoTracker Red CMXRos selective probe (#M7512, Invitrogen, Carlsbad, CA, USA). C2C12 myotubes were exposed to unconditioned medium (UCM) or ES‐2 CM for up to 48 h and subsequently incubated in serum‐free medium containing 100 nM MitoTracker at 37°C for 30 min. The cells were then washed with Phosphate‐buffered saline (PBS) and fixed with 3.7% formaldehyde in serum‐free medium at 37°C for 15 min. 4′,6‐diamidino‐2‐phenylindole (DAPI) staining was then performed to stain the nuclei. Images were captured using an Axio Observer.Z1 motorized microscope (Zeiss). Quantitative measurement of red fluorescence intensity was analysed using the ImageJ 1.43 software.40
Body composition assessment by dual‐energy X‐ray absorptiometry
Lean and adipose tissue, as well as whole body bone mineral density (BMD) and bone mineral content (BMC) were assessed by means of dual‐energy X‐ray absorptiometry (DXA) scanning of formalin‐fixed carcasses. According to the manufacturer's guidelines, in order to calibrate and validate the apparatus for its performance, a spine phantom was scanned using the Lunar PIXImus densitometer (PIXImus, Fitchburg, WI, USA) before scanning the first carcass. Animal carcasses were placed in a prone position with the limbs outstretched. From the whole‐body scans, areal BMD and BMC were calculated for the entire body minus head region of interest (ROI).
Microcomputed tomography analysis of femurs and vertebrae bone morphometry
Microcomputed tomography (μCT) scanning was performed to measure morphological indices of metaphyseal regions of femurs and vertebrae, as described in Bouxsein et al.41 After euthanasia, the mouse carcasses were fixed for 2 days in 10% neutral buffered formalin, transferred into 70% ethanol, the right femurs and the fifth lumbar vertebrae dissected, and prepared for μCT scanning on a high‐throughput μCT specimen scanner. Bone samples were rotated around their long axes, and images were acquired using a Bruker Skyscan 1176 (Bruker, Kontich, Belgium) with the following parameters: pixel size = 9 μm3; peak tube potential = 50 kV; X‐ray intensity = 500 μA; 0.9° rotation step. Raw images were reconstructed using SkyScan reconstruction software (NRecon; Bruker) to three‐dimensional cross‐sectional image data sets using a three‐dimensional cone beam algorithm. Structural indices were calculated on reconstructed images using the SkyScan CT Analyser software (CTAn; Bruker). Cortical and trabecular bone were separated using a custom processing algorithm in CTAn, based on the different thicknesses of the structures. Cortical bone was analysed by threshold 160–255 in the femoral mid‐shaft. Cortical bone parameters included bone volume fraction (Ct.BV/TV), thickness (Ct.Th), and porosity. Trabecular bone was analysed between 2.0 and 3.0 mm under the femoral distal growth plate and 1 mm equally between the distal and proximal growth plates in the fifth lumbar vertebra using a threshold of 80–255. Trabecular parameters included bone volume fraction (Tb.BV/TV), number (Tb.N), thickness (Tb.Th), separation (Tb.Sp), and connectivity (Conn.Dn).
Western blotting
Total protein extracts were obtained by lysing cell layers or homogenizing 100 mg quadriceps muscle tissue in radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 1.0% NP‐40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), and 50 mM Tris, pH 8.0) completed with protease (Roche, Indianapolis, IN, USA) and phosphatase (Thermo Scientific, Rockford, IL, USA) inhibitor cocktails. Cell debris were removed by centrifugation (15 min, 14 000 g), and the supernatant was collected and stored at −80°C. Protein concentration was determined using the bicinchoninic acid (BCA) protein assay method (Thermo Scientific). Protein extracts (30 μg) were then electrophoresed in 4–15% gradient SDS Criterion TGX precast gels (Bio‐Rad, Hercules, CA, USA). Gels were transferred to nitrocellulose membranes (Bio‐Rad). Membranes were blocked with SEA BLOCK blocking reagent (Thermo Scientific) at room temperature for 1 h, followed by an overnight incubation with diluted antibody in SEA BLOCK buffer containing 0.2% Tween‐20 at 4°C with gentle shaking. After washing with PBS containing 0.2% Tween‐20, the membrane was incubated at room temperature for 1 h with either Anti‐rabbit IgG (H + L) DyLight 800 or Anti‐mouse IgG (H + L) DyLight 600 (Cell Signaling Technologies, Danvers, MA, USA). Blots were then visualized with Odyssey Infrared Imaging System (LI‐COR Biosciences, Lincoln, NE, USA). Optical density measurements were taken using the Gel‐Pro Analyser software. Antibodies used were pSTAT3‐Y705 (#9145), STAT3 (#8768), p‐Akt‐S473 (#4060), Akt (#9272), p‐P38 MAPK (T180/Y182) (#4511), P38 MAPK (#9212), p‐ERK1/2 (p‐p44/42 MAPK, T202/Y204) (#4370), ERK1/2 (p44/42 MAPK) (#4695), OPA1 (#80471), Mitofusin‐2 (#9482), DRP1 (#8570), Cytochrome C (#11940), and Ubiquitin (#3933) from Cell Signaling Technologies, PGC1α (#ab3242) from Abcam, and α‐Tubulin (#12G10) from Developmental Studies Hybridoma Bank.
Masson trichrome staining
For direct visualization of collagen and histological assessment of collagen deposition, trichrome staining was performed on tibialis anterior muscle sections using the Masson Trichrome Staining Kit (Richard‐Allan Scientific, San Diego, CA, USA) and according to the instructions provided by the manufacturer. Briefly, 8 μm thick muscle sections were fixed in 10% neutral buffered formalin for 1 h at room temperature. The sections were then re‐fixed in Bouins solution for overnight at room temperature and then stained with Weigerts Haematoxylin (for nuclei staining), followed by Biebrich Scarlet‐Acid Fuchsin (for cytoplasm staining) and Alanine blue (for collagen staining). Samples were then dehydrated and observed under an Axio Observer.Z1 motorized microscope (Zeiss).
Succinate dehydrogenase staining
Tibialis anterior cross sections (10 μm) were cut on a cryostat and incubated for 40 min at 37°C with 0.5 mg/mL nitroblue tetrazoliumand, 50 mM Na‐succinate, and 0.08 mM phenazine methosulfate in PBS. The slide was then incubated in acetone, rinsed in deionized water, mounted with PBS‐glycerol, and photographed. The ratio between oxidative [succinate dehydrogenase (SDH)‐positive, dark purple] and glycolytic (SDH‐negative, light purple) fibres (expressed as percentage of the total fibres) was determined using the ImageJ software on randomly chosen fields (n = 100–150 fibres).
Succinate dehydrogenase enzymatic activity
The total SDH enzymatic activity was measured using a SDH Colorimetric Assay Kit (#MAK197; Sigma‐Aldrich, St. Louis, MO, USA), as per manufacturer's instruction. Briefly, 10 mg of quadriceps muscle tissue were homogenized in 100 μL ice‐cold assay buffer and subsequently centrifuged. The supernatant was collected, and the total protein content was measured using the BCA protein assay (Thermo Fisher Scientific, Waltham, MA, USA). Ten microlitres of homogenate were then added to a 96‐well plate, along with an appropriate volume of reaction mix, and then incubated at 37°C. The absorbance at 600 nm was measured after 0, 3, and 20 min. The rate of absorbance decrease between 3 and 20 min was corrected for the protein concentration and used to calculate the SDH content.
Extracellular flux analysis
The mitochondrial oxygen consumption rate (OCR) was measured using a Mito‐Stress Test kit in a Seahorse XF Extracellular Flux Analyser (Agilent, Santa Clara, CA, USA), according to the manufacturer's instructions. C2C12 myoblasts were plated in an XF cell culture microplate (30 000 cells/cm2) and then differentiated to myotubes by exposing the myoblasts to DMEM containing 2% horse serum and replacing the medium every other day for 5 days. The cells were then exposed to UCM or 50% ES‐2 CM for up to 48 h. Prior to the assay, the cells were incubated in Seahorse XF Base Medium containing 1 mM Na‐Pyruvate, 2 mM glutamine, and 25 mM glucose in a non‐CO2 incubator for 1 h at 37°C. Basal respiration was monitored prior to the addition of 2 μM oligomycin. The OCR values were obtained at baseline and after the addition of 2 μM oligomycin, 1 μM carbonyl cyanide‐4(trifluoromethoxy)phenylhydrazone (FCCP), and 0.5 μM Rotenone. OCR was automatically calculated and recorded by the Seahorse XF software, and data were expressed as pmol/min.
Real‐time quantitative polymerase chain reaction
RNA from quadriceps was isolated using the miRNeasy Mini kit (Qiagen, Valencia, CA, USA) and following the protocol provided by the manufacturer. RNA was quantified by using a Synergy H1 spectrophotometer (BioTek, Winooski, VT, USA). RNA integrity was checked by electrophoresis on 1.2% agarose gel containing 0.02 mol/L morpholinopropanesulfonic acid and 18% formaldehyde. Total RNA was reverse transcribed to cDNA by using the Verso cDNA kit (Thermo Fisher Scientific, Waltham, MA, USA). Transcript levels were measured by Real‐Time PCR (Light Cycler 96, Roche) taking advantage of the TaqMan gene expression assay system (Life Technologies, Carlsbad, CA, USA). Expression levels for Atrogin‐1 (Mm00499523_m1), MuRF‐1 (Mm01185221_m1), Fbxo21 (SMART; Mm01208074_m1), and Fbxo31 (Mm00505343_m1) were detected. Gene expression was normalized to TATA‐binding protein (TBP) (Mm01277042_m1) levels using the standard 2−ΔCT methods.
Quantification of IL‐6 in plasma and ascites
The circulating levels of human IL‐6 were measured in mouse poor‐platelet plasma and ascites from control and ES‐2 bearing mice by using a specific enzyme‐linked immunosorbent assay (ELISA) kit (#D6050; Bio‐Techne Corporation, Minneapolis, MN, USA), according to the manufacturer's protocol.
Statistical analysis
Results are presented as means ± standard deviation. Significance of the differences was determined by either unpaired t‐test or analysis of variance followed by Tukey's post‐test. Differences were considered significant when P < 0.05.
Results
The ES‐2 tumours cause significant body and muscle weight loss
In order to assess the effects of OC development on skeletal muscle mass in in vivo conditions, NSG female mice were intraperitoneally injected with 1 × 107 ES‐2 cells.4 Both control and tumour hosts were assessed for changes in body weight over the duration of the entire experiment. After an initial lag phase, the ES‐2‐bearing mice displayed significant accumulation of ascites, consistent with increased body weight compared with the control animals (4.7 ± 1.5 mL) (Figure 1A). In line with previous data, the ascites was highly cellular and milky in appearance, with tumour cells existing mostly as single‐cell suspension and with limited infiltration into diaphragm, pancreas (Figure S1), mesentery, or omentum to form solid tumours.37 However, the tumour‐free body weight was significantly decreased in the tumour hosts (−12% vs. Control, P < 0.01) (Figure 1B). Additionally, skeletal muscles were markedly impacted by tumour growth, with the quadriceps showing a 38% reduction in weight with respect to the Controls (P < 0.001). Similarly, the heart was markedly smaller in the tumour hosts (−35% vs. Control, P < 0.001) (Figure 2). The liver was also affected by the growth of the ES‐2 tumours (−28%, P < 0.01), while the spleen was unchanged compared with the control animals (Figure 3). Notably, the gonadal adipose tissue was massively depleted in the tumour hosts (P < 0.001), therefore suggesting the occurrence of severe tumour‐associated wasting (Figure 3).
Figure 1.
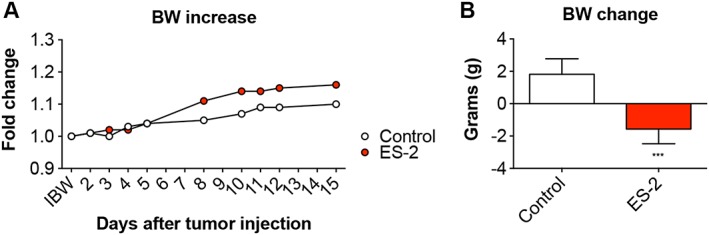
The ES‐2 tumours cause significant body weight (BW) loss. Body weight curves (A) and body weight change (i.e. body weight at time of sacrifice vs. initial body weight) (B) in mice bearing the ES‐2 ovarian cancer (n = 6–10). Data expressed as means ± standard deviation. Significance of the differences: *** P < 0.001 vs. Control.
Figure 2.
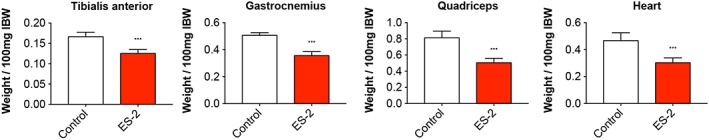
Growth of the ES‐2 ovarian cancer causes marked muscle depletion. Muscle weights in ES‐2 tumour hosts (n = 6–10). Weights were normalized to the initial body weight (IBW) and expressed as weight/100 mg IBW. Data expressed as means ± standard deviation. Significance of the differences: *** P < 0.001 vs. Control.
Figure 3.
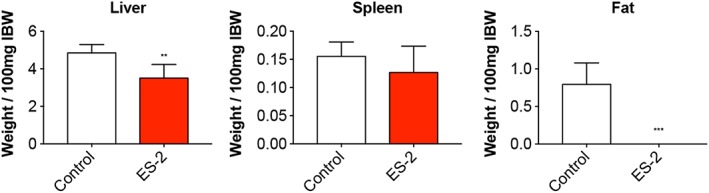
Organ size is affected by the occurrence of ES‐2 ovarian cancer. Liver, spleen, and gonadal fat weights in mice bearing the ES‐2 ovarian cancer (n = 6–10). Weights were normalized to the initial body weight (IBW) and expressed as weight/100 mg IBW. Data expressed as means ± standard deviation. Significance of the differences: ** P < 0.01, *** P < 0.001 vs. Control.
Ovarian cancer growth affects muscle size and function
Along with the depletion of skeletal muscle mass, the growth of the ES‐2 OC cells was associated with the occurrence of severe muscle atrophy (−34% vs. Control, P < 0.01) (Figure 4A). No other apparent changes in muscle histology, including fibrosis, were detected ( Figure S2A). Similarly, the number of central nuclei was comparable between control and tumour hosts, thus suggesting the absence of muscle regeneration ( Figure S2B). Consistent with the reduced fibre size, progressive loss of muscle strength was observed over the experimental period, and severe muscle weakness was apparent at time of sacrifice (−50% vs. Control, P < 0.001) (Figure 4B).
Figure 4.
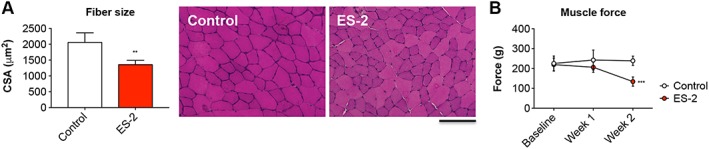
Development of ovarian cancer is accompanied by muscle atrophy and progressive muscle weakness. Muscle morphology (haematoxylin and eosin staining) and quantification of the cross‐sectional area (CSA) in the tibialis anterior muscle of ES‐2 hosts. Scale bar: 100 μm (A). Whole body grip strength in animals bearing the ES‐2 tumours, reported as peak force, was measured weekly by taking advantage of a grip strength metre and expressed as the average of the three top pulls from each animal (n = 6–10) (B). Data expressed as means ± standard deviation. Significance of the differences: ** P < 0.01, *** P < 0.001 vs. Control.
The growth of the ES‐2 tumour affects the overall body composition
In order to understand the effects of ES‐2 tumour development on body composition, the carcasses from both control and tumour‐bearing animals were assessed by means of DXA. The amount of total fat and lean tissue was significantly reduced in the tumour hosts (−12% and −23% vs. Control, respectively) (Figure 5). Interestingly, the bone tissue was affected by ES‐2 growth, with BMD and BMC significantly decreased with respect to the control animals (−8% and −19%, respectively) (Figure 5).
Figure 5.
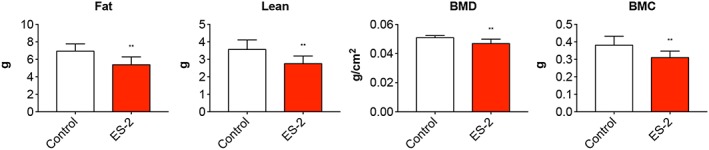
Body composition is affected by the growth of ES‐2 tumours. Body composition was assessed by means of dual‐energy X‐ray absorptiometry scanning of carcasses from ovarian cancer hosts (n = 6–10). Fat and lean tissue, total bone mineral density (BMD), and bone mineral content (BMC) measurements of the whole body are shown. Data (means ± standard deviation) are expressed as g (for fat, lean, and BMC) or g/cm2 (for BMD). Significance of the differences: * P < 0.05, ** P < 0.01, *** P < 0.001 vs. Control.
Ovarian cancer promotes the loss of trabecular bone
In order to determine whether the reduction in BMC and BMD was also consistent with loss of bone mass, morphometry of sections from femurs and vertebrae extracted from animals bearing OCs was analysed by means of μCT and the amount of trabecular bone quantified. The ES‐2 tumour hosts exhibited a severe loss of trabecular bone in the femur (Figure 6A–B). This was evidenced by markedly reduced trabecular bone volume fraction (BV/TV; −39%, P < 0.01), thickness (Tb.Th; −16%, P < 0.05), number (Tb.N; −23%, P < 0.01), and connectivity density (Conn.Dn; −23%, P < 0.05), as well as increased trabecular separation (Tb.Sp; +11%, P = 0.10) and pattern factor (Tb.pf; +43%, P < 0.01) vs. Control (Figure 6A). The vertebral trabecular bone was also affected by the development of OC, as shown by reduced BV/TV (−27%, P < 0.001), Tb.Th (−15%, P < 0.001), Tb.N (−14%; P < 0.01), and Conn.Dn (−10%, P < 0.001) and by increased Tb.Sp (+15%, P < 0.001) and Tb.pf (+41%, P < 0.01) with respect to the control mice (Figure 6B).
Figure 6.
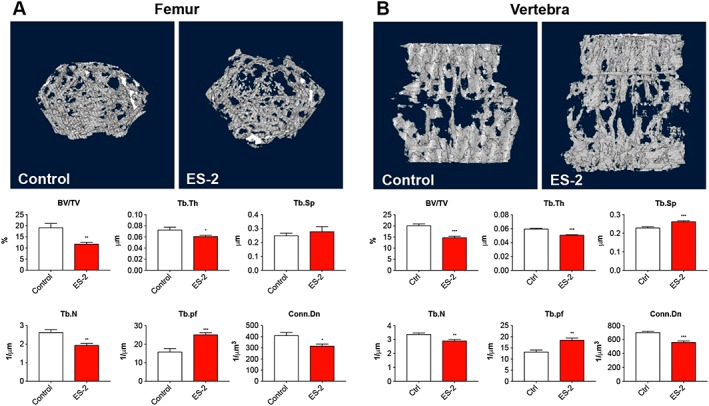
Growth of the ES‐2 tumour affects bone quality. Representative three‐dimensional rendering of microcomputed tomography scan images and quantification of bone volume fraction (BV/TV), trabecular thickness (Tb.Th), trabecular separation (Tb.Sp), trabecular number (Tb.N), and trabecular connectivity density (Conn.Dn) in the femurs (A) and vertebrae (B) of mice bearing the ES‐2 tumour (n = 6–10). Data are expressed as means ± standard deviation. Significance of the differences: * P < 0.05, ** P < 0.01, *** P < 0.001 vs. Control.
Pro‐atrophic signalling pathways are activated in the muscle of mice bearing ovarian cancer
In order to investigate whether the muscle phenotype observed following ES‐2 tumour growth was also associated with modulation of muscle‐specific signalling pathways, we assessed the levels of a number of proteins known to be involved in the regulation of skeletal muscle growth. Interestingly, we observed modulation of several mediators of muscle atrophy and markers of mitochondrial biogenesis using western blotting analysis performed on whole muscle protein extracts from mice bearing OC. In particular, the tumour hosts showed increased phospho‐STAT3/STAT3 ratio (+274% vs. Control, P < 0.001) and decreased activation of AKT1/2 (−44% vs. Control, P < 0.001), in line with previous data generated in animal models characterized by muscle atrophy15, 42 (Figure 7A). As expected, expression of total STAT3 was also increased (+71% vs. Control, P < 0.001), in accordance with published evidence from our laboratory.14 The growth of the ES‐2 tumour was also associated with increased phospho‐P38 (+156% vs. Control, P < 0.05) and unchanged phospho‐ERK1/2 levels (Figure 7A). Interestingly, mitochondrial proteins, such as OPA1 (−78%, P < 0.001), Mitofusin‐2 (−40%, P < 0.05), PGC1α (−79%, P < 0.001), and Cytochrome C (−78%, P < 0.001), were also markedly decreased in the muscle of the ES‐2 hosts compared with the control animals, thereby suggesting that mitochondrial alterations may play a role in the development of muscle wasting and muscle weakness (Figure 7B). Accordingly, myofibre oxidative capacity was significantly reduced in the skeletal muscle of ES‐2 hosts, as confirmed by the analysis of the SDH enzymatic activity (Figure 7C). In a similar manner, based on the quantification of the muscle SDH staining conducted in the tibialis muscle of ES‐2 bearing mice (Figure 7D), HGS‐OC growth was also shown to drive an oxidative‐to‐glycolytic fibre shift, characterized by a reduction in oxidative fibres (dark purple) and a significant increase in the number of fibres associated with a more glycolytic metabolism (light purple) (P < 0.001 vs. Control; Figure 7E).
Figure 7.
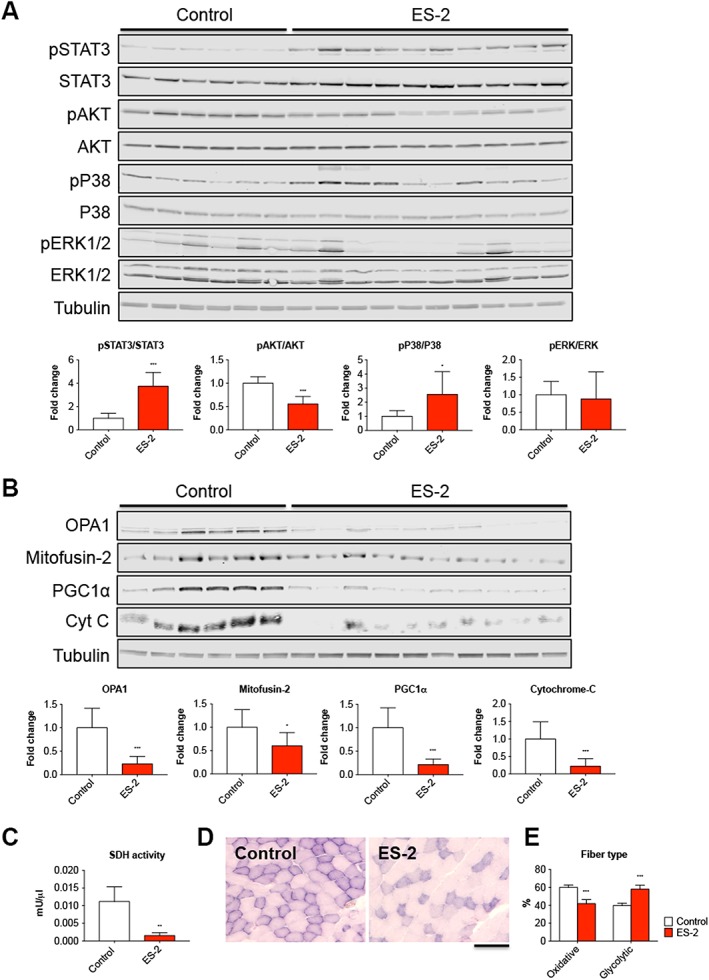
Development of ovarian cancer is accompanied by changes in the expression of key regulators of muscle size. Representative western blotting and quantification for pSTAT3, pAKT, pP38, and pERK1/2 (A) and OPA1, Mitofusin‐2, PGC1α, and Cytochrome C (Cyt C) (B) in whole muscle protein extracts from mice bearing ovarian cancers (n = 6–10). Levels of phosphorylated proteins were normalized to the respective total protein expression. Tubulin was used as loading control. Data are expressed as fold change vs. control and reported as means ± standard deviation. Succinate dehydrogenase (SDH) enzymatic activity (expressed as mU/μL) in quadriceps muscle from control mice and ES‐2 hosts (n = 6–10) (C). Succinate dehydrogenase staining performed on 8 μm thick sections from tibialis anterior muscles excised from controls and ES‐2 (n = 6–10) (D). The number of oxidative (dark purple) and glycolytic (light purple) fibres was assessed and reported as % of total fibres per field (E). Scale bar: 100 μm. Data are expressed as means ± standard deviation. Significance of the differences: * P < 0.05, ** P < 0.01, *** P < 0.001 vs. Control.
Protein catabolism is exacerbated in the muscle of ES‐2 tumour hosts
In order to determine whether protein catabolism was associated with the loss of muscle mass in animals bearing OC, markers of protein catabolism, as well as protein ubiquitination, were investigated. The total ubiquitination was more elevated in the muscle of ES‐2 hosts (+42% vs. Control, P < 0.001) (Figure 8A), in line with the overexpression of the ubiquitin ligases Atrogin‐1 (+3.9‐fold vs. Control, P < 0.001), MuRF‐1 (+8.4‐fold vs. Control, P < 0.001), and Fbxo31 (+3.0‐fold vs. Control, P < 0.001), normally overexpressed in conditions associated with muscle atrophy, while Fbxo21 (SMART) was not affected by tumour development43, 44 (Figure 8B).
Figure 8.
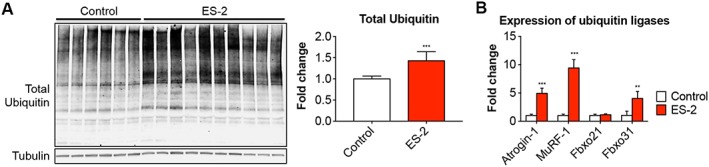
Elevated muscle protein catabolism is a hallmark of ovarian cancer cachexia. Representative western blotting and quantification for total ubiquitin (A) in whole muscle protein extracts from mice bearing ovarian cancers (n = 6–10). Tubulin was used as loading control. Gene expression levels for Atrogin‐1, MuRF‐1, Fbxo21, and Fbxo31 ubiquitin ligases (B) were performed by quantitative real‐time PCR. Gene expression was normalized to TBP levels. Data (fold change vs. control) were expressed as means ± standard deviation.
The IL‐6/STAT3 signalling is associated with muscle atrophy in the ES‐2 model of ovarian cancer
In order to investigate whether cancer cell‐derived factors were directly responsible for muscle wasting, we exposed C2C12 murine myotubes to ES‐2 CM for up to 48 h, while control cells were exposed to UCM. No effects associated with the exposure of C2C12 myotubes to McCoys medium (either 25% or 50% in differentiation medium) were observed ( Figure S3). On the other hand, the ES‐2 CM determined concentration‐dependent effects on myotube size (−11% and −16% vs. UCM with 25% and 50% CM, respectively) (Figure 9A). Notably, the phosphorylation of STAT3, downstream of IL‐6 binding to its receptor, was significantly higher in the tumour medium‐treated C2C12 myotubes compared with the control cells exposed to UCM (+1.4‐fold in 25% CM vs. UCM, P < 0.001) (Figure 9B). In order to understand whether counteraction of the IL‐6/STAT3 signalling was sufficient to preserve the myofibre size in the presence of OC‐derived factors, we exposed C2C12 myotubes to 50% ES‐2 CM, with or without INCB018424, a JAK1/2 inhibitor previously shown to promote body weight gain in patients with myelofibrosis36 and to preserve fibre size in C2C12 cells exposed to high IL‐615 ( Figure S4). As shown in Figure 9C, INCB018424 was sufficient to protect the C2C12 myotubes from undergoing atrophy in the presence of ES‐2 CM, also consistent with abolished STAT3 phosphorylation (Figure 9D). To further support the idea that activation of the IL‐6/STAT3 pathway plays a pivotal role in determining the cachectic phenotype in the ES‐2 model, elevated human IL‐6 was detected at high levels in plasma and ascites from ES‐2 bearing mice (26.3 and 279.6 pg/mL, respectively) compared with the control animals (not detected) (Figure 10A), thus suggesting that the OC tumours were the main source of IL‐6 in this animal model. To further corroborate our findings, IL‐6 was also found expressed in abdominal solid tumours collected from OC hosts (Figure 10B).
Figure 9.
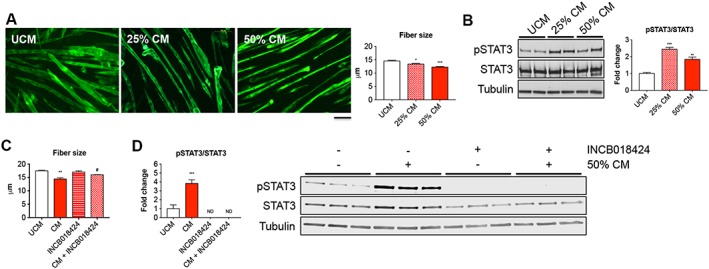
ES‐2‐derived factors directly affect fibre size in C2C12 myotube cultures. Assessment of myofibre size in C2C12 murine myotube cultures exposed for up to 48 h to 25% and 50% ES‐2 unconditioned medium (UCM) or conditioned medium (CM) in 2% horse serum‐containing differentiation medium (n = 250–350). Green staining: myosin heavy chain. Scale bar: 100 μm (A). Representative western blotting for pSTAT3 and STAT3 in whole protein extracts from C2C12 myotubes exposed to different concentrations of ES‐2 CM (n = 3) (B). Representative western blotting and quantification for pSTAT3 (C) and myotubes size (D) in C2C12 myotubes exposed to 50% ES‐2 CM, with or without INCB018424 (400 nM). pSTAT3 levels were normalized to STAT3 expression. Tubulin was used as loading control. ND, not detected. Data (means ± standard deviation) are expressed as fold change vs. UCM. Significance of the differences: * P < 0.05, ** P < 0.01, *** P < 0.001 vs. UCM; # P < 0.05 vs. CM.
Figure 10.
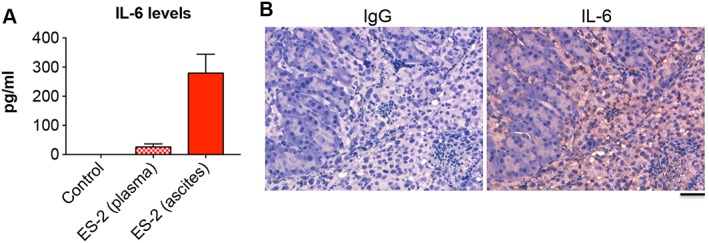
IL‐6 is elevated in ES‐2 hosts. IL‐6 levels assessed in plasma and ascites from control and ES‐2 bearing mice (n = 6–10) by means of ELISA (A). Ascites was collected at time of euthanasia and subjected to centrifugation to remove cells and cellular debris. Data (pg/mL) are expressed as means ± standard deviation. Immunohistochemistry for IL‐6 in two representative sections from ES‐2 solid tumours infiltrating murine pancreas (n = 10) (B). Normal rabbit IgG was used as negative control. Scale bar: 50 μm.
Mitochondrial homoeostasis and respiration are impaired in C2C12 myotubes exposed to ES‐2 conditioned medium
To assess whether tumour‐derived factors were directly responsible for the changes in the expression of mitochondrial proteins, as also described in the muscle of OC hosts, C2C12 myotubes were exposed to 25% and 50% CM from ES‐2 cultures for up to 48 h. Expression of proteins associated with mitochondrial biogenesis (PGC1α, Cytochrome C), fusion (OPA1), and fission (DRP1) was evaluated by means of western blotting. Exposure to 50% CM was associated with significant reduction in PGC1α levels (−45% vs. UCM, P < 0.05), Cytochrome C (−46% vs. UCM, P < 0.05), and OPA1 (−35% vs. UCM, P < 0.05), while DRP1 was substantially unchanged (Figure 11A). Consistently, the amount of polarized mitochondria, as measured by means of MitoTracker staining (Figure 11B), was significantly decreased in C2C12 cells exposed to 50% ES‐2 CM (−50% vs. UCM, P < 0.01). Additionally, the extracellular flux analysis performed by means of Seahorse XF (Figure 11C) showed significant reduction in mitochondrial respiration (−27% vs. UCM, P < 0.01), spare respiratory capacity (−31% vs. UCM, P < 0.01), and overall ATP production (−25% vs. UCM, P < 0.01). On the contrary, the proton leak was markedly increased (+214% vs. UCM, P < 0.05).
Figure 11.
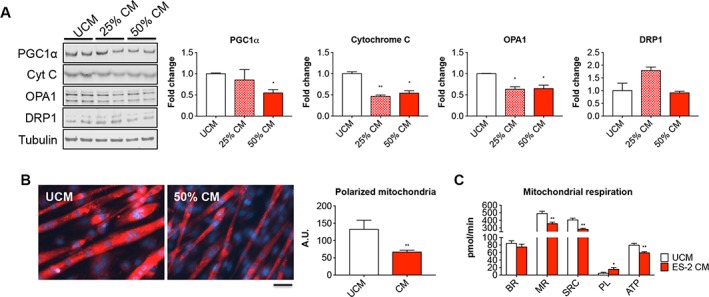
Mitochondrial homoeostasis and respiration are impaired in C2C12 myotubes exposed to ES‐2 conditioned medium (CM). Representative western blotting and quantification for PGC1α, Cytochrome C (Cyt C), OPA1, and DRP1 in protein extracts from C2C12 myotubes exposed to different concentrations of ES‐2 CM (n = 3). Tubulin was used as loading control (A). Mitochondrial polarization assessed by means of MitoTracker red CMXRos staining in C2C12 exposed to 50% ES‐2 CM (n = 3). Red signal intensity was quantified by using the ImageJ software (five fields per sample). Scale bar: 100 μm (B). Basal respiration (BR), mitochondrial respiration (MR), spare respiratory capacity (SRC), proton leak (PL), and ATP production (data expressed in pmol/min) determined by extracellular flux analysis in C2C12 myotubes exposed to 50% ES‐2 CM (n = 3) (C). Data are reported as means ± standard deviation. Significance of the differences: * P < 0.05, ** P < 0.01, *** P < 0.001 vs. unconditioned medium (UCM).
Discussion
Ovarian cancer is the deadliest amongst gynaecologic malignancies in the USA.1 Advanced OC patients often experience the symptoms of cachexia, an extremely debilitating condition mainly responsible for body weight loss, severe muscle and fat wasting, fatigue, decreased response to antineoplastic therapies, and overall reduced survival.45, 46 We and others showed that counteraction of cachexia by means of pro‐anabolic agents prevents chemotherapy side toxicities in skeletal muscle and promotes better survival in cancer hosts, despite limited or no effects on tumour size.24, 30, 47, 48, 49 This also suggests that preservation of skeletal muscle mass per se may benefit quality of life and survival in patients with cancer. Hence, determining the mechanisms responsible for muscle wasting in OC represents a task of the utmost importance. Unfortunately, cachexia is not easily detected in women with OC, due to bulky tumour deposits or to the development of ascites that could mask changes in body weight.34, 50
Research on OC‐related cachexia is currently limited by the paucity of molecular data on the development of cachexia in the available animal models. These models remain poorly characterized in terms of body composition as well as cytokine and gene expression data.3 Zhou et al. previously reported that the growth of the TOV‐21G clear cell carcinoma, a distinct pathologic subtype of epithelial OC, only accounting for less than 5% of all ovarian malignancies, promotes progressive body and muscle weight loss, although no functional data were shown.30 Administration of ACVR2B/Fc was shown to completely restore muscle wasting in this model.30 ACVR2B/Fc is an inhibitor of the Activin Receptor 2B signalling known to preserve muscle mass and prolong survival in tumour hosts and to increase muscle and bone mass following chemotherapy treatment.48 However, based on the results of a recent genomic profiling, the TOV‐21G tumours were found to possess few copy number alterations and a ‘hyper‐mutated’ genome, thus setting this cell type apart from the rest of the OC cell lines and from the HGS‐OC tissue samples.4
In order to establish an animal model that resembles the human phenotype as close as possible, in the present study, we characterized the pro‐cachectic effects associated with the in vivo growth of ES‐2 OC xenografts, both at the functional and molecular level. The ES‐2 cells were originally classified as ovarian clear cell carcinoma.51 After comparison with the genomic profiles of the most extensively studied HGS‐OC cell types, such as SK‐OV‐3, A2780, OVCAR‐3, and CAOV‐3, the ES‐2 cells were recently classified as a suitable model for the study of HGS‐OC, the most aggressive subtype of OCs, accounting for up to two‐thirds of all deaths among women affected with this kind of malignancy.3, 4
The implantation of OC tumours in immune‐deficient animals is an accepted method to model and study the development, the progression, and the response to anti‐proliferative treatments of OC cells, in line with the first report describing the use of xenografts of human tumours.52 However, none of these models had been previously characterized in terms of effects on muscle size, muscle function, and bone mass, thus representing a critical gap to be addressed. In our study, human ES‐2 cells were inoculated intraperitoneally in immune‐deficient mice, similarly to what previously described.51
One of the main limitations associated with the use of xenograft models relates to the difficulty in monitoring intraperitoneal disease formation and tumour progression in vivo. Indeed, in the absence of validated exogenous serum markers, the assessment of tumour growth, especially at early time points, is possible only by means of magnetic resonance imaging or by using cells that are stably expressing fluorescent markers, such as green fluorescent protein (GFP).53 However, these analyses can often be cost‐prohibitive or very stressful for the experimental animals. An additional limitation is often associated with the site of tumour implantation, e.g. subcutaneous vs. intraperitoneal vs. orthotopic. The majority of OC xenograft models described in the literature displays subcutaneous or intraperitoneal implantation of OC cells, while only recently orthotopic tumour implantation has been reported as a novel approach that more closely resembles the human disease.54 Some patient‐derived xenograft (PDX) models for the study of OC are also available that likely recapitulate the primary disease and the patient phenotype, as well as important features such as tumour histotype and response to chemotherapy. However, Ricci et al.55 advised on the difficulty in developing these models, associated with an overall low take rate of about 25%, and raised concerns about the higher genomic variability found in the PDX models compared with patient tumours. Moreover, these PDX models have not been investigated for cachexia development.
Importantly, it is well accepted that the intraperitoneal implantation of ovarian tumours is generally an appropriate and clinically relevant way to model the development and the progression of OC.56 In our experimental approach, we chose to implant ES‐2 cells intraperitoneally in order to simulate disseminated abdominal disease and ascites formation, as frequently observed in women with advanced HGS‐OC. Our choice was also supported by the evidence that the ES‐2 tumours were shown to maintain similar undifferentiated phenotype and proliferative rates, regardless of the injection site.37 In the present study, we found that approximately 2 weeks after tumour inoculation, the NSG mice demonstrated significant ascites accumulation, along with severe muscle weight loss and increased muscle weakness. These features are consistent with advanced disease in patients with OC and are normally referred to as cancer cachexia.32
Despite differences in the site of tumour implantation, gender, or mouse strain, most of the cachexia models that are currently utilized for investigations in the field (e.g. C26 or LLC) present similar phenotypes, although the molecular mechanisms may differ significantly. One of the advantages of our animal model is that the intraperitoneal implantation of ES‐2 cells, recapitulating the clinical presentation of HGS‐OC (e.g. TP53 mutations, ascites formation, and hyper‐inflammatory state), also allows the study of the direct muscle effects associated with the development of ovarian tumours. Moreover, along with the fact that the ES‐2 cells exhibit moderate chemoresistance to a number of chemotherapeutic agents, including doxorubicin, cisplatin, and etoposide,51 our model may also represent a suitable tool for studies aimed at investigating the development of chemoresistance and the occurrence of muscle toxicity that follows to chemotherapy administration.24, 49
Our data showed markedly elevated STAT3 phosphorylation in the skeletal muscle of ES‐2 hosts and in myotubes exposed to ES‐2 CM. Interestingly, the levels of total STAT3 were also increased in the muscle of tumour hosts, in line with our previous evidence reporting STAT3 itself among the known STAT3 targets in cachectic muscles.14 Therefore, our data suggest that muscle wasting in the presence of OC may follow to activation of the IL‐6/STAT3 signalling pathway, in accordance with our previous findings.14, 15 Indeed, the manifestation of an overall inflammatory state is one of the hallmarks of cachexia.57 In particular, the systemic metabolic derangements that follow the occurrence of cancer cachexia have been described in association with high levels of pro‐inflammatory cytokines, including tumor necrosis factor (TNF), IL‐1α, IL‐1β, interferon‐γ, and IL‐6.14, 58, 59 Strong evidence indicates that pro‐inflammatory cytokines such as IL‐6 are markedly elevated in OC due to combined systemic response to tumourigenesis and antineoplastic therapies. In our animal model, making use of immune deficient NSG mice, markedly elevated human IL‐6 levels were detected in plasma and ascites. In a similar manner, high IL‐6 was detected in ES‐2 abdominal tumours isolated from OC‐bearers, therefore suggesting that the ES‐2 cells were the main source of this cytokine. Of note, IL‐6 in ascites was previously shown to correlate with reduced survival as well as cachexia‐related symptoms (muscle depletion, increased fatigue, disability, and depression) in OC patients.13, 60 Moreover, published studies suggest that pharmacologic inhibition of the IL‐6 signalling both reduces OC growth and enhances the efficacy of chemo‐therapy and radio‐therapy,12, 28, 61, 62 and also improves muscle mass and restores muscle strength in tumour‐bearing mice.15 In line with these observations, here we showed that pharmacologic inhibition of the IL‐6/STAT3 signalling by means of INCB018424 was sufficient to prevent OC‐induced myofibre atrophy in C2C12 cultures.
Evidence from the literature supports the idea that mitochondrial alterations and enhanced protein catabolism are among the most relevant factors associated with the development of muscle atrophy in cancer. In our study, the levels of regulators of mitochondrial fusion, such as OPA1 and Mitofusin‐2, and of mitochondrial biogenesis, such as PGC1α and Cytochrome‐C, were significantly reduced in the muscle of tumour hosts compared with the control animals. This suggests that OC growth may affect the muscle mitochondrial homoeostasis, thereby promoting metabolic derangements that, in turn, could lead to occurrence of muscle atrophy and increased fatigue. Our hypothesis was further supported by the fact that, along with myofibre atrophy, C2C12 myotubes exposed to CM from ES‐2 cells also showed modulation of mitochondrial protein expression, reduced number of polarized mitochondria, and abnormalities in mitochondrial respiration and energy production. These findings are consistent with our and other previous reports obtained by characterizing experimental models of cancer‐induced or chemotherapy‐induced cachexia.15, 24, 63, 64 Notably, White et al. suggested that altered mitochondrial homoeostasis is an early event in the initiation of IL‐6 induced cachexia, which may precede the loss of muscle mitochondria.65 Similarly, the ES‐2 bearers show the signs of markedly upregulated ubiquitin ligases expression, such as Atrgin‐1, MuRF‐2, and Fbxo31, and elevated muscle protein ubiquitination, previously described as molecular alterations consistent with cancer cachexia.43, 66
In the present study, we also demonstrated that the overall body composition assessment by DXA was significantly affected in the ES‐2 hosts, showing marked depletion of lean and adipose tissue, as well as reduction in BMD and BMC. These findings were consistent with the bone morphometry analyses conducted by means of μCT scanning, reporting significant depletion of bone mass, along with reduced trabecular bone in both femurs and vertebrae. Ours is the first report showing involvement of bone tissue following growth of experimental OC. Although no functional measurements were performed on the bones from ES‐2 hosts, our previous evidence suggests that bone content generally correlates with the overall bone strength in cancer cachexia.67 Although the molecular mechanism(s) responsible for bone depletion in association with the occurrence of OC are not known, high levels of IL‐6, as shown in the animals bearing the ES‐2 OC, have been previously reported in association not only with the development of paraneoplastic complications, such as cachexia, leucocytosis, and thrombocytosis, but also with the occurrence of hypercalcaemia, a condition responsible for severe loss of bone mass,68 or juvenile idiopathic arthritis, a systemic inflammatory condition characterized by clinical features that include stunted growth and skeletal abnormalities.69, 70 In this regard, elevated IL‐6 seems to play a relevant role by primarily affecting bone turnover, along with increased bone resorption and reduced bone formation.71
Unlike our previous reports describing the occurrence of bone loss following development of colorectal tumours or after prolonged administration of chemotherapy,48, 67 bone tissue is not generally investigated in the experimental models utilized for the study of HGS‐OCs. In the clinical setting, OC is not frequently associated with enhanced bone fractures per se or described as a risk factor for the development of osteoporosis, although elevated bone turnover markers were detected in women undergoing risk‐reducing salpingo‐oophorectomy to decrease the risk for breast cancer and OC.72, 73 Surgery represents the preferred treatment for OC and frequently involves the removal of both ovaries as a part of treatment, eventually followed by radiation therapy, intraperitoneal chemotherapy with platinum and a taxane, or hormone replacement treatment.74 Consistently, women treated for their gynaecologic cancers are often affected with hypogonadism, along with reduced BMD as a consequence of surgical or chemotherapy treatments.75, 76, 77 Indeed, bone fragility represents a condition notoriously associated with the occurrence of hypogonadism in a large percentage of patients and contributes to worsen the overall outcomes. While in the present study we did not evaluate the presence of bone tumours in the animals inoculated with ES‐2 cells, the formation of bone metastases is a rare, but well‐recognized event in OC, representing a factor predictive of poor prognosis and increased morbidity.78, 79
In conclusion, we provided evidence that the growth of ES‐2 intraperitoneal xenografts causes progressive decline in muscle weight and muscle function, depletion of adipose tissue, and bone degeneration in association with high levels of tumour‐derived IL‐6. In accordance with our previous findings, systemic inflammation, muscle protein hypercatabolism, and mitochondrial alterations concur to the development of cachexia. Overall, the in vivo implantation of ES‐2 cells resembles the clinical presentation of HGS‐OCs and provides an accurate in vivo platform for the study of OC‐associated cachexia and for future testing of targeted treatments. Moreover, our observations describe for the first time the occurrence of bone loss in association with OC and support the importance of screening for bone quality in patients receiving treatment for OC, especially in the long term.
Conflict of interest
None declared.
Author contributions
F. P., R. B., T. A. Z., and A. B. conceived and designed the experiments; F. P., R. B., Y. K., C. E. E., and A. B. performed the in vivo experiments, the body composition assessment, the muscle function analysis, and the molecular characterization of cachexia; Y. K. and L. F. B. performed and analysed the bone morphometry analysis; L. N., S. M., M. E. C., L. F. B., and A. B. wrote and edit the paper.
Supporting information
Figure S1. Morphology of ES‐2 solid tumours.
Figure S2: Growth of the ES‐2 Tumour does not affect muscle fibrosis or regeneration.
Figure S3: Exposure to McCoys medium does not affect fibre size.
Figure S4: INCB018424 rescues OC‐induced fibre atrophy.
Acknowledgements
The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle.80 This study was supported by the Department of Surgery and the Department of Otolaryngology – Head&Neck Surgery at Indiana University and by grants from the National Institutes of Health (R21CA190028 to A. B., 1PO1AG039355 to L. F. B., and R01CA194593 and R01CA122596 to T. A. Z.), the V Foundation for Cancer Research (V2017‐021 to A. B.), the Indiana University Simon Cancer Center (IUSCC Associate Member Pilot Funding Mechanism to A. B.), the Veterans Administration (I01BX002764‐01A1 to T. A. Z), and the Commonwealth of Pennsylvania Health Research Formula Fund (2012F‐RFA67‐16 and 080‐37038‐AI0801 to T. A. Z.). We thank the Cancer Center at Indiana University School of Medicine funded by the IU Simon Cancer Center Support Grant (P30CA082709) for the use of the In Vivo Therapeutics Core Facility, which generated the NSG mice used in this study. The #12G10 anti‐Tubulin monoclonal antibody developed by Frankel J and Nelsen EM at University of Iowa and the #MF‐20 anti‐Myosin Heavy Chain antibody developed by Donald A. Fischman at Cornell University were obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA, USA. The authors would also like to thank John Spence, PhD, for his precious contribution in editing the manuscript.
Pin, F. , Barreto, R. , Kitase, Y. , Mitra, S. , Erne, C. E. , Novinger, L. J. , Zimmers, T. A. , Couch, M. E. , Bonewald, L. F. , and Bonetto, A. (2018) Growth of ovarian cancer xenografts causes loss of muscle and bone mass: a new model for the study of cancer cachexia. Journal of Cachexia, Sarcopenia and Muscle, 9: 685–700. 10.1002/jcsm.12311.
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- 2. Gershenson DM, Sun CC, Lu KH, Coleman RL, Sood AK, Malpica A, et al. Clinical behavior of stage II‐IV low‐grade serous carcinoma of the ovary. Obstet Gynecol 2006;108:361–368. [DOI] [PubMed] [Google Scholar]
- 3. Bowtell DD. The genesis and evolution of high‐grade serous ovarian cancer. Nat Rev Cancer 2010;10:803–808. [DOI] [PubMed] [Google Scholar]
- 4. Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun 2013;4:2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Anglesio MS, Wiegand KC, Melnyk N, Chow C, Salamanca C, Prentice LM, et al. Type‐specific cell line models for type‐specific ovarian cancer research. PLoS One 2013;8:e72162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30. [DOI] [PubMed] [Google Scholar]
- 7. Baldwin LA, Huang B, Miller RW, Tucker T, Goodrich ST, Podzielinski I, et al. Ten‐year relative survival for epithelial ovarian cancer. Obstet Gynecol 2012;120:612–618. [DOI] [PubMed] [Google Scholar]
- 8. Bristow RE, Tomacruz RS, Armstrong DK, Trimble EL, Montz FJ. Survival effect of maximal cytoreductive surgery for advanced ovarian carcinoma during the platinum era: a meta‐analysis. J Clin Oncol Off J Am Soc Clin Oncol 2002;20:1248–1259. [DOI] [PubMed] [Google Scholar]
- 9. Winter WE 3rd, Maxwell GL, Tian C, Sundborg MJ, Rose GS, Rose PG, et al. Tumor residual after surgical cytoreduction in prediction of clinical outcome in stage IV epithelial ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol Off J Am Soc Clin Oncol 2008;26:83–89. [DOI] [PubMed] [Google Scholar]
- 10. Armstrong DK, Bundy B, Wenzel L, Huang HQ, Baergen R, Lele S, et al. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med 2006;354:34–43. [DOI] [PubMed] [Google Scholar]
- 11. Han Z, Feng J, Hong Z, Chen L, Li W, Liao S, et al. Silencing of the STAT3 signaling pathway reverses the inherent and induced chemoresistance of human ovarian cancer cells. Biochem Biophys Res Commun 2013;435:188–194. [DOI] [PubMed] [Google Scholar]
- 12. Stone RL, Nick AM, McNeish IA, Balkwill F, Han HD, Bottsford‐Miller J, et al. Paraneoplastic thrombocytosis in ovarian cancer. N Engl J Med 2012;366:610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lane D, Matte I, Rancourt C, Piche A. Prognostic significance of IL‐6 and IL‐8 ascites levels in ovarian cancer patients. BMC Cancer 2011;11:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bonetto A, Aydogdu T, Kunzevitzky N, Guttridge DC, Khuri S, Koniaris LG, et al. STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PLoS One 2011;6:e22538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bonetto A, Aydogdu T, Jin X, Zhang Z, Zhan R, Puzis L, et al. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL‐6 and in experimental cancer cachexia. Am J Physiol Endocrinol Metab 2012;303:E410–E421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bronger H, Hederich P, Hapfelmeier A, Metz S, Noel PB, Kiechle M, et al. Sarcopenia in advanced serous ovarian cancer. International Journal of Gynecological Cancer: Official Journal of the International Gynecological Cancer Society 2017;27:223–232. [DOI] [PubMed] [Google Scholar]
- 17. Partridge EE, Barnes MN. Epithelial ovarian cancer: prevention, diagnosis, and treatment. CA Cancer J Clin 1999;49:297–320. [DOI] [PubMed] [Google Scholar]
- 18. Maccio A, Madeddu C. Inflammation and ovarian cancer. Cytokine 2012;58:133–147. [DOI] [PubMed] [Google Scholar]
- 19. Gadducci A, Cosio S, Fanucchi A, Genazzani AR. Malnutrition and cachexia in ovarian cancer patients: pathophysiology and management. Anticancer Res 2001;21:2941–2947. [PubMed] [Google Scholar]
- 20. Hess LM, Barakat R, Tian C, Ozols RF, Alberts DS. Weight change during chemotherapy as a potential prognostic factor for stage III epithelial ovarian carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol 2007;107:260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Laky B, Janda M, Bauer J, Vavra C, Cleghorn G, Obermair A. Malnutrition among gynaecological cancer patients. Eur J Clin Nutr 2007;61:642–646. [DOI] [PubMed] [Google Scholar]
- 22. Gil KM, Frasure HE, Hopkins MP, Jenison EL, von Gruenigen VE. Body weight and composition changes in ovarian cancer patients during adjuvant chemotherapy. Gynecol Oncol 2006;103:247–252. [DOI] [PubMed] [Google Scholar]
- 23. Damrauer JS, Stadler ME, Acharyya S, Baldwin AS, Couch ME, Guttridge DC. Chemotherapy‐induced muscle wasting: association with NF‐kB and cancer cachexia. Basic Applied Myology 2008;18:139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barreto R, Waning DL, Gao H, Liu Y, Zimmers TA, Bonetto A. Chemotherapy‐related cachexia is associated with mitochondrial depletion and the activation of ERK1/2 and p38 MAPKs. Oncotarget 2016;7:43442–43460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thoresen L, Frykholm G, Lydersen S, Ulveland H, Baracos V, Prado CM, et al. Nutritional status, cachexia and survival in patients with advanced colorectal carcinoma. Different assessment criteria for nutritional status provide unequal results. Clin Nutr 2013;32:65–72. [DOI] [PubMed] [Google Scholar]
- 26. Prado CM, Antoun S, Sawyer MB, Baracos VE. Two faces of drug therapy in cancer: drug‐related lean tissue loss and its adverse consequences to survival and toxicity. Curr Opin Clin Nutr Metab Care 2011;14:250–254. [DOI] [PubMed] [Google Scholar]
- 27. Antoun S, Baracos VE, Birdsell L, Escudier B, Sawyer MB. Low body mass index and sarcopenia associated with dose‐limiting toxicity of sorafenib in patients with renal cell carcinoma. Ann Oncol 2010;21:1594–1598. [DOI] [PubMed] [Google Scholar]
- 28. Cohen S, Bruchim I, Graiver D, Evron Z, Oron‐Karni V, Pasmanik‐Chor M, et al. Platinum‐resistance in ovarian cancer cells is mediated by IL‐6 secretion via the increased expression of its target cIAP‐2. J Mol Med 2013;91:357–368. [DOI] [PubMed] [Google Scholar]
- 29. Kanabrocki EL, Hermida RC, Haseman MB, Bettis K, Young RM, Mathew JP, et al. Chronotherapy of ovarian cancer: effect on blood variables and serum cytokines. A case report. Clin Ter 2006;157:349–354. [PubMed] [Google Scholar]
- 30. Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010;142:531–543. [DOI] [PubMed] [Google Scholar]
- 31. Kim TY, Kim MY, Sohn JH, Kim SM, Ryu JA, Lim S, et al. Sarcopenia as a useful predictor for long‐term mortality in cirrhotic patients with ascites. J Korean Med Sci 2014;29:1253–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Torres ML, Hartmann LC, Cliby WA, Kalli KR, Young PM, Weaver AL, et al. Nutritional status, CT body composition measures and survival in ovarian cancer. Gynecol Oncol 2013;129:548–553. [DOI] [PubMed] [Google Scholar]
- 33. Aust S, Knogler T, Pils D, Obermayr E, Reinthaller A, Zahn L, et al. Skeletal muscle depletion and markers for cancer cachexia are strong prognostic factors in epithelial ovarian cancer. PLoS One 2015;10:e0140403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kumar A, Moynagh MR, Multinu F, Cliby WA, McGree ME, Weaver AL, et al. Muscle composition measured by CT scan is a measurable predictor of overall survival in advanced ovarian cancer. Gynecol Oncol 2016;142:311–316. [DOI] [PubMed] [Google Scholar]
- 35. Rutten IJ, Ubachs J, Kruitwagen RF, van Dijk DP, Beets‐Tan RG, Massuger LF, et al. The influence of sarcopenia on survival and surgical complications in ovarian cancer patients undergoing primary debulking surgery. Eur J Surg Oncol 2017;43:717–724. [DOI] [PubMed] [Google Scholar]
- 36. Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes‐Franco J, Thomas DA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med 2010;363:1117–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shaw TJ, Senterman MK, Dawson K, Crane CA, Vanderhyden BC. Characterization of intraperitoneal, orthotopic, and metastatic xenograft models of human ovarian cancer. Mol Ther 2004;10:1032–1042. [DOI] [PubMed] [Google Scholar]
- 38. Fontes‐Oliveira CC, Busquets S, Toledo M, Penna F, Paz Aylwin M, Sirisi S, et al. Mitochondrial and sarcoplasmic reticulum abnormalities in cancer cachexia: altered energetic efficiency? Biochim Biophys Acta 2013;1830:2770–2778. [DOI] [PubMed] [Google Scholar]
- 39. Bonetto A, Andersson DC, Waning DL. Assessment of muscle mass and strength in mice. Bonekey Rep 2015;4:732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012;9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Muller R. Guidelines for assessment of bone microstructure in rodents using micro‐computed tomography. J Bone Miner Res 2010;25:1468–1486. [DOI] [PubMed] [Google Scholar]
- 42. Fanzani A, Zanola A, Rovetta F, Rossi S, Aleo MF. Cisplatin triggers atrophy of skeletal C2C12 myotubes via impairment of Akt signalling pathway and subsequent increment activity of proteasome and autophagy systems. Toxicol Appl Pharmacol 2011;250:312–321. [DOI] [PubMed] [Google Scholar]
- 43. Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, et al. Foxo transcription factors induce the atrophy‐related ubiquitin ligase atrogin‐1 and cause skeletal muscle atrophy. Cell 2004;117:399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Milan G, Romanello V, Pescatore F, Armani A, Paik JH, Frasson L, et al. Regulation of autophagy and the ubiquitin‐proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun 2015;6:6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Muscaritoli M, Anker SD, Argiles J, Aversa Z, Bauer JM, Biolo G, et al. Consensus definition of sarcopenia, cachexia and pre‐cachexia: joint document elaborated by Special Interest Groups (SIG) “cachexia‐anorexia in chronic wasting diseases” and “nutrition in geriatrics”. Clin Nutr 2010;29:154–159. [DOI] [PubMed] [Google Scholar]
- 46. Fearon KC, Glass DJ, Guttridge DC. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 2012;16:153–166. [DOI] [PubMed] [Google Scholar]
- 47. Benny Klimek ME, Aydogdu T, Link MJ, Pons M, Koniaris LG, Zimmers TA. Acute inhibition of myostatin‐family proteins preserves skeletal muscle in mouse models of cancer cachexia. Biochem Biophys Res Commun 2010;391:1548–1554. [DOI] [PubMed] [Google Scholar]
- 48. Barreto R, Kitase Y, Matsumoto T, Pin F, Colston KC, Couch KE, et al. ACVR2B/Fc counteracts chemotherapy‐induced loss of muscle and bone mass. Sci Rep 2017;7:14470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nissinen TA, Degerman J, Rasanen M, Poikonen AR, Koskinen S, Mervaala E, et al. Systemic blockade of ACVR2B ligands prevents chemotherapy‐induced muscle wasting by restoring muscle protein synthesis without affecting oxidative capacity or atrogenes. Sci Rep 2016;6:32695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rutten IJG, van Dijk DPJ, Kruitwagen RFPM, Betts‐Tan RGH, Olde Damink SWM, van Gorp T. Loss of skeletal muscle during neoadjuvant chemotherapy is related to decreased survival in ovarian cancer patients. J Cachexia Sarcopenia Muscle 2016;7:458–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lau DH, Lewis AD, Ehsan MN, Sikic BI. Multifactorial mechanisms associated with broad cross‐resistance of ovarian carcinoma cells selected by cyanomorpholino doxorubicin. Cancer Res 1991;51:5181–5187. [PubMed] [Google Scholar]
- 52. Rygaard J, Povlsen CO. Heterotransplantation of a human malignant tumour to "Nude" mice. Acta Pathol Microbiol Scand 1969;77:758–760. [DOI] [PubMed] [Google Scholar]
- 53. Gossmann A, Helbich TH, Mesiano S, Shames DM, Wendland MF, Roberts TP, et al. Magnetic resonance imaging in an experimental model of human ovarian cancer demonstrating altered microvascular permeability after inhibition of vascular endothelial growth factor. Am J Obstet Gynecol 2000;183:956–963. [DOI] [PubMed] [Google Scholar]
- 54. Shao M, Cao L, Shen C, Satpathy M, Chelladurai B, Bigsby RM, et al. Epithelial‐to‐mesenchymal transition and ovarian tumor progression induced by tissue transglutaminase. Cancer Res 2009;69:9192–9201. [DOI] [PubMed] [Google Scholar]
- 55. Ricci F, Bizzaro F, Cesca M, Guffanti F, Ganzinelli M, Decio A, et al. Patient‐derived ovarian tumor xenografts recapitulate human clinicopathology and genetic alterations. Cancer Res 2014;74:6980–6990. [DOI] [PubMed] [Google Scholar]
- 56. Mitra AK, Davis DA, Tomar S, Roy L, Gurler H, Xie J, et al. In vivo tumor growth of high‐grade serous ovarian cancer cell lines. Gynecol Oncol 2015;138:372–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Onesti JK, Guttridge DC. Inflammation based regulation of cancer cachexia. Biomed Res Int 2014;2014:168407–168407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Argiles JM, Busquets S, Lopez‐Soriano FJ. Cytokines in the pathogenesis of cancer cachexia. Curr Opin Clin Nutr Metab Care 2003;6:401–406. [DOI] [PubMed] [Google Scholar]
- 59. Costelli P, Carbo N, Tessitore L, Bagby GJ, Lopez‐Soriano FJ, Argiles JM, et al. Tumor necrosis factor‐alpha mediates changes in tissue protein turnover in a rat cancer cachexia model. J Clin Invest 1993;92:2783–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schrepf A, Clevenger L, Christensen D, DeGeest K, Bender D, Ahmed A, et al. Cortisol and inflammatory processes in ovarian cancer patients following primary treatment: relationships with depression, fatigue, and disability. Brain Behav Immun 2013;30:S126–S134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang Y, Niu XL, Qu Y, Wu J, Zhu YQ, Sun WJ, et al. Autocrine production of interleukin‐6 confers cisplatin and paclitaxel resistance in ovarian cancer cells. Cancer Lett 2010;295:110–123. [DOI] [PubMed] [Google Scholar]
- 62. Ji T, Gong D, Han Z, Wei X, Yan Y, Ye F, et al. Abrogation of constitutive Stat3 activity circumvents cisplatin resistant ovarian cancer. Cancer Lett 2013;341:231–239. [DOI] [PubMed] [Google Scholar]
- 63. Barreto R, Mandili G, Witzmann FA, Novelli F, Zimmers TA, Bonetto A. Cancer and chemotherapy contribute to muscle loss by activating common signaling pathways. Front Physiol 2016;7:472, 10.3389/fphys.2016.00472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pin F, Busquets S, Toledo M, Camperi A, Lopez‐Soriano FJ, Costelli P, et al. Combination of exercise training and erythropoietin prevents cancer‐induced muscle alterations. Oncotarget 2015;6:43202–43215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. White JP, Puppa MJ, Sato S, Gao S, Price RL, Baynes JW, et al. IL‐6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the ApcMin/+ mouse. Skelet Muscle 2012;2:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kwak KS, Zhou X, Solomon V, Baracos VE, Davis J, Bannon AW, et al. Regulation of protein catabolism by muscle‐specific and cytokine‐inducible ubiquitin ligase E3alpha‐II during cancer cachexia. Cancer Res 2004;64:8193–8198. [DOI] [PubMed] [Google Scholar]
- 67. Bonetto A, Kays JK, Parker VA, Matthews RR, Barreto R, Puppa MJ, et al. Differential bone loss in mouse models of colon cancer cachexia. Front Physiol 2017;7:679, 10.3389/fphys.2016.00679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Black K, Garrett IR, Mundy GR. Chinese hamster ovarian cells transfected with the murine interleukin‐6 gene cause hypercalcemia as well as cachexia, leukocytosis and thrombocytosis in tumor‐bearing nude mice. Endocrinology 1991;128:2657–2659. [DOI] [PubMed] [Google Scholar]
- 69. MacRae VE, Farquharson C, Ahmed SF. The pathophysiology of the growth plate in juvenile idiopathic arthritis. Rheumatology (Oxford) 2006;45:11–19. [DOI] [PubMed] [Google Scholar]
- 70. Lien G, Selvaag AM, Flato B, Haugen M, Vinje O, Sorskaar D, et al. A two‐year prospective controlled study of bone mass and bone turnover in children with early juvenile idiopathic arthritis. Arthritis Rheum 2005;52:833–840. [DOI] [PubMed] [Google Scholar]
- 71. De Benedetti F, Rucci N, Del Fattore A, Peruzzi B, Paro R, Longo M, et al. Impaired skeletal development in interleukin‐6‐transgenic mice: a model for the impact of chronic inflammation on the growing skeletal system. Arthritis Rheum 2006;54:3551–3563. [DOI] [PubMed] [Google Scholar]
- 72. Fakkert IE, Abma EM, Westrik IG, Lefrandt JD, Wolffenbuttel BH, Oosterwijk JC, et al. Bone mineral density and fractures after risk‐reducing salpingo‐oophorectomy in women at increased risk for breast and ovarian cancer. Eur J Cancer 2015;51:400–408. [DOI] [PubMed] [Google Scholar]
- 73. Fakkert IE, van der Veer E, Abma EM, Lefrandt JD, Wolffenbuttel BH, Oosterwijk JC, et al. Elevated bone turnover markers after risk‐reducing salpingo‐oophorectomy in women at increased risk for breast and ovarian cancer. PLoS One 2017;12:e0169673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ottanelli S. Prevention and treatment of bone fragility in cancer patient. Clin Cases Miner Bone Metab 2015;12:116–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Stavraka C, Maclaran K, Gabra H, Agarwal R, Ghaem‐Maghami S, Taylor A, et al. A study to evaluate the cause of bone demineralization in gynecological cancer survivors. Oncologist 2013;18:423–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Pfeilschifter J, Diel IJ. Osteoporosis due to cancer treatment: pathogenesis and management. J Clin Oncol Off J Am Soc Clin Oncol 2000;18:1570–1593. [DOI] [PubMed] [Google Scholar]
- 77. Michaud LB, Goodin S. Cancer‐treatment‐induced bone loss, part 1. Am J Health Syst Pharm 2006;63:419–430. [DOI] [PubMed] [Google Scholar]
- 78. Petty RD, Gilbert F, Hutcheon AW, Sarkar TK, McNally O, Parkin D. Bone metastasis from epithelial ovarian carcinoma. Lancet Oncol 2002;3:513. [DOI] [PubMed] [Google Scholar]
- 79. Zhang M, Sun J. Bone metastasis from ovarian cancer. Clinical analysis of 26 cases. Saudi Med J 2013;34:1270–1273. [PubMed] [Google Scholar]
- 80. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Morphology of ES‐2 solid tumours.
Figure S2: Growth of the ES‐2 Tumour does not affect muscle fibrosis or regeneration.
Figure S3: Exposure to McCoys medium does not affect fibre size.
Figure S4: INCB018424 rescues OC‐induced fibre atrophy.