

Abstract
Significance: Aging is a complex trait that is influenced by a combination of genetic and environmental factors. Although many cellular and physiological changes have been described to occur with aging, the precise molecular causes of aging remain unknown. Given the biological complexity and heterogeneity of the aging process, understanding the mechanisms that underlie aging requires integration of data about age-dependent changes that occur at the molecular, cellular, tissue, and organismal levels.
Recent Advances: The development of high-throughput technologies such as next-generation sequencing, proteomics, metabolomics, and automated imaging techniques provides researchers with new opportunities to understand the mechanisms of aging. Using these methods, millions of biological molecules can be simultaneously monitored during the aging process with high accuracy and specificity.
Critical Issues: Although the ability to produce big data has drastically increased over the years, integration and interpreting of high-throughput data to infer regulatory relationships between biological factors and identify causes of aging remain the major challenges. In this review, we describe recent advances and survey emerging omics approaches in aging research. We then discuss their limitations and emphasize the need for the further development of methods for the integration of different types of data.
Future Directions: Combining omics approaches and novel methods for single-cell analysis with systems biology tools would allow building interaction networks and investigate how these networks are perturbed with aging and disease states. Together, these studies are expected to provide a better understanding of the aging process and could provide insights into the pathophysiology of many age-associated human diseases. Antioxid. Redox Signal. 29, 985–1002.
Keywords: : systems biology, aging, next-generation sequencing, translational regulation, proteomics, metabolomics
Introduction
Aging is a complex biological process that is characterized by a progressive decline in physiological functions at multiple levels, including changes at the molecular, organelle, cell, tissue, and, finally, the whole organism level (Fig. 1A) (116). Over the past few decades, we have also learned that almost every biological process in the cell can contribute to physiological decline and progressive damage accumulation that eventually result in aging and death (47). Genome instability, DNA mutations, errors in RNA transcription, and protein translation, among other factors, can all contribute to organelle and cell dysfunction with aging (116, 137, 175, 177), but different macromolecules also interact with each other.
FIG. 1.

Systems biology approaches in aging research. (A) Understanding the molecular mechanisms that underlie aging requires integration of multi-layered data about age-dependent changes that occur at the molecular, cellular, tissue, and organismal levels. (B) A scheme showing the Central Dogma of Molecular Biology and different “omics” approaches that are currently available for the high-throughput quantitative analysis of molecular changes associated with aging.
A number of genes have been shown to change their expression with aging. Although transcriptional regulation plays an important role in the control of gene expression, multiple factors beyond transcription factors, including the epigenetic modifications and the rate of mRNA degradation, contribute to determining protein concentration in the cell (Fig. 1B) (131). Additional processes, such as alternative pre-mRNA splicing and post-translational modifications (PTMs), add further complexity to the organism's proteome. Moreover, protein degradation through the autophagy and ubiquitin-proteasome pathways affects protein turnover and degradation, whereas spatial location and PTMs may influence activity of proteins. Finally, changes in protein expression and their activity can influence the patterns of metabolites and change the metabolic state of the cell. Together, a gradual decline in all of the functions cited earlier may contribute to the aging process. Therefore, understanding of the mechanisms of aging is not possible without taking into account all components of the biological system, interactions between individual components, and the system's hierarchy (18, 200).
Recent advances in the development of high-throughput sequencing and different “omics” technologies now allow quantitative analyses of biological molecules at multiple levels, and how they change with aging, with high specificity and sensitivity. Despite this progress, integration of high-throughput data remains a challenging problem. In this review, we describe recent advances and discuss emerging omics approaches in aging research. We then highlight current challenges in the field and point to the need for the further development of approaches for the integration of multiple omics data. Integration of multiple types of data would make it possible to investigate the cause-consequence relationships and understand the interactions between multiple aging factors, and understand how these factors can be manipulated to develop new therapeutic targets for age-associated diseases.
Genome-Wide Approaches in Aging Research
Transcriptomics
Aging is controlled by a combination of genetic, environmental, and stochastic factors. A number of diverse genes have been shown to modulate aging. Studies in invertebrate organisms have led to the identification of hundreds of genes, whose deletion or knockdown can dramatically increase lifespan (27, 57, 129, 158). For example, a recent genome-wide analysis of yeast replicative lifespan in a non-essential gene deletion set revealed that more than 5% of the yeast gene deletions can significantly extend lifespan (129). Many of these genes were grouped into several functional categories (Fig. 2) that represent conserved biological processes, suggesting that they might be involved in the modulation of mammalian aging (40, 55). It has been also reported that the aging process can be delayed by environmental manipulations, including dietary restriction (DR) and various stress stimuli (e.g., hypoxia) (107, 123). Given that there are multiple ways by which the lifespan of an organism can be extended, it is critical to understand how the various longevity-associated genetic and environmental manipulations are related to each other, and how the different aging genes and pathways are coordinately regulated.
FIG. 2.
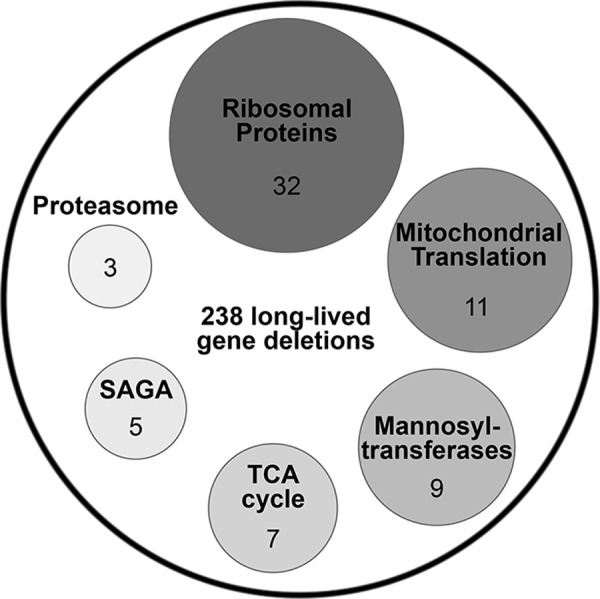
Functional groups enriched among 238 long-lived gene deletions that were identified by the genome-wide analysis of replicative lifespan in 4698 yeast deletions strains. Number of genes is indicated for each functional category (129). SAGA, components of the Spt-Ada-Gcn5-acetyltransferase complex.
Transcriptional regulation
Expression of many genes is altered with aging, and these changes may have causative effect on aging and the development of age-associated pathologies. A number of research groups used microarrays and RNA sequencing (RNA-Seq) to analyze age-dependent changes in gene expression in different model organisms (108, 113, 149, 189) as well as in humans (117, 152, 185). In addition, multiple transcriptome analyses have been performed to identify genome-wide transcriptional changes associated with increased longevity (21, 30, 130, 136, 186). These studies revealed profound changes in the transcriptome with age, which reflect age-associated degenerative processes, but might also result from transcriptional response or adaptation to aging (33). Moreover, aging is accompanied by increased cell-to-cell and increased inter-individual variation in gene expression, which was attributed to stochastic variation or transcriptional noise (4).
To identify changes in mRNA transcript levels with aging, several bioinformatics tools and statistical methods have been developed for multiple comparison analysis and functional enrichment analysis of RNA-Seq data [reviewed in Hou et al. (72)]. In addition, gene expression clustering and dimension reduction approaches, for example, principal component analysis, are commonly used to identify genes with similar expression patterns, which could help identify potential regulators. However, it is hard to establish which of the differentially expressed genes are the regulatory factors and which are the consequences of regulatory changes based only on expression patterns.
To identify upstream regulatory factors, one of the commonly used methods is to search for enrichment of binding sites in the promoter regions of genes with a similar transcription pattern (37, 155). This approach relies on information about the promoter sequence binding sites identified by chromatin immunoprecipitation followed by deep sequencing (ChIP-Seq) analysis. A similar approach, named eDM_BN (for extended Deletion Mutant Bayesian Network inference algorithm), has been recently applied to identify clusters of genes and their upstream regulatory transcription factors and signaling pathways that mediate the effects of DR on lifespan in Caenorhabditis elegans (71). As opposed to other methods, this algorithm allows integration of transcriptome data with information about known regulatory relationships between transcription factors and their targets from the modENCODE database (24) as well as the incorporation of real valued fold-change data for gene expression changes. However, care should be taken when interpreting results of these studies. Transcription factors often recognize overlapping motifs and may have overlapping sets of targets, which could present difficulty for establishing causative links.
In addition to changes in gene expression, aging is characterized by systemic decline in fidelity of transcriptional machinery, leading to transcription errors and loss of protein homeostasis (169, 175). Transcription is one of the fundamental biological processes in living organisms. Due to the inherent imperfectness of biological systems, numerous transcription errors are inevitably made during mRNA synthesis and processing. Although organisms evolved dedicated systems for degradation of aberrant RNA, increasing evidence suggests that some stable RNA intermediates can accumulate in cells, including fragments derived from intronic lariat RNA, tRNA, rRNA, as well as small noncoding RNA (43, 89, 198). Aberrant transcripts could also arise from genetic mutations, defects in transcription, premature polyadenylation, or errors in pre-mRNA splicing (174, 190).
Errors in pre-mRNA splicing
In a recent study, the link between RNA splicing machinery and longevity was established by using the C. elegans model system (62). Using transcriptomics and analysis of splicing in young and old worms in vivo with fluorescent alternative splicing reporters, researchers observed aberrant patterns of pre-mRNA splicing in old animals. Old worms were characterized by significantly increased levels of exon skipping, intron retention, and unannotated splice junctions compared with young animals, suggesting a dysregulation of splicing with aging (Fig. 3). Interestingly, DR and inhibition of mechanistic target of rapamycin (mTOR) signaling pathway, which are known to extend lifespan, shifted signatures of splicing to a more youthful state.
FIG. 3.

A model for the role of pre-mRNA splicing in aging. Errors in pre-mRNA splicing lead to damage accumulation and aging. DR promotes pre-mRNA splicing efficiency, leading to lifespan extension. DR, dietary restriction.
These findings also correlate with recent reports showing that the number of alternatively spliced genes is significantly increased with aging in mice and humans (104, 151), and suggest that manipulating RNA splicing could be used as a target for potential aging interventions in humans. Further analysis of the RNA splicing machinery in worms revealed that increased lifespan by DR and modulation of mTOR signaling is dependent on the functional SFA-1 splicing factor (62). The exact mechanisms linking DR- and mTOR-mediated longevity with SFA-1 remain unknown. Reversible protein phosphorylation has been shown to modulate the assembly and activity of the spliceosome (161). It is an attractive possibility that dysregulation of mTOR kinase activity or mTOR pathway downstream targets with aging may directly affect phosphorylation of spliceosome components and lead to altered patterns of pre-mRNA splicing. Additional genomics and proteomics studies are needed to characterize changes in spliceosome composition with aging. Integration of the results of these studies with the global analysis of PTMs (i.e., phosphorylation) would shed light on how mTOR signaling modulates pre-mRNA splicing machinery.
Translatomics
Despite the importance of transcriptional regulation in the control of gene expression during aging, many of the genes are also regulated at the level of translation. However, the mechanistic details by which translational control affects the aging process remain poorly understood. Recent technological advances in proteomics and next-generation sequencing now enabled researchers to quantitatively measure the rates of protein translation at the genome-wide level that could facilitate investigation of translational control of gene expression during aging.
The rates of protein translation can be quantitatively measured by pulse labeling of proteins using Stable Isotope Labeling by Amino acids in Cell culture (SILAC) (139). Pulsed SILAC allows labeling of newly synthesized proteins with “heavy” 13C- or 15N-labeled amino acids, which can then be distinguished from pre-existing proteins containing “light” amino acids by using mass spectrometry (MS). Among the limitations of SILAC is sensitivity of detection, which is common for MS-based methods. Insufficient “heavy” isotope incorporation into proteins expressed at low levels may produce mass spectrometric signals below the detection limit. To overcome this limitation, an alternative method has been developed and is named puromycin-associated nascent chain proteomics (PUNCH-P) (2). This method involves separation of translating ribosomes by ultracentrifugation followed by enrichment of newly synthesized proteins using biotinylated translation inhibitor.
Recently, a novel approach based on next-generation sequencing, named ribosome profiling or Ribo-Seq, has emerged as a powerful technology to study mRNA translation and mechanisms of translational regulation at the genome-wide level. This technique uses deep sequencing to quantitatively analyze genome-wide ribosome occupancy and allows monitoring the rate of protein translation in vivo at codon resolution (78). Because ribosome occupancy is determined by overall mRNA abundance and translation efficiency, Ribo-Seq combined with RNA-Seq measurements can be used to identify genes that are specifically changed at the level of translation. Despite its advantages, Ribo-Seq has several technical limitations (77). For example, the choice of ribonuclease (44) and the use of translation inhibitors (76) in ribosome profiling experiments have been shown to affect the data interpretation. Furthermore, standardized bioinformatics protocols for analysis of the Ribo-Seq data are lacking. Although multiple reports have been published on utilizing Ribo-Seq in various model systems, which investigated translational regulation in different physiological and disease states, application of this technique to studying aging has been limited.
To elucidate the role of translational regulation in aging, our group has recently used ribosome profiling to investigate genome-wide translational changes in several long-lived yeast mutants, previously identified in yeast replicative lifespan screens (10). By integrating transcriptional (RNA-Seq) and translational (Ribo-Seq) gene expression data, we identified common and unique patterns of protein synthesis associated with increased longevity, and we uncovered regulatory factors from gene and protein expression signatures. Our study revealed that the long-lived single gene deletions coordinately activate several common transcription factors and mRNA-binding proteins (RBPs), which specifically enhance the translation of proteins involved in cellular response to stress (Fig. 4) (10). At the same time, the long-lived mutants also contained adaptations that were unique for each of the strains, suggesting that there are multiple ways by which the lifespan of an organism can be extended. Consistent with the putative role of RBPs in translational regulation, we identified multiple targets of RBPs that were activated in the long-lived mutants at the level of translation.
FIG. 4.
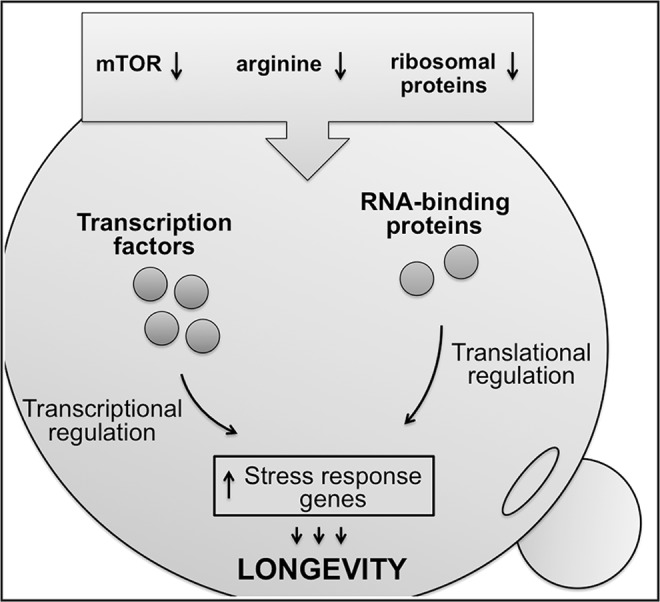
A model for the role of translational regulation in yeast aging. Lifespan-extending manipulations, such as inhibition of mTOR signaling, impaired amino acid import, and ribosome deficiency, extend yeast lifespan by activating several transcription factors and RNA-binding proteins that coordinately regulate the expression of stress-response genes. mTOR, mechanistic target of rapamycin.
Hundreds of putative RBPs that may play a role in translational regulation and can influence stability, transport, and sub-cellular localization of specific mRNAs have been identified (5, 17). However, the functions of most of the RBPs in translational regulation during aging remain unknown. To characterize the role of these RBPs in translational control, there is a need to identify their downstream targets and mRNA-specific cis-regulatory sequences. Several methods have been developed to identify targets of RBPs that are based on immunoprecipitation, such as CLIP-Seq (cross-linking immunoprecipitation sequencing) (172) and RIP-Seq (RNA immunoprecipitation sequencing) (199). Integrating genome-wide translational profiling data obtained by using Ribo-Seq with information about RBP targets will allow researchers to identify genes that are translationally regulated during aging and characterize the role of RBPs in post-transcriptional gene regulation in aging.
In addition to RBPs, non-coding RNAs such as microRNAs (miRNAs) and long non-coding RNAs (lncRNAs) have been shown to play important roles in the post-transcriptional regulation of various biological processes (7, 8). Recent advances in omics techniques allowed identification of multiple regulatory non-coding RNAs and facilitated studies of their role in aging. In particular, miRNAs have been recently shown to modulate lifespan in C. elegans (13, 32) and Drosophila model organisms (114). Moreover, genome-wide profiling of non-coding RNAs revealed that many miRNAs and lncRNAs are up- or downregulated during mammalian aging (109, 122, 159) and may regulate cellular senescence (91, 97). However, the specific functions of many of the non-coding RNAs remain unknown. Similar to RBPs, miRNAs and lncRNAs may target multiple genes and have a potential to coordinate multiple pathways implicated in aging and longevity (79).
Proteomics
Although the rate of mRNA synthesis determines the bulk of the specific protein's concentration in the cell, and the regulation at the level of transcription for the most part governs protein expression, regulation at the level of protein translation and turnover appears to fine-tune protein concentration to the cellular needs (36). Current techniques for high-throughput analysis of protein concentration include liquid chromatography coupled with mass spectrometry (LC-MS/MS), antibody-based immunohistochemistry, protein arrays, and fluorescence-based imaging using fluorescent reporters (e.g., GFP-tagged proteins). However, one of the common limitations for the proteomics measurements is sensitivity of detection. Currently, only a small fraction of the proteome can be quantitatively measured in a single experiment. Also, compared with transcriptomics that can detect different transcript isoforms produced as a result of alternative splicing, proteomics analysis is restricted to a limited number of protein variants.
Proteomics has been particularly useful for studying pathways regulating protein homeostasis and their role in aging. Loss of protein homeostasis, or proteostasis, has been linked to aging and multiple age-related diseases, but the impact of aging on different proteostasis mechanisms remains understudied. In a recent study, proteomic profiling of more than 5000 proteins at multiple time points during the lifespan of C. elegans revealed that the proteome of an animal undergoes extensive remodeling during aging (178). Although the concentration of many of the proteins declined with aging, a large portion of the proteins increased their abundance, leading to protein imbalance and aggregation. Besides the loss of protein stoichiometry, increased protein aggregation can be induced by protein modifications and other covalent damage to proteins (e.g., glycation, oxidation, nitration) accumulating with increasing age that may influence protein conformation and stability (92). Future proteomics studies in mammalian organisms are required to test whether similar changes in proteome are contributing to the loss of protein homeostasis during mammalian aging.
This study also revealed that proteostasis systems form a complex network containing multiple pathways that function in a coordinated manner. Although levels of ribosomal proteins declined with aging, the abundance of components of the proteasome system and small heat-shock proteins increased (178). These findings are in line with several reports demonstrating the crosstalk between proteasome and autophagy (87). For example, loss of proteostasis due to inhibition of the proteasome is compensated by an upregulation of macroautophagy (96). In addition, the inhibition of chaperone-mediated autophagy or macroautophagy leads to activation of a different form of autophagy (88, 127). This complexity allows cells to preserve protein homeostasis even if one of the systems fails by activating alternative mechanisms. The crosstalk between the protein degradation pathways should be taken into account when interpreting results from proteomic experiments. In future studies, it would be interesting to see how protein homeostasis network is changed with aging, and whether aging has a negative effect on the crosstalk between different protein degradation pathways.
Tissue-specific aging
One of the limitations of studies in invertebrate organisms, such as yeast and worms, is the extent to which these findings are relevant to the biology of aging in higher eukaryotes, including humans. Previous studies have shown that aging affects tissues and organs of the same organism in different ways, and different tissues age at different rates (63). For example, comparative transcriptome and proteome analyses between young and old rats revealed major organ-specific differences in ways that different organs (i.e., brain and liver) age (142). Although changes in gene expression and protein translation signatures were subtle with aging (usually less than 5% of the genes were changed at the transcriptional and translational levels with aging within the same tissue), the changes in the transcriptome and proteome between different organs were more pronounced (142, 179).
This fact highlights the need to account for different physiological functions and proliferation rates between tissues when analyzing high-throughput transcriptomics and proteomics data. It is expected that tissues with low proliferation rates, such as brain, are more susceptible to damage accumulation compared with highly proliferative tissues that can regenerate, for example, liver. Consistent with a different capacity for cellular regeneration, brain and liver are characterized by different contribution of transcriptional and translational regulation to proteomic changes with aging (142). Interestingly, the majority of age-dependent changes in the brain proteome could be explained by changes at the level of translation, whereas changes in age-dependent proteome signatures in the liver were mostly regulated by changes in mRNA abundance (142).
Together, these findings imply that biological age of an organ and the way different organs age depend on its unique physiological function and specific cellular properties. Moreover, one tissue or organ can affect aging of other tissues and even the whole organism. It is important to emphasize the need to survey multiple organs and cell types and to integrate different omics data. Integration of multiple omics approaches such as transcriptomics, proteomics, metabolomics, as well as global analysis of pre-mRNA splicing, and methylation could reveal an in-depth picture of how different components deteriorate with aging, contribution of specific organs to aging, and a better understanding of aging at the level of the whole organism.
Post-translational modifications
Among other factors, the activity of the protein is determined by its expression, rate of protein synthesis and protein turnover, as well as protein localization within cells and regulatory PTMs. In turn, PTMs can affect the biochemical properties of the protein by changing its structure, enzyme active site environment, and protein-protein interactions. The most common PTMs of proteins include addition of small chemical groups (such as phosphorylation, acetylation, and oxidation), attachment of large sugar molecule oligosaccharides (e.g., glycosylation) or lipid chains (e.g, palmitoylation), and covalent attachment of ubiquitin protein moieties (ubiquitination), among others. However, it is important to distinguish between true functional PTMs and protein damage, for example, protein oxidation or dysfunctional adduct formation by sugars (glycation), methylglyoxal, nitric oxide etc., that accumulates with aging (160). For example, oxidative modifications that are commonly considered as protein damage can be also used by cells in redox-regulated signaling pathways (28).
Similar to other reversible protein modifications, such as phosphorylation, oxidation of cysteine residues has been shown to play regulatory roles, and dysregulation of these processes has been linked to aging (99). Although detection of such modifications at the genome-wide scale is difficult, recently several large-scale proteomic approaches were developed to identify functional cysteines that can be oxidized in vivo, including OxiTMT (157), OxICAT (106), and isoTOP-ABPP (182). More recently, targeted oxidation/reduction of methionine residues in proteins has been also implicated in signaling and redox regulation of diverse cellular functions (35, 124). Although the role of methionine sulfoxide-dependent signaling in aging awaits further investigation, future studies will benefit from newly developed proteomic methods (143) and fluorescent probes (168) for detection of methionine oxidation products.
One of the examples that illustrates multi-layered regulation of protein activity includes the 26S proteasome. Impaired 26S proteasome system has been implicated in the aging process (116). Consistent with the importance of the 26S proteasome in protein homeostasis and its multi-subunit composition, the activity of the 26S proteasome is regulated at multiple levels. In addition to transcriptional control, PTMs have been shown to regulate its activity (135, 150, 171). Previous large-scale studies of the phosphoproteome and other PTMs in yeast (93) and high eukaryotes (49) revealed that multiple proteasome subunits are subject to phosphorylation. In addition, proteasome activity might be modulated by other PTMs, such as acetylation, ubiquitination, addition of O-linked N-acetyl-glucosamine, S-glutathionylation, and oxidation (16, 154, 196). Notably, aging is associated with the decline in proteasome function that occurs at multiple levels, including reduced expression of genes encoding proteasome subunits (105), reduced proteasome activity due to oxidative damage (16, 181), and disassembly of the 26S proteasome complex (181).
Most of the prior studies that investigated age-dependent changes in PTMs have focused on one of the specific modifications that may regulate protein function (e.g., phosphorylation or protein oxidative damage), but comprehensive and quantitative characterization of these complex factors at the genome-wide level and integration with transcriptional/protein translation data in the context of aging lagged behind. By combining genomics and proteomics approaches Ori et al. (142) have recently simultaneously analyzed changes in gene transcription, levels of protein translation, and protein phosphorylation status in the same tissue samples isolated from young and old rats. Although the majority of identified proteins that changed their protein abundance between young and old animals, specifically in the brain and liver, were primarily changed due to changes in the rates of protein synthesis, a fraction of the proteins also demonstrated changes in protein phosphorylation status. Moreover, several kinases have been found to be differentially expressed between young and old animals, which could potentially explain these differences in phosphorylation (142). At the same time, researchers identified a decreased level of phosphorylation in several proteins. Together, these findings highlight the complexity of the changes that occur with aging that could be missed by focusing on one aspect of age-associated gene expression studies.
Epigenomics
Recent studies revealed an important role of the chromatin-based epigenomic changes during aging (14). These epigenomic changes include DNA methylation and multiple histone modifications that can influence transcription of genes through inhibiting binding of transcription factors, heterochromatin formation, and affecting genome stability. The environmental and experimental perturbations of the epigenome and chromatin remodelers have been shown to directly affect lifespan in a variety of organisms. Further, epigenetic modifications have been recently proposed to be useful biomarkers of aging.
Methylation of cytosine bases in CpG dinucleotides (5-methylcytosine; 5mC) is a conserved epigenetic modification that is commonly associated with repression of transcription. Global hypomethylation of CpG sites with aging has been observed in humans in multiple tissues (56, 66). However, some CpG sites, which are preferentially located near promoters of transcription factors, tissue-specific genes, and genes involved in differentiation and development, become hypermethylated with the advanced age (11, 31, 121). Currently, the consequences of DNA methylation changes during aging on cellular function are not completely understood. One of the possibilities is that these methylation changes could influence binding of the transcription factors to the promoter regions and lead to altered expression of genes with aging (3, 195). Recently, several novel forms of DNA methylation have been identified in metazoa, including hydroxymethylation of cytosines (5-hydroxymethyl cytosine; 5 hmC) (34, 145) and N6-methylation of adenines (6-methyl adenine; 6 mA) (41, 51, 197). Interestingly, N6-methylation of adenines has been implicated in regulation of transcription and activity of transposable elements as well as in transgenerational epigenetic inheritance (118).
Notably, genome-wide analyses of 5mC DNA methylation profiles have been used to predict chronological and biological age in human tissues. Recently, several methylation clocks have been developed that could predict the chronological age based on methylation changes at distinct sets of CpG sites in the human genome (56, 66, 183). These methylation sites are consistently changed with aging across different tissues and cell types (66) and can also predict age in a tissue-specific manner (56). Notably, epigenetic changes can be used to estimate not only chronological age but also biological age. The human DNA methylation clock is predictive of functional decline with aging (19, 22, 67, 70), increased mortality (125), and time to death (19). Moreover, accelerated epigenetic aging was associated with HIV infection (69) and was observed in patients with Down syndrome (68).
Beyond accelerated aging, an important question is whether epigenetic clock can be slowed and whether the organism's lifespan can be extended by reversing epigenetic modifications. Although answering these questions in humans is challenging, recently an epigenetic clock has been developed based on mouse blood DNA methylation profiles (147). Using reduced representation bisulfite sequencing of mouse blood samples, a set of CpG methylation sites has been identified in the mouse genome that could reliably predict mouse biological age. These CpG sites are scattered across the genome and are distinct from CpG sites observed in the human epigenetic clock. Importantly, the mouse DNA methylation clocks can robustly predict modulation of lifespan by different dietary and genetic interventions, for example, DR, rapamycin and gene knockouts known to extend lifespan (147, 180) as well as high fat diet and ovariectomy leading to shortened lifespan (166).
Because mice have a much shorter lifespan and are commonly used to study mammalian aging, the epigenetic clock in mice is an attractive biomarker of aging that could provide a valuable tool for researchers to test the effect of novel interventions and drugs to delay aging. However, precise assessment of biological age and comparison of data between different studies could be challenging. Among the limitations of early reports is a high prediction error due to the low number of samples available and the limited range of ages analyzed. Moreover, the lack of fully standardized procedures to quantitatively measure DNA methylation status and different genetic backgrounds could influence the estimation of the exact age. Recent technological advances in whole-genome bisulfate sequencing (WGBS) could further improve the resolution and predictive power of the DNA methylation clocks by surveying an increased number of CpG sites (100). In future studies, it would be also important to investigate to what extent different epigenetic clocks are able to predict biological age in different mouse tissues. Similar to human DNA methylation clocks (56, 66), the composition of the mouse epigenetic clock may change in a tissue-specific manner as a result of different developmental programs. These studies could help to explain differences between rates of aging among organs and may provide better understanding of molecular mechanisms that underlie aging in different organs and tissues (see the discussion on the Tissue-Specific Aging section).
In addition to changes in DNA methylation, other epigenomic changes such as histone modifications have been associated with aging. Histone methylation is dynamically regulated by histone methyltransferases and demethylases and can affect patterns of gene expression by regulating chromatin structure (54). Experimental perturbation of histone methylation has been found to modulate longevity in model organisms (52, 81, 128). Similarly, histone acetylation, which is regulated by histone acetyltransferases and histone deacetylases, has been shown to directly affect lifespan (29). More recently, studies in C. elegans revealed that histone methylation might have a long-lasting effect on lifespan that can be transferred through several generations (50, 53). Together, these studies suggest that multiple epigenomic changes are associated with the aging process, and that modulation of epigenetic modifiers can be used as a therapeutic strategy to slow and possibly reverse aging.
Metabolomics
Gradual accumulation of molecular damage, which is produced by numerous metabolic processes, has been proposed to be the principal driver of aging (48, 94, 193). Such cumulative damage is not limited to oxidative damage and may include byproducts of enzymatic reactions, alterations of gene expression, errors in RNA and protein synthesis, and accumulation of DNA mutations, among others (46). Moreover, the diversity of these damage forms and byproducts of various metabolic processes is expected to increase with advanced age (47). Therefore, analysis of metabolic signatures associated with the old age could provide a better understanding of the mechanisms of aging.
Two commonly used methods to quantitatively measure low-molecular-weight molecules, or metabolites, include the LC-MS/MS and nuclear magnetic resonance (NMR) spectroscopy. Currently, Human Metabolome Database (HMDB) contains more than 52,000 entries (187), including both water-soluble and lipid-soluble small molecule metabolites. However, out of this large number, only ∼1500 can be identified by non-targeted metabolite profiling, and less than 500 by targeted MS-based metabolomics. Although fewer metabolites can be analyzed by using targeted metabolomics, this approach provides highly sensitive absolute quantification of metabolites with predefined structures. Despite lower sensitivity, the advantages of NMR spectroscopy include high reproducibility and ability to quantitatively measure metabolites over a wide range of concentrations. NMR can be also used to determine the structure of unknown compounds, and it offers advantages for compounds that are difficult to ionize or require derivatization for MS.
Recently, both targeted and non-targeted metabolome-wide profiling was used to characterize changes in levels of small-molecule metabolites associated with aging in several organisms, including nematodes (42), fruit flies (1, 64), mice (73, 170), and humans (83, 102, 194). These studies revealed significant changes in metabolic profiles with age across different species and organs. Most metabolites demonstrate organ-specific patterns, whereas some of the small-molecule metabolites correlate with aging across different organs. In addition to organ-specific changes, significant differences in metabolite levels were observed between men and women in humans (83). However, a set of small-molecule metabolites consistently correlates with age independently of gender, suggesting that metabolite signatures could be used for the development of reliable biomarkers to predict biological age. Importantly, by using non-targeted metabolomics, researchers found that the diversity of metabolites (total number of distinct metabolites) was increased with aging (1). The appearance of new small-molecule metabolites with aging implies that some of them might represent byproducts of metabolism or other damage forms that accumulate in old organisms. Further advances in sensitivity of metabolomics techniques and structural identification of non-targeted hits that accumulate with aging may provide insights into causes of aging.
In addition to characterizing metabolic changes associated with aging, metabolomics has been applied to investigate patterns of small-molecule metabolites associated with increased longevity in model organisms (90, 146) as well as in humans (23, 84). Analysis of metabolic profiles in a group of centenarians demonstrated distinctive signatures in amino acid and lipid levels (23, 84). In particular, extreme longevity in centenarians was associated with significantly decreased levels of tryptophan, high sphingomyelin levels, a higher ratio of monounsaturated to polyunsaturated lipids, and decreased levels of lipid peroxidation markers in plasma. Decreased levels of polyunsaturated lipids may reflect enhanced resistance to peroxidation damage in centenarians that is consistent with the high sensitivity of polyunsaturated fatty acids to oxidative stress (23, 84).
Interestingly, lifespan-extending manipulations, such as DR, may reverse age-dependent metabolic signatures to a more “youthful” metabolic state (1, 64). Moreover, different metabolic processes are affected by DR in different tissues, suggesting that metabolic reprogramming by DR occurs in a tissue-specific manner (103). However, the exact mechanism by which DR induces the shift in metabolite concentrations is still not completely understood. More recently, targeted high-throughput analysis of metabolites in Drosophila revealed that among the most significantly changed metabolites with age are metabolites involved in methionine metabolism (144). Interestingly, deficiency of two S-adenosyl-homocysteine (SAH) hydrolase-like proteins dAhcyL1 and dAhcyL2, which are involved in regulation of methionine metabolism, leads to significant lifespan extension and is associated with suppressed H3K4 trimethylation (H3K4me3) resembling methionine restriction (144).
These findings highlight the importance to identify metabolites and/or factors that play a causative role in metabolic reprogramming with aging. Similar to metabolites involved in the methionine metabolism, other small molecules, for example, nicotinamide adenine dinucleotide (NAD), flavin adenine dinucleotide (FAD), acetyl coenzyme A (acetyl-CoA), and α-ketoglutarate, serve as cofactors or regulators of multiple enzymes that could affect aging through remodeling of chromatin, transcriptional regulation of gene expression, and other processes. Another level of complexity is based on the observations that increased levels of metabolites in one tissue can affect aging in a non-cell-autonomous manner and extend the lifespan of the whole organism (38).
However, many unresolved questions regarding the link between metabolome and aging remain. Because metabolome is influenced by both genetic and environmental factors at multiple levels, integration of transcriptomic, proteomic, and metabolomic data can provide important insights into causative relationships between metabolome and longevity phenotypes. Further, it is not known how the patterns of metabolites change in species with diverse maximum lifespans. A detailed comparative analysis of metabolomes in different species could reveal the mechanisms by which the nature reprograms metabolomes to achieve a different range of lifespans (119, 120).
Automated Systems for Yeast Saccharomyces cerevisiae and worm C. elegans Lifespan Studies
Another important direction of research is how aging differs among individual organisms within the same species. For example, even in the population of genetically identical organisms, there is a huge variation in lifespans, which is determined by the stochastic nature of the aging process. To understand the mechanisms for phenotypic variation in aging and longevity, there is a need to quantitatively analyze age-associated phenotypes and molecular changes at the single-cell level. Yeast S. cerevisiae and worm C. elegans historically have been widely used for genome-wide aging studies. Recently, several microfluidic systems have been developed for these models that allow automation and performing aging experiments in a high-throughput manner. Here, we discuss advantages of microfluidic platforms for S. cerevisiae and C. elegans, current challenges and potential application of microfluidics for aging research.
Although manual microdissection of old yeast mother cells on agar plates is still the gold standard for analysis of replicative lifespan in yeast (162), it is a very labor- and time-intensive technique, which limits its application in high-throughput studies. Recently, several groups have developed microfluidic devices that allow monitoring of yeast cells with the single-cell resolution [reviewed in Chen et al. (20)]. Because microfluidic chambers allow trapping of individual yeast cells, when combined with time-lapse microscopy, these microfluidic systems can be used to observe multiple physiological parameters and monitor how they change with aging. For example, by using fluorescently labeled protein reporters, gene expression, protein expression, subcellular protein localization, organelle structure, and internal pH can be monitored throughout the aging process. Similarly, fluorescent dyes can be added to the media flow to track other physiological changes such as redox state and mitochondrial membrane potential (192). It is important to note that physiological changes measured by using microfluidic systems are obtained at the single-cell resolution, allowing to assess the penetrance of these changes within the overall population of mother cells. Moreover, they could be directly correlated with the life expectancy and the mother cell lifespan, which is monitored by scoring the periodic appearance and disappearance of daughter cells that are being constantly washed away by the fluid media (20).
To measure yeast replicative lifespan in real time, several designs of microfluidic systems have been used to trap mother cells. One of the major requirements to these devices is that they should trap and retain mother cells in the field of view through their lifespan, allowing automatic separation of the daughter cells from their mothers by media flow. Different designs have their strengths and weaknesses. Although none of the currently published designs are yet optimal, the devices containing a small set of polydimethylsiloxane columns/pillars (usually two or three) that form the trap (Fig. 5) are particularly attractive due to the ease of fabrication and the low manufacturing costs (26, 82, 115, 153). Among the limitation of currently available microfluidic devices is the lack of data on how different designs and experimental conditions affect the robustness and reproducibility of lifespan measurements. In addition, care should be taken when interpreting the results of such microfluidic lifespan measurements in cases when genetic and environmental interventions under study can directly affect the yeast cell size (20). In future studies, it will be important to establish which microfluidic device design, and under what experimental conditions, most closely recapitulates the results obtained by using traditional replicative lifespan measurements.
FIG. 5.

Design of microfluidics systems for automated yeast replicative lifespan analysis. (A–C) At the beginning of the experiment, mother cells are trapped by using a two-column trap in the form of V shape (A) (26), two L-shaped PDMS pillars (B) (82), or a three-column jail (C) (115, 153). (D) Smaller daughter cells are constantly removed by media flow. Yeast lifespan is monitored by counting the number of daughter cells produced by each mother cell before it undergoes senescence.
As discussed earlier, the role of transcriptional and translational regulation in aging is not completely understood. Although several key regulatory factors, including transcription factors and regulatory RBPs, that coordinate transcriptional and translational responses have been identified, how their activity is modulated and how transcriptional and translational signatures are changed with aging remain unknown. Using fluorescence microscopy combined with automated microfluidic systems would allow researchers to monitor changes in gene expression and protein translation in yeast cells over their entire life. For example, quantitative single-cell tracking of the TEF2 translation elongation factor revealed a surprising increase in its expression in old mother cells near the end of their lifespan compared with young cells, suggesting a dysregulation of translational control with aging (192). In addition, microfluidic devices have been used to validate molecular markers, for example, Hsp104, and to assess their ability to predict lifespan in individual cells (192). Future studies using microfluidics systems will broaden our understanding of the aging process by quantitative analysis of the genetic and phenotypic factors that drive or accompany the aging process with the single-cell resolution.
Similar to yeast, the lifespan analysis in worms is a labor- and time-intensive procedure. In addition, worm longitudinal lifespan studies suffer from low resolution and reproducibility, and can be influenced by human bias and multiple experimental variables. To overcome these limitations, several microfluidic devices have been developed to automate lifespan analysis in worms (75, 148, 191). However, currently such systems suffer from limited throughput and are not completely automated. Nevertheless, microfluidics systems are capable of monitoring physiological changes throughout the entire animal's lifespan, including body size, movement rates, age pigment accumulation, as well as fluorescent protein reporters. These microfluidic devices also allow for exposing the worms to different external stimuli by adding reagents directly to the media flow.
In addition to measuring lifespan, microfluidic systems could be applied to study reproductive aging. Recently, a new microfluidic platform combined with time-lapse imaging has been developed that allows quantitative real-time analysis of progeny from multiple animals and provides high-resolution data on reproductive age (110).
In addition to microfluidics systems, which analyze worm aging in liquid media, an alternative approach called the “Lifespan Machine” utilizes high-throughput imaging and tracking of movement in worms placed on plates with solid media (165). The Lifespan Machine allows monitoring a large number of individual worms by combining imaging using flatbed scanners with a novel image-analysis pipeline. This system was successfully used to compare the effects of different interventions, such as high temperature, oxidative stress, DR, and different genetic manipulations, to assess lifespan dynamics and high-precision mortality statistics in the large populations of worms (164).
Combining large-scale omics technologies with systems for automated lifespan analysis in S. cerevisiae and C. elegans could provide a better understanding of the mechanisms that underlie aging and has the potential to transform the aging research field.
Data Integration
Although the development of high-throughput techniques led to production of a huge amount of data, integration of the omics data has been one of the biggest challenges in the field. Importantly, to perform direct correlation between parameters using multi-omics techniques, these analyses have to be performed and datasets have to be collected for the same sets of samples in a controlled laboratory environment. Due to variability of conditions, comparison and integration of omics data collected by different labs remains a challenge. Therefore, care should be taken when interpreting and integrating the data, taking into account experimental variables. Also, due to high technical variation, high-throughput approaches require proper data normalization and data quality control. Despite these challenges, the unbiased genome-wide analysis and monitoring how complex biological networks change with age are critical for understanding the mechanisms that underlie aging.
Specifically, the aging field will benefit from applying systems biology approaches to efficiently integrate the dynamic transcriptional, translational, and metabolic changes with aging and to reliably infer regulatory factors from multi-layered data (Fig. 6). Moreover, understanding of the aging process is not possible without taking into account molecular interactions (or “interactome”) occurring among different components of the biological system (proteins, lipids, nucleic acids, etc.) as well as how they affect physiological parameters associated with aging (i.e., “physiome”). In fact, aging is indicated as one of the goals of the “Human Physiome project”, which is focused on precisely defining interactions that occur in the human body as a whole by using quantitative computer modeling (173).
FIG. 6.
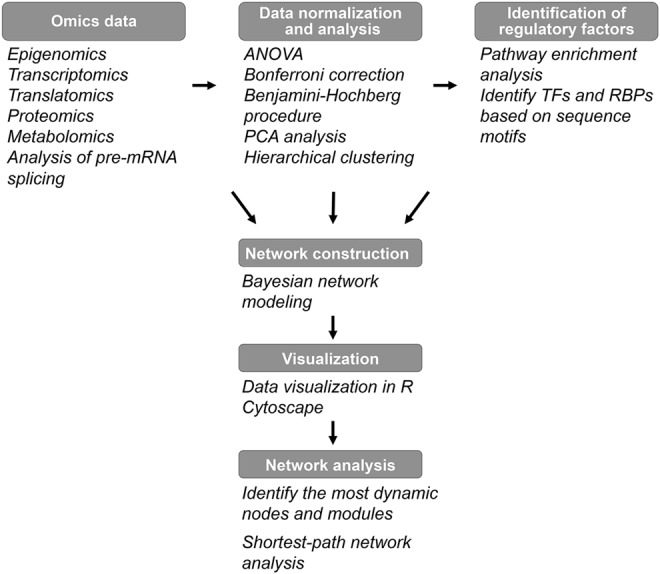
Workflow for data integration. RBP, mRNA-binding protein.
Perspectives
Although significant progress has been made in studying changes associated with aging over the past decades, the mechanisms that underlie the aging process are not completely understood and many fundamental questions remain (Box 1). Answering these questions will allow researchers to build regulatory aging networks and help identify new targets for the development of new lifespan interventions to delay the aging process.
Box 1. Major Unsolved Questions.
Which molecular changes associated with the aging process are causes of aging and which are consequences?
Given that there are multiple ways by which the lifespan of an organism can be extended, how are the various longevity-associated genetic and environmental manipulations related to each other? How are different aging genes and pathways interconnected and coordinately regulated?
How do aging regulatory networks dynamically change with age?
Is aging programmed to lead to death at a species-specific age or is it a random process?
Considering significant diversity (more than 100-fold difference) in lifespan among mammalian species, what are the genes and pathways that determine the differences among organisms' lifespan?
What are the common gene expression and metabolite signatures associated with increased longevity?
What are the mechanisms that underlie differences in lifespan among organisms within the same species?
Can human aging be slowed or reversed therapeutically? Is there a therapeutic window in which age therapeutic interventions in humans will be the most beneficial?
What is the role of epigenomic modifications and epigenome-modifying enzymes in the transgenerational effects on lifespan?
Recent advances in high-throughput “omics” methods have enabled detailed studies of physiological changes that occur with aging across different species as well as in different tissues and organs. The increasing number of available datasets that describe dynamic changes in transcription, translation, levels of metabolites, and PTMs with aging have prompted researchers to integrate these datasets. As the field progresses, additional datasets obtained from different organisms and multiple tissues, together with increased sensitivity of “omics” methods, will allow a more detailed investigation (e.g., multiple time points, single-cell sequencing, and quantitative analysis of physiological changes in vivo using microfluidic platforms), and will be instrumental to identify causative relationships.
In addition to age-dependent changes, several studies analyzed changes in gene expression, protein translation, DNA methylation state, PTMs, and other factors in response to dietary interventions, genetic manipulations, and environmental conditions known to extend lifespan, which allowed to identify a number or regulatory factors and proteins (Table 1). In future studies, it would be important to examine the common and unique mechanisms of lifespan extension, and to investigate how different longevity-associated environmental and genetic manipulations are related to each other. These studies will enable the identification of risk factors and allow making predictions, which individuals will benefit the most from specific interventions based on their gene expression signatures or genetic background. For example, several bioinformatics and computational methods have been developed for identifying and predicting biologically active small molecules based on gene expression profiles (80, 101). However, these methods have not been extensively tested to predict drugs or small molecules with anti-aging properties and would be of interest in future studies (6).
Table 1.
Major Interventions and Targets of Genetic Manipulations That Increase Lifespan
| Intervention/signaling pathway | Molecular function/regulatory factors | Species | References |
|---|---|---|---|
| 17-α-estradiol | Estrogen agonist | Mus musculus | 59 |
| L-deprenyl | Monoamine oxidase inhibitor | Rattus norvegicus | 95 |
| Acarbose | Inhibitor of intestinal α-glucosidase | M. musculus | 59 |
| Aspirin | Anti-inflammatory agent | M. musculus | 163 |
| Ibuprofen | Anti-inflammatory agent | Saccharomyces cerevisiae | 61 |
| Caenorhabditis elegans | |||
| Drosophila melanogaster | |||
| Metformin | Antidiabetic drug, mTOR inhibitor, AMPK signaling pathway activator | M. musculus | 126, 140 |
| C. elegans | |||
| Methylene blue | Mitochondrial complex IV activator | M. musculus | 59 |
| Nordihydroguaiaretic acid | Lipoxygenase inhibitor, anti-inflammatory agent | M. musculus | 59, 163 |
| Rapamycin | mTOR inhibitor | M. musculus | 12, 60, 133, 134 |
| D. melanogaster | |||
| Resveratrol | SIRT1 activator | M. musculus | 9, 74, 188 |
| C. elegans | |||
| D. melanogaster | |||
| S. cerevisiae | |||
| Dietary restriction | mTOR, AMPK, IGF-1 | M. musculus | 71, 112, 149, 184 |
| C. elegans | |||
| D. melanogaster | |||
| S. cerevisiae | |||
| GH signaling | GH, GHR | M. musculus | 25, 39 |
| Insulin/IGF-1 signaling | IGF-1, IGF-1R, PI3K, AKT, FOXO | M. musculus | 15, 65, 138 |
| C. elegans | |||
| D. melanogaster | |||
| mTOR signaling | mTOR, S6K | M. musculus | 85, 86, 111, 156 |
| C. elegans | |||
| D. melanogaster | |||
| S. cerevisiae |
GH, growth hormone; mTOR, mechanistic target of rapamycin.
Another important area of investigation is the mechanisms that regulate species lifespan. Mammals are characterized by more than 100-fold difference in lifespan. For example, the bowhead whale, the longest-lived mammal, is reported to live >200 years. Using comparative “omics” approaches to identify differences in genome sequences, gene expression profiles, and metabolic signatures among short- and long-lived organisms would allow researchers to identify key regulators of lifespan.
Scientists have been trying to understand aging for centuries, but what is aging is still not completely understood. Although many distinct, often contradicting each other, theories of aging have been proposed, at present no single unifying theory of aging exists that could explain all discordant observations (47, 94, 116, 176). Many of these theories assume that only one primary cause of aging exists or plays a major role, for example, oxidative damage (free radical theory of aging) (58), errors in protein synthesis (Orgel's error catastrophe theory) (141), somatic mutations (Szilard's model of somatic mutation accumulation) (167), or epigenomic changes (14). We argue that all biological processes contribute to aging, and none of these senescence factors individually is more important than others, rather the biological systems' inherent susceptibility to errors lies in the center of the aging process (45). The advances in next-generation sequencing, proteomics, and metabolomics may finally allow analyses of how different theories of aging relate to each other. Indeed, there have been considerable efforts in recent years to reconcile different theories and develop new theories based on the latest advances in “omics” research (45, 132).
For example, Orgel's error catastrophe hypothesis, which implies a positive feedback between errors in proteins and the introduction of new errors in protein synthesis, has recently been re-examined and extended to somatic mutations to create a new catastrophe theory of aging (132). In this theory, a primary role is played by somatic mutations with a contribution from other mechanisms. Because mutations in genes encoding enzymes involved in DNA repair are expected to introduce more somatic mutations, they would lead to an exponential increase in DNA damage, forming a feedback loop. This model is consistent with Szilard's theory, but it also attempts to incorporate interactions between cellular processes (DNA repair, mRNA transcription, protein translation) and the system's hierarchy. As opposed to errors in protein synthesis, DNA mutations may not only lead to changes in protein sequences but also affect gene expression if they occur in regulatory regions or genes encoding transcription factors. Further experimental investigation is still needed to test this hypothesis, and it would require integration of genomic, transcriptomic, and proteomic data. Combining different omics approaches with systems biology tools and mathematical modeling has the potential to elucidate causality among multiple aging mechanisms and may also resolve the controversies between competing aging theories (e.g., is aging a programmed or stochastic process?) (98).
Understanding a complex biological trait such as aging requires analysis of cause-consequence relationships between multiple, often influencing each other, factors in the biological system rather than studying individual components. Therefore, applying systems biology tools to integrate the data from different “omics” approaches obtained at multiple levels and at multiple time points throughout the organism's lifespan is necessary to gain a global understanding of the aging process.
Abbreviations Used
- acetyl-CoA
acetyl coenzyme A
- DR
dietary restriction
- FAD
flavin adenine dinucleotide
- GH
growth hormone
- HMDB
Human Metabolome Database
- lncRNA
long non-coding RNA
- miRNA
microRNA
- MS
mass spectrometry
- mTOR
mechanistic target of rapamycin
- NAD
nicotinamide adenine dinucleotide
- PTM
post-translational modification
- RBP
mRNA-binding protein
- RNA-Seq
RNA sequencing
- SAGA
Spt-Ada-Gcn5-acetyltransferase
- SAH
S-adenosyl-homocysteine
Acknowledgments
The authors would like to thank the reviewers for their suggestions and insightful comments on this article. This work was supported by NIH grants AG040191 and AG054566 to V.M.L. This research was conducted while V.M.L. was an AFAR Research Grant recipient from the American Federation for Aging Research.
References
- 1.Avanesov AS, Ma S, Pierce KA, Yim SH, Lee BC, Clish CB, and Gladyshev VN. Age- and diet-associated metabolome remodeling characterizes the aging process driven by damage accumulation. Elife 3: e02077, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aviner R, Geiger T, and Elroy-Stein O. Genome-wide identification and quantification of protein synthesis in cultured cells and whole tissues by puromycin-associated nascent chain proteomics (PUNCH-P). Nat Protoc 9: 751–760, 2014 [DOI] [PubMed] [Google Scholar]
- 3.Avrahami D, Li C, Zhang J, Schug J, Avrahami R, Rao S, Stadler MB, Burger L, Schubeler D, Glaser B, and Kaestner KH. Aging-dependent demethylation of regulatory elements correlates with chromatin state and improved beta cell function. Cell Metab 22: 619–632, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bahar R, Hartmann CH, Rodriguez KA, Denny AD, Busuttil RA, Dolle ME, Calder RB, Chisholm GB, Pollock BH, Klein CA, and Vijg J. Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature 441: 1011–1014, 2006 [DOI] [PubMed] [Google Scholar]
- 5.Baltz AG, Munschauer M, Schwanhausser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold-Brown D, Drew K, Milek M, Wyler E, Bonneau R, Selbach M, Dieterich C, and Landthaler M. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell 46: 674–690, 2012 [DOI] [PubMed] [Google Scholar]
- 6.Barardo DG, Newby D, Thornton D, Ghafourian T, Pedro de Magalhães J, and Freitas AA. Machine learning for predicting lifespan-extending chemical compounds. Aging 9: 1721–1737, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281–297, 2004 [DOI] [PubMed] [Google Scholar]
- 8.Batista PJ. and Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell 152: 1298–1307, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, Pistell PJ, Poosala S, Becker KG, Boss O, Gwinn D, Wang M, Ramaswamy S, Fishbein KW, Spencer RG, Lakatta EG, Le Couteur D, Shaw RJ, Navas P, Puigserver P, Ingram DK, de Cabo R, and Sinclair DA. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444: 337–342, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beaupere C, Wasko BM, Lorusso J, Kennedy BK, Kaeberlein M, and Labunskyy VM. CAN1 arginine permease deficiency extends yeast replicative lifespan via translational activation of stress response genes. Cell Rep 18: 1884–1892, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benayoun BA, Pollina EA, and Brunet A. Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat Rev Mol Cell Biol 16: 593–610, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bjedov I, Toivonen JM, Kerr F, Slack C, Jacobson J, Foley A, and Partridge L. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab 11: 35–46, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boehm M. and Slack F. A developmental timing microRNA and its target regulate life span in C. elegans. Science 310: 1954–1957, 2005 [DOI] [PubMed] [Google Scholar]
- 14.Booth LN. and Brunet A. The aging epigenome. Mol Cell 62: 728–744, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Broughton SJ, Piper MD, Ikeya T, Bass TM, Jacobson J, Driege Y, Martinez P, Hafen E, Withers DJ, Leevers SJ, and Partridge L. Longer lifespan, altered metabolism, and stress resistance in Drosophila from ablation of cells making insulin-like ligands. Proc Natl Acad Sci U S A 102: 3105–3110, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bulteau AL, Lundberg KC, Humphries KM, Sadek HA, Szweda PA, Friguet B, and Szweda LI. Oxidative modification and inactivation of the proteasome during coronary occlusion/reperfusion. J Biol Chem 276: 30057–30063, 2001 [DOI] [PubMed] [Google Scholar]
- 17.Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM, Krijgsveld J, and Hentze MW. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 149: 1393–1406, 2012 [DOI] [PubMed] [Google Scholar]
- 18.Cevenini E, Bellavista E, Tieri P, Castellani G, Lescai F, Francesconi M, Mishto M, Santoro A, Valensin S, Salvioli S, Capri M, Zaikin A, Monti D, de Magalhaes JP, and Franceschi C. Systems biology and longevity: an emerging approach to identify innovative anti-aging targets and strategies. Curr Pharm Des 16: 802–813, 2010 [DOI] [PubMed] [Google Scholar]
- 19.Chen BH, Marioni RE, Colicino E, Peters MJ, Ward-Caviness CK, Tsai PC, Roetker NS, Just AC, Demerath EW, Guan W, Bressler J, Fornage M, Studenski S, Vandiver AR, Moore AZ, Tanaka T, Kiel DP, Liang L, Vokonas P, Schwartz J, Lunetta KL, Murabito JM, Bandinelli S, Hernandez DG, Melzer D, Nalls M, Pilling LC, Price TR, Singleton AB, Gieger C, Holle R, Kretschmer A, Kronenberg F, Kunze S, Linseisen J, Meisinger C, Rathmann W, Waldenberger M, Visscher PM, Shah S, Wray NR, McRae AF, Franco OH, Hofman A, Uitterlinden AG, Absher D, Assimes T, Levine ME, Lu AT, Tsao PS, Hou L, Manson JE, Carty CL, LaCroix AZ, Reiner AP, Spector TD, Feinberg AP, Levy D, Baccarelli A, van Meurs J, Bell JT, Peters A, Deary IJ, Pankow JS, Ferrucci L, and Horvath S. DNA methylation-based measures of biological age: meta-analysis predicting time to death. Aging (Albany NY) 8: 1844–1865, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen KL, Crane MM, and Kaeberlein M. Microfluidic technologies for yeast replicative lifespan studies. Mech Ageing Dev 161: 262–269, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng C, Fabrizio P, Ge H, Longo VD, and Li LM. Inference of transcription modification in long-live yeast strains from their expression profiles. BMC Genomics 8: 219, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Christiansen L, Lenart A, Tan Q, Vaupel JW, Aviv A, McGue M, and Christensen K. DNA methylation age is associated with mortality in a longitudinal Danish twin study. Aging Cell 15: 149–154, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Collino S, Montoliu I, Martin FP, Scherer M, Mari D, Salvioli S, Bucci L, Ostan R, Monti D, Biagi E, Brigidi P, Franceschi C, and Rezzi S. Metabolic signatures of extreme longevity in northern Italian centenarians reveal a complex remodeling of lipids, amino acids, and gut microbiota metabolism. PLoS One 8: e56564, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Contrino S, Smith RN, Butano D, Carr A, Hu F, Lyne R, Rutherford K, Kalderimis A, Sullivan J, Carbon S, Kephart ET, Lloyd P, Stinson EO, Washington NL, Perry MD, Ruzanov P, Zha Z, Lewis SE, Stein LD, and Micklem G. modMine: flexible access to modENCODE data. Nucleic Acids Res 40: D1082–1088, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coschigano KT, Holland AN, Riders ME, List EO, Flyvbjerg A, and Kopchick JJ. Deletion, but not antagonism, of the mouse growth hormone receptor results in severely decreased body weights, insulin, and insulin-like growth factor I levels and increased life span. Endocrinology 144: 3799–3810, 2003 [DOI] [PubMed] [Google Scholar]
- 26.Crane MM, Clark IB, Bakker E, Smith S, and Swain PS. A microfluidic system for studying ageing and dynamic single-cell responses in budding yeast. PLoS One 9: e100042, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Curran SP. and Ruvkun G. Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet 3: e56, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D'Autreaux B. and Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol 8: 813–824, 2007 [DOI] [PubMed] [Google Scholar]
- 29.Dang W, Steffen KK, Perry R, Dorsey JA, Johnson FB, Shilatifard A, Kaeberlein M, Kennedy BK, and Berger SL. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 459: 802–807, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dang W, Sutphin GL, Dorsey JA, Otte GL, Cao K, Perry RM, Wanat JJ, Saviolaki D, Murakami CJ, Tsuchiyama S, Robison B, Gregory BD, Vermeulen M, Shiekhattar R, Johnson FB, Kennedy BK, Kaeberlein M, and Berger SL. Inactivation of yeast Isw2 chromatin remodeling enzyme mimics longevity effect of calorie restriction via induction of genotoxic stress response. Cell Metab 19: 952–966, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Day K, Waite LL, Thalacker-Mercer A, West A, Bamman MM, Brooks JD, Myers RM, and Absher D. Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape. Genome Biol 14: R102, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Lencastre A, Pincus Z, Zhou K, Kato M, Lee SS, and Slack FJ. MicroRNAs both promote and antagonize longevity in C. elegans. Curr Biol 20: 2159–2168, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Magalhaes JP, Curado J, and Church GM. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics 25: 875–881, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Delatte B, Deplus R, and Fuks F. Playing TETris with DNA modifications. EMBO J 33: 1198–1211, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Drazic A. and Winter J. The physiological role of reversible methionine oxidation. Biochim Biophys Acta 1844: 1367–1382, 2014 [DOI] [PubMed] [Google Scholar]
- 36.Edfors F, Danielsson F, Hallstrom BM, Kall L, Lundberg E, Ponten F, Forsstrom B, and Uhlen M. Gene-specific correlation of RNA and protein levels in human cells and tissues. Mol Syst Biol 12: 883, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ernst J, Vainas O, Harbison CT, Simon I, and Bar-Joseph Z. Reconstructing dynamic regulatory maps. Mol Syst Biol 3: 74, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Finkel T. The metabolic regulation of aging. Nat Med 21: 1416–1423, 2015 [DOI] [PubMed] [Google Scholar]
- 39.Flurkey K, Papaconstantinou J, Miller RA, and Harrison DE. Lifespan extension and delayed immune and collagen aging in mutant mice with defects in growth hormone production. Proc Natl Acad Sci U S A 98: 6736–6741, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fontana L, Partridge L, and Longo VD. Extending healthy life span—from yeast to humans. Science 328: 321–326, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fu Y, Luo GZ, Chen K, Deng X, Yu M, Han D, Hao Z, Liu J, Lu X, Dore LC, Weng X, Ji Q, Mets L, and He C. N6-methyldeoxyadenosine marks active transcription start sites in Chlamydomonas. Cell 161: 879–892, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fuchs S, Bundy JG, Davies SK, Viney JM, Swire JS, and Leroi AM. A metabolic signature of long life in Caenorhabditis elegans. BMC Biol 8: 14, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gardner EJ, Nizami ZF, Talbot CC, Jr., and Gall JG. Stable intronic sequence RNA (sisRNA), a new class of noncoding RNA from the oocyte nucleus of Xenopus tropicalis. Genes Dev 26: 2550–2559, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gerashchenko MV. and Gladyshev VN. Ribonuclease selection for ribosome profiling. Nucleic Acids Res 45: e6, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gladyshev VN. Aging: progressive decline in fitness due to the rising deleteriome adjusted by genetic, environmental, and stochastic processes. Aging Cell 15: 594–602, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gladyshev VN. The free radical theory of aging is dead. Long live the damage theory! Antioxid Redox Signal 20: 727–731, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gladyshev VN. The origin of aging: imperfectness-driven non-random damage defines the aging process and control of lifespan. Trends Genet 29: 506–512, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Golubev A, Hanson AD, and Gladyshev VN. Non-enzymatic molecular damage as a prototypic driver of aging. J Biol Chem 292: 6029–6038, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gomes AV, Zong C, Edmondson RD, Li X, Stefani E, Zhang J, Jones RC, Thyparambil S, Wang GW, Qiao X, Bardag-Gorce F, and Ping P. Mapping the murine cardiac 26S proteasome complexes. Circ Res 99: 362–371, 2006 [DOI] [PubMed] [Google Scholar]
- 50.Greer EL, Becker B, Latza C, Antebi A, and Shi Y. Mutation of C. elegans demethylase spr-5 extends transgenerational longevity. Cell Res 26: 229–238, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Greer EL, Blanco MA, Gu L, Sendinc E, Liu J, Aristizabal-Corrales D, Hsu CH, Aravind L, He C, and Shi Y. DNA methylation on N6-adenine in C. elegans. Cell 161: 868–878, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Greer EL, Maures TJ, Hauswirth AG, Green EM, Leeman DS, Maro GS, Han S, Banko MR, Gozani O, and Brunet A. Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature 466: 383–387, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Greer EL, Maures TJ, Ucar D, Hauswirth AG, Mancini E, Lim JP, Benayoun BA, Shi Y, and Brunet A. Transgenerational epigenetic inheritance of longevity in Caenorhabditis elegans. Nature 479: 365–371, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Greer EL. and Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet 13: 343–357, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guarente L. and Kenyon C. Genetic pathways that regulate ageing in model organisms. Nature 408: 255–262, 2000 [DOI] [PubMed] [Google Scholar]
- 56.Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, Deconde R, Chen M, Rajapakse I, Friend S, Ideker T, and Zhang K. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell 49: 359–367, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hansen M, Hsu AL, Dillin A, and Kenyon C. New genes tied to endocrine, metabolic, and dietary regulation of lifespan from a Caenorhabditis elegans genomic RNAi screen. PLoS Genet 1: 119–128, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol 11: 298–300, 1956 [DOI] [PubMed] [Google Scholar]
- 59.Harrison DE, Strong R, Allison DB, Ames BN, Astle CM, Atamna H, Fernandez E, Flurkey K, Javors MA, Nadon NL, Nelson JF, Pletcher S, Simpkins JW, Smith D, Wilkinson JE, and Miller RA. Acarbose, 17-alpha-estradiol, and nordihydroguaiaretic acid extend mouse lifespan preferentially in males. Aging Cell 13: 273–282, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, and Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460: 392–395, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.He C, Tsuchiyama SK, Nguyen QT, Plyusnina EN, Terrill SR, Sahibzada S, Patel B, Faulkner AR, Shaposhnikov MV, Tian R, Tsuchiya M, Kaeberlein M, Moskalev AA, Kennedy BK, and Polymenis M. Enhanced longevity by ibuprofen, conserved in multiple species, occurs in yeast through inhibition of tryptophan import. PLoS Genet 10: e1004860, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Heintz C, Doktor TK, Lanjuin A, Escoubas CC, Zhang Y, Weir HJ, Dutta S, Silva-Garcia CG, Bruun GH, Morantte I, Hoxhaj G, Manning BD, Andresen BS, and Mair WB. Splicing factor 1 modulates dietary restriction and TORC1 pathway longevity in C. elegans. Nature 541: 102–106, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Herndon LA, Schmeissner PJ, Dudaronek JM, Brown PA, Listner KM, Sakano Y, Paupard MC, Hall DH, and Driscoll M. Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature 419: 808–814, 2002 [DOI] [PubMed] [Google Scholar]
- 64.Hoffman JM, Soltow QA, Li S, Sidik A, Jones DP, and Promislow DE. Effects of age, sex, and genotype on high-sensitivity metabolomic profiles in the fruit fly, Drosophila melanogaster. Aging Cell 13: 596–604, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Holzenberger M, Dupont J, Ducos B, Leneuve P, Geloen A, Even PC, Cervera P, and Le Bouc Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 421: 182–187, 2003 [DOI] [PubMed] [Google Scholar]
- 66.Horvath S. DNA methylation age of human tissues and cell types. Genome Biol 14: R115, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Horvath S, Erhart W, Brosch M, Ammerpohl O, von Schonfels W, Ahrens M, Heits N, Bell JT, Tsai PC, Spector TD, Deloukas P, Siebert R, Sipos B, Becker T, Rocken C, Schafmayer C, and Hampe J. Obesity accelerates epigenetic aging of human liver. Proc Natl Acad Sci USA 111: 15538–15543, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Horvath S, Garagnani P, Bacalini MG, Pirazzini C, Salvioli S, Gentilini D, Di Blasio AM, Giuliani C, Tung S, Vinters HV, and Franceschi C. Accelerated epigenetic aging in Down syndrome. Aging Cell 14: 491–495, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Horvath S. and Levine AJ. HIV-1 infection accelerates age according to the epigenetic clock. J Infect Dis 212: 1563–1573, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Horvath S, Pirazzini C, Bacalini MG, Gentilini D, Di Blasio AM, Delledonne M, Mari D, Arosio B, Monti D, Passarino G, De Rango F, D'Aquila P, Giuliani C, Marasco E, Collino S, Descombes P, Garagnani P, and Franceschi C. Decreased epigenetic age of PBMCs from Italian semi-supercentenarians and their offspring. Aging (Albany NY) 7: 1159–1170, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hou L, Wang D, Chen D, Liu Y, Zhang Y, Cheng H, Xu C, Sun N, McDermott J, Mair WB, and Han JD. A systems approach to reverse engineer lifespan extension by dietary restriction. Cell Metab 23: 529–540, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hou L, Wang D, Cheng H, Xian B, and Han JD. Systems approaches to understanding aging. In: Handbook of the Biology of Aging, edited by Kaeberlein M. and Martin GM. London, UK: Elsevier, 2016, pp. 241–261 [Google Scholar]
- 73.Houtkooper RH, Argmann C, Houten SM, Canto C, Jeninga EH, Andreux PA, Thomas C, Doenlen R, Schoonjans K, and Auwerx J. The metabolic footprint of aging in mice. Sci Rep 1: 134, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, and Sinclair DA. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425: 191–196, 2003 [DOI] [PubMed] [Google Scholar]
- 75.Hulme SE, Shevkoplyas SS, McGuigan AP, Apfeld J, Fontana W, and Whitesides GM. Lifespan-on-a-chip: microfluidic chambers for performing lifelong observation of C. elegans. Lab Chip 10: 589–597, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hussmann JA, Patchett S, Johnson A, Sawyer S, and Press WH. Understanding biases in ribosome profiling experiments reveals signatures of translation dynamics in yeast. PLoS Genet 11: e1005732, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ingolia NT. Ribosome profiling: new views of translation, from single codons to genome scale. Nat Rev Genet 15: 205–213, 2014 [DOI] [PubMed] [Google Scholar]
- 78.Ingolia NT, Ghaemmaghami S, Newman JR, and Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324: 218–223, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Inukai S. and Slack F. MicroRNAs and the genetic network in aging. J Mol Biol 425: 3601–3608, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Iorio F, Bosotti R, Scacheri E, Belcastro V, Mithbaokar P, Ferriero R, Murino L, Tagliaferri R, Brunetti-Pierri N, Isacchi A, and di Bernardo D. Discovery of drug mode of action and drug repositioning from transcriptional responses. Proc Natl Acad Sci U S A 107: 14621–14626, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jin C, Li J, Green CD, Yu X, Tang X, Han D, Xian B, Wang D, Huang X, Cao X, Yan Z, Hou L, Liu J, Shukeir N, Khaitovich P, Chen CD, Zhang H, Jenuwein T, and Han JD. Histone demethylase UTX-1 regulates C. elegans life span by targeting the insulin/IGF-1 signaling pathway. Cell Metab 14: 161–172, 2011 [DOI] [PubMed] [Google Scholar]
- 82.Jo MC, Liu W, Gu L, Dang W, and Qin L. High-throughput analysis of yeast replicative aging using a microfluidic system. Proc Natl Acad Sci U S A 112: 9364–9369, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jove M, Mate I, Naudi A, Mota-Martorell N, Portero-Otin M, De la Fuente M, and Pamplona R. Human aging is a metabolome-related matter of gender. J Gerontol A Biol Sci Med Sci 71: 578–585, 2016 [DOI] [PubMed] [Google Scholar]
- 84.Jove M, Naudi A, Gambini J, Borras C, Cabre R, Portero-Otin M, Vina J, and Pamplona R. A stress-resistant lipidomic signature confers extreme longevity to humans. J Gerontol A Biol Sci Med Sci 72: 30–37, 2017 [DOI] [PubMed] [Google Scholar]
- 85.Kaeberlein M, Powers RW, 3rd, Steffen KK, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S, and Kennedy BK. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 310: 1193–1196, 2005 [DOI] [PubMed] [Google Scholar]
- 86.Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, and Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol 14: 885–890, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kaushik S. and Cuervo AM. Proteostasis and aging. Nat Med 21: 1406–1415, 2015 [DOI] [PubMed] [Google Scholar]
- 88.Kaushik S, Massey AC, Mizushima N, and Cuervo AM. Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol Biol Cell 19: 2179–2192, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kawaji H, Nakamura M, Takahashi Y, Sandelin A, Katayama S, Fukuda S, Daub CO, Kai C, Kawai J, Yasuda J, Carninci P, and Hayashizaki Y. Hidden layers of human small RNAs. BMC genomics 9: 157, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kaya A, Ma S, Wasko B, Lee M, Kaeberlein M, and Gladyshev VN. Defining molecular basis for longevity traits in natural yeast isolates. NPJ Aging Mech Dis 112: 10685–10690, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Khanna A, Muthusamy S, Liang R, Sarojini H, and Wang E. Gain of survival signaling by down-regulation of three key miRNAs in brain of calorie-restricted mice. Aging (Albany NY) 3: 223–236, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kikis EA, Gidalevitz T, and Morimoto RI. Protein homeostasis in models of aging and age-related conformational disease. Adv Exp Med Biol 694: 138–159, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kikuchi J, Iwafune Y, Akiyama T, Okayama A, Nakamura H, Arakawa N, Kimura Y, and Hirano H. Co- and post-translational modifications of the 26S proteasome in yeast. Proteomics 10: 2769–2779, 2010 [DOI] [PubMed] [Google Scholar]
- 94.Kirkwood TB. and Austad SN. Why do we age? Nature 408: 233–238, 2000 [DOI] [PubMed] [Google Scholar]
- 95.Knoll J. The striatal dopamine dependency of life span in male rats. Longevity study with (-)deprenyl. Mech Ageing Dev 46: 237–262, 1988 [DOI] [PubMed] [Google Scholar]
- 96.Korolchuk VI, Menzies FM, and Rubinsztein DC. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett 584: 1393–1398, 2010 [DOI] [PubMed] [Google Scholar]
- 97.Kour S. and Rath PC. Long noncoding RNAs in aging and age-related diseases. Ageing Res Rev 26: 1–21, 2016 [DOI] [PubMed] [Google Scholar]
- 98.Kowald A. and Kirkwood TB. Can aging be programmed? A critical literature review. Aging Cell 15: 986–998, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Labunskyy VM. and Gladyshev VN. Role of reactive oxygen species-mediated signaling in aging. Antioxid Redox Signal 19: 1362–1372, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet 11: 191–203, 2010 [DOI] [PubMed] [Google Scholar]
- 101.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, Reich M, Hieronymus H, Wei G, Armstrong SA, Haggarty SJ, Clemons PA, Wei R, Carr SA, Lander ES, and Golub TR. The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science 313: 1929–1935, 2006 [DOI] [PubMed] [Google Scholar]
- 102.Lawton KA, Berger A, Mitchell M, Milgram KE, Evans AM, Guo L, Hanson RW, Kalhan SC, Ryals JA, and Milburn MV. Analysis of the adult human plasma metabolome. Pharmacogenomics 9: 383–397, 2008 [DOI] [PubMed] [Google Scholar]
- 103.Laye MJ, Tran V, Jones DP, Kapahi P, and Promislow DE. The effects of age and dietary restriction on the tissue-specific metabolome of Drosophila. Aging Cell 14: 797–808, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lee BP, Pilling LC, Emond F, Flurkey K, Harrison DE, Yuan R, Peters LL, Kuchel GA, Ferrucci L, Melzer D, and Harries LW. Changes in the expression of splicing factor transcripts and variations in alternative splicing are associated with lifespan in mice and humans. Aging Cell 15: 903–913, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lee CK, Klopp RG, Weindruch R, and Prolla TA. Gene expression profile of aging and its retardation by caloric restriction. Science 285: 1390–1393, 1999 [DOI] [PubMed] [Google Scholar]
- 106.Leichert LI, Gehrke F, Gudiseva HV, Blackwell T, Ilbert M, Walker AK, Strahler JR, Andrews PC, and Jakob U. Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc Natl Acad Sci U S A 105: 8197–8202, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Leiser SF, Fletcher M, Begun A, and Kaeberlein M. Life-span extension from hypoxia in Caenorhabditis elegans requires both HIF-1 and DAF-16 and is antagonized by SKN-1. J Gerontol A Biol Sci Med Sci 68: 1135–1144, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lesur I. and Campbell JL. The transcriptome of prematurely aging yeast cells is similar to that of telomerase-deficient cells. Mol Biol Cell 15: 1297–1312, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Li N, Bates DJ, An J, Terry DA, and Wang E. Up-regulation of key microRNAs, and inverse down-regulation of their predicted oxidative phosphorylation target genes, during aging in mouse brain. Neurobiol Aging 32: 944–955, 2011 [DOI] [PubMed] [Google Scholar]
- 110.Li S, Stone HA, and Murphy CT. A microfluidic device and automatic counting system for the study of C. elegans reproductive aging. Lab Chip 15: 524–531, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Libina N, Berman JR, and Kenyon C. Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell 115: 489–502, 2003 [DOI] [PubMed] [Google Scholar]
- 112.Lin SJ, Defossez PA, and Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science 289: 2126–2128, 2000 [DOI] [PubMed] [Google Scholar]
- 113.Lin SS, Manchester JK, and Gordon JI. Enhanced gluconeogenesis and increased energy storage as hallmarks of aging in Saccharomyces cerevisiae. J Biol Chem 276: 36000–36007, 2001 [DOI] [PubMed] [Google Scholar]
- 114.Liu N, Landreh M, Cao K, Abe M, Hendriks GJ, Kennerdell JR, Zhu Y, Wang LS, and Bonini NM. The microRNA miR-34 modulates ageing and neurodegeneration in Drosophila. Nature 482: 519–523, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liu P, Young TZ, and Acar M. Yeast replicator: a high-throughput multiplexed microfluidics platform for automated measurements of single-cell aging. Cell Rep 13: 634–644, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, and Kroemer G. The hallmarks of aging. Cell 153: 1194–1217, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, and Yankner BA. Gene regulation and DNA damage in the ageing human brain. Nature 429: 883–891, 2004 [DOI] [PubMed] [Google Scholar]
- 118.Luo GZ, Blanco MA, Greer EL, He C, and Shi Y. DNA N(6)-methyladenine: a new epigenetic mark in eukaryotes? Nat Rev Mol Cell Biol 16: 705–710, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ma S, Upneja A, Galecki A, Tsai YM, Burant CF, Raskind S, Zhang Q, Zhang ZD, Seluanov A, Gorbunova V, Clish CB, Miller RA, and Gladyshev VN. Cell culture-based profiling across mammals reveals DNA repair and metabolism as determinants of species longevity. Elife 5: e1930, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ma S, Yim SH, Lee SG, Kim EB, Lee SR, Chang KT, Buffenstein R, Lewis KN, Park TJ, Miller RA, Clish CB, and Gladyshev VN. Organization of the mammalian metabolome according to organ function, lineage specialization, and longevity. Cell Metab 22: 332–343, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Maegawa S, Hinkal G, Kim HS, Shen L, Zhang L, Zhang J, Zhang N, Liang S, Donehower LA, and Issa JP. Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res 20: 332–340, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Maes OC, An J, Sarojini H, and Wang E. Murine microRNAs implicated in liver functions and aging process. Mech Ageing Dev 129: 534–541, 2008 [DOI] [PubMed] [Google Scholar]
- 123.Mair W. and Dillin A. Aging and survival: the genetics of life span extension by dietary restriction. Annu Rev Biochem 77: 727–754, 2008 [DOI] [PubMed] [Google Scholar]
- 124.Manta B. and Gladyshev VN. Regulated methionine oxidation by monooxygenases. Free Radic Biol Med 109: 141–155, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Marioni RE, Shah S, McRae AF, Chen BH, Colicino E, Harris SE, Gibson J, Henders AK, Redmond P, Cox SR, Pattie A, Corley J, Murphy L, Martin NG, Montgomery GW, Feinberg AP, Fallin MD, Multhaup ML, Jaffe AE, Joehanes R, Schwartz J, Just AC, Lunetta KL, Murabito JM, Starr JM, Horvath S, Baccarelli AA, Levy D, Visscher PM, Wray NR, and Deary IJ. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol 16: 25, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Martin-Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye-Knudsen M, Gomes AP, Ward TM, Minor RK, Blouin MJ, Schwab M, Pollak M, Zhang Y, Yu Y, Becker KG, Bohr VA, Ingram DK, Sinclair DA, Wolf NS, Spindler SR, Bernier M, and de Cabo R. Metformin improves healthspan and lifespan in mice. Nat Commun 4: 2192, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Massey AC, Kaushik S, Sovak G, Kiffin R, and Cuervo AM. Consequences of the selective blockage of chaperone-mediated autophagy. Proc Natl Acad Sci U S A 103: 5805–5810, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Maures TJ, Greer EL, Hauswirth AG, and Brunet A. The H3K27 demethylase UTX-1 regulates C. elegans lifespan in a germline-independent, insulin-dependent manner. Aging Cell 10: 980–990, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.McCormick MA, Delaney JR, Tsuchiya M, Tsuchiyama S, Shemorry A, Sim S, Chou AC, Ahmed U, Carr D, Murakami CJ, Schleit J, Sutphin GL, Wasko BM, Bennett CF, Wang AM, Olsen B, Beyer RP, Bammler TK, Prunkard D, Johnson SC, Pennypacker JK, An E, Anies A, Castanza AS, Choi E, Dang N, Enerio S, Fletcher M, Fox L, Goswami S, Higgins SA, Holmberg MA, Hu D, Hui J, Jelic M, Jeong KS, Johnston E, Kerr EO, Kim J, Kim D, Kirkland K, Klum S, Kotireddy S, Liao E, Lim M, Lin MS, Lo WC, Lockshon D, Miller HA, Moller RM, Muller B, Oakes J, Pak DN, Peng ZJ, Pham KM, Pollard TG, Pradeep P, Pruett D, Rai D, Robison B, Rodriguez AA, Ros B, Sage M, Singh MK, Smith ED, Snead K, Solanky A, Spector BL, Steffen KK, Tchao BN, Ting MK, Vander Wende H, Wang D, Welton KL, Westman EA, Brem RB, Liu XG, Suh Y, Zhou Z, Kaeberlein M, and Kennedy BK. A comprehensive analysis of replicative lifespan in 4,698 single-gene deletion strains uncovers conserved mechanisms of aging. Cell Metab 22: 895–906, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.McElwee J, Bubb K, and Thomas JH. Transcriptional outputs of the Caenorhabditis elegans forkhead protein DAF-16. Aging Cell 2: 111–121, 2003 [DOI] [PubMed] [Google Scholar]
- 131.McManus J, Cheng Z, and Vogel C. Next-generation analysis of gene expression regulation—comparing the roles of synthesis and degradation. Mol Biosyst 11: 2680–2689, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Milholland B, Suh Y, and Vijg J. Mutation and catastrophe in the aging genome. Exp Gerontol 94: 34–40, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, Fernandez E, Flurkey K, Javors MA, Nelson JF, Orihuela CJ, Pletcher S, Sharp ZD, Sinclair D, Starnes JW, Wilkinson JE, Nadon NL, and Strong R. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci 66: 191–201, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Miller RA, Harrison DE, Astle CM, Fernandez E, Flurkey K, Han M, Javors MA, Li X, Nadon NL, Nelson JF, Pletcher S, Salmon AB, Sharp ZD, Van Roekel S, Winkleman L, and Strong R. Rapamycin-mediated lifespan increase in mice is dose and sex dependent and metabolically distinct from dietary restriction. Aging Cell 13: 468–477, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Murata S, Yashiroda H, and Tanaka K. Molecular mechanisms of proteasome assembly. Nat Rev Mol Cell Biol 10: 104–115, 2009 [DOI] [PubMed] [Google Scholar]
- 136.Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, and Kenyon C. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424: 277–283, 2003 [DOI] [PubMed] [Google Scholar]
- 137.Oberdoerffer P. and Sinclair DA. The role of nuclear architecture in genomic instability and ageing. Nat Rev Mol Cell Biol 8: 692–702, 2007 [DOI] [PubMed] [Google Scholar]
- 138.Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, and Ruvkun G. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 389: 994–999, 1997 [DOI] [PubMed] [Google Scholar]
- 139.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, and Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 1: 376–386, 2002 [DOI] [PubMed] [Google Scholar]
- 140.Onken B. and Driscoll M. Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans Healthspan via AMPK, LKB1, and SKN-1. PLoS One 5: e8758, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Orgel LE. The maintenance of the accuracy of protein synthesis and its relevance to ageing. Proc Natl Acad Sci U S A 49: 517–521, 1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ori A, Toyama BH, Harris MS, Bock T, Iskar M, Bork P, Ingolia NT, Hetzer MW, and Beck M. Integrated transcriptome and proteome analyses reveal organ-specific proteome deterioration in old rats. Cell Syst 1: 224–237, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Pan C, Valente JJ, LoBrutto R, Pickett JS, and Motto M. Combined application of high resolution and tandem mass spectrometers to characterize methionine oxidation in a parathyroid hormone formulation. J Pharm Sci 99: 1169–1179, 2010 [DOI] [PubMed] [Google Scholar]
- 144.Parkhitko AA, Binari R, Zhang N, Asara JM, Demontis F, and Perrimon N. Tissue-specific down-regulation of S-adenosyl-homocysteine via suppression of dAhcyL1/dAhcyL2 extends health span and life span in Drosophila. Genes Dev 30: 1409–1422, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pastor WA, Aravind L, and Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol 14: 341–356, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Patti GJ, Tautenhahn R, Johannsen D, Kalisiak E, Ravussin E, Bruning JC, Dillin A, and Siuzdak G. Meta-analysis of global metabolomic data identifies metabolites associated with life-span extension. Metabolomics 10: 737–743, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Petkovich DA, Podolskiy DI, Lobanov AV, Lee SG, Miller RA, and Gladyshev VN. Using DNA methylation profiling to evaluate biological age and longevity interventions. Cell Metab 25: 954–960 e956, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Pincus Z, Smith-Vikos T, and Slack FJ. MicroRNA predictors of longevity in Caenorhabditis elegans. PLoS Genet 7: e1002306, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Pletcher SD, Macdonald SJ, Marguerie R, Certa U, Stearns SC, Goldstein DB, and Partridge L. Genome-wide transcript profiles in aging and calorically restricted Drosophila melanogaster. Curr Biol 12: 712–723, 2002 [DOI] [PubMed] [Google Scholar]
- 150.Radhakrishnan SK, Lee CS, Young P, Beskow A, Chan JY, and Deshaies RJ. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol Cell 38: 17–28, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Rodriguez SA, Grochova D, McKenna T, Borate B, Trivedi NS, Erdos MR, and Eriksson M. Global genome splicing analysis reveals an increased number of alternatively spliced genes with aging. Aging Cell 15: 267–278, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Rodwell GE, Sonu R, Zahn JM, Lund J, Wilhelmy J, Wang L, Xiao W, Mindrinos M, Crane E, Segal E, Myers BD, Brooks JD, Davis RW, Higgins J, Owen AB, and Kim SK. A transcriptional profile of aging in the human kidney. PLoS Biol 2: e427, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ryley J. and Pereira-Smith OM. Microfluidics device for single cell gene expression analysis in Saccharomyces cerevisiae. Yeast 23: 1065–1073, 2006 [DOI] [PubMed] [Google Scholar]
- 154.Schmidt M. and Finley D. Regulation of proteasome activity in health and disease. Biochim Biophys Acta 1843: 13–25, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Segal E, Shapira M, Regev A, Pe'er D, Botstein D, Koller D, and Friedman N. Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nat Genet 34: 166–176, 2003 [DOI] [PubMed] [Google Scholar]
- 156.Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, Woods A, Robinson IC, Schuster E, Batterham RL, Kozma SC, Thomas G, Carling D, Okkenhaug K, Thornton JM, Partridge L, Gems D, and Withers DJ. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 326: 140–144, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Shakir S, Vinh J, and Chiappetta G. Quantitative analysis of the cysteine redoxome by iodoacetyl tandem mass tags. Anal Bioanal Chem 409: 3821–3830, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Smith DL, Jr., Maharrey CH, Carey CR, White RA, and Hartman JL., 4th Gene-nutrient interaction markedly influences yeast chronological lifespan. Exp Gerontol 86: 113–123, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Smith-Vikos T, Liu Z, Parsons C, Gorospe M, Ferrucci L, Gill TM, and Slack FJ. A serum miRNA profile of human longevity: findings from the Baltimore Longitudinal Study of Aging (BLSA). Aging (Albany NY) 8: 2971–2987, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Stadtman ER. Protein oxidation and aging. Science 257: 1220–1224, 1992 [DOI] [PubMed] [Google Scholar]
- 161.Stamm S. Regulation of alternative splicing by reversible protein phosphorylation. J Biol Chem 283: 1223–1227, 2008 [DOI] [PubMed] [Google Scholar]
- 162.Steffen KK, Kennedy BK, and Kaeberlein M. Measuring replicative life span in the budding yeast. J Vis Exp 28: e1209, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Strong R, Miller RA, Astle CM, Floyd RA, Flurkey K, Hensley KL, Javors MA, Leeuwenburgh C, Nelson JF, Ongini E, Nadon NL, Warner HR, and Harrison DE. Nordihydroguaiaretic acid and aspirin increase lifespan of genetically heterogeneous male mice. Aging Cell 7: 641–650, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Stroustrup N, Anthony WE, Nash ZM, Gowda V, Gomez A, Lopez-Moyado IF, Apfeld J, and Fontana W. The temporal scaling of Caenorhabditis elegans ageing. Nature 530: 103–107, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Stroustrup N, Ulmschneider BE, Nash ZM, Lopez-Moyado IF, Apfeld J, and Fontana W. The Caenorhabditis elegans lifespan machine. Nat Methods 10: 665–670, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Stubbs TM, Bonder MJ, Stark AK, Krueger F, Team BIAC, von Meyenn F, Stegle O, and Reik W. Multi-tissue DNA methylation age predictor in mouse. Genome Biol 18: 68, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Szilard L. On the nature of the aging process. Proc Natl Acad Sci U S A 45: 30–45, 1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Tarrago L, Peterfi Z, Lee BC, Michel T, and Gladyshev VN. Monitoring methionine sulfoxide with stereospecific mechanism-based fluorescent sensors. Nat Chem Biol 11: 332–338, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Taylor RC. and Dillin A. Aging as an event of proteostasis collapse. Cold Spring Harb Perspect Biol 3: pii: , 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Tomas-Loba A, Bernardes de Jesus B, Mato JM, and Blasco MA. A metabolic signature predicts biological age in mice. Aging Cell 12: 93–101, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Tomko RJ, Jr, and Hochstrasser M. Molecular architecture and assembly of the eukaryotic proteasome. Annu Rev Biochem 82: 415–445, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, and Darnell RB. CLIP identifies Nova-regulated RNA networks in the brain. Science 302: 1212–1215, 2003 [DOI] [PubMed] [Google Scholar]
- 173.van Beek JH, Kirkwood TB, and Bassingthwaighte JB. Understanding the physiology of the ageing individual: computational modelling of changes in metabolism and endurance. Interface Focus 6: 20150079, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.van Hoof A, Frischmeyer PA, Dietz HC, and Parker R. Exosome-mediated recognition and degradation of mRNAs lacking a termination codon. Science 295: 2262–2264, 2002 [DOI] [PubMed] [Google Scholar]
- 175.Vermulst M, Denney AS, Lang MJ, Hung CW, Moore S, Moseley MA, Thompson JW, Madden V, Gauer J, Wolfe KJ, Summers DW, Schleit J, Sutphin GL, Haroon S, Holczbauer A, Caine J, Jorgenson J, Cyr D, Kaeberlein M, Strathern JN, Duncan MC, and Erie DA. Transcription errors induce proteotoxic stress and shorten cellular lifespan. Nat Commun 6: 8065, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Vijg J. and Suh Y. Genetics of longevity and aging. Annu Rev Med 56: 193–212, 2005 [DOI] [PubMed] [Google Scholar]
- 177.Vijg J. and Suh Y. Genome instability and aging. Annu Rev Physiol 75: 645–668, 2013 [DOI] [PubMed] [Google Scholar]
- 178.Walther DM, Kasturi P, Zheng M, Pinkert S, Vecchi G, Ciryam P, Morimoto RI, Dobson CM, Vendruscolo M, Mann M, and Hartl FU. Widespread proteome remodeling and aggregation in aging C. elegans. Cell 161: 919–932, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Walther DM. and Mann M. Accurate quantification of more than 4000 mouse tissue proteins reveals minimal proteome changes during aging. Mol Cell Proteomics 10: M110.004523, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wang T, Tsui B, Kreisberg JF, Robertson NA, Gross AM, Yu MK, Carter H, Brown-Borg HM, Adams PD, and Ideker T. Epigenetic aging signatures in mice livers are slowed by dwarfism, calorie restriction and rapamycin treatment. Genome Biol 18: 57, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Wang X, Yen J, Kaiser P, and Huang L. Regulation of the 26S proteasome complex during oxidative stress. Sci Signal 3: ra88, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MB, Bachovchin DA, Mowen K, Baker D, and Cravatt BF. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468: 790–795, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Weidner CI, Lin Q, Koch CM, Eisele L, Beier F, Ziegler P, Bauerschlag DO, Jockel KH, Erbel R, Muhleisen TW, Zenke M, Brummendorf TH, and Wagner W. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol 15: R24, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Weindruch R. and Walford RL. Retardation of Aging and Disease by Dietary Restriction. Springfield, IL: CC Thomas, 1988 [Google Scholar]
- 185.Welle S, Brooks AI, Delehanty JM, Needler N, Bhatt K, Shah B, and Thornton CA. Skeletal muscle gene expression profiles in 20–29 year old and 65–71 year old women. Exp Gerontol 39: 369–377, 2004 [DOI] [PubMed] [Google Scholar]
- 186.Wierman MB, Matecic M, Valsakumar V, Li M, Smith DL, Jr, Bekiranov S, and Smith JS. Functional genomic analysis reveals overlapping and distinct features of chronologically long-lived yeast populations. Aging (Albany NY) 7: 177–194, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wishart DS, Jewison T, Guo AC, Wilson M, Knox C, Liu Y, Djoumbou Y, Mandal R, Aziat F, Dong E, Bouatra S, Sinelnikov I, Arndt D, Xia J, Liu P, Yallou F, Bjorndahl T, Perez-Pineiro R, Eisner R, Allen F, Neveu V, Greiner R, and Scalbert A. HMDB 3.0—the human metabolome database in 2013. Nucleic Acids Res 41: D801–D807, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, and Sinclair D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 430: 686–689, 2004 [DOI] [PubMed] [Google Scholar]
- 189.Wood SH, Craig T, Li Y, Merry B, and de Magalhaes JP. Whole transcriptome sequencing of the aging rat brain reveals dynamic RNA changes in the dark matter of the genome. Age (Dordr) 35: 763–776, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Wyers F, Rougemaille M, Badis G, Rousselle JC, Dufour ME, Boulay J, Regnault B, Devaux F, Namane A, Seraphin B, Libri D, and Jacquier A. Cryptic pol II transcripts are degraded by a nuclear quality control pathway involving a new poly(A) polymerase. Cell 121: 725–737, 2005 [DOI] [PubMed] [Google Scholar]
- 191.Xian B, Shen J, Chen W, Sun N, Qiao N, Jiang D, Yu T, Men Y, Han Z, Pang Y, Kaeberlein M, Huang Y, and Han JD. WormFarm: a quantitative control and measurement device toward automated Caenorhabditis elegans aging analysis. Aging Cell 12: 398–409, 2013 [DOI] [PubMed] [Google Scholar]
- 192.Xie Z, Zhang Y, Zou K, Brandman O, Luo C, Ouyang Q, and Li H. Molecular phenotyping of aging in single yeast cells using a novel microfluidic device. Aging Cell 11: 599–606, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Yin D. and Chen K. The essential mechanisms of aging: irreparable damage accumulation of biochemical side-reactions. Exp Gerontol 40: 455–465, 2005 [DOI] [PubMed] [Google Scholar]
- 194.Yu Z, Zhai G, Singmann P, He Y, Xu T, Prehn C, Romisch-Margl W, Lattka E, Gieger C, Soranzo N, Heinrich J, Standl M, Thiering E, Mittelstrass K, Wichmann HE, Peters A, Suhre K, Li Y, Adamski J, Spector TD, Illig T, and Wang-Sattler R. Human serum metabolic profiles are age dependent. Aging Cell 11: 960–967, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Yuan T, Jiao Y, de Jong S, Ophoff RA, Beck S, and Teschendorff AE. An integrative multi-scale analysis of the dynamic DNA methylation landscape in aging. PLoS Genet 11: e1004996, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Zhang F, Su K, Yang X, Bowe DB, Paterson AJ, and Kudlow JE. O-GlcNAc modification is an endogenous inhibitor of the proteasome. Cell 115: 715–725, 2003 [DOI] [PubMed] [Google Scholar]
- 197.Zhang G, Huang H, Liu D, Cheng Y, Liu X, Zhang W, Yin R, Zhang D, Zhang P, Liu J, Li C, Liu B, Luo Y, Zhu Y, Zhang N, He S, He C, Wang H, and Chen D. N6-methyladenine DNA modification in Drosophila. Cell 161: 893–906, 2015 [DOI] [PubMed] [Google Scholar]
- 198.Zhang W, Liu Y, Sun N, Wang D, Boyd-Kirkup J, Dou X, and Han JD. Integrating genomic, epigenomic, and transcriptomic features reveals modular signatures underlying poor prognosis in ovarian cancer. Cell Rep 4: 542–553, 2013 [DOI] [PubMed] [Google Scholar]
- 199.Zhao J, Ohsumi TK, Kung JT, Ogawa Y, Grau DJ, Sarma K, Song JJ, Kingston RE, Borowsky M, and Lee JT. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol Cell 40: 939–953, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Zierer J, Menni C, Kastenmuller G, and Spector TD. Integration of ‘omics’ data in aging research: from biomarkers to systems biology. Aging Cell 14: 933–944, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]