

Abstract
Cochlear synaptopathy produced by exposure to noise levels that cause only transient auditory threshold elevations is a condition that affects many people and is believed to contribute to poor speech discrimination in noisy environments. These functional deficits in hearing, without changes in sensitivity, have been called hidden hearing loss (HHL). It has been proposed that activity of the medial olivocochlear (MOC) system can ameliorate acoustic trauma effects. Here we explore the role of the MOC system in HHL by comparing the performance of two different mouse models: an α9 nicotinic receptor subunit knock-out (KO; Chrna9 KO), which lacks cholinergic transmission between efferent neurons and hair cells; and a gain-of-function knock-in (KI; Chrna9L9′T KI) carrying an α9 point mutation that leads to enhanced cholinergic activity. Animals of either sex were exposed to sound pressure levels that in wild-type produced transient cochlear threshold shifts and a decrease in neural response amplitudes, together with the loss of ribbon synapses, which is indicative of cochlear synaptopathy. Moreover, a reduction in the number of efferent contacts to outer hair cells was observed. In Chrna9 KO ears, noise exposure produced permanent auditory threshold elevations together with cochlear synaptopathy. In contrast, the Chrna9L9′T KI was completely resistant to the same acoustic exposure protocol. These results show a positive correlation between the degree of HHL prevention and the level of cholinergic activity. Notably, enhancement of the MOC feedback promoted new afferent synapse formation, suggesting that it can trigger cellular and molecular mechanisms to protect and/or repair the inner ear sensory epithelium.
SIGNIFICANCE STATEMENT Noise overexposure is a major cause of a variety of perceptual disabilities, including speech-in-noise difficulties, tinnitus, and hyperacusis. Here we show that exposure to noise levels that do not cause permanent threshold elevations or hair cell death can produce a loss of cochlear nerve synapses to inner hair cells as well as degeneration of medial olivocochlear (MOC) terminals contacting the outer hair cells. Enhancement of the MOC reflex can prevent both types of neuropathy, highlighting the potential use of drugs that increase α9α10 nicotinic cholinergic receptor activity as a pharmacotherapeutic strategy to avoid hidden hearing loss.
Keywords: acoustic trauma, cholinergic receptor, hearing loss, medial olivocochlear system, ribbon synapse
Introduction
Noise-induced hearing loss (NIHL) is growing as one of the most prevalent types of noncongenital hearing loss. It has recently been shown that exposure to loud sounds, causing only transient cochlear threshold elevations, can produce a loss of synapses between inner hair cells (IHCs) and auditory nerve fibers (Kujawa and Liberman, 2009). This cochlear synaptopathy, also known as hidden hearing loss (HHL; Schaette and McAlpine, 2011), occurs within hours after acoustic exposure, and it is proposed to be a type of glutamate excitotoxicity (Liberman and Mulroy, 1982; Pujol et al., 1993; Pujol and Puel, 1999). Recent studies have demonstrated that auditory nerve fibers with high-threshold and low spontaneous rates (SRs) are most vulnerable to noise damage compared with low-threshold high-SR fibers (Furman et al., 2013; Liberman et al., 2015). These observations provide an explanation for the absence of auditory threshold elevations in cochlear synaptopathy models. Due to the wider dynamic range and threshold distribution, it was suggested that these low-SR fibers are critical for signal coding in noisy backgrounds (Costalupes, 1985; Young and Barta, 1986).
Medial olivocochlear (MOC) efferent neurons form a-negative feedback gain-control system that inhibits amplification of sounds by outer hair cells (OHCs) (Galambos, 1955; Wiederhold and Kiang, 1970; Guinan, 2011). Activation of the MOC pathway reduces cochlear sensitivity through the action of acetylcholine on α9α10 cholinergic nicotinic ACh receptors (nAChRs) at the base of OHCs (Elgoyhen et al., 1994, 2001; Ballestero et al., 2011; Guinan, 2011). Several lines of evidence have demonstrated that the MOC system has an important role in the protection from NIHL: (1) stimulation of MOC fibers during sound overexposure produces a reduction of sensitivity loss (Reiter and Liberman, 1995); (2) chronic sectioning of the olivocochlear bundle renders the ear more vulnerable to permanent acoustic injury (Handrock and Zeisberg, 1982; Kujawa and Liberman, 1997; Maison et al., 2013); (3) the strength of the olivocochlear reflex is inversely correlated with the degree of NIHL (Maison and Liberman, 2000); and (4) genetically modified mice, in which the magnitude and duration of efferent cholinergic effects are increased, exhibit higher tolerance to noise-induced trauma (Taranda et al., 2009). However, the precise role of the MOC reflex on HHL remains mostly undefined. Recent studies show enhanced cochlear synaptopathy after olivocochlear bundle lesion, which removes much of the MOC innervation to OHCs with almost no alteration in the lateral olivocochlear (LOC) neurons (Maison et al., 2013). A variety of data indicates that MOC neurons release not only acetylcholine but also GABA and calcitonin gene-related peptide (Maison et al., 2003; Wedemeyer et al., 2013). Thus, cochlear de-efferentation produced by surgical lesion to the olivocochlear pathway removes all the different neurotransmitter/neuromodulator components and possibly some LOC neurons. Here we explored the effect of the strength of the cholinergic component of the MOC reflex on the cochlear synaptopathy that occurs in HHL by using genetically modified mice with different levels of α9α10 nAChR activity. In addition, we analyzed for the first time the effect of acoustic trauma (AT) in ears exposed at sound levels well below those that cause hair cell damage and permanent threshold shifts on efferent synaptic terminals contacting the OHCs. We used mice at the early onset of puberty (i.e., at 3 weeks of age), a period of enhanced sensitivity to NIHL (Henry, 1984; Ohlemiller et al., 2000; Kujawa and Liberman, 2006). Our results show that the degree of cochlear synaptopathy is negatively correlated to the level of α9α10 nAChR activity. Notably, the enhancement of cholinergic activity not only prevented cochlear synaptopathy but also promoted new afferent synapse formation after acoustic trauma. Moreover, exposure to loud noise produced plastic changes leading to the degeneration of MOC terminals synapsing on OHCs. We propose that both cochlear synaptopathy and the loss of MOC terminals contribute to HHL, compromising the performance of complex listening tasks such as understanding speech in a noisy environment. Therefore, our findings unequivocally show that strengthening of the MOC feedback by enhancement of α9α10 nAChR activity can prevent the development of HHL symptoms after noise exposure.
Materials and Methods
Animals.
Chrna9 KO and Chrna9L9′T KI mice have been previously described (Vetter et al., 1999; Taranda et al., 2009) and were backcrossed with a congenic FVB.129P2-Pde6bþ Tyrc-ch/AntJ strain (https://www.jax.org/strain/004828; RRID:IMSR_JAX:004828) for 17 generations (i.e., N-17). We used a similar male/female ratio in all the experimental groups in the different genotypes. All experimental protocols were performed in accordance with the American Veterinary Medical Association Guidelines for the Euthanasia of Animals (June 2013) as well as Instituto de Investigaciones en Ingeniería Genética y Biologıa Molecular Institutional Animal Care and Use Committee guidelines, and best practice procedures.
Cochlear function tests.
Inner ear physiology, including auditory brainstem responses (ABRs) and distortion-product otoacoustic emissions (DPOAEs), was performed in mice of either sex anesthetized with xylazine (10 mg/kg, i.p.) and ketamine (100 mg/kg, i.p.) and placed in a soundproof chamber maintained at 30°C. The first recording was performed at postnatal day 21 (P21), followed by noise exposure and the additional measurements 1, 7, 8, and 14 d postexposure. Sound stimuli were delivered through a custom acoustic system with two dynamic earphones used as sound sources (CDMG15008–03A, CUI) and an electret condenser microphone (FG-23329-PO7, Knowles) coupled to a probe tube to measure sound pressure near the eardrum (for details, see https://www.masseyeandear.org/research/otolaryngology/investigators/laboratories/eaton-peabody-laboratories/epl-engineering-resources/epl-acoustic-system). Digital stimulus generation and response processing were handled by digital input-output boards from National Instruments driven by custom software written in LabVIEW (given by Dr. M. Charles Liberman, Eaton-Peabody Laboratories, Massachusetts Eye & Ear Infirmary, Boston, MA). For ABRs, needle electrodes were placed into the skin at the dorsal midline close to the neural crest and pinna with a ground electrode near the tail. ABR potentials were evoked with 5 ms tone pips (0.5 ms rise–fall, with a cos2 envelope at 40/s) delivered to the eardrum at log-spaced frequencies from 5.6 to 45.25 kHz. The response was amplified 10,000× with a 0.3–3 kHz passband. Sound level was raised in 5 dB steps from 20 to 80 dB sound pressure level (SPL). At each level, 1024 responses were averaged with stimulus polarity alternated. The threshold for ABR was defined as the lowest stimulus level at which a repeatable peak 1 could be identified in the response waveform. The ABR peak 1 amplitude was computed by off-line analysis of the peak to baseline amplitude of stored waveforms. The DPOAEs in response to two primary tones of frequency f1 and f2 were recorded at 2f1–f2, with f2/f1 = 1.2, and the f2 level 10 dB lower than the f1 level. Ear-canal sound pressure was amplified and digitally sampled at 4 μs intervals. The DPOAE threshold was defined as the lowest f2 level in which the signal-to-noise floor ratio is >1.
Noise exposure.
Animals were exposed under anesthesia to a 1–16 kHz noise at 100 dB SPL for 1 h in the same acoustic chamber used for cochlear function tests. Noise calibration to target SPL was performed immediately before each acoustic overexposure.
Cochlear processing and immunostaining.
For the immunolabeling and quantification, we divided animals for each genotype into the following two groups: “control” and “7 d after AT” (AT + 7d). First, we tested the auditory function at P21 in both control and AT + 7d groups. Immediately after, the control group was introduced into the acoustic chamber under anesthesia for 1 h for a “sham” condition, while the AT + 7d group was exposed to 100 dB SPL for 1 h in the same chamber. Acoustic thresholds in both groups were measured again at 1 and 7 d after acoustic trauma and after the final auditory test (P28), tissues were collected for immunostaining. Cochleae were perfused intralabyrinthly with 4% paraformaldehyde (PFA) in PBS, after being fixed with 4% PFA overnight, and decalcified in 0.12 m EDTA. Cochlear tissues were then microdissected and permeabilized by freeze/thawing in 30% sucrose (for CtBP2/GluA2 immunostaining) or directly blocked (for synaptophysin immunostaining). The microdissected pieces were blocked in 5% normal goat serum with 1% Triton X-100 in PBS for 1 h, followed by incubation in primary antibodies (diluted in blocking buffer) at 37°C for 16 h (for CtBP2/GluA2 immunostaining) or 4°C for 16 h (for synaptophysin immunostaining). The primary antibodies used in this study were as follows: (1) mouse anti-synaptophysin antibody (1:1000; MAB5258, Millipore; RRID:AB_2313839) to reveal the efferent terminals; (2) anti-C-terminal binding protein 2 (mouse anti-CtBP2 IgG1; 1:200; catalog #612044, BD Biosciences; RRID:AB_399431) to label the presynaptic ribbon; and (3) anti-glutamate receptor 2 (mouse anti-GluA2 IgG2a; 1:2000; MAB397, Millipore; RRID:AB_11212990) to label the postsynaptic receptor plaques. Tissues were then incubated with the appropriate Alexa Fluor-conjugated fluorescent secondary antibodies (1:1000 in blocking buffer; Invitrogen) for 2 h at room temperature (for CtBP2/GluA2 immunostaining) or processed with biotinylated secondary, ABC Reagent (Vector Laboratories; RRID:AB_2336827), and diaminobenzidine for standard light microscopy (for synaptophysin immunostaining). Finally, tissues were mounted on microscope slides in FluorSave mounting media (Millipore). For IHC synaptic counts, confocal z-stacks (0.1 μm step size) of the apical, medial, and basal regions from each cochlea were taken using a Leica TCS SPE Microscope equipped with 63× (1.5× digital zoom) oil-immersion lens. Image stacks were imported to Fiji software (RRID:SCR_002285; Schindelin et al., 2012), where IHCs were identified based on their CtBP2-stained nuclei. Each image usually contained 10–20 IHCs. For each stack, a custom Fiji plugin was developed to automate the quantifications of synaptic ribbons, glutamate receptor patches, and colocalized synaptic puncta. Briefly, each channel was analyzed separately, and maximum projections were generated to quantify the number of CtBP2 or GluA2 puncta. Additionally, a composite between the two channels was produced to draw the different ROIs that correspond to each IHC taking the CtBP2-stained nuclei as a reference. The maximum projections from the single channels were multiplied to generate a merged 32-bit image of the two channels. Then, they were converted to binary images after a custom thresholding procedure. Automatic counting of the number of particles on each ROI was performed.
Statistical analysis.
Data were analyzed with R Statistical Software (RRID:SCR_001905). The Shapiro–Wilks test was used for testing the normal distribution of the residuals. We used nonparametric tests because our data were non-normally distributed. For one-group comparisons that involved repeated measures (ABR thresholds, peak 1 amplitudes and latencies, and DPOAE thresholds), Friedman tests were used followed by a post hoc test (Pohlert, 2014). For group comparisons that do not involve repeated measures (ABR comparisons of unexposed animals, IHC synaptic counts), Kruskal–Wallis nonparametric ANOVA followed by Dunn's post-tests were used to determine statistical significance. For independent, two-group comparisons (MOC terminals per OHC), a two-sample Kolmogorov–Smirnov test was applied. Statistical significance was set to p < 0.05.
Results
Auditory function in mice with different degrees of efferent inhibition after acoustic trauma
To evaluate cochlear function, we recorded ABRs, the sound-evoked potentials generated by neuronal circuits in the ascending auditory pathways. We used genetically modified mice with different levels of α9α10 nAChR activity: Chrna9 KO mice, which lack cholinergic transmission between MOC neurons and hair cells (Vetter et al., 1999); and a gain-of-function Chrna9L9′T KI mice carrying an α9 point mutation that leads to enhanced responses to MOC activity (Taranda et al., 2009). We examined ABR responses at P21 in WT, Chrna9 KO, and Chrna9L9′T KI unexposed mice and found no difference in the median ABR thresholds between WT and Chrna9 KO mice except at the highest frequency tested (Kruskal–Wallis test: df = 2, p = 0.04 at 45.25 kHz). However, median ABR thresholds were slightly elevated by 5–10 dB in Chrna9L9′T KI mice at all frequencies, except for high-frequency stimuli (Kruskal–Wallis test: df = 2, p < 0.05 at 5.6, 8, 11.33, and 16 kHz; Fig. 1a). ABR peak 1 amplitudes at 80 dB, the summed activity of the cochlear nerve, were not modified in the three genotypes (Kruskal–Wallis test: df = 2, p > 0.05 at all the frequencies tested; Fig. 1b).
Figure 1.

Auditory function in unexposed WT, Chrna9 KO, and Chrna9L9′T KI mice. a, ABR thresholds for control WT (n = 12), Chrna9 KO (n = 14), and Chrna9L9′T KI (n = 15) mice at P21. b, ABR peak 1 amplitudes at 80 dB SPL. c, DPOAE thresholds in the same unexposed mice in the different genotypes. Cochlear thresholds are elevated in Chrna9L9′T KI ears, as measured by either ABRs (a) or DPOAEs (c). In all cases, median and interquartile ranges are shown, and the comparisons were made by a Kruskal–Wallis nonparametric ANOVA followed by a Dunn's post-test. Dark gray asterisks represent the statistical significance between Chrna9 KO and WT mice, and light gray asterisks between Chrna9L9′T KI and WT mice (*p < 0.05 and **p < 0.01).
We then exposed WT, Chrna9 KO, and Chrna9L9′T KI mice to a 1–16 kHz noise at 100 dB SPL for 1 h and tested the animals 1 and 7 d after acoustic trauma (Fig. 2). As shown in Figure 2b, large auditory threshold shifts from 10 to 35 dB SPL were found 1 d after exposure in WT and Chrna9 KO mice (Friedman test: df = 2 p < 0.05 at all the frequencies tested for both genotypes). One week later, auditory thresholds returned to pre-exposure values in WT mice (Friedman test: df = 2, p > 0.05 at all the frequencies tested), a result known to occur in mice with clear signs of HHL (Kujawa and Liberman, 2009). However, in Chrna9 KO ears, which lack a functional MOC system feedback, auditory thresholds did not recover (Friedman test: df = 2, p < 0.05 at all the frequencies tested), indicating a persistence of damage (Fig. 2b, middle). In contrast, the Chrna9L9′T KI gain-of-function mouse model with an enhanced α9α10 nAChRs activity was completely resistant to the same acoustic exposure protocol (Friedman test: df = 2, p > 0.05 at all the frequencies tested; Fig. 2b, right).
Figure 2.
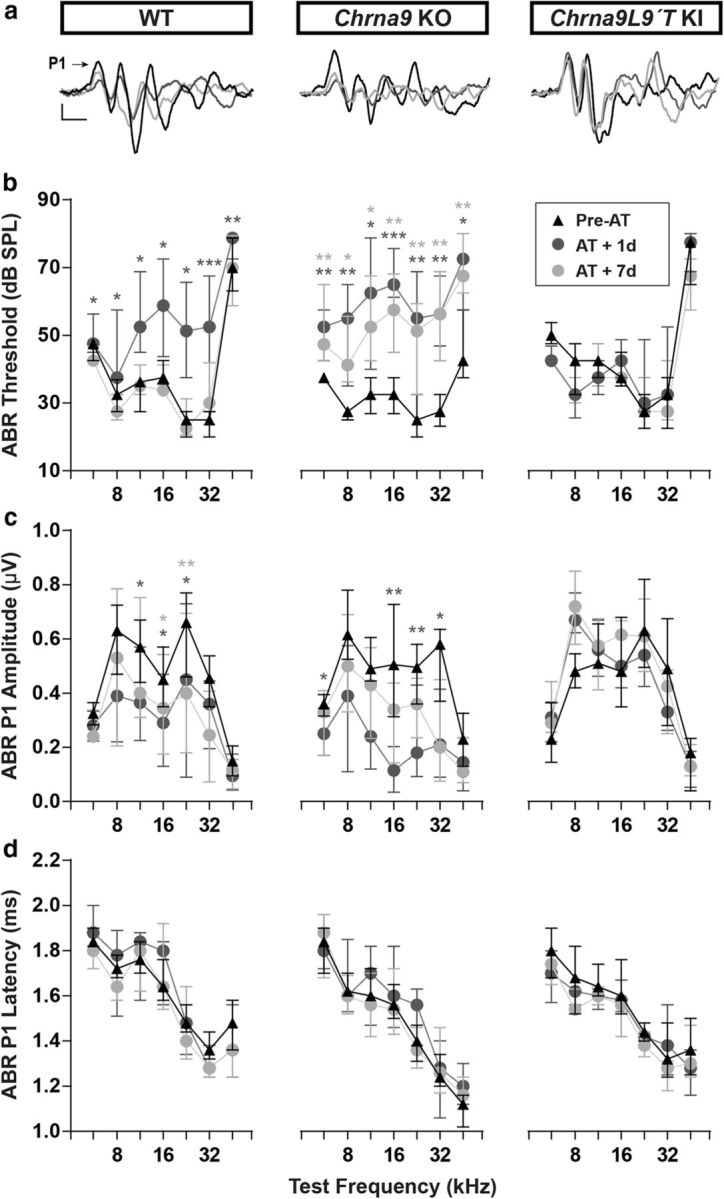
ABR measurements before and after AT. a, Representative ABR traces from WT, Chrna9 KO, and Chrna9L9′T KI mice at P21 before trauma (Pre-AT, black trace), 1 d after AT (AT + 1d, dark gray trace), and AT + 7d (light gray trace). Arrow indicates peak 1 amplitude. Calibration: vertical, 0.4 μV; horizontal, 1 ms. b, ABR thresholds in WT (n = 12), Chrna9 KO (n = 14), and Chrna9L9′T KI (n = 15) mice at P21 before trauma, 1 d after AT, and 7 d after AT. WT and Chrna9 KO mice showed a significant increase in ABR thresholds 1 d after AT. A recovery of ABR thresholds was observed a week after exposure only in WT mice. Chrna9L9′T KI mice did not present any changes in ABR thresholds at any time after AT. c, ABR peak 1 amplitudes at 80 dB SPL in WT, Chrna9 KO, and Chrna9L9′T KI mice at the same time points shown in b. WT and Chrna9 KO mice displayed a reduction in amplitudes 1 d after AT and only a partial recovery a week after exposure. Peak 1 amplitudes in Chrna9L9′T KI mice were unaffected by noise exposure. d, ABR peak 1 latencies in WT, Chrna9 KO, and Chrna9L9′T KI mice at the same time points shown in b. Latencies were not affected in any of the genotypes after AT. Median and interquartile ranges are shown, and the comparisons were made by Friedman tests followed by a post hoc test. Dark gray asterisks represent the statistical significance of AT + 1d values compared with Pre-AT, and light gray asterisks represent AT + 7d values compared with Pre-AT controls. *p < 0.05; **p < 0.01; ***p < 0.001.
One signature of cochlear synaptopathy observed in HHL in mice is a reduction of ABR peak 1 amplitudes without permanent changes in ABR thresholds (Kujawa and Liberman, 2009). This peak represents the summed sound-evoked spike activity at the first synapse between IHCs and afferent nerve fibers (Buchwald and Huang, 1975; Antoli-Candela and Kiang, 1978). As shown in Figure 2, a and c, a large reduction in amplitudes was observed at 11.33, 16, and 22.65 kHz in WT mice 1 d after acoustic trauma (Friedman test: df = 2, p = 0.048 at 11.33 kHz; p = 0.026 at 16 kHz; and p = 0.018 at 22.65 kHz), which did not completely recover after 7 d (Friedman test: df = 2, p = 0.048 at 16 kHz; and p = 0.003 at 22.65 kHz). Similarly, in Chrna9 KO mice, ABR peak 1 amplitudes were reduced 1 d after trauma at 5.6, 16, 22.65, and 32 kHz (Friedman test: df = 2, p = 0.048 at 5.6 kHz; p = 0.007 at 16 kHz; p = 0.01 at 22.65 kHz; and p = 0.026 at 32 kHz). Seven days after acoustic overexposure, ABR peak 1 amplitudes had not completely recovered; however, it was not significantly different (Friedman test: df = 2, p > 0.05 at all the frequencies tested; Fig. 2a,c). Interestingly, ABR peak 1 amplitudes were not modified in the Chrna9L9′T KI mice after acoustic trauma (Friedman test: df = 2, p > 0.05 at all the frequencies tested), suggesting that enhanced α9α10 nAChRs activity might prevent the loss of afferent synapses. The ABR peak 1 latencies were not altered after acoustic trauma in any of the evaluated genotypes (Friedman test: df = 2 p < 0.05 at all the frequencies tested for all the genotypes; Fig. 2d), indicating that variation in the MOC strength does not affect the conduction velocity of cochlear nerve fibers.
Noise-induced cochlear synaptopathy in mice with different levels of MOC feedback
Cochleae were harvested and fixed for histological analysis at P28 in all of the different groups. Hair cell count showed no loss of IHCs or OHCs in the different genotypes in control and exposed groups (data not shown). To analyze the IHC–auditory nerve fiber synapses, organ of Corti whole-mounts were immunostained with antibodies against CtBP2-Ribeye, a critical protein present at the presynaptic ribbon (Khimich et al., 2005), and GluA2 AMPA-type glutamate receptors, which are expressed at the postsynaptic afferent terminal (Matsubara et al., 1996; Liberman et al., 2011; Maison et al., 2013). IHC–afferent synapses were identified by colocalization of a pair of CtBP2/GluA2 puncta at the base of the IHC (Liberman et al., 2011). In each whole-mount organ of Corti, counting was performed at three different cochlear locations: apical, medial, and basal. The number of prelocalized, postlocalized, or colocalized synaptic markers was averaged from 10 to 20 IHCs per each imaged cochlear section (4–14 animals/genotype) to generate the synaptic density per IHC.
First, we compared the number of prelocalized, postlocalized, and colocalized synaptic markers per IHC in unexposed WT, Chrna9 KO, and Chrna9L9′T KI mice (Fig. 3). Synaptic counts were lower in Chrna9 KO mice compared with WT mice in the apical cochlear end (Kruskal–Wallis test, apical: χ2 = 17.4, df = 1, p < 0.001; Fig. 3, right). However, there was a slight, but a significant increase in the number of ribbon synapses in the medial cochlear region (Kruskal–Wallis test; medial: χ2 = 6.1, df = 1, p = 0.01; Fig. 3, right). It is important to note that synaptic counts showed the same spatial distribution in Chrna9 KO mice compared with WT mice: the number of ribbon synapses peaks in the medial region where the cochlea is most sensitive to sound (Fig. 3). Nevertheless, in Chrna9L9′T KI mice synaptic counts were comparable to those of WT mice in low- and high-frequency areas, but in the middle cochlear region a large reduction in synaptic counts of ∼15% was observed (Kruskal–Wallis test, χ2 = 19.3, df = 1, p < 0.0001; Fig. 3, right). Thus, the spatial distribution from base to apex seen in the ears of WT and Chrna9 KO mice seems to be lost in the Chrna9L9′T KI mice.
Figure 3.

Analysis of IHC ribbon synapses in unexposed WT, Chrna9 KO, and Chrna9L9′T KI mice. Quantitative data obtained from unexposed WT, Chrna9 KO, and Chrna9L9′T KI mice at P28. For each IHC, we analyzed the number of CtBP2 puncta, postsynaptic GluA2 receptor patches, and putative ribbon synapses, defined as juxtaposed CtBP2 and GluA2 positive puncta. Horizontal lines inside the boxplots represent the median and whiskers correspond to percentiles 10–90. Comparisons were made by a Kruskal–Wallis nonparametric ANOVA followed by a Dunn's post-test. Dark gray asterisks represent the statistical significance between Chrna9 KO and WT mice, and light gray asterisks between Chrna9L9′T KI and WT mice (*p < 0.05; **p < 0.01; ***p < 0.001).
The impact of noise exposure on the number of auditory nerve synapses is summarized in Figure 4). Ribbon (CtBP2) counts in exposed ears of WT mice were significantly reduced in the three cochlear regions (Fig. 4a). Reduction was more pronounced in the low-frequency (apical-end) and high-frequency (basal-end) regions with a 10% reduction compared with controls (Kruskal–Wallis test; apical: χ2 = 23.6, df = 1, p < 0.0001 and basal: χ2 = 8.9, df = 1, p = 0.002). Similarly, GluA2 postsynaptic receptors were diminished in the whole cochlea of traumatized WT mice with a bigger reduction at the apical and medial turns (Kruskal–Wallis test; apical: χ2 = 42.2, df = 1, p < 0.0001; and medial: χ2 = 5.8, df = 1, p = 0.01). Putative ribbon synapse counts, defined as juxtaposed CtBP2- and GluA2-positive puncta, showed a 10% reduction only in the apical turn after acoustic trauma in WT mice (Kruskal–Wallis test, χ2 = 26.4, df = 1, p < 0.0001; Fig. 4a, right).
Figure 4.

Analysis of the degree of IHC synaptopathy a week after AT. Left, Representative confocal images of IHC synapses from cochleae immunolabeled for presynaptic ribbons (CtBP2-red) and postsynaptic receptor patches (GluA2-green). Scale bar, 10 μm. The dashed lines show the approximate outline of one IHC. CtBP2 antibody also weakly stains IHC nuclei. Right, Quantitative data obtained from WT, Chrna9 KO, and Chrna9L9′T KI mice at P28. For each IHC, we analyzed the number of CtBP2 puncta, postsynaptic GluA2 receptor patches, and putative ribbon synapses. a, In traumatized WT mice, there was a reduction in the number of CtBP2 puncta, GluA2 receptor patches, and putative synapse, depending on cochlear frequency/location (nWTcontrol = 360 IHCs at the apical, 350 IHCs at the medial, and 185 IHCs at the basal region from 8 to 10 animals; nWTAT+7d = 394 IHCs at the apical, 492 IHCs at the medial, and 166 IHCs at the basal region from 6 to 11 animals). b, In traumatized Chrna9 KO mice, there was a reduction in the number of prelocalized, postlocalized, and colocalized puncta that was more pronounced at the basal turn (nKOcontrol = 234 IHCs at the apical, 200 IHCs at the medial, and 218 IHCs at the basal region from 6 to 8 animals; nKOAT+7d = 291 IHCs at the apical, 271 IHCs at the medial, and 130 IHCs at the basal region from 4 to 7 animals). c, In traumatized Chrna9L9′T KI mice, we found a significant increase in the number of presynaptic ribbons, postsynaptic AMPA receptors, and colocalization puncta for the three regions of the cochlea (nKIcontrol = 262 IHCs at the apical, 309 IHCs at the medial, and 135 IHCs at the basal region from 7 to 12 animals; nKIAT+7d = 179 IHCs at the apical, 406 IHCs at the medial, and 119 IHCs at the basal region from 5 to 14 animals). Horizontal lines inside the boxplots represent the median, and whiskers correspond to percentiles 10–90. Comparisons were made by a Kruskal–Wallis nonparametric ANOVA followed by a Dunn's post-test (*p < 0.05; **p < 0.01; ***p < 0.001).
In Chrna9 KO mice, there was also a reduction in the number of prelocalized, postlocalized, and colocalized puncta, depending on cochlear frequency/location after exposure to noise (Fig. 4b). Ribbon puncta in exposed ears of Chrna9 KO mice were reduced up to 20% of the control at the basal cochlear end after acoustic trauma (Kruskal–Wallis test, χ2 = 53.8, df = 1, p < 0.0001). GluA2 postsynaptic receptors were reduced by 13% in both the apical and basal turns of traumatized mice (Kruskal–Wallis test; apical: χ2 = 25.4, df = 1, p < 0.0001; and basal: χ2 = 24.4, df = 1, p < 0.0001). Synaptic puncta showed a 20% reduction only in the basal turn after acoustic trauma (Kruskal–Wallis test, χ2 = 41.7, df = 1, p < 0.0001; Fig. 4b, right).
Surprisingly, after 7 d of acoustic exposure, the higher density (∼10–15% increase) of presynaptic ribbons, postsynaptic GluA2 receptor patches, and putative synapses at all cochlear regions were observed in the ears of Chrna9L9′T KI mice (Fig. 4c; synaptic counts, Kruskal–Wallis test; apical: χ2 = 14.9, df = 1, p = 0.0001; medial: χ2 = 64.8, df = 1, p < 0.0001; and basal: χ2 = 6.7, df = 1, p = 0.009; Fig. 4c, right). These results suggest that potentiation of α9α10 nAChRs responses cannot only prevent the noise-induced functional signs of HHL and afferent synaptic loss but also might promote synapse formation after acoustic trauma.
OHC function and efferent innervation pattern after noise exposure
OHC function was assessed through DPOAEs, which can be measured from the external auditory canal (Shera and Guinan, 1999). When two tones are presented, electrical distortions are created, amplified, and conducted back into mechanical motion of the sensory epithelium at the distortion frequencies by normally functioning OHCs and can be detected by a microphone in close proximity to the tympanic membrane. As shown in Figure 1c, DPOAE responses at P21 in unexposed WT and Chrna9 KO mice were similar except at 45.25 kHz (Kruskal–Wallis test: df = 2, p = 0.004 at 45.25 kHz). However, there was a small elevation of ∼5 dB of mean DPOAE thresholds in unexposed Chrna9L9′T KI mice at some frequencies (Kruskal–Wallis test: χ2 = 7.3, df = 2, p = 0.020 at 11.33 kHz; χ2 = 5.7, df = 2, p = 0.004 at 16 kHz; and χ2 = 10.8, df = 2, p = 0.030 at 22.65 kHz; Fig. 1c). Threshold elevations in Chrna9L9′T KI mice were similar in magnitude whether measured by ABRs (Fig. 1a) or DPOAEs (Fig. 1c), as already reported by Taranda et al., 2009. Thus, compared with Chrna9 KO mice, in which baseline cochlear thresholds were normal, elevated thresholds in unexposed Chrna9L9′T KI mice suggest that they may arise from the enhancement of cholinergic activity on OHCs, revealing cholinergic MOC effects under resting conditions. However, we cannot disregard that unexposed Chrna9L9′T KI mice have fewer afferent synapses per IHC at the medial cochlear region (Fig. 3), and this may also contribute to generating higher auditory thresholds in these mice.
In ears of WT mice, with normal MOC feedback, exposure to loud sounds produced DPOAE threshold shifts from 5 to 20 dB 1 d after acoustic trauma at some frequencies (Friedman test: df = 2, p = 0.019 at 11.33 kHz; p = 0.012 at 16 kHz; p = 0.001 at 22.65 kHz; and p = 0.012 at 32 kHz) that returned to almost normal at day 7 (Friedman test: df = 2, p > 0.05 at all the frequencies tested; Fig. 5, left). In contrast, in Chrna9 KO mice, which are functionally de-efferented, noise exposure produced DPOAE threshold shifts from 5 to 35 dB 1 d after trauma at some frequencies (Friedman test: df = 2, p = 0.022 at 11.33 kHz; p = 0.003 at 16 kHz; p = 0.017 at 22.65 kHz; p = 0.014 at 32 kHz; and p = 0.014 at 45.25 kHz). DPOAE thresholds remained elevated for at least 7 d after exposure (Friedman test: df = 2, p = 0.009 at 11.33 kHz; p = 0.009 at 16 kHz; p = 0.030 at 22.65 kHz; p = 0.014 at 32 kHz; and p = 0.043 at 45.25 kHz; Fig. 5, middle). Remarkably, there were no changes in DPOAE thresholds after acoustic trauma in Chrna9L9′T KI mice with enhanced MOC feedback (Friedman test: df = 2, p > 0.05 at all the frequencies tested; Fig. 5, right). These results show that the degree of functional OHC damage depends on the level of α9α10 nAChRs activity.
Figure 5.

Evaluation of OHC functional integrity before and after AT. DPOAE thresholds for WT (n = 12), Chrna9 KO (n = 13), and Chrna9L9′T KI (n = 11) mice at the same time points as in Figure 2. DPOAE thresholds showed a significant increase 1 d after AT in WT and Chrna9 KO mice, but not in Chrna9L9′T KI mice. Seven days after AT, DPOAE thresholds recovered only in WT mice, whereas DPOAE thresholds remain elevated in Chrna9 KO mice. Median and interquartile ranges are shown, and the comparisons were made by Friedman tests followed by a post hoc test. Dark gray asterisks represent the statistical significance of AT + 1d values compared with Pre-AT values, and light gray asterisks represent AT + 7d values compared with Pre-AT controls. *p < 0.05; **p < 0.01; ***p < 0.001.
To evaluate whether exposure to loud sounds can alter the distribution of MOC terminals in OHCs, a quantitative analysis of whole-mount organ of Corti immunostained for synaptophysin, an integral protein of the synaptic vesicle membrane, was performed (Fig. 6). In WT mice, MOC terminals typically occur in clusters under OHCs, along the entire cochlea from base to apex (Vetter et al., 1999). As seen in Figure 6b, left, in the middle cochlear turn, the number of terminals per OHC in WT mice ranged from 1 to 5, with most of the OHCs contacted by two to three synaptic terminals (39.8% and 41.7%, respectively). However, 7 d after acoustic trauma, there was a reduction in the number of MOC terminals per OHC with almost 40% of terminals occurring as singlets rather than as clusters. The mean number of terminals under OHCs before and 7 d after acoustic overexposure was reduced from 2.60 ± 0.02 to 1.80 ± 0.01, respectively. Thus, the distribution of MOC contacts under OHC was significantly changed after noise exposure (two-sample Kolmogorov–Smirnov test, D = 0.3, p < 0.0001).
Figure 6.
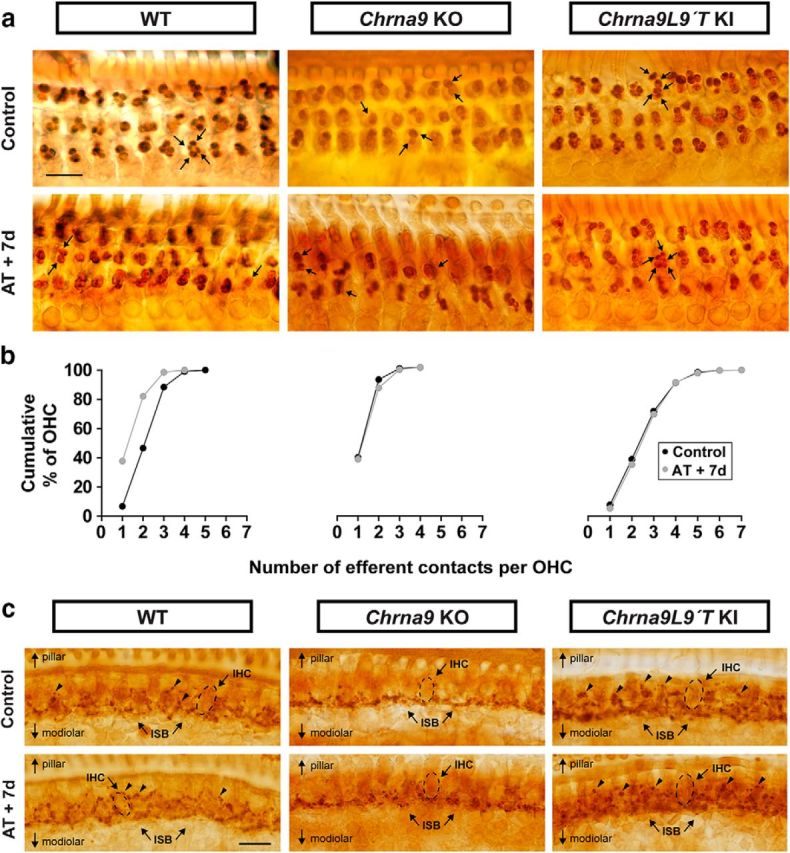
Olivocochlear synaptic boutons a week after AT. a, Synaptophysin immunostaining of the OHC region in control and traumatized WT, Chrna9 KO, and Chrna9L9′T KI mice at P28 reveals MOC terminals under OHCs. Scale bar, 10 μm. In WT ears, MOC terminals regularly occurred as clusters, such as the quadruplet, which is indicated by four arrows. After AT, there was a reduction in the number of MOC terminals per OHC. In Chrna9 KO mice, most terminals occurred as a singlet or doublet, which is indicated by one or two arrows, respectively. After AT, there was no alteration in the number of efferent contacts. In Chrna9L9′T KI mice, each OHC is contacted by a larger than normal contingent of MOC terminals (arrows) with no modification after AT. b, Cumulative frequency histograms of the efferent innervation pattern to OHCs in control and 7 d after AT in the different genotypes (nWTcontrol = 1418 OHCs from 8 animals, nWTAT+7d = 2226 OHCs from 6 animals, nKOcontrol = 2017 OHCs from 8 animals, nKOAT+7d = 1319 from 7 animals, nKIcontrol = 2860 OHCs from 10 animals, and nKIAT+7d = 1795 OHCs from 8 animals). c, Synaptophysin immunostaining of the inner spiral bundle (ISB) region in control and traumatized WT, Chrna9 KO, and Chrna9L9′T KI mice. Scale bar, 10 μm. The dashed lines show the approximate outline of one IHC. WT mice exhibit a regular progression of efferent terminals along the modiolar side of the IHCs (ISB; arrows). These boutons are larger than those on the pillar side of the IHC (arrowheads). After AT, there was no alteration in the efferent innervation pattern. In Chrna9 KO mice, there was a reduction in efferent synapses on the pillar side that was not modified after AT. In Chrna9L9′T KI mice, there was an increase of efferent terminals on the pillar side with no modification after AT.
As previously reported (Vetter et al., 1999), quantification of MOC endings in cochlear sections of Chrna9 KO mice revealed abnormalities in the number of terminals. Most OHCs were contacted by one to two terminals (39.3% and 52.4%, respectively), and very few by >2. After acoustic trauma, there were no changes in the pattern of labeled terminals in Chrna9 KO mice (Fig. 6a,b, middle panels). In contrast, the Chrna9L9′T KI gain-of-function mouse model already showed a basal increase in the mean number of efferent terminals per OHC, with some cells contacted by as many as seven terminals (Fig. 6a, right). Previous studies in Chrna9L9′T KI mice have shown the same increase in the mean number of efferent terminals per OHC (Murthy et al., 2009). Notably, no changes were observed in the number and distribution of efferent terminals under OHCs in the Chrna9L9′T KI mice after exposure to loud sounds (Fig. 6a,b, right panels). Altogether, these observations suggest that α9α10 nAChR activity influences olivocochlear presynaptic terminal differentiation required for proper synapse assembly.
Although healthy adult IHCs lack MOC innervation, it has been suggested recently that MOC contacts return to IHCs in the damaged and aged cochlea (Ruel et al., 2007; Lauer et al., 2012; Zachary and Fuchs, 2015). To determine whether the present noise exposure protocol can produce changes in MOC synapses on IHCs, we analyzed synaptophysin-stained cochlea in the area of the IHCs. Figure 6c shows light microscopy micrographs of the inner spiral bundle that reveal the olivocochlear innervation. Synaptophysin labels both MOC and LOC fibers, which cannot be differentiated. As previously demonstrated by Vetter et al., 1999, in unexposed WT mice the spiraling plexus of efferent terminals consist of a dense matrix at levels well below the IHC base, on the modiolar side, where terminals contact dendrites of auditory nerve fibers. Moreover, a sporadic plexus of terminals positioned around the IHC soma on the pillar side was observed (Fig. 6c, top left, arrowheads). Seven days after noise exposure, there was no apparent change in the efferent terminals of the IHC area (Fig. 6c, bottom left). Thus, similar to unexposed ears, the dense matrix of efferent terminals at the base of the IHCs showed no alteration and a much sparser innervation in the IHC somata region was observed. Interestingly, as previously shown (Vetter et al., 1999), under control conditions the population of efferent terminals on the pillar side of IHCs appears to be absent in Chrna9 KO mice, whereas the terminals on the modiolar side were not modified (Fig. 6c, top middle). After acoustic trauma, there was no overt change in the pattern of labeled terminals in Chrna9 KO mice (Fig. 6c, bottom middle). Finally, in the organ of Corti of unexposed Chrna9L9′T KI mice, synaptophysin immunostaining revealed an intense cholinergic innervation (Fig. 6c, top right) with an apparent increase in the number of terminals in the pillar side. Furthermore, efferent terminals appeared disorganized compared with WT mice. Seven days after acoustic overexposure, there was no overt change in the pattern of efferent innervation in Chrna9L9′T KI mice (Fig. 6c, bottom right).
The reduction in the number of efferent contacts in WT OHCs after noise exposure (Fig. 6a,b, left panels) led us to analyze whether further de-efferentation would occur when applying a second noise challenge of 1 h at 100 dB SPL, which could render the organ of Corti totally devoid of efferent feedback. At P28 (i.e., 7 d after the first noise exposure), we exposed WT mice again and tested their cochlear function the next day and 7 d after. This second exposure caused the same ABR threshold elevations 1 d after acoustic trauma (Friedman test: df = 4, p = 0.0375 at 11.33 kHz; p = 0.016 at 16 kHz; p = 0.049 at 22.65 kHz; p = 0.009 at 32 kHz; and p = 0.022 at 45.25 kHz), which completely recovered to pre-exposure values by 7 d after the second noise challenge (Friedman test: df = 4, p > 0.05 at all the frequencies tested; Fig. 7a). The distribution pattern of MOC terminals in the OHC area, analyzed by synaptophysin immunostaining, showed no further de-efferentation (Fig. 7b). The mean numbers of MOC terminals under OHCs after the first and second acoustic overexposures were 1.80 ± 0.01 and 2.09 ± 0.01, respectively. These results suggest that with a consecutive noise exposure there is no additional degeneration of MOC terminals and that their cochlear sensitivity was not altered.
Figure 7.

Auditory function and OHC connectivity after a second noise exposure in WT mice. Seven days after AT, WT mice were traumatized again with the same acoustic protocol (n = 6 animals). a, ABR thresholds of WT control, 1 d after AT (AT + 1d), AT + 7d, 1 d after a second noise exposure (2AT + 1d), and 7 d after the second trauma (2AT + 7d). There were large threshold elevations both the day after the first and the second AT with a complete recovery 7 d after either acoustic overexposure. Median and interquartile ranges are shown, and the comparisons were made by Friedman tests followed by a post hoc test. Gray asterisks represent the statistical significance of AT + 7d values compared with 2AT + 1d values (*p < 0.05; **p < 0.01). b, Cumulative frequency histograms of MOC innervation patterns after the first and the second noise exposure. Data corresponding to P21 control mice (Ctrl P21) and AT + 7d are the same as shown in Figure 6. After the second noise exposure, there was not any further reduction in the number of MOC synapses to OHC. We also showed the quantification of MOC terminals in unexposed P35 control (Ctrl p35) mice and found no difference compared with unexposed ctrl P28 mice.
Discussion
HHL was recently described as an auditory neuropathy believed to contribute to perceptual abnormalities, including tinnitus, hyperacusis, and speech discrimination, in noisy environments (Kujawa and Liberman, 2009; Schaette and McAlpine, 2011). The causative contribution of cochlear synaptopathy to HHL has become the most suitable explanation since in the noise-exposed and/or aging ear significant IHC de-afferentation takes place well before elevation of auditory thresholds (Kujawa and Liberman, 2009). Here we show an inverse correlation between the activity of the α9α10 nAChRs and the HHL phenotype, which is suggestive of the critical role of the MOC system in cochlear synaptopathy.
Until now, the delivery of neurotrophins appeared as the only way of triggering the repair of noise-induced damage to cochlear synapses (Wan et al., 2014; Suzuki et al., 2016). Our study shows that the enhancement of the MOC feedback can counteract noise-induced cochlear synaptopathy and the loss of MOC terminals. Thus, the potentiation of α9α10 nAChR-mediated responses by a pharmacologically positive modulator could have a potential therapeutic use in the prevention or treatment of HHL.
Noise-induced cochlear synaptopathy and MOC feedback
Noise-induced cochlear synaptopathy was initially described in adult mice with up to 40% of synaptic loss (Kujawa and Liberman, 2009) and afterward validated in guinea pigs with a smaller reduction of 30% (Furman et al., 2013). A more recent study (Jensen et al., 2015) in mice revealed a similar degree of cochlear synaptopathy at 6 weeks of age. Similarly, in aging mice, there is a 25–30% loss of cochlear nerve synapses, well before there is any loss of hair cells or significant threshold elevation (Sergeyenko et al., 2013). Our work shows noise-induced cochlear synaptopathy at P21 (i.e., just at the early onset of puberty and also an early stage of the development of the hearing system). The smaller reduction in our ribbon synapses counts (up to 20%) compared with that described by Kujawa and Liberman (2009) can derive from differences in mice strains and/or from a reduced time of the noise exposure protocol. Although most studies have been performed in rodents, a recent work has described noise-induced loss of ribbon synapses in nonhuman primates (Valero et al., 2017). Several lines of evidence indicate that humans are less vulnerable to noise injury (Dobie and Humes, 2017). Nonetheless, emerging information in humans show that auditory nerve fibers are more susceptible than hair cells, as in rodents (Viana et al., 2015). Together, these results suggest that cochlear synaptopathy is a general phenomenon that occurs in different species and at different stages of development and might be a more important factor in noise-induced and age-related hearing loss than previously valued.
It has been proposed that the MOC reflex in adults controls the dynamic range of hearing (Guinan, 1996), improves signal detection in background noise (Kawase et al., 1993), is involved in selective attention (Delano et al., 2007), and protects from acoustic injury (Liberman, 1991). Before hearing onset, MOC neurons establish transient synapses on IHC somata mediated by α9α10 nAChRs (Glowatzki and Fuchs, 2000; Katz et al., 2004; Gómez-Casati et al., 2005). This transient MOC activity modulates the temporal fine structure of spontaneous activity and plays a role in the maturation of the IHC synaptic machinery and central synapse formation (Glowatzki and Fuchs, 2002; Johnson et al., 2013; Clause et al., 2014). However, the importance of the MOC system on the maintenance of cochlear nerve synapses after damage has not been deeply studied. Recent work in mice with cochlear de-efferentation induced by surgical lesion, showed a dramatic loss of ribbon synapses after exposure to noise (Maison et al., 2013). Although the surgery spares most of the LOC neurons, some of them can be affected. The present work provides evidence that MOC-mediated protection of cochlear synaptopathy occurs via the α9α10 nAChR complexes on OHCs. However, a developmental effect of an altered α9α10 nAChR activity on the susceptibility to damage in IHCs and auditory nerve fibers cannot be precluded. The magnitude of cochlear synaptopathy is inversely correlated with the activity of α9α10 nAChRs: high in Chrna9 KO mice and undetectable in Chrna9L9′T KI mice. Previous work with Chrna9L9′T KI mice showed that increasing the magnitude of MOC effects rendered mice more resistant to acoustic trauma (Taranda et al., 2009). This is consistent with studies showing that overexpressing the α9 channels also increased the resistance of the ear to noise injury (Maison et al., 2002). We now show that the enhanced α9α10 nAChR activity also prevents cochlear synaptopathy and points to the importance of the integrity of MOC synapses as a feedback pathway to protect the inner ear from everyday acoustic environments.
The reduction in synaptic counts can account for the decrease in neural response amplitudes after acoustic trauma in WT and Chrna9 KO mice. A decrease in the number of ribbon synapses could reduce the amount of synchronous EPSPs in response to sounds altering the amplitude of ABR peak 1 (Kiang et al., 1976; Kujawa and Liberman, 2009). It will be interesting to investigate whether the remaining synapses are equally capable of releasing glutamate as before trauma. Similarly, the lack of reduction in ABR amplitudes in Chrna9L9′T KI mice correlates with the absence of synaptic loss after acoustic overexposure, indicating that the enhancement of the MOC reflex prevents the neuronal loss.
The observation in Chrna9L9′T KI mice of an increase in synaptic counts after noise exposure, is intriguing. In the ears of unexposed WT mice, synaptic counts show a spatial distribution from base to apex: the number of ribbon synapses per IHC peaks at the medial region, where the cochlea is most sensitive to sound (Meyer et al., 2009). However, in Chrna9L9′T KI mice, this distribution is lost: synaptic counts were comparable to those in WT mice at low- and high-frequency regions, except in the middle turn where a significant reduction was observed. One possibility to explain the abnormal cochlear distribution of ribbon synapses in the Chrna9L9′T KI mouse model is that before the onset of hearing, the enhanced transient MOC activity could modify the pattern of spontaneous IHC firing, leading to an alteration in the definitive number of auditory nerve synapses. Nevertheless, after acoustic trauma there was an increase in synaptic puncta across the cochlea, suggesting that the enhancement of MOC activity together with sound overexposure prompted new afferent synapse formation. An alternative explanation may be that the cochlea already contains synaptic contacts, but without presynaptic and postsynaptic specializations necessary for neurotransmission, which then gain function following noise trauma. The functionality of these new synapses is still an open question, since there were no changes in ABR peak 1 amplitudes in Chrna9L9′T KI mice after acoustic trauma. Currently, we can speculate that acoustic overexposure in this gain-of-function mouse model could lead to longer lasting increases in intracellular calcium concentration (Wedemeyer et al., 2018), serving as a second messenger to increase the expression of, for example, neurotrophic factors, which might finally allow the formation of new synapses. Together, the present results show for the first time that potentiation of the MOC system not only strengthens cochlear suppression in vivo, but also triggers cellular and molecular pathways that protect and/or repair the inner ear sensory epithelium after injury.
OHCs function after noise exposure and MOC feedback
DPOAE threshold measurements show the same inverse correlation between the strength of MOC activity and the degree of acoustic damage. WT mouse ears presented a transient elevation after acoustic trauma, indicating a full recovery of OHCs function throughout the dynamic range of the ear. In contrast, Chrna9 KO mice thresholds remain elevated, suggesting OHC dysfunction after noise overexposure. It is interesting to note that after acoustic trauma we did not observe any loss of OHCs in the Chrna9 KO mice, suggesting that the elevation of DPOAE thresholds is not caused by the death of OHCs. Instead, it could indicate that acoustic overexposure without a functional inhibitory MOC reflex can alter the OHC electromotility by changing the mechanotransduction and/or prestin properties. It remains to be shown whether OHCs are more prone to degeneration in Chrna9 KO mice more than a week after exposure. In agreement with our observations, it has been shown that cochlear de-efferentation produces permanent DPOAE threshold elevations without or with minimal OHC loss in aged and noise-exposed mice, respectively (Maison et al., 2013; Liberman et al., 2014). Conversely, DPOAE thresholds in Chrna9L9′T KI mice were not modified with the same noise protocol, showing that enhanced MOC activity can protect OHC damage after acoustic overexposure.
Noise-induced degeneration of MOC terminals
The present work shows for the first time that noise exposure leading to temporary threshold shifts and cochlear synaptopathy also causes a reduction of MOC terminals to OHCs. There is a sparse literature dealing with the degeneration of MOC terminals after exposure to noise. Only a few studies have shown acute damage to efferent nerve endings immediately following noise exposure, but they used an acoustic trauma protocol that causes permanent auditory threshold elevations concomitantly with the loss of OHCs in some cochlear regions (Omata et al., 1992; Canlon et al., 1999). It has been suggested that the degeneration of MOC neurons could be a contributing factor to age-related hearing loss (Liberman et al., 2014; Chumak et al., 2016). However, recent work by Lauer (2017) with Chrna9 KO mice did not find accelerated onset of hearing loss up to 15 months of age. Considering that MOC neurons regulate several aspects of auditory processing like the dynamic range of hearing and detection of relevant auditory signals in background noise (Maison et al., 2001; Guinan, 2011), we propose that the interruption in synaptic communication between MOC terminals and OHCs after acoustic trauma contributes, together with cochlear synaptopathy, to the reported symptoms of HHL.
Footnotes
This research was supported in part by Agencia Nacional de Promoción Científica y Técnica (Argentina) to J.D.G. and M.E.G.-C., Pew Charitable Trust (USA) to M.E.G.-C., National Organization for Hearing Research (USA) to M.E.G.-C. and J.D.G. and NIH Grant R01-DC-001508 (Paul A. Fuchs, Department of Otolaryngology-Head and Neck Surgery, the Center for Hearing and Balance and the Center for Sensory Biology, Institute for Basic Biomedical Sciences, The Johns Hopkins University School of Medicine, Baltimore, 21205 MD, and A.B.E.).
The authors declare no competing financial interests.
References
- Antoli-Candela FJ, Kiang NYS (1978) Unit activity underlying the N1 potential. In: Evoked electrical activity in the auditory nervous system (Naunton R, Fernandez C, eds), pp 165–191. New York, NY: Academic. [Google Scholar]
- Ballestero J, Zorrilla de San Martín J, Goutman J, Elgoyhen AB, Fuchs PA, Katz E (2011) Short-term synaptic plasticity regulates the level of olivocochlear inhibition to auditory hair cells. J Neurosci 31:14763–14774. 10.1523/JNEUROSCI.6788-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchwald JS, Huang C (1975) Far-field acoustic response: origins in the cat. Science 189:382–384. 10.1126/science.1145206 [DOI] [PubMed] [Google Scholar]
- Canlon B, Fransson A, Viberg A (1999) Medial olivocochlear efferent terminals are protected by sound conditioning. Brain Res 850:253–260. 10.1016/S0006-8993(99)02091-0 [DOI] [PubMed] [Google Scholar]
- Chumak T, Bohuslavova R, Macova I, Dodd N, Buckiova D, Fritzsch B, Syka J, Pavlinkova G (2016) Deterioration of the medial olivocochlear efferent system accelerates age-related hearing loss in Pax2-Isl1 transgenic mice. Mol Neurobiol 53:2368–2383. 10.1007/s12035-015-9215-1 [DOI] [PubMed] [Google Scholar]
- Clause A, Kim G, Sonntag M, Weisz CJ, Vetter DE, Rűbsamen R, Kandler K (2014) The precise temporal pattern of prehearing spontaneous activity is necessary for tonotopic map refinement. Neuron 82:822–835. 10.1016/j.neuron.2014.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costalupes JA. (1985) Representation of tones in noise in the responses of auditory nerve fibers in cats. I. Comparison with detection thresholds. J Neurosci 5:3261–3269. 10.1523/JNEUROSCI.05-12-03261.1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delano PH, Elgueda D, Hamame CM, Robles L (2007) Selective attention to visual stimuli reduces cochlear sensitivity in chinchillas. J Neurosci 27:4146–4153. 10.1523/JNEUROSCI.3702-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobie RA, Humes LE (2017) Commentary on the regulatory implications of noise-induced cochlear neuropathy. Int J Audiol 56:74–78. 10.1080/14992027.2016.1255359 [DOI] [PubMed] [Google Scholar]
- Elgoyhen AB, Johnson DS, Boulter J, Vetter DE, Heinemann S (1994) Alpha 9: an acetylcholine receptor with novel pharmacological properties expressed in rat cochlear hair cells. Cell 79:705–715. 10.1016/0092-8674(94)90555-X [DOI] [PubMed] [Google Scholar]
- Elgoyhen AB, Vetter DE, Katz E, Rothlin CV, Heinemann SF, Boulter J (2001) Alpha10: a determinant of nicotinic cholinergic receptor function in mammalian vestibular and cochlear mechanosensory hair cells. Proc Natl Acad Sci U S A 98:3501–3506. 10.1073/pnas.051622798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman AC, Kujawa SG, Liberman MC (2013) Noise-induced cochlear neuropathy is selective for fibers with low spontaneous rates. J Neurophysiol 110:577–586. 10.1152/jn.00164.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galambos R. (1955) Suppresion of auditory nerve activity by stimulation of efferent fibers to cochlea. J Neurophysiol 5:424–437. [DOI] [PubMed] [Google Scholar]
- Glowatzki E, Fuchs PA (2000) Cholinergic synaptic inhibition of inner hair cells in the neonatal mammalian cochlea. Science 288:2366–2368. 10.1126/science.288.5475.2366 [DOI] [PubMed] [Google Scholar]
- Glowatzki E, Fuchs PA (2002) Transmitter release at the hair cell ribbon synapse. Nat Neurosci 5:147–154. 10.1038/nn796 [DOI] [PubMed] [Google Scholar]
- Gómez-Casati ME, Fuchs PA, Elgoyhen AB, Katz E (2005) Biophysical and pharmacological characterization of nicotinic cholinergic receptors in rat cochlear inner hair cells. J Physiol 566:103–118. 10.1113/jphysiol.2005.087155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinan JJ. (1996) Physiology of olivocochlear efferents. In: The cochlea (Dallos P, Popper AN, Fay RR, eds), pp 435–502. New York: Springer. [Google Scholar]
- Guinan JJ. (2011) Physiology of the medial and lateral olivocochlear systems. In: Auditory and vestibular efferents (Ryugo DK, Fay RR, eds), pp 39–81. New York, NY: Springer; 10.1007/978-1-4419-7070-1_3 [DOI] [Google Scholar]
- Handrock M, Zeisberg J (1982) The influence of the effect system on adaptation, temporary and permanent threshold shift. Arch Otorhinolaryngol 234:191–195. 10.1007/BF00453630 [DOI] [PubMed] [Google Scholar]
- Henry KR. (1984) Noise and the young mouse: genotype modifies the sensitive period for effects on cochlear physiology and audiogenic seizures. Behav Neurosci 98:1073–1082. 10.1037/0735-7044.98.6.1073 [DOI] [PubMed] [Google Scholar]
- Jensen JB, Lysaght AC, Liberman MC, Qvortrup K, Stankovic KM (2015) Immediate and delayed cochlear neuropathy after noise exposure in pubescent mice. PLoS One 10:e0125160. 10.1371/journal.pone.0125160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Wedemeyer C, Vetter DE, Adachi R, Holley MC, Elgoyhen AB, Marcotti W (2013) Cholinergic efferent synaptic transmission regulates the maturation of auditory hair cell ribbon synapses. Open Biol 3:130163. 10.1098/rsob.130163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz E, Elgoyhen AB, Gómez-Casati ME, Knipper M, Vetter DE, Fuchs PA, Glowatzki E (2004) Developmental regulation of nicotinic synapses on cochlear inner hair cells. J Neurosci 24:7814–7820. 10.1523/JNEUROSCI.2102-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawase T, Delgutte B, Liberman MC (1993) Antimasking effects of the olivocochlear reflex. II. enhancement of auditory-nerve response to masked tones. J Neurophysiol 70:2533–2549. 10.1152/jn.1993.70.6.2533 [DOI] [PubMed] [Google Scholar]
- Khimich D, Nouvian R, Pujol R, Tom Dieck S, Egner A, Gundelfinger ED, Moser T (2005) Hair cell synaptic ribbons are essential for synchronous auditory signalling. Nature 434:889–894. 10.1038/nature03418 [DOI] [PubMed] [Google Scholar]
- Kiang NYS, Moxon E, Kahn A (1976) The relationship of gross potentials recorded from the cochlea to single unit activity in the auditory nerve. In: Electrocochleography (Ruben R, Eberling C, Solomon G, eds). Baltimore, MD: University Park. [Google Scholar]
- Kujawa SG, Liberman MC (1997) Conditioning-related protection from acoustic injury: effects of chronic deefferentation and sham surgery. J Neurophysiol 78:3095–3106. 10.1152/jn.1997.78.6.3095 [DOI] [PubMed] [Google Scholar]
- Kujawa SG, Liberman MC (2006) Acceleration of age-related hearing loss by early noise exposure: evidence of a misspent youth. J Neurosci 26:2115–2123. 10.1523/JNEUROSCI.4985-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujawa SG, Liberman MC (2009) Adding insult to injury: cochlear nerve degeneration after “temporary” noise-induced hearing loss. J Neurosci 29:14077–14085. 10.1523/JNEUROSCI.2845-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer AM. (2017) Minimal effects of age and exposure to a noisy environment on hearing in alpha9 nicotinic receptor knockout mice. Front Neurosci 11:304. 10.3389/fnins.2017.00304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer AM, Fuchs PA, Ryugo DK, Francis HW (2012) Efferent synapses return to inner hair cells in the aging cochlea. Neurobiol Aging 33:2892–2902. 10.1016/j.neurobiolaging.2012.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberman LD, Wang H, Liberman MC (2011) Opposing gradients of ribbon size and AMPA receptor expression underlie sensitivity differences among cochlear-nerve/hair-cell synapses. J Neurosci 31:801–808. 10.1523/JNEUROSCI.3389-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberman LD, Suzuki J, Liberman MC (2015) Dynamics of cochlear synaptopathy after acoustic overexposure. J Assoc Res Otolaryngol 16:205–219. 10.1007/s10162-015-0510-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberman M, Mulroy M (1982) Acute and chronic effects of acoustic trauma: Cochlear pathology and auditory nerve pathophysiology. In (Hamernik R, Henderson D, Salvi R, eds) New perspectives on noise-induced hearing loss, pp 105–136. New York, NY: Raven Press. [Google Scholar]
- Liberman MC. (1991) The olivocochlear efferent bundle and susceptibility of the inner ear to acoustic injury. J Neurophysiol 65:123–132. 10.1152/jn.1991.65.1.123 [DOI] [PubMed] [Google Scholar]
- Liberman MC, Liberman LD, Maison SF (2014) Efferent feedback slows cochlear aging. J Neurosci 34:4599–4607. 10.1523/JNEUROSCI.4923-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maison SF, Liberman MC (2000) Predicting vulnerability to acoustic injury with a noninvasive assay of olivocochlear reflex strength. J Neurosci 20:4701–4707. 10.1523/JNEUROSCI.20-12-04701.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maison SF, Luebke AE, Liberman MC, Zuo J (2002) Efferent protection from acoustic injury is mediated via alpha9 nicotinic acetylcholine receptors on outer hair cells. J Neurosci 22:10838–10846. 10.1523/JNEUROSCI.22-24-10838.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maison SF, Adams JC, Liberman MC (2003) Olivocochlear innervation in the mouse: immunocytochemical maps, crossed versus uncrossed contributions, and transmitter colocalization. J Comp Neurol 455:406–416. 10.1002/cne.10490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maison SF, Usubuchi H, Liberman MC (2013) Efferent feedback minimizes cochlear neuropathy from moderate noise exposure. J Neurosci 33:5542–5552. 10.1523/JNEUROSCI.5027-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maison S, Micheyl C, Collet L (2001) Influence of focused auditory attention on cochlear activity in humans. Psychophysiology 38:35–40. 10.1111/1469-8986.3810035 [DOI] [PubMed] [Google Scholar]
- Matsubara A, Laake JH, Davanger S, Usami S, Ottersen OP (1996) Organization of AMPA receptor subunits at a glutamate synapse: a quantitative Immunogold analysis of hair cell synapses in the rat organ of Corti. J Neurosci 16:4457–4467. 10.1523/JNEUROSCI.16-14-04457.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer AC, Frank T, Khimich D, Hoch G, Riedel D, Chapochnikov NM, Yarin YM, Harke B, Hell SW, Egner A, Moser T (2009) Tuning of synapse number, structure and function in the cochlea. Nat Neurosci 12:444–453. 10.1038/nn.2293 [DOI] [PubMed] [Google Scholar]
- Murthy V, Taranda J, Elgoyhen AB, Vetter DE (2009) Activity of nAChRs containing alpha9 subunits modulates synapse stabilization via bidirectional signaling programs. Dev Neurobiol 69:931–949. 10.1002/dneu.20753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlemiller KK, Wright JS, Heidbreder AF (2000) Vulnerability to noise-induced hearing loss in “middle-aged” and young adult mice: a dose–response approach in CBA, C57BL, and BALB inbred strains. Hear Res 149:239–247. 10.1016/S0378-5955(00)00191-X [DOI] [PubMed] [Google Scholar]
- Omata T, Omata E, Wilhelms HJ, Schätzle W (1992) Neural and infranuclear region changes in outer hair cells in acoustically exposed rabbits. Eur Arch Otorhinolaryngol 249:287–292. 10.1007/BF00714495 [DOI] [PubMed] [Google Scholar]
- Pohlert T. (2014) The pairwise multiple comparison of mean ranks package (PMCMR). Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Pujol R, Puel JL (1999) Excitotoxicity, synaptic repair, and functional recovery in the mammalian cochlea: a review of recent findings. Ann N Y Acad Sci 884:249–254. 10.1111/j.1749-6632.1999.tb08646.x [DOI] [PubMed] [Google Scholar]
- Pujol R, Puel JL, Gervais d'Aldin C, Eybalin M (1993) Pathophysiology of the glutamatergic synapses in the cochlea. Acta Otolaryngol 113:330–334. 10.3109/00016489309135819 [DOI] [PubMed] [Google Scholar]
- Reiter ER, Liberman MC (1995) Efferent-mediated protection from acoustic overexposure: relation to slow effects of olivocochlear stimulation. J Neurophysiol 73:506–514. 10.1152/jn.1995.73.2.506 [DOI] [PubMed] [Google Scholar]
- Ruel J, Wang J, Rebillard G, Eybalin M, Lloyd R, Pujol R, Puel JL (2007) Physiology, pharmacology and plasticity at the inner hair cell synaptic complex. Hear Res 227:19–27. 10.1016/j.heares.2006.08.017 [DOI] [PubMed] [Google Scholar]
- Schaette R, McAlpine D (2011) Tinnitus with a normal audiogram: physiological evidence for hidden hearing loss and computational model. J Neurosci 31:13452–13457. 10.1523/JNEUROSCI.2156-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeyenko Y, Lall K, Liberman MC, Kujawa SG (2013) Age-related cochlear synaptopathy: an early-onset contributor to auditory functional decline. J Neurosci 33:13686–13694. 10.1523/JNEUROSCI.1783-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shera CA, Guinan JJ Jr (1999) Evoked otoacoustic emissions arise by two fundamentally different mechanisms: a taxonomy for mammalian OAEs. J Acoust Soc Am 105:782–798. 10.1121/1.426948 [DOI] [PubMed] [Google Scholar]
- Suzuki J, Corfas G, Liberman MC (2016) Round-window delivery of neurotrophin 3 regenerates cochlear synapses after acoustic overexposure. Sci Rep 6:24907. 10.1038/srep24907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taranda J, Maison SF, Ballestero JA, Katz E, Savino J, Vetter DE, Boulter J, Liberman MC, Fuchs PA, Elgoyhen AB (2009) A point mutation in the hair cell nicotinic cholinergic receptor prolongs cochlear inhibition and enhances noise protection. PLoS Biol 7:e18. 10.1371/journal.pbio.1000018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valero MD, Burton JA, Hauser SN, Hackett TA, Ramachandran R, Liberman MC (2017) Noise-induced cochlear synaptopathy in rhesus monkeys (Macaca mulatta). Hear Res 353:213–223. 10.1016/j.heares.2017.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter DE, Liberman MC, Mann J, Barhanin J, Boulter J, Brown MC, Saffiote-Kolman J, Heinemann SF, Elgoyhen AB (1999) Role of α9 nicotinic ACh receptor subunits in the development and function of cochlear efferent innervation. Neuron 23:93–103. 10.1016/S0896-6273(00)80756-4 [DOI] [PubMed] [Google Scholar]
- Viana LM, O'Malley JT, Burgess BJ, Jones DD, Oliveira CA, Santos F, Merchant SN, Liberman LD, Liberman MC (2015) Cochlear neuropathy in human presbycusis: confocal analysis of hidden hearing loss in post-mortem tissue. Hear Res 327:78–88. 10.1016/j.heares.2015.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan G, Gómez-Casati ME, Gigliello AR, Liberman MC, Corfas G (2014) Neurotrophin-3 regulates ribbon synapse density in the cochlea and induces synapse regeneration after acoustic trauma. Elife 3:e03564. 10.7554/eLife.03564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedemeyer C, Zorrilla de San Martín J, Ballestero J, Gómez-Casati ME, Torbidoni AV, Fuchs PA, Bettler B, Elgoyhen AB, Katz E (2013) Activation of presynaptic GABA(B(1a,2)) receptors inhibits synaptic transmission at mammalian inhibitory cholinergic olivocochlear-hair cell synapses. J Neurosci 33:15477–15487. 10.1523/JNEUROSCI.2554-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedemeyer C, Vattino LG, Moglie MJ, Ballestero J, Maison SF, Di Guilmi MN, Taranda J, Liberman MC, Fuchs PA, Katz E, Elgoyhen AB (2018) A gain-of-function mutation in the α9 nicotinic acetylcholine receptor alters medial olivocochlear efferent short-term synaptic plasticity. J Neurosci 38:3939–3954. 10.1523/JNEUROSCI.2528-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederhold ML, Kiang NY (1970) Effects of electric stimulation of the crossed olivocochlear bundle on single auditory-nerve fibers in the cat. J Acoust Soc Am 48:950–965. 10.1121/1.1912234 [DOI] [PubMed] [Google Scholar]
- Young ED, Barta PE (1986) Rate responses of auditory nerve fibers to tones in noise near masked threshold. J Acoust Soc Am 79:426–442. 10.1121/1.393530 [DOI] [PubMed] [Google Scholar]
- Zachary SP, Fuchs PA (2015) Re-emergent inhibition of cochlear inner hair cells in a mouse model of hearing loss. J Neurosci 35:9701–9706. 10.1523/JNEUROSCI.0879-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]