

Abstract
Gorlin-Goltz syndrome (GGS) is an autosomal dominant disorder with a high degree of penetrance and variable expressivity. It is a rare phakomatosis characterized by multiple odontogenic keratocysts (OKCs), bifid ribs, and other abnormalities. The incidence of the GGS is estimated at 1 in 57,000–1 in 256,000 in the general population. The OKC is frequently the presenting manifestation of this syndrome. We report a case of a 25-year-old male patient, presenting with a swelling in the right side of the face which was diagnosed as GGS by correlating the clinical findings, histological findings, and evaluating the various tools of imaging. In the case of GGS, it is of great importance to make an early diagnosis since the severity of complications such as maxillofacial deformities related to the jaw cysts can be avoided.
Keywords: Gorlin-Goltz syndrome, odontogenic keratocysts, pericoronal radiolucency
Introduction
Gorlin-Goltz syndrome (GGS) is an autosomal dominant inherent condition with multisystemic involvement and variable presenting symptoms.[1] It was first reported in 1894 when Jarisch and White discovered it in a patient who posed multiple basal cell carcinomas (BCCs), scoliosis, and learning disability.[2] The fusillade of events hit a milestone in 1960 with the revelation of the classical triad (multiple basocellular epitheliomas, keratocysts in the jaws, and bifid ribs) by Robert James Gorlin and William Goltz, thereby establishing the diagnosis of this syndrome.[3] The earliest suggestion regarding the diagnostic criteria for GGS came from Evans et al. in 1993.[4] This triad was modified by Rayner et al., who established that, for giving the diagnosis, at least cysts had to appear in combination with calcification of the falx cerebri or palmar and plantar pits.[5] It was later modified by Kimonis et al. in 1997 who went on to state that the diagnosis can be established only when two major or one major and two minor criteria are present.[6]
The main characteristic features of GGS are multiple odontogenic keratocysts (OKCs) (75%), BCC (50%–97%), bifid ribs (40%), palmar and plantar pits (60%–90%), and ectopic calcification of falx cerebri (37%–79%).[7] The estimated prevalence of the ailment shuttles between 1/57,000 and 1/256,000, with a male-to-female ratio of 1:1. The clinical features arise in the first, second, or third decades of life.[2] This syndrome includes a wide spectrum of defects encompassing the skin, eyes, central nervous and endocrine system, and bones.[8] The principal cause preceding the GGS is the germline mutations of the PTCH gene.[9]
The early diagnosis of GGS is of great importance as it may even progress to a condition which may cause the maxillofacial deformities and hamper the normal functioning of different parts of our body.[7] We hereby report a case of GGS, in which the new imaging modalities of radiology emphasized our diagnosis.
Case Report
A male patient of 25 years old came to our dental outpatient department with a chief complaint of swelling in the right face region since 2–3 months and reduced mouth opening since 10–12 days. Swelling slowly progressed into the present size which caused facial asymmetry and the reduction in mouth opening hindered the masticatory functions.
On general examination, the patient was moderately built and nourished and all the vital signs were normal. On extraoral examination, there was a diffuse swelling involving right lower and middle one-third of the face, which have caused the facial asymmetry [Figure 1]. On palpation, the swelling was firm in consistency, nontender, and there was no local rise in temperature. On intraoral examination, mouth opening was reduced to 21 mm. Multiple missing teeth in relation to 13, 22, 23, 33, 37, 43, 44, and 48 and multiple retained deciduous teeth in relation to 62, 63, 83, and 84 [Figure 2] were present which put us in a diagnostic dilemma.
Figure 1.

Diffuse swelling involving right lower and middle one-third of the face
Figure 2.

Intraoral figure showing multiple retained deciduous teeth (black arrow)
Based on the clinical findings of clinically missing 48 and reduced mouth opening, we have given a provisional diagnosis of infected dentigerous cyst secondary to impacted 48. Moreover, as a differential diagnosis, OKC was given.
Panoramic radiograph [Figure 3] and reformatted panoramic cone-beam computed tomography (CT) image of maxilla and mandible shows multiple impacted teeth with the pericoranal radiolucency, with a displacement of impacted teeth and adjacent roots of erupted teeth. Retained deciduous canine and first molar were noted in the maxillary left and mandibular right quadrants [Figure 4]. There were no cortical plate expansions in relation to mandible and maxilla, although there was perforation of lingual cortical plate in the right mandibular posterior region; buccal and palatal cortical plate perforation in the right maxillary posterior region [Figure 5]. Reformatted panoramic image of left ramus shows inverted and superiorly displaced mandibular left third molar with pericoronal radiolucency [Figure 6].
Figure 3.

Panoramic radiograph showing multiple impacted teeth and multiple unilocular as well as multilocular well-defined radiolucencies
Figure 4.

Cone-beam computed tomography–reformatted panoramic image
Figure 5.

Axial section of maxilla and mandible
Figure 6.

Coronal section through the mandible and left reformatted ramus view
As multiple jaw cysts were present we were of a suspicion whether this is associated with a syndrome; hence, further investigations were carried out, which revealed the presence of bifid rib [Figure 7] in AP chest view and bridging of sella turcica in lateral skull view [Figure 8]. CT brain was advised to rule out calcification in the falx cerebri, which revealed the absence of any calcification. Thus, we arrived at a final diagnosis of GGS.
Figure 7.
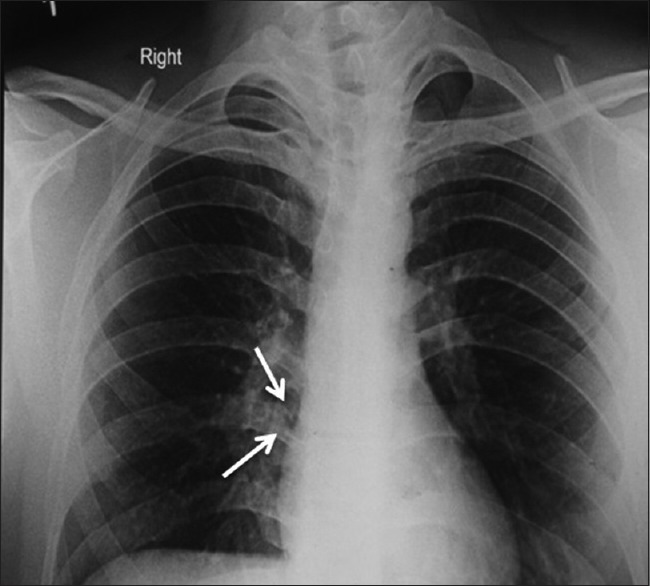
Presence of bifid rib in anteroposterior chest view (white arrow)
Figure 8.

Bridging of sella tursica in lateral skull view
The enucleation of the jaw cysts in the maxillary and mandibular region was done [Figure 9]. Histological examination revealed epithelial proliferation of the stratified squamous epithelium and luminal proliferation. Multiple daughter cysts are present. ×40 magnification revealed the presence of parakeratinized epithelium, with an increased number of cell thickness of 8–12 layers. Tombstone appearance of the basal cell layers was also seen [Figures 10-12]. All these findings revealed the presence of multiple OKC which emphasized our diagnosis of GGS.
Figure 9.

Intraoperative picture of enucleation of cyst
Figure 10.

Stratified squamous epithelium, multiple daughter cysts
Figure 12.

Tombstone appearance of the basal cell layers
Figure 11.

8–12 layers of parakeratinised epithelium
Discussion
GGS, is an autosomal dominant disorder characterized by a predisposition to neoplasms and other developmental abnormalities.[10] The synonyms of GGS are Nevoid BCC syndrome (NBCCS), Gorlin syndrome, basal cell nevus syndrome, and fifth phacomatosis.[4] There is evidence showing that this syndrome existed during times of Egyptian Dynasty, as findings compatible with the syndrome were found in mummies dating back to 1000 BC.[9]
Incidence of GGS has been estimated to be 1 in 50,000–150,000 in general population but may vary with region. In the United Kingdom, Evans et al. reported the prevalence about 1 in 150,000 in the year 1993. Shanley et al. in 1994 estimated the prevalence of 1 in 164,000 in Australia. In Italy, incidence found to be 1 in 256,000 (Lo Muzio et al. in 1999), whereas in North West of England, it was found to be1 in 55,600. Least incidence was found in Korea (Ahn et al. in 2004) about 1 in 13,939,393. Though there is difference in the reported incidence of GGS in different population of the world, all the data indicate that it is a very rare syndrome.[4]
The principal cause preceding the GGS is the germline mutations of the PTCH gene.[9] This hereditary condition is caused by mutations in the PTCH1 (Patched1) gene which is mapped to the long arm of chromosome 9q22.3-q31.[10] In about 50% of NBCCS patients, allelic losses including this site have been reported.[4] The evaluation of the data extracted from the studies shows that the product of this gene acts as a tumor suppressor.[10] This reveals the capability of these genes to alter the fundamental functioning that controls growth and development of normal tissues.[4] In 1996, the human homolog of the Drosophila PTCH1 gene was isolated simultaneously in Australia and in the USA.[4]
One of the prominent features of 75% of GGS cases is OKC that is often the first sign in these instances. The male-to-female ratio of OKC associated with this syndrome is 1:1 with usual development in the second and third decades of life.[3] In young patients, the cysts may be associated with unerupted teeth and cause tooth displacement and root resorption. These cysts even foster the development of ameloblastoma and squamous cell carcinoma.[2] The major differences between OKCs associated with GGS and solitary OKCs are listed in Table 1.[11]
Table 1.
Difference between Syndromic and solitary odontogenic keratocysts

Some of the other clinical manifestations associated with the GGS are cutaneous anomalies, skeletal anomalies, dental anomalies, cardiac anomalies, craniofacial anomalies, neurological anomalies, sexual anomalies, and ophthalmic anomalies.[2] Among all these, the most commonly presenting manifestations are basal cell nevus/carcinoma, palmar and plantar pitting and keratosis, multiple OKC, impacted teeth or agenesis, calcification of falx cerebra, bridging of sella turcica, bifid ribs, syndactyly, hypertelorism, etc.[7]
The early diagnosis of this syndrome is of great importance to hinder the further progression to threatening conditions. For the same, the diagnosis criteria put forward by Evans et al. and modified by Kimonis et al. in 1973, forms a milestone.[4,6] According to them, diagnosis of GGS can be established when two major or one major and two minor are present which are described below:[12]
Major criteria are as follows:
More than 2 BCCs or one under age of 20 years
OKC
Three or more palmar pits
Bilamellar calcification of falx cerebri
Bifid, fused, or splayed ribs
First-degree relative with NBCCS.
Minor criteria are as follows:
Macrocephaly adjusted for height
Fontal bossing, cleft lip/palate, and hypertelorism
Sprengel deformity, pectus, and syndactyly of digits
Bridging of sella turcica, hemivertebrae, and flame-shaped radiolucencies
Ovarian fibroma
Medulloblastoma.
The presence of two major criteria (multiple OKCs and bifid ribs) and minor criteria (bridging of sella turcica) emphasized our diagnosis.[11] The usual radiological features associated with the GGS are multiple OKCs, bilamellar calcification of the falx cerebri, rib anomalies such as bifid rib, fused, splayed, bridging of the sella turcica,[13] hemivertebrae, and flame-shaped osseous radiolucencies.
OKCs often present as the first diagnostic clue of this syndrome. Clinically, these lesions show an aggressive growth and they have high tendency to recur after surgical treatment. The increased mitotic activity of the epithelial cells in the basal layer, its potential for budding, and the presence of daughter cysts in the wall, all these form the characteristic histological feature of this syndrome.[4] The presence of daughter cysts was found to be one of the reasons for the recurrence of OKC.
The major histopathological features of this syndrome are (Woolgar et al. and Dominiguez et al.)[14]
More number of satellite cysts
Solid islands of epithelial proliferation
Intramural epithelial remnants
Odontogenic rests within the capsule
Increased parakeratinization
Mitotic figures in the epithelium
Shorter epithelial height and smaller nuclei when compared with solitary OKCs.
Investigations
Investigation protocol suggested by Muzio[2]
Family history – Past medical and dental history
Clinical examinations – Oral, skin, central nervous system, head circumference, interpupillary distance, eyes, genitourinary system, cardiovascular system, respiratory system, and skeletal system
Genetic testing
Radiographs – Chest, anteroposterior (AP) and lateral skull, panoramic radiograph, cervical and thoracic spine (AP and lateral), hands (for pseudocysts), pelvic (female), ovarian ultrasound (female) for ovarian fibroma, and echocardiogram (children) for cardiac fibroma.
However, confirmation is by ultrasound and DNA analysis.
Laboratory findings which show notable changes in the syndrome are increased serum uric acid level (3%), increased levels of alkaline phosphate, and cyclic adenosine monophosphate.[12] Ellsworth Howard test (absence of significant phosphorus diuresis after an intravenous injection of the hormone) confirms the GGS.[15]
Mainly two treatment approaches are carried out for the treatment of multiple OKCs associated with this syndrome. They are conservative methods, i.e. simple enucleation with or without curettage and marsupialization and aggressive method peripheral ostectomy, chemical curettage with Carnoy's solution, and resection.[9]
Conclusion
GGS is a rare autosomal dominant disorder that involves multiple organ systems including the skin, skeleton, and jaws. Knowledge of the maxillofacial manifestations is important for early diagnosis of this syndrome so that optimal treatment can be rendered and thus the progression to a threatening condition can be curbed.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Agrawal A, Murari A, Vutukuri S, Singh A. Gorlin-Goltz syndrome: Case report of a rare hereditary disorder. Case Rep Dent 2012. 2012 doi: 10.1155/2012/475439. 475439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ramesh M, Krishnan R, Chalakkal P, Paul G. Gorlin-Goltz syndrome: Case report and literature review. J Oral Maxillofac Pathol. 2015;19:267. doi: 10.4103/0973-029X.164557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Namdeoraoji Bahadure R, Surendraji Jain E, Badole GP. Gorlin and Goltz syndrome: A case report with surgical review. Int J Clin Pediatr Dent. 2013;6:104–8. doi: 10.5005/jp-journals-10005-1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lo Muzio L. Nevoid basal cell carcinoma syndrome (Gorlin syndrome) Orphanet J Rare Dis. 2008;3:32. doi: 10.1186/1750-1172-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kiran NK, Tilak Raj TN, Mukunda KS, Rajashekar Reddy V. Nevoid basal cell carcinoma syndrome (Gorlin-Goltz syndrome) Contemp Clin Dent. 2012;3:514–8. doi: 10.4103/0976-237X.107459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bijjaragi SC, Suragimath A, Sangle VA, Patil VS. Gorlin-goltz syndrome: A clinicopathological case report. J Indian Acad Oral Med Radiol. 2014;26:85–8. [Google Scholar]
- 7.Chandran S, Marudhamuthu K, Riaz R, Balasubramaniam S. Odontogenic keratocysts in Gorlin-Goltz syndrome: A Case report. J Int Oral Health. 2015;7(Suppl 1):76–9. [PMC free article] [PubMed] [Google Scholar]
- 8.Mehta D, Raval N, Patadiya H, Tarsariya V. Gorlin-Goltz syndrome. Ann Med Health Sci Res. 2014;4:279–82. doi: 10.4103/2141-9248.129064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Casaroto AR, Loures DC, Moreschi E, Veltrini VC, Trento CL, Gottardo VD, et al. Early diagnosis of Gorlin-Goltz syndrome: Case report. Head Face Med. 2011;7:2. doi: 10.1186/1746-160X-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mohan RP, Verma S, Agarwal N, Singh U. Gorlin-Goltz syndrome: A rare case report. BMJ Case Rep 2013. 2013:pii: bcr2013010409. doi: 10.1136/bcr-2013-010409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arshad F. Syndromic odontogenic keratocyst: A case report and review of literature. J Int Soc Prev Community Dent. 2016;6:84–8. doi: 10.4103/2231-0762.175414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kannan KS, Sundharam SB, Manikandan R. Nevoid basal cell carcinoma syndrome. Indian J Dent Res. 2006;17:50–3. doi: 10.4103/0970-9290.29891. [DOI] [PubMed] [Google Scholar]
- 13.Leonardi R, Farella M, Cobourne MT. An association between sella turcica bridging and dental transposition. Eur J Orthod. 2011;33:461–5. doi: 10.1093/ejo/cjq106. [DOI] [PubMed] [Google Scholar]
- 14.Woolgar JA, Rippin JW, Browne RM. “A comparative histological study of odontogenic keratocysts in basal cell naevus syndrome and control patients”. J Oral Pathol Med. 1987;16:75–80. doi: 10.1111/j.1600-0714.1987.tb00691.x. [DOI] [PubMed] [Google Scholar]
- 15.Wood NK, Goaz PW, editors. 5th ed. Philadelphia: Mosby; 1997. Multiple separate well-defined radiolucencies. Origins of Differential Diagnosis of Oral and Maxillofacial Lesions; pp. 382–4. [Google Scholar]