

Abstract
Epigenetic mechanisms, especially DNA methylation, are suggested to play a role in the age-of-onset in Huntington's disease (HD) based on studies on patient brains, and cellular and animal models. Methylation is tissue-specific and it is not clear how HD specific methylation in the brain correlates with the blood compartment, which represents a much more clinically accessible sample. Therefore, we explored the presence of HD specific DNA methylation patterns in whole blood on a cohort of HDM and healthy controls from Slovenia. We compared CpG site-specific DNA methylation in whole blood of 11 symptomatic and 9 pre-symptomatic HDM (HDM), and 15 healthy controls, by using bisulfite converted DNA on the Infinium® Human Methylation27 BeadChip microarray (Illumina) covering 27,578 CpG sites and 14,495 genes. Of the examined 14,495 genes, 437 were differentially methylated (p < 0.01) in pre-symptomatic HDM compared to controls, with three genes (CLDN16, DDC, NXT2) retaining statistical significance after the correction for multiple testing (false discovery rate, FDR < 0.05). Comparisons between symptomatic HDM and controls, and the comparison of symptomatic and pre-symptomatic HDM further identified 260 and 198 differentially methylated genes (p < 0.01), respectively, whereas the comparison of all HDM (symptomatic and pre-symptomatic) and healthy controls identified 326 differentially methylated genes (p < 0.01), however, none of these changes retained significance (FDR < 0.05) after the correction for multiple testing. The results of our study suggest that methylation signatures in the blood compartment are not robust enough to prove as valuable biomarkers for predicting HD progression, but recognizable changes in methylation deserve further research.
Keywords: Huntington's disease, 5mC methylation, differential DNA methylation, epigenetics, whole blood
Introduction
Huntington's disease (HD) is a fatal autosomal dominant neurodegenerative disorder manifested by progressive impairment of motor function, cognitive decline, and various psychiatric symptoms, beginning typically after 45 years of age (1). HD is a monogenic disease and is caused by a pathological CAG triplet expansion in the HTT gene coding for the Huntingtin protein. Pathological expansion lengths below 40 are not always fully penetrant, and while longer lengths show some inverse correlation with the HD age-of-onset, additional environmental factors, genetics, and epigenetics likely also play a role (2–5).
Up to date, epigenetic changes in HD have been studied predominantly in the brains of patients, as well as in cell and animal models (6). In these studies, epigenetic changes have been identified to occur both in general processes such as histone acetylation and methylation (7–9), as well as in specific epigenetic changes, such as DNA methylation (10–16). Cytosine DNA methylation (5-mC) is one of the most studied specific modifications. It typically occurs at CpG sites which are enriched in regions called CpG islands (17) and is involved in gene expression silencing and regulation of splicing. Furthermore, this tissue-specific process is thought to be one of the main mechanisms regulating tissue-specific gene expression (18, 19).
The DNA methylation studies performed so far on the brains of HD patients have not been conclusive and have suggested that while 5-mC methylation in the cortex may have minimal association with HD status, its level may nevertheless be associated with the disease age-of-onset (14). This is important as intermediate-length pathological expansions are not fully penetrant in HD and the DNA methylation status may, therefore, have some predictive value in regard to the age-of-onset. Furthermore, even in the case of larger pathological expansions, which are considered to be fully penetrant, the age-of-onset and HD progression cannot be accurately predicted from the length of CAG repeat alone due to various influencing factors (3, 20, 21). Therefore, the identification of disease-modifying genetic factors for HD presents an important priority, and biomarkers from a more easily obtainable tissue than the CNS, such as whole blood, are needed for any future clinical use.
In order to identify any HD specific epigenetic changes in whole blood, we performed a whole-genome study of DNA methylation status in peripheral blood of 11 symptomatic and 9 pre-symptomatic HD mutation carriers (HDM), and 15 healthy controls.
Materials and methods
Cohort characteristics
Neurological status of HD patients was assessed by an experienced HD neurologist using Unified Huntington's Disease Rating Scale (UHDRS) (22) using total functional capacity score. All samples were obtained in accordance with institutional and national review boards (Republic of Slovenia National Medical Ethics Committee, Permit No. 138/03/11), and written informed consent was given by the participants. All pre-symptomatic individuals and patients were confirmed as carriers of the pathological CAG triplet expansions in the HTT gene. Only one HD patient received an angiotensin II receptor blocker, with the rest being drug-naïve at the time of blood withdrawal. Thirty-five samples were included in the study. Twenty mutation carriers were different in age due to pre-symptomatic (9 samples−4 males, 5 females, age 33.6 ± 7.26; UHDRS 0-3) and symptomatic (11 samples−5 males, 6 females, age 58.0 ± 15.00; UHDRS above 50) stage of the disease and 15 healthy controls (7 males, 8 females, age 38.1 ± 11.05) with no HD family history (all listed in Supplementary Table 8).
Blood samples were taken using EDTA blood collection tubes (BD Vacutainer® Blood Collection Tube) and DNA isolation from whole blood was performed using FlexiGene DNA Kit (Qiagen GmbH, Hilden, Germany), all according to the manufacturer's protocol.
The quality of DNA was analyzed using Thermo Scientific NanoDrop 2000c Spectrophotometer (Nanodrop Technologies, USA). The starting concentration of DNA for methylation analysis was 50 ng/μL. One microliters of DNA was used for the microarray analysis.
DNA methylation profiling
Microarray methylation analysis was performed on bisulfite converted DNA using Infinium® Human Methylation27 BeadChip microarray (Illumina Inc, San Diego, California, USA), according to the manufacturer's protocol. DNA methylation datasets in matrix format were obtained by Bead Array Reader (Illumina, USA) (23).
Bioinformatics and statistical analysis
Quality control and quantile normalization were performed using Lumi package (24). Batch effect correction was assessed by ComBat tool using non-parametric empirical Bayes statistical method (25). Statistical comparison of methylation values was performed using MethyLumi package from Bioconductor v2.8 project in R statistical environment version 2.13.1. (26). Statistical comparisons were performed between all pairs of the three groups of subjects: symptomatic HDM (S), pre-symptomatic HDM (PS), and controls (K). Additionally, methylation levels of all HDM (symptomatic and pre-symptomatic) were compared against the controls. Statistical significance cut-off value was set to p < 0.01 and a false discovery rate (FDR) < 0.05 to correct for multiple testing. In order to better portray actual DNA methylation differences between groups we calculated the Δ% of the average 5-mC for each position.
Comparison of DNA methylation and transcriptomic changes in HD
In order to identify an overlap between methylation patterns and transcriptomic changes in HD in whole blood, a comparison was performed between the results of the methylation of all HDM and controls (326 genes reaching p < 0.01) and the 740 previously identified differentially expressed (DE) transcripts (p < 0.01, FDR < 0.1) in the same HD cohort (27), as well as the 15 consistent DE transcripts identified by three independent studies (27). Shared genes identified by methylation and transcriptomic comparisons were visualized using an online VENN tool (http://bioinformatics.psb.ugent.be/cgi-bin/liste/Venn/calculate_venn.htpl).
Gene phenotype, disease, and GO annotation
Top 30 differentially methylated genes from the comparison of all HDM and controls, and 12 genes with overlapping differential DNA methylation and differential expression determined previously (27), were annotated for associated phenotypes, diseases, and GO_biological process using the online PANTHER™ tool version 12.0 (28) and the ENSEMBL Biomart tool (29).
Results
Microarray methylation analysis of 27,578 CpG sites covering 14,495 genes was performed on bisulfite converted DNA from whole blood of 11 symptomatic, 9 pre-symptomatic HDM, and 15 healthy controls. Paired comparisons between pre-symptomatic HDM and controls showed 437 genes to be differentially methylated (p < 0.01), of which three (CLDN16, DDC, NXT2) retained statistical significance (FDR < 0.05) after the correction for multiple testing. Paired comparisons between symptomatic HDM and controls, and symptomatic and pre-symptomatic HDM further identified 260 and 198 differentially methylated genes (p < 0.01), respectively, however none retained statistical significance (FDR < 0.05). Similarly, while the comparison of all HDM (symptomatic and pre-symptomatic) and healthy controls identified 326 differentially methylated genes (p < 0.01), none remained significant (FDR < 0.05). The top 30 hyper- and hypo-methylated genes for each of the comparisons are shown in Figure 1 and the associated diseases/phenotypes are given in Supplementary Table 1. The complete methylation data and comparisons are provided in Supplementary Tables 3–7.
Figure 1.

Top 30 differentially methylated genes (p < 0.01) from the comparison of (A) pre-symptomatic carriers and controls, (B) symptomatic HDM and controls, (C) pre-symptomatic and symptomatic HDM, and (D) pooled HDM and controls. K, controls; PS, pre-symptomatic HDM; S, symptomatic HDM. **FDR < 0.05.
In order to identify any global links between methylation and transcriptomic changes in whole blood in HD, we performed a comparison of the 327 differential methylated genes between all HDM and controls, and 740 DE transcripts previously identified in the same cohort (27). The comparison showed 12 genes (FBXL5, S100P, PRDX1, COPS7B, SP1, SEC24C, PDIA6, USP5, GRAP, POP5, WRB, PCSK7) with differential methylation and transcription in the same HD cohort (27) (Supplementary Table 2), however, none of the overlapping genes were supported by FDR in the case of differential methylation. We also compared the differentially methylated genes and DE transcripts with the 15 consistently DE transcripts identified in independent cohorts (27), and we found an overlap in a single gene, F-box/LRR-repeat protein 5 (FBXL5), a protein involved in chaperonin-mediated protein folding and the innate immune system (Figure 2).
Figure 2.
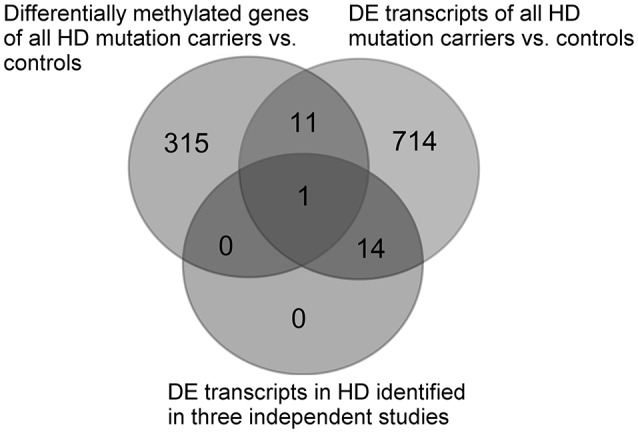
Comparison between differentially methylated genes of all HDM and controls and previously identified DE transcripts in the same cohort (27), as well as the consistently identified DE transcripts identified in whole blood by three independent HD studies (27). DE, differentially expressed.
Discussion
In our various group comparisons of CpG methylation in whole blood, we identified several hundred differentially methylated genes (p < 0.01), of which only three [claudin 16 (CLDN16), dopa decarboxylase (DDC), and nuclear transport factor 2 like export factor 2 (NXT2)] retained significance after the correction for multiple testing (FDR < 0.05) in the comparison between pre-symptomatic HDM and controls, but not in any of the other group comparisons. DDC has so far been implicated in aromatic L-amino acid decarboxylase deficiency disorder, while CLDN16 has been implicated in hypomagnesemia, hypercalciuria, and nephrocalcinosis, whereas NXT2 is involved in mRNA and protein transport, however, none of the three genes have been previously linked with HD. Additionally, we were unable to confirm differential methylation in whole blood, of the three genes previously observed as being differentially methylated in brain and other tissues: the adenosine A (2A) receptor gene (ADORA2A) (15), CCCTC-binding factor (CTCF) (14), or Huntingtin (HTT) (14, 16). This likely reflects the differences in their tissue-specific methylation and expression between blood and other tissues, as well as methodological differences in detection in the case of HTT, for which a different methylation site was examined in the original study (16).
Similarly, our comparison of the differentially methylated genes between all HDM and controls with previously analyzed DE transcripts from the same cohort (27) showed 12 overlapping genes, none of which achieved a statistically significant FDR. This may in part reflect the differences in cell populations contributing to the pool of transcripts and DNA in whole blood, however, it was an expected result given the low concordance observed between HD transcriptomic studies and the high FDR observed in the case of methylation. One of the 12 overlapping genes, F-box and leucine rich repeat protein 5 (FBXL5), was also detected as being DE in whole blood in HD in three independent transcriptomic studies (27). FBXL5 is involved in iron homeostasis and protein ubiquitination, as well as the innate immune system pathway, and may therefore be involved in the systemic response to HD; however, it has not been directly linked with HD pathology. Furthermore, of the 12 overlapping genes, the four genes (POP5, GRAP, USP5, and SEC24C) showing an expected inverse correlation between methylation and RNA expression levels are involved in RNA metabolism, the innate immune response, ubiquitination, and intracellular protein transport and antigen presentation, respectively, also supporting their involvement in systemic response to HD, however, they have not been previously linked with HD and none of them achieved FDR < 0.05 in the case of differential methylation.
Although the lack of statistical significance in our study may be the result of the relatively low number of samples and the large number of simultaneously examined CpG sites (27,578), our results are consistent with recent findings showing no change in the overall or global (not site-specific) level of 5-methylcytosine in whole blood from HD patients using an ELISA test (30), as well as the recent study where the comparison between HD and control cortex samples failed to identify statistically significant differentially methylated regions (14). Nevertheless, while 5-mC methylation in the cortex was shown to have minimal association with HD status, the study found its level to be associated with age-of-onset (14).
In other neurodegenerative disorders, where epigenetic factors are also suggested to play an important role (7, 12, 30–33), there have been mixed findings in regard to global differential methylation in whole blood. In Alzheimer's disease (AD), elevated global, not site-specific, DNA methylation has been observed in mononuclear cells from whole blood (34), and correlated with the up-regulation of plasma homocysteine. Plasma homocysteine is responsible for decreased DNA methylase activity, and is an independent risk factor for AD, known to occur at disease onset (35). Similarly, global DNA methylation in blood was shown in amyotrophic lateral sclerosis (30, 36), as well as spinocerebellar ataxias 1 and 2 (30). However, in Parkinson's disease, similarly to HD, differential DNA methylation of the alfa-synuclein gene was not confirmed in blood (37), despite being shown to be differentially methylated in brain cells of patients and healthy controls (38).
Together, the recent results including our study, indicate that whole blood is unlikely to prove robust epigenetic biomarkers predictive of HD progression and age-of-onset, however, since the ELISA approach does not enable identification of specific CpG sites, while our study has, for the first time, examined the methylation of 27,578 of the estimated 28 million distinct CpG sites present in the whole human genome, it remains possible that additional differentially methylated sites may be identified through further research on larger cohorts.
Conclusions
Our results based on the examined 27,578 CpG sites suggest that HD methylation signatures in the blood compartment are not sufficiently strong to prove as valuable biomarkers for predicting age-of-onset or HD progression, however, recognizable changes in methylation in HD deserve further research.
Author contributions
MZ and BP conceived the study. MZ and AM collected patients/controls and performed the experiments. AM and AK analyzed data and performed the bioinformatics analyses. All authors contributed substantially to the final manuscript.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge and thank all patients and controls participating in this study.
Glossary
Abbreviations
- 5-mC
cytosine DNA methylation
- AD
Alzheimer's disease
- DE
differentially expressed
- FDR
false discovery rate
- HD
Huntington's disease
- HDM
Huntington's disease mutation carriers
- UHDRS
Unified Huntington's Disease Rating Scale.
Footnotes
Funding. This work was supported by the Slovenian Research Agency (ARRS) grant no. J3-2377.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2018.00655/full#supplementary-material
References
- 1.Huntington's Disease Collaborative Research Group . A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's disease Collaborative Research Group. Cell (1993) 72:971–83. [DOI] [PubMed] [Google Scholar]
- 2.Winder JY, Roos RAC. Premanifest Huntington's disease: examination of oculomotor abnormalities in clinical practice. PLoS ONE (2018) 13:e0193866. 10.1371/journal.pone.0193866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wexler NS, Lorimer J, Porter J, Gomez F, Moskowitz C, Shackell E, et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. Proc Natl Acad Sci USA. (2004) 101:3498–503. 10.1073/pnas.0308679101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duyao M, Ambrose C, Myers R, Novelletto A, Persichetti F, Frontali M, et al. Trinucleotide repeat length instability and age of onset in Huntington's disease. Nat Genet. (1993) 4:387–92. [DOI] [PubMed] [Google Scholar]
- 5.Rubinsztein DC, Leggo J, Coles R, Almqvist E, Biancalana V, Cassiman JJ, et al. Phenotypic characterization of individuals with 30-40 CAG repeats in the Huntington disease (HD) gene reveals HD cases with 36 repeats and apparently normal elderly individuals with 36-39 repeats. Am J Hum Genet. (1996) 59:16–22. [PMC free article] [PubMed] [Google Scholar]
- 6.Thomas EA. DNA methylation in Huntington's disease: implications for transgenerational effects. Neurosci Lett. (2016) 625:34–9. 10.1016/j.neulet.2015.10.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee J, Hwang YJ, Kim KY, Kowall NW, Ryu H. Epigenetic Mechanisms of Neurodegeneration in Huntington's disease. Neurotherapeutics (2013) 10:664–76. 10.1007/s13311-013-0206-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ryu H, Lee J, Hagerty SW, Soh BY, McAlpin SE, Cormier KA, et al. ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington's disease. Proc Natl Acad Sci USA. (2006) 103:19176–81. 10.1073/pnas.0606373103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jia H, Morris CD, Williams RM, Loring JF, Thomas EA. HDAC inhibition imparts beneficial transgenerational effects in Huntington's disease mice via altered DNA and histone methylation. Proc Natl Acad Sci USA. (2015) 112:E56–64. 10.1073/pnas.1415195112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sugars KL, Rubinsztein DC. Transcriptional abnormalities in Huntington disease. Trends Genet. (2003) 19:233–8. 10.1016/S0168-9525(03)00074-X [DOI] [PubMed] [Google Scholar]
- 11.Mollica PA, Reid JA, Ogle RC, Sachs PC, Bruno RD, MacDonald ME, et al. DNA methylation leads to dna repair gene down-regulation and trinucleotide repeat expansion in patient-derived huntington disease cells. Am J Pathol. (2016) 186:971–83. 10.1016/j.ajpath.2016.03.014 [DOI] [PubMed] [Google Scholar]
- 12.Lu H, Liu X, Deng Y, Qing H. DNA methylation, a hand behind neurodegenerative diseases. Front Aging Neurosci. (2013) 5:85. 10.3389/fnagi.2013.00085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ng CW, Yildirim F, Yap YS, Dalin S, Matthews BJ, Velez PJ, et al. Extensive changes in DNA methylation are associated with expression of mutant huntingtin. Proc Natl Acad Sci USA. (2013) 110:2354–9. 10.1073/pnas.1221292110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Souza RAG, Islam SA, McEwen LM, Mathelier A, Hill A, Mah SM, et al. DNA methylation profiling in human Huntington's disease brain. Hum Mol Genet. (2016) 25:2013–30. 10.1093/hmg/ddw076 [DOI] [PubMed] [Google Scholar]
- 15.Villar-Menéndez I, Blanch M, Tyebji S, Pereira-Veiga T, Albasanz JL, Martín M, et al. Increased 5-methylcytosine and decreased 5-hydroxymethylcytosine levels are associated with reduced striatal A2AR levels in Huntington's disease. NeuroMolecular Med. (2013) 15:295–309. 10.1007/s12017-013-8219-0 [DOI] [PubMed] [Google Scholar]
- 16.Reik W, Maher ER, Morrison PJ, Harding AE, Simpson SA. Age at onset in Huntington's disease and methylation at D4S95. J Med Genet. (1993) 30:185–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones PA. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat Rev Genet. (2012) 13:484–92. 10.1038/nrg3230 [DOI] [PubMed] [Google Scholar]
- 18.Gutierrez-Arcelus M, Ongen H, Lappalainen T, Montgomery SB, Buil A, Yurovsky A, et al. Tissue-specific effects of genetic and epigenetic variation on gene regulation and splicing. PLoS Genet. (2015) 11:e1004857. 10.1371/journal.pgen.1004857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wan J, Oliver VF, Wang G, Zhu H, Zack DJ, Merbs SL, et al. Characterization of tissue-specific differential DNA methylation suggests distinct modes of positive and negative gene expression regulation. BMC Genomics (2015) 16:49. 10.1186/s12864-015-1271-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Djoussé L, Knowlton B, Hayden M, Almqvist EW, Brinkman R, Ross C, et al. Interaction of normal and expanded CAG repeat sizes influences age at onset of Huntington disease. Am J Med Genet. (2003) 119A:279–82. 10.1002/ajmg.a.20190 [DOI] [PubMed] [Google Scholar]
- 21.Gusella JF, MacDonald ME, Lee J-M. Genetic modifiers of Huntington's disease. Mov Disord. (2014) 29:1359–65. 10.1002/mds.26001 [DOI] [PubMed] [Google Scholar]
- 22.Kieburtz K. Unified Huntington's disease rating scale: reliability and consistency. Mov Disord. (1996) 11:136–42. [DOI] [PubMed] [Google Scholar]
- 23.Dunning MJ, Smith ML, Ritchie ME, Tavaré S. Beadarray: R classes and methods for Illumina bead-based data. Bioinformatics (2007) 23:2183–4. 10.1093/bioinformatics/btm311 [DOI] [PubMed] [Google Scholar]
- 24.Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illumina microarray. Bioinformatics (2008) 24:1547–8. 10.1093/bioinformatics/btn224 [DOI] [PubMed] [Google Scholar]
- 25.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics (2007) 8:118–27. 10.1093/biostatistics/kxj037 [DOI] [PubMed] [Google Scholar]
- 26.Gentlean RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. (2004) 5:R80. 10.1186/gb-2004-5-10-r80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zadel M, Maver A, Kovanda A, Peterlin B. Transcriptomic biomarkers for Huntington's disease: are gene expression signatures in whole blood reliable biomarkers? Omi J Integr Biol. (2018) 22:1–4. 10.1089/omi.2017.0206 [DOI] [PubMed] [Google Scholar]
- 28.Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, et al. PANTHER version 11: expanded annotation data from gene ontology and reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. (2017) 45:D183–9. 10.1093/nar/gkw1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, et al. Ensembl 2018. Nucleic Acids Res. (2018) 46:D754–61. 10.1093/nar/gkx1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamzeiy H, Savaş D, Tunca C, Sen NE, Gündogdu Eken A, Sahbaz I, et al. Elevated global DNA methylation is not exclusive to amyotrophic lateral sclerosis and is also observed in spinocerebellar ataxia types 1 and 2. Neurodegener Dis. (2018) 18:38–48. 10.1159/000486201 [DOI] [PubMed] [Google Scholar]
- 31.Lardenoije R, Iatrou A, Kenis G, Kompotis K, Steinbusch HWM, Mastroeni D, et al. The epigenetics of aging and neurodegeneration. Prog Neurobiol. (2015) 131:21–64. 10.1016/j.pneurobio.2015.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Christopher MA, Kyle SM, Katz DJ. Neuroepigenetic mechanisms in disease. Epigenetics Chromatin (2017) 10:47. 10.1186/s13072-017-0150-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Agrawal M, Biswas A. Molecular diagnostics of neurodegenerative disorders. Front Mol Biosci. (2015) 2:54. 10.3389/fmolb.2015.00054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Di Francesco A, Arosio B, Falconi A, Micioni Di Bonaventura MV, Karimi M, Mari D, et al. Global changes in DNA methylation in Alzheimer's disease peripheral blood mononuclear cells. Brain Behav Immun. (2015) 45:139–44. 10.1016/j.bbi.2014.11.002 [DOI] [PubMed] [Google Scholar]
- 35.Seshadri Sudha, Beisar Alexa SJ. Plasma homocysteine as a risk factor for dementia and Alzheimer's disease. N Engl J Med. (2002) 346:476–83. 10.1056/NEJMoa011613 [DOI] [PubMed] [Google Scholar]
- 36.Tremolizzo L, Messina P, Conti E, Sala G, Cecchi M, Airoldi L, et al. Whole-blood global DNA methylation is increased in amyotrophic lateral sclerosis independently of age of onset. Amyotroph Lateral Scler Front Degener. (2014) 15:98–105. 10.3109/21678421.2013.851247 [DOI] [PubMed] [Google Scholar]
- 37.Richter J, Appenzeller S, Ammerpohl O, Deuschl G, Paschen S, Brüggemann N, et al. No evidence for differential methylation of α-synuclein in leukocyte DNA of Parkinson's disease patients. Mov Disord. (2012) 27:590–1. 10.1002/mds.24907 [DOI] [PubMed] [Google Scholar]
- 38.de Boni L, Tierling S, Roeber S, Walter J, Giese A, Kretzschmar HA. Next-generation sequencing reveals regional differences of the alpha-synuclein methylation state independent of Lewy body disease. Neuromolecular Med. (2011) 13:310–20. 10.1007/s12017-011-8163-9 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.