

To the editor:
Cartilage-hair hypoplasia (CHH) is a pleiotropic autosomal recessive disorder caused by mutations in the RMRP gene. Clinical features include short stature and metaphyseal bone dysplasia, as well as a range of extraskeletal manifestations such as hypotrichosis, bone marrow dysplasia, Hirschsprung disease, increased risk of cancer, and immune defects [1, 2]. The degree of immunodeficiency is highly variable, ranging from mild to severe forms such as severe combined immunodeficiency and Omenn syndrome [2]. Defective cellular immunity is common in CHH; however, impaired humoral immune responses have also been described [2, 3]. Patients frequently suffer from recurrent infections and immune dysregulation. Reported autoimmune sequelae of CHH include autoimmune hemolytic anemia, arthritis, immune thrombocytopenic purpura, as well as autoimmune hypothyroidism and enteropathy [1, 2]. The mechanisms behind the immune deficiency and dysregulation associated with CHH are still incompletely understood. Decreased AIRE expression and absence of FOXP3+ T cells in the thymus have been implicated in the pathogenesis of Omenn syndrome in a CHH patient [4]. Cell cycle abnormalities, reduced thymic output, impaired lymphocyte proliferation, dysfunctional telomere machinery, and increased T cell apoptosis have also been associated with the immune defects seen in CHH [5, 6]. The role of autoantibody-mediated immune dysregulation, however, has not been determined.
In order to evaluate whether autoantibodies contribute to CHH-related immune dysfunction, a protein microarray was performed on CHH patients in comparison to both healthy controls and patients with autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED). APECED is an autosomal recessive disorder caused by mutations in the AIRE gene, leading to various autoimmune disorders and chronic mucocutaneous candiasis [7]. Autoantibodies are a known contributor to APECED disease pathogenesis [7]. All individuals who participated, or their guardians, signed an informed consent, and the study was approved by the Institutional Review Boards at the referring institutions. Clinical and laboratory features of the evaluated CHH patients are reported in Supplemental Table S1. A panel of IgG autoantibodies was screened using an autoantibody array (University of Texas Southwestern Medical Center, Genomic and Microarray Core Facility) as previously described with minor modifications [8, 9]. Briefly, serum samples were diluted to 1:33 and incubated with the autoantigen array. Autoantibodies binding to antigens on the array were detected with Cy3 labeled anti-IgG at a laser wavelength of 532 nm. The array was scanned with GenePix® 4400A Microarray Scanner, and images analyzed using GenePixPro 6.0 software to generate GenePix Results (GPR) files. Averaged net fluorescent intensity (NFI) of each autoantigen was normalized using a background normalization factor. The average normalized signal intensity obtained from healthy controls (N ≥ 2) was determined for each antigen. Relative autoantibody reactivity (RAR) ratios were then calculated between each sample and the average of healthy controls plus 2 SD, with ratios greater than 1 considered positive [8, 9]. Statistical analysis was performed using significance analysis of microarray (SAM) [8, 9]. Hierarchical clustering of the data was performed using Euclidean distance. A heat map of the ratio values was generated using MultiExperiment Viewer software (MeV, DFCI Boston, MA) and values were coded as follows: 0 blue, 1 black, 5 yellow. To validate the microarray results, an ELISA was performed to a selected antigen (gliadin IgG, Eagle Biosciences) according to the manufacturer’s protocol using serum samples from patients with CHH (N = 16), healthy controls (N = 4), and patients with APECED (N = 5). Protein concentrations were obtained using a standard curve.
Results of the protein microarray revealed broad autoantibody reactivity in both CHH and APECED patients compared to healthy controls, with variable autoantibody antigen specificity observed between patients (Fig. 1a). Samples were evaluated for multireactivity, defined as positive autoantibody ratios to at least 20% of the self-antigens present on the array [8, 9]. Multireactivity was seen in 10 of the 16 CHH samples assayed. SAM performed on the entire group of CHH samples in comparison to healthy controls did not reach statistical significance, which may reflect the heterogeneity in autoantibody specificity observed between CHH patients. Cluster analysis revealed unique groupings of CHH and APECED patients within the cohort (Fig. 1b). When SAM was repeated comparing a distinct cluster of five CHH patients to healthy controls, nine autoantibodies reached statistical significance with a mean false discovery rate of less than 1% (Fig. 1c). Interestingly, four of the nine autoantibodies are implicated in inflammatory myopathies, including myositis-specific antibodies Mi-2, SRP54, and PL-7, as well as an additional autoantibody targeting myosin itself. The remainder consisted of autoantibodies against thyroglobulin, gliadin, nuclear antigens Sm/RNP and PCNA, as well as vitronectin, a glycoprotein important in tissue remodeling and repair. The presence of IgG autoantibodies to gliadin was validated using ELISA (Fig. S1).
Fig. 1.
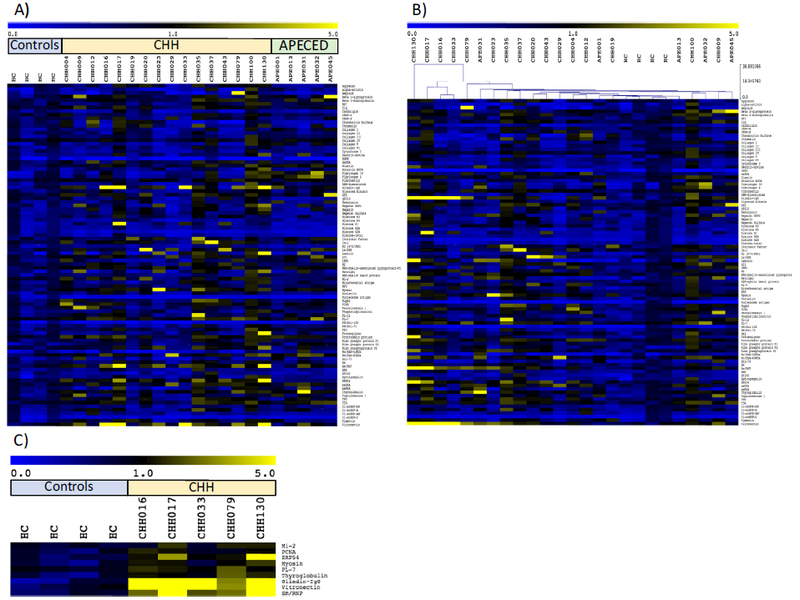
a Heat-map of IgG autoantibodies in patients with cartilage-hair hypoplasia (CHH) and autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED) compared to healthy controls (HC). Relative antibody autoreactivity compared to healthy controls is depicted in colorimetric form according to the scale shown at the top. b Heat-map rearranged according to hierarchical clustering. c Autoantibodies with significantly increased reactivity in a particular cluster of CHH patients identified in panel b
This study is the first to identify broad autoantibody reactivity in a cohort of CHH patients. This finding highlights the importance of considering B cell immune dysregulation as a potential contributing factor in the autoimmune manifestations of CHH. The significant autoantibody ratios identified in a specific cluster within the CHH cohort demonstrate intriguing clinical correlates, including myositis-specific antibodies, as well as autoantibodies important in autoimmune manifestations implicated in CHH, such as hypothyroidism and autoimmune enteropathy [2]. Future work will focus on determining the mechanism by which mutant RMRP leads to the generation of autoreactive antibodies, as well as investigating the pathophysiologic connections between the autoantibodies identified and CHH clinical manifestations.
Supplementary Material
Acknowledgments
Financial Support This work was supported in part with funds from the NIAID Division of Intramural Research. CMB received support from NIH grant T32AI007512 and is currently supported by the CAAIF/AllerGen Research Fellowship and the AllerGen Emerging Clinician-Scientist Research Fellowship. SK is supported by the Doctoral School in Health Sciences at the University of Helsinki. MT is supported by the Foundation for Pediatric Research. OM is supported by the Sigrid Jusélius Foundation, the Academy of Finland, the Folkhälsan Research Foundation, the Helsinki University Hospital Research Funds, and the Swedish Childhood Cancer Foundation.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10875–017-0408-4) contains supplementary material, which is available to authorized users.
Compliance with Ethical Standards All individuals who participated, or their guardians, signed an informed consent, and the study was approved by the Institutional Review Boards at the referring institutions.
Conflict of Interest The authors declare that they have no conflict of interest.
References
- 1.Rider NL, Morton DH, Puffenberger E, Hendrickson CL, Robinson DL, Strauss KA Immunologic and clinical features of 25 Amish patients with RMRP 70 A–>G cartilage hair hypoplasia. Clin Immunol. 2009;131(1):119–28. [DOI] [PubMed] [Google Scholar]
- 2.Bordon V, Gennery AR, Slatter MA, Vandecruys E, Laureys G, Veys P, et al. Clinical and immunologic outcome of patients with cartilage hair hypoplasia after hematopoietic stem cell transplantation. Blood. 2010;116(1):27–35. [DOI] [PubMed] [Google Scholar]
- 3.Makitie O Kaitila I Cartilage-hair hypoplasia—clinical manifestations in 108 Finnish patients. Eur J Pediatr. 1993;152(3):211–7. [DOI] [PubMed] [Google Scholar]
- 4.Poliani PL, Facchetti F, Ravanini M, Gennery AR, Villa A, Roifman CM, et al. Early defects in human T-cell development severely affect distribution and maturation of thymic stromal cells: possible implications for the pathophysiology of Omenn syndrome. Blood. 2009;114(1):105–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de la Fuente MA, Recher M, Rider NL, Strauss KA, Morton DH, Adair M, et al. Reduced thymic output, cell cycle abnormalities, and increased apoptosis of T lymphocytes in patients with cartilage-hair hypoplasia. J Allergy Clin Immunol. 2011;128(1):139–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kostjukovits S, Degerman S, Pekkinen M, Klemetti P, Landfors M, Roos G, et al. Decreased telomere length in children with cartilage-hair hypoplasia. J Med Genet. 2017;54(5):365–70. [DOI] [PubMed] [Google Scholar]
- 7.Kisand K, Peterson P Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy: known and novel aspects of the syndrome. Ann N Y Acad Sci. 2011;1246:77–91. [DOI] [PubMed] [Google Scholar]
- 8.Crestani E, Volpi S, Candotti F, Giliani S, Notarangelo LD, Chu J, et al. Broad spectrum of autoantibodies in patients with Wiskott-Aldrich syndrome and X-linked thrombocytopenia. J Allergy Clin Immunol. 2015;136(5):1401–4 e1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walter JE, Rosen LB, Csomos K, Rosenberg JM, Mathew D, Keszei M, et al. Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J Clin Investig. 2015;125(11):4135–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.