

Abstract
Cancer cells rely on the chaperone, heat shock protein 70 (Hsp70), for survival and proliferation. Recently, benzothiazole-rhodacyanines have been shown to bind an allosteric site on Hsp70, interrupting its binding to nucleotide-exchange factors (NEFs) and promoting cell death in breast cancer cell lines. However, proof-of-concept molecules, such as JG-98, have relatively modest potency (EC50 ~0.7 to 0.4 μM) and are rapidly metabolized in animals. Here, we explored this chemical series through structure- and property-based design of ~300 analogs, showing that the most potent had >10-fold improved EC50 values (~0.05 to 0.03 μM) against two breast cancer cells. Biomarkers and whole genome CRISPRi screens confirmed members of the Hsp70 family as cellular targets. Based on these results, JG-231 was found to reduce tumor burden in an MDA-MB-231 xenograft model (4 mg/kg; i.p.). Together, these studies support the hypothesis that Hsp70 may be a promising target for anti-cancer therapeutics.
Keywords: chaperone, allostery, structure-activity relationships, breast cancer, non-oncogene addiction, proteostasis
INTRODUCTION
Cancer cells depend on molecular chaperones, including heat shock protein 70 (Hsp70), for their proliferation and survival, in a process termed non-oncogene addiction.1 Briefly, these chaperone seem to buffer cancer cells from the proteotoxic stress caused by high mutational burden, rapid proliferation, altered metabolism and aneuploidy. For example, Hsp70 is known to bind and stabilize many oncogenes and pro-survival “client” proteins,2–4 with the net effect of suppressing multiple cell death pathways.5–8 Consistent with this idea, simultaneous knockdown of the two major cytoplasmic Hsp70 paralogs, Hsc70 (HSPA8) and Hsp72 (HSPA1A), in HCT116 cells leads to degradation of client proteins and apoptosis. In contrast, knockdown of Hsc70 and Hsp72 is well tolerated in non-tumorigenic PNT2 epithelial cells.9 supporting the idea that chaperone function is disproportionately required in cancer. In part, this selectivity seems to be because the chaperones of cancer cells are assembled in dedicated multi-protein complexes.10 Moreover, Hsp72 is commonly overexpressed in many cancers, where its levels correlate with poor patient prognosis, especially in cancers of the breast.2, 3, 11 Together, these observations have suggested that Hsp70 family members may be good targets for anti-cancer therapy.
Efforts by multiple groups have started to produce chemical inhibitors of Hsp70s. All members of the Hsp70 family are composed of a nucleotide binding domain (NBD) and a substrate-binding domain (SBD), which are connected by a short linker.12–16 The chaperone function of Hsp70 requires a dynamic conformational change that involves hydrolysis of ATP in the NBD and subsequent re-positioning of the SBD.17, 18 These observations have guided the design of chemical inhibitors targeting distinct pockets. For example, VER-15500819, 20 and apoptozole21,22 compete with nucleotide for binding the NBD. However, Hsp70 binds tightly to nucleotides (Kd ~100 to 500 nM), making it difficult to compete with millimolar concentrations of ATP in the cytosol. This issue motivated the recent discovery of a VER-155008 analog that covalently modifies an active site lysine residue.23 Other approaches have tackled this problem by focusing on allosteric sites. The Chiosis group used a computational strategy to identify YK5 and its analogues24–26, which bind a cryptic, allosteric site in the NBD. Similarly, pifithrin-μ (2-phenylethynesulfonamide, PES)27 and novalactone28, target allosteric sites in the SBD28, 29. Finally, other reported Hsp70 inhibitors have been designed to target its protein-protein interactions (PPIs) with co-chaperones.30 For example, members of the BAG family of nucleotide exchange factors (NEFs), such as BAG3, bind to Hsp70’s NBD and promote its ATP cycling.31, 32 Indeed, Hsp70 has a strikingly poor turnover rate (less than 1 ATP per minute) in the absence of co-chaperones, suggesting that PPIs might be good drug targets. In support of this idea, knockdown of BAG3 leads to loss of important, pro-survival clients in MCF7 cells.33 Further, it has been shown that blocking the interaction with BAG proteins “traps” Hsp70 in an ADP-bound state and that this conformation favors degradation of some bound clients.34 These features led us to focus on the development of molecules that inhibit the PPI between Hsp70 and BAG family co-chaperones. This effort started with MKT-077, a benzothiazole-rhodacyanine first described by Wadwha and coworkers,35 that binds to an allosteric site within Hsp70’s NBD and disrupts the PPI with BAG proteins.36, 37 Binding of MKT-077 seems to reposition the IB and IIB subdomains of Hsp70’s NBD to interrupt electrostatic interactions with BAGs. Indeed, deleting mutating a residue in BAG3 (R480A) required for the PPI mimics treatment with the Hsp70 inhibitors in breast cancer cells33. Consistent with a role for Hsp70-BAG in cancer, MKT-077 has anti-proliferative activity35 and was advanced to a Phase I clinical trial for solid tumors38. However, this molecule has relatively modest potency (EC50 ~ 2.2 μM) in MCF7 breast cancer cells and, importantly, it is rapidly metabolized in vivo or in the presence of liver microsomes (< 5 min).37 In pilot studies, we measured the metabolite profile and completed a series of ~100 analogs designed to improve its stability. This effort identified JG-98 as a more potent (EC50 ~0.4 μM) analog that had anti-tumor activity in MCF7 xenograft models.37, 39, 40 Molecules in this chemical series have been important chemical probes of Hsp70 in multiple disease models, including Dengue virus,41 hepatitis C virus,42 tauopathy34 and others43. Moreover, compounds in this series appear to be relatively selective for members of the Hsp70 family, including Hsc70/HSPA8 and mtHsp70/HSPA9, as judged by pulldowns35, 37 and mutagenesis44. Importantly, the binding site of JG-98 is highly conserved in the major human Hsp70 paralogs (Hsc70/HSPA8, Hsp72/HSPA1, BiP/HSPA5, mtHsp70/HSPA9), suggesting the possibility that these allosteric inhibitors may display pan-activity for multiple family members. This property might be beneficial in some settings, because, as mentioned above, it is known that knockdown of both Hsc70 and Hsp72 is required to block cancer cell proliferation.9 Moreover, both mtHsp70/HSPA945 and BiP/HSPA546 have also been reported to be promising anti-cancer targets.
Further advancement of these compounds as chemical probes is limited by their modest potency and relatively poor pharmacokinetics in animal models. In this study, we took advantage of new structural insights13, 36 and the recent description of Hsp70 biomarkers47 to design analogs of JG-98 From a series of ~300 molecules, we report structure-activity relationships (SAR) and explore target engagement in vitro and in cells, including a whole genome CRISPRi screen. From these studies, we selected four compounds for maximum tolerated dose (MTD) and pharmacokinetics (PK) experiments in mice. The best of these molecules, JG-231, was modestly active in MDA-MB-231 xenograft models of triple negative breast cancer (TNBC). We anticipate that these analogs, with additional optimization of the solubility and formulation, may become important chemical probes for the study of Hsp70-BAG biology in cellular and animal models.
RESULTS AND DISCUSSION
Design and chemistry.
JG-98 is a non-competitive inhibitor of Hsp70 that binds to an allosteric site in the NBD (Fig. 1A).36, 37 To guide our medicinal chemistry campaign, we first performed induced fit docking of JG-98 into HSPA8’s NBD (PDB 3HSC) using Schrodinger software, which allows flexibility in both the protein and ligand. In the best-predicted models, JG-98 was anchored in a deep pocket formed by residues R72, K71, T13, F150, P147, T204 and G12 (Fig 1B). The carbonyl in the central ring was predicted to make electrostatic interactions with a hydrogen bonding network involving D86, H227 and T226, while the N-ethyl group in that same ring was predicted to make limited hydrophobic contact with V82. In this orientation, the delocalized cation in JG-98’s thiazolium ring was largely solvent-exposed, but it positioned the benzyl group into an adjacent, secondary pocket formed by Y149, T222, T226 and backbone atoms of T223-T226, with pi stacking interactions predicted with the side chain of Y149. Based on this low energy model, we performed additional docking studies using a rigid-receptor, but flexible-ligand, to inspire the design of putative, new analogs. The resulting structures suggested that better binding to the pocket might be achieved by adding small substituents at the R1 position of the benzothiazole, to expand into gaps in the area around R72, K71, T13, F150, P147 and T204 (Fig 1C; deep pocket). In addition, it seemed that making substituents on the benzyl ring might create better contacts with a groove located between Y149 and the b-sheet composed of residues 222 through 226 (Fig 1D; 2nd pocket).
Figure 1:
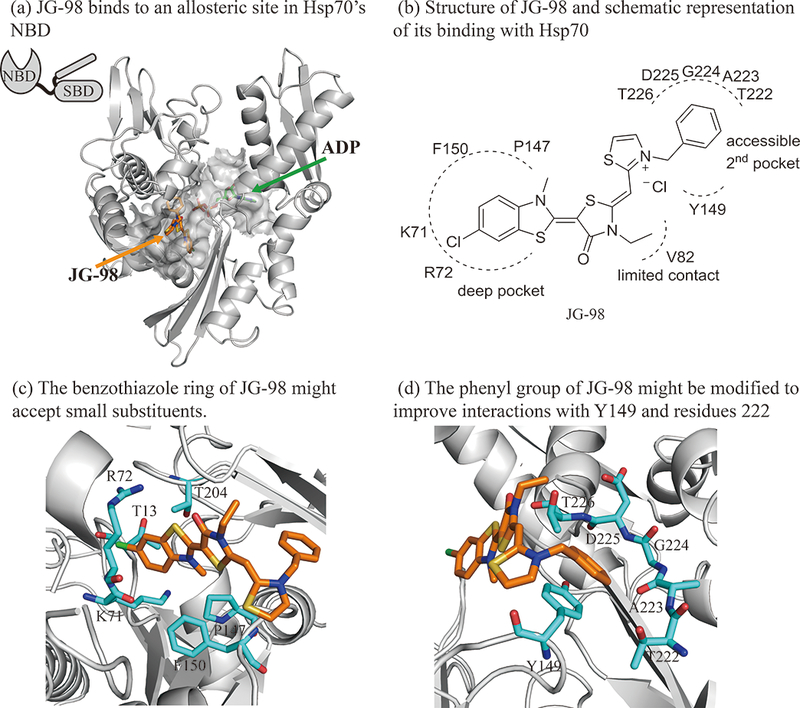
Structure guided design of Hsp70 inhibitor JG-98 analogs. (a) JG-98 binds to an allosteric site in the nucleotide binding domain (NBD) of Hsp70, rather than its necleotide binding site. JG-98 was docked into HSPA8 using flexible-receptor flexible-ligand (InducedFit) docking. The carbon atoms of the protein, JG-98 and ADP are shown as gray, orange and green, respectively. JG-98 and ADP are shown as stick representations. (b) Schematic representation of JG-98 interacting with Hsp70. (c) The benzothiaole ring of JG-98 is located in a deep hydrophobic pocket formed by R72, K71, F150, P147, T204 and T13. (d) The phenyl ring forms favorable interactions with Y149 and the backbone atoms of residues 223 through 226. Selected residues are shown in cyan as stick representation.
Guided by the molecular docking, we assembled ~ 300 analogs of JG-98 using a synthetic route that was reported previously (Scheme 1). Briefly, the synthesis starts with cyclization of substituted anilines with potassium ethyl xanthate, followed by methylation with iodomethane under mildly basic conditions. The resulting benzothiazoles are treated with methyl p-toluenesulfonate and coupled with N-substituted rhodanines. These products were methylated by methyl p-toluenesulfonate, followed by coupling with substituted thiazoliums in mild basic condition. The products were then dissolved in a methanol/DCM mixture and passed through a chloride exchange column to yield the final compounds as chloride salts. Overall yields ranged from 20– 60% and purity was > 95% by HPLC (see the Supporting Information).
Scheme 1.
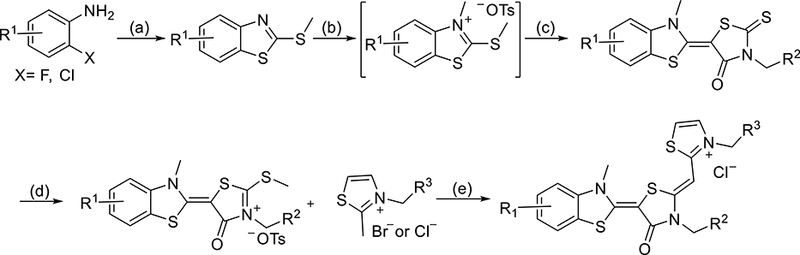
Reaction Conditions: (a) 1) potassium ethylxanthate, DMF, 140 °C, 4h 2) Mel Net3, EtOH, 80 °C, 1h; 40 - 80%; (b) p-TsOMe, anisole, 125 °C, 4 h, used directly for next step; (c) 3-substituted rhodanine, NEt3, MeCN, 25 °C, 4 h, 30-75%; (d) p-TsOMe, DMF, 135 °C 3 h, 50 - 85%; (e) NEt3, MeCN, 70 °C, 3 h; 2) Cl−-ion exchange resin; 20 - 60%.
Structure activity relationships (SAR).
A major objective of this campaign was to improve potency against breast cancer cells, while maintaining selectivity over non-transformed cells. Accordingly, the anti-proliferative activity (EC50) of each molecule was calculated from MTT viability assays after treating two breast cancer cell lines (MCF-7, MDA-MB-231) and normal, mouse embryonic fibroblasts (MEFs). As a benchmark, we first measured the activity of JG-98, which had EC50 values of 0.71 ± 0.22 μM against MCF-7 cells and 0.39 ± 0.03 μM against MDA-MB-231 cells (Table 1). Consistent with previous reports, 37 this compound was somewhat less toxic to MEFs, with an EC50 of 4.2 ± 1 μM.
Table 1:
Structure and Activity Relationship of Allosteric Hsp70 inhibitors
| Compd | R1 | R2 | R3 | MCF-7 IC50/μM |
MDA-MB-231 IC50/μM |
MEF IC50/μM |
|---|---|---|---|---|---|---|
| JG-98 | 5-Cl | ethyl | H | 0.71 ± 0.22 | 0.39 ± 0.03 | 4.2 ± 1 |
| 1 | 5-Br | ethyl | H | 0.44 ± 0.03 | 0.33 ± 0.04 | 6.1 ± 0.3 |
| 2 | 4-Br | ethyl | H | 0.27 ± 0.03 | 0.11 ± 0.01 | 0.66 ± 0.04 |
| 3 | 5-NO2 | ethyl | H | >5 | 4.8 ± 0.4 | 2.8 ± 0.1 |
| 4 | 4-SO2Me | ethyl | H | >5 | >20 | 2.5 ± 0.6 |
| 5 | 5-CF3 | ethyl | H | 0.53 ± 0.11 | 0.36 ± 0.03 | 2.4 ± 0.2 |
| 6 | 4-OCF3 | ethyl | H | 0.26 ± 0.02 | 1.2 ± 0.1 | 0.34 ± 0.02 |
| 7 | 5-OCF3 | ethyl | H | 0.4 ± 0.04 | 0.8 ± 0.1 | 1.2 ± 0.1 |
| 8 | 4-Me | ethyl | H | 0.21 ± 0.01 | 0.078 ± 0.01 | 1.3 ± 0.1 |
| 9 (JG-194) | 5-Me | ethyl | H | 0.16 ± 0.02 | 0.15 ± 0.01 | 1.9 ± 0.1 |
| 10 | 3-Me | ethyl | H | 0.25 ± 0.02 | 0.21 ± 0.04 | 2.3 ± 0.2 |
| 11 | 6-Me | ethyl | H | 0.29 ± 0.03 | 0.25 ± 0.03 | 1.6 ± 0.1 |
| 12 | 5-Ethyl | ethyl | H | 0.080 ± 0.008 | 0.14 ± 0.01 | 1.0 ± 0.1 |
| 13 | 4-OCH3 | ethyl | H | 0.37 ± 0.02 | 0.36 ± 0.05 | 1.0 ± 0.1 |
| 14 | 5-OCH3 | ethyl | H | 0.13 ± 0.01 | 0.16 ± 0.01 | 2.0 ± 0.1 |
| 15 | 5-SCH3 | ethyl | H | 0.13 ± 0.01 | 0.12 ± 0.01 | 1.7 ± 0.1 |
| 16 | 5-Cl | allyl | H | 0.17 ± 0.02 | 0.10 ± 0.01 | 2.4 ± 0.1 |
| 17 | 5-Cl | cyclopropyl | H | 0.33 ± 0.03 | 0.56 ± 0.08 | 3.4 ± 0.2 |
| 18 | 5-Cl | 3-methoxy-3- oxopropyl) | H | 0.66 ± 0.07 | 0.24 ± 0.03 | 4.9 ± 0.3 |
| 19 | 5-Cl | ethyl | o-F | 0.16 ± 0.02 | 0.29 ± 0.04 | 1.2 ± 0.1 |
| 20 | 5-Cl | ethyl | m-F | 0.48 ± 0.04 | 1.0 ± 0.2 | 1.0 ± 0.1 |
| 21 | 5-Cl | ethyl | p-F | 0.61 ± 0.03 | 1.1 ± 0.2 | 1.9 ± 0.1 |
| 22 | 5-Cl | ethyl | o-Cl | 0.19 ± 0.01 | 0.18 ± 0.02 | 0.70 ± 0.03 |
| 23 | 5-Cl | ethyl | m-Cl | 0.26 ± 0.02 | 0.51 ± 0.06 | 0.69 ± 0.03 |
| 24 | 5-Cl | ethyl | p- Cl | 1.45 ± 0.13 | 0.41 ± 0.06 | 3.1 ± 0.3 |
| 25 | 5-Cl | ethyl | o-CF3 | 0.13 ± 0.02 | 0.13 ± 0.01 | 1.2 ± 0.1 |
| 26 | 5-Cl | ethyl | o-Br | 0.19 ± 0.01 | 0.20 ± 0.03 | 1.6 ± 0.1 |
| 27 | 5-Cl | ethyl | o-methoxy carbonyl | 0.17 ± 0.02 | 0.17 ± 0.02 | 2.9 ± 0.1 |
| 28 | 5-Cl | ethyl | o-CH3 | 0.19 ± 0.01 | 0.20 ± 0.03 | 1.6 ± 0.1 |
| 29 | 5-Cl | ethyl | o-OCH3 | 0.40 ± 0.03 | 0.12 ± 0.01 | 1.5 ± 0.1 |
| 30 | 5-Cl | ethyl | o-SCF3 | 0.13 ± 0.01 | 0.16 ± 0.01 | 3.7 ± 0.2 |
| 31 | 5-Cl | ethyl | o-OCF3 | 0.22 ± 0.01 | 0.12 ± 0.009 | 2.7 ± 0.1 |
| 32(JG-258) | - | - | - | > 5 | > 20 | > 20 |
In the exploration of SAR, we first explored the influence of substitutions to the benzothiazole (R1). This region is relatively buried in the bound structure, so we largely restricted modifications to small groups (e.g. halogens, methyl) but also tried to test these limits. We found that replacing the chlorines in JG-98 for bromine (compound 1) or moving it to the 4-position (compound 2) improved potency (EC50 values 0.11 to 0.44 μM). This result supported the idea that slightly larger groups could be favored. However, consistent with the hydrophobicity of the pocket, nitro (compound 3) and methanesulfonyl group (compound 4,) substantially decreased activity (EC50 values >1 μM). Next, we turned our attention to understanding the effects of electron withdrawing/donating character of the substitutions. Replacing the 5-chlorine with 5-trifluoromethyl (compound 5) did not significantly improve upon JG-98 (EC50 ~ 0.53 to 0.36 μM). However, introducing methyl groups in the 3, 4, 5, or 6 positions (compounds 8–11) improved cellular potency about 2 to 4-fold with EC50 values ~0.2 μM. For example, compound 8, with a methyl at the 4 position, was the most potent molecule in the series (EC50 value = 0.078 ± 0.01 μM). Replacing methyl (compound 9, JG-194) for ethyl (compound 12) at the 5-position also improved potency about 3-fold (EC50 = 0.08 ± 0.008 μM) in MCF7 cells although activity in MDA-MB-231 cells was unchanged. Overall, these results suggest that small, electron-donating substitutions on the benzothiazole ring improve anti-proliferative activity, likely by better filling the allosteric pocket on Hsp70 (see below). As a control, we also assembled JG-258 (compound 32), in which the benzothiazole was truncated. As expected, this compound was inactive (EC50 > 5 μM).
After probing the SAR at position R1, we made substitutions at the central rhodacyanine ring (R2). We found that changing the ethyl group to an allyl (compound 16) improved cellular activity by ~4-fold compared with JG-98, while a cyclopropyl (compound 17) at this position led to slightly increased potency against MCF-7 cells (EC50 0.33 ± 0.03 μM) but reduced activity against MDA-MB-231 cells (EC50 value 0.56 ± 0.08 μM). Finally, increasing the polarity of this substituent to 3-methoxy-3-oxopropyl (compound 18) had little effect (EC50 0.66 ± 0.07 and 0.24 ± 0.03 μM). Together, these results suggest that small hydrophobic substitutions are allowed at R2. However, the substitutions tested here had a relatively minor influence on the overall activity.
Next, we explored substitutions and replacements of JG-98’s benzyl ring. Docking predicted that additional contacts might be made with a region between Y149 and a b-sheet (see Fig 1D). To first probe this possibility, we found that a fluorine at the ortho-position (compound 19) enhanced activity against both cell lines (MCF-7, EC50 0.16 ± 0.02 μM; MDA-MB-231, EC50 = 0.29 ± 0.04 μM), while fluorine at the meta- or para-positions (compounds 20 and 21) had little effect. This relationship was also true when R3 was chlorine instead of fluorine (compounds 22, 23 and 24). After identifying the importance of the ortho position, we compared electron-withdrawing groups (compounds 25, 26 and 27) to electron-donating ones (compounds 28, 29, 30 and 31). We found that these analogs were similarly active (EC50 values ~0.12 to 0.20 μM), suggesting that sterics might be important. Because of the strong effect of ortho-substitutions to the benzyl group, we further explored the SAR at this position. As a starting point, we focused on compound 22, which features an ortho-chloro (Table 2). Installation of a second chlorine at the other ortho position (compound 33) did not further improve activity. Next, we tried different heterocycles as replacements of the benzyl group, but found that substituted 3- or 4- pyridyl groups (compounds 34 and 35) significantly reduced activity (e.g. compound 35; EC50 >5 μM). When we revisited the docked binding mode of JG-98 with this data in mind, we noted that the corresponding pocket is rather narrow; therefore, we hypothesized that contracting the ring size might allow for a wider range of modifications and better contact with the b-sheet. Indeed, a 3-trifluoromethyl substituted furan (compound 36) was ~3-fold more active in both cell lines (MCF-7, EC50 = 0.21 ± 0.01 μM; MDA-MB-231, EC50 = 0.12 ± 0.02 μM). However, the larger, more polar methoxycarbonyl at the 3-position (compound 37) reduced activity ~7-fold in both cell lines, suggesting limits to the size of this pocket (see below). However, we were able to improve anti-proliferative activity in MCF-7 cells by 2- to 7-fold (MCF7, EC50 0.81 ± 0.09 μM; MDA-MB-231, EC50 0.13 ± 0.01 μM) by moving the methoxycarbonyl from the 3- to 5-position of the furan (compound 38). This result, consistent with the docking orientation, suggests that the furan might set the 5-position at the proper angle for engaging the pocket. To test this idea, we replaced the furan with a thiophene (compound 39). This compound had different connectivity to the heterocycle (see Table 2), but had similar potency (MCF7 EC50 = 0.73 ± 0.06 μM; MDA - MB-231 EC50 = 0.11 ± 0.01 μM). Consistent with the model, the equivalent substitution with a methoxycarbonyl (compound 40) had similar potency (MCF7, EC50 0.16 ± 0.02 μM; MDA-MB-231, EC50 0.22 ± 0.02 μM) compared to compound 38. In the context of the thiophene, we also replaced the methoxycarbonyl with a bromine (compound 41) at the 2-position, but found that it was not dramatically improved (MCF7 cells EC50 0.52 ± 0.09 μM; MDA-MB-231; EC50 0.20 ± 0.02 μM). However, at other positions around the thiophene, bromo or methyl substitutions tended to have increased potency. For example, the 2,4-dimethyl (compound 43) was the most active (MCF-7, EC50 0.098 ± 0.008 μM; MDA-MB-231, EC50 0.080 ± 0.008 μM) of the thiazole-containing analogs. In contrast, thiazole and isoxazole analogues had significantly reduced activity, with compounds 46 and 47 being inactive (EC50 > 5 μM) in both cell lines.
Table 2:
Substituents on the Thiazolium Ring are Important for Bioactivity
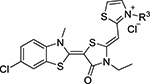 | ||||
|---|---|---|---|---|
| Compd | R3 | MCF-7 IC50/μM |
MDA-MB-231 IC50/μM |
MEF IC50/μM |
| JG-98 |  |
0.71 ± 0.22 | 0.39 ± 0.03 | 4.2 ± 1 |
| 22 | 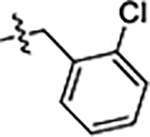 |
0.19 ± 0.01 | 0.18 ± 0.02 | 0.70 ± 0.03 |
| 33 | 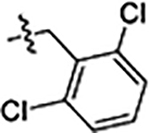 |
0.16 ± 0.01 | 0.16 ± 0.01 | 1.4 ± 0.1 |
| 34 | 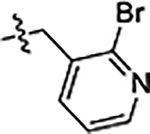 |
1.5 ± 0.1 | 1.6 ± 0.2 | 1.7 ± 0.1 |
| 35 | 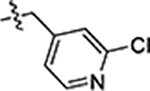 |
>5 | 7.2 ± 0.8 | > 5 |
| 36 | 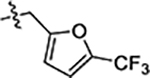 |
0.21 ± 0.01 | 0.12 ± 0.02 | 5.5 ± 0.5 |
| 37 | 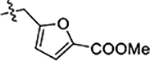 |
1.5 ± 0.1 | 0.97 ± 0.10 | 2.0 ± 0.2 |
| 38 | 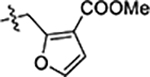 |
0.81 ± 0.09 | 0.13 ± 0.01 | 2.7 ± 0.1 |
| 39 | 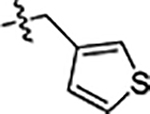 |
0.73 ± 0.06 | 0.11 ± 0.01 | 4.4 ± 0.3 |
| 40 | 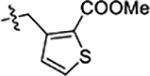 |
0.16 ± 0.02 | 0.22 ± 0.02 | 3.2 ± 0.3 |
| 41 | 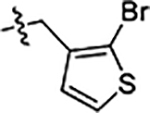 |
0.52 ± 0.09 | 0.20 ± 0.02 | 1.3 ± 0.1 |
| 42(JG-231) | 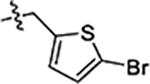 |
0.12 ± 0.01 | 0.25 ± 0.02 | 2.5 ± 0.1 |
| 43 | 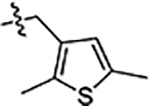 |
0.098 ± 0.008 | 0.080 ± 0.008 | 4.2 ± 0.4 |
| 44 | 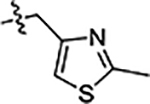 |
0.96 ± 0.08 | 0.74 ± 0.07 | 4.9 ± 0.3 |
| 45 | 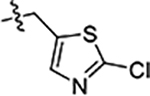 |
>5 | 2.3 ± 0.3 | 8.9 ± 0.8 |
| 46 | 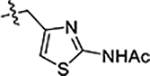 |
>5 | >20 | 4.2 ± 0.6 |
| 47 | 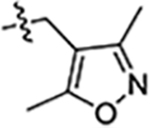 |
>5 | 10 ± 2 | 7.6 ± 0.3 |
After studying the individual effects of substitutions at R1, R2 and R3, we combined these features. Indeed, combining the electron-donating substituent at R1 with the ortho substituted benzyl ring at R3 (compounds 48, 49, 50, 51 and 52) significantly improved potency compared to JG-98 (Table 3), with EC50 values between 0.060 and 0.21 μM. In this series, the analogs were consistently less potent when R1 was a methyl or methoxyl group in the 4- position instead of the 5-position. Thus, in the context of the 5-substituted scaffold, we further explored optimization of R3. Replacing the ortho-trifluoromethoxy with an ortho-chlorine (compounds 53 and 54) did not further enhance activity (EC50 values ~ 0.03 to 0.15 μM). The same outcomes were observed when electron-donating groups at R1 were combined with substitution of the phenyl group with ortho-methoxy (compounds 55 and 56) or ortho-methoxycarbonyl groups (compound 57). Introducing an extra chlorine in the ortho positions (compounds 58 and 59) did not dramatically affect cellular activity, but we hoped that it might change the planarity of the molecule and improve solubility (see below). Finally, we wanted to combine substituted furans at R3 with electron-donating (compound 60) or electron-withdrawing substituents (compound 61) at R1. As predicted, these molecules were more potent that JG-98, with EC50 values around 0.10 μM. Overall, this campaign improved activity by 11- to 14-fold, yielding compounds with ~30 to 50 nM potency in cultured breast cancer cells.
Table 3:
Combinations of optimal substituents creats more potent Hsp70 inhibitors
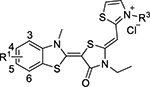 | |||||
|---|---|---|---|---|---|
| Compd | R1 | R3 | MCF-7 IC50/μM |
MDA-MB-231 IC50/μM |
MEF IC50/μM |
| JG-98 | 5-Cl | 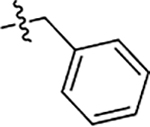 |
0.71 ± 0.22 | 0.39 ± 0.03 | 4.2 ± 1 |
| 48 | 4-CH3 |  |
0.089 ± 0.006 | 0.18 ± 0.01 | 1.4 ± 0.1 |
| 49 | 5-CH3 |  |
0.064 ± 0.005 | 0.077 ± 0.009 | 0.75 ± 0.05 |
| 50 | 4-OCH3 | 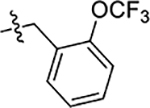 |
0.16 ± 0.02 | 0.16 ± 0.01 | 2.2 ± 0.1 |
| 51 | 5-OCH3 | 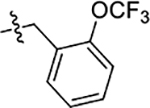 |
0.070 ± 0.003 | 0.053 ± 0.005 | 0.70 ± 0.01 |
| 52 | 5-isopropyl | 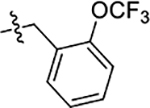 |
0.090 ± 0.005 | 0.21 ± 0.01 | 0.48 ± 0.02 |
| 53 | 5-OCH3 |  |
0.048 ± 0.003 | 0.033 ± 0.005 | 0.66 ± 0.01 |
| 54 | 5-SCH3 | 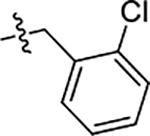 |
0.15 ± 0.01 | 0.13 ± 0.01 | 0.75 ± 0.05 |
| 55 | 5-OCH3 | 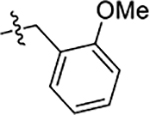 |
0.076 ± 0.005 | 0.062 ± 0.006 | 0.77 ± 0.03 |
| 56 | 5-CH3 | 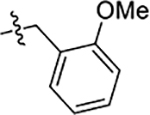 |
0.071 ± 0.006 | 0.082 ± 0.010 | 0.96 ± 0.03 |
| 57(JG-345) | 5-CH2CH3 | 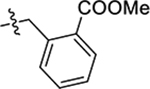 |
0.049 ± 0.003 | 0.046 ± 0.006 | 1.0 ± 0.1 |
| 58 | 4-CH3 | 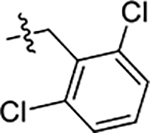 |
0.088 ± 0.008 | 0.054 ± 0.006 | 0.55 ± 0.03 |
| 59 | 5-CH2CH3 | 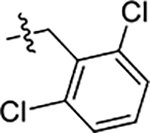 |
0.093 ± 0.007 | 0.10 ± 0.01 | 0.30 ± 0.10 |
| 60(JG-294) | 5-Br | 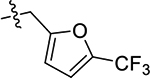 |
0.10 ± 0.01 | 0.18 ± 0.01 | 3.5 ± 0.4 |
| 61 | 5-CH2CH3 | 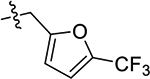 |
0.11 ± 0.01 | 0.10 ± 0.01 | 1.4 ± 0.06 |
Selection of candidate molecules for further exploration.
Based on the SAR, we selected the most potent molecules for additional studies. In the first experiments, thirteen of the most potent analogs, plus JG-98 for comparison, were tested for aqueous solubility and mouse liver microsome stability (Table 4). Dramatic improvements in solubility were not observed, so molecules with solubility near that of JG-98 (between 16 and 31 μM ) were considered for additional studies. In contrast, some substantial improvements in microsomal stability were observed (> 60 min for compound 31, 42 (JG-231), 51 and 60 (JG-294)). Finally, we tested these analogs for anti-proliferative activity in an additional cell line: the IMR90 normal, human lung fibroblasts. Comparing the resulting EC50 values (along with the previous data on MEFs) confirmed that many of the new analogs were less toxic to normal fibroblasts (Table 4). For example, JG-294 was 35-fold selective for MCF7 cells and more than 98-fold selective compared to IMR-90 cells. Although we did not obtain substantial increases in solubility, we selected JG-194 (compound 9), JG-231 (compound 42), JG-294 (compound 60) and JG-345 (compound 57) for further biological testing and mechanistic studies, based on the combination of potency, selectivity and stability.
Table 4:
Selectivity Index, Solubility and Microsomal Stability of Selected Compounds
| Compd | Selectivity Index (MEF/MCF7) |
Selectivity Index (IMR-90/MCF7) |
Solubility (uM) | Microsomal Stability (T1/2, min) |
|---|---|---|---|---|
| JG-98 | 6 | 2 | 31 | 37 |
| 9(JG-194) | 11 | 11 | 16 | 40 |
| 31 | 12 | 7 | 31 | >60 |
| 42 (JG-231) | 20 | 38 | 16 | >60 |
| 43 | 42 | 45 | <8 | 25 |
| 49 | 11 | 10 | 8 | 53 |
| 51 | 10 | 14 | <8 | >60 |
| 53 | 13 | 27 | <8 | 27 |
| 55 | 10 | 18 | 8 | 13 |
| 56 | 13 | 29 | 31 | 21 |
| 57 (JG-345) | 20 | 65 | 31 | 25 |
| 58 | 6 | 7 | 31 | 33 |
| 60 (JG-294) | 35 | 98 | 31 | >60 |
| 61 | 12 | 30 | 16 | 37 |
Molecular docking and confirmation of the binding site.
First, we docked all four compounds, plus JG-258, to Hsp70’s allosteric site. As expected, each active analog was predicted to bind in a similar overall orientation as JG-98 (Fig 2A and Suppl. Fig 1), while JG-258 was not predicted to bind. The biggest differences between the bound compounds were in the second pocket, near Y149. Namely, while the benzyl of JG-194, the bromothiophene in JG-231 and trifluromethylfuran in JG-294 were predicted to engage in favorable π stacking interactions with this amino acid residue’s sidechain, the ester group of compound JG-345 seemed to shift this interaction to favor contacts between a backbone nitrogen instead.
Figure 2:
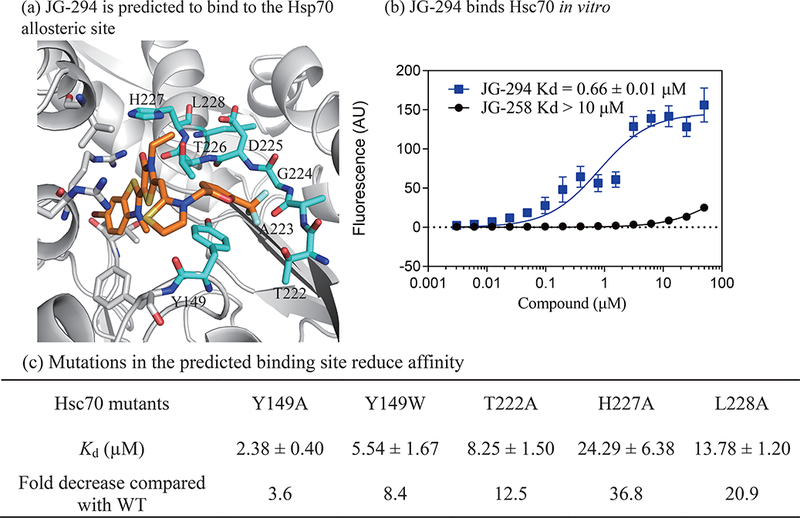
JG-294 binds to an allosteric site in the nucleotide binding domain (NBD) of Hsp70. (a) The docked pose of JG-294. JG-294 and the residues within 5 Å are shown as stick representations. The carbon atoms of the protein and JG-294 are shown in gray and orange, respectively. (b) JG-294 binds to Hsc70 with a Kd value as 0.6 ± 0.01μM in fluorescence shift assays. Results are the average of at least three independent replicates performed in triplicate each. Error bars represent standard deviation (SD). Some error bars are smaller than the symbol. (c) Mutations in Hsc70 weaken binding of JG-294. The experiments were performed as in b.
To further test binding in the predicted pocket, we measured the affinity of a representative compound, JG-294, for purified Hsp70 in vitro using a fluorescence shift assay (see Methods). We found that JG-294 bound to human Hsc70 (HSPA8), with an apparent affinity value of ~0.6 μM (Fig. 2B), while the negative control, JG-258, did not bind (Kd >10 μM). The affinity value for JG-294 was slightly larger than we expected from the cellular potency values (~0.1 μM), but MKT-077 is known to accumulate by up to 100-fold in cells.48 Importantly, this assay allowed us to test the prediction that JG-294 binds in the allosteric pocket. Accordingly, we mutated residues of the putative pocket: Y149W, T222A, H227A or L228A and measured the affinity of JG-294 for these proteins. Consistent with the predicted model, each mutation reduced the apparent affinity by ~4- to 20-fold (Fig 2C).
Analogs interrupt the Hsp70-BAG PPIs.
Binding of JG-98 to Hsp70 is known to suppress its PPIs with the BAG family of co-chaperones.34, 39 To determine whether the new analogs share this activity, we used a flow cytometry protein interaction assay (FCPIA)49. Briefly, purified Hsp70 is immobilized on beads and binding to fluorescently labeled BAG1 is measured by flow cytometry. We found that all of the analogs could, like JG-98, suppress binding of Hsp70 to BAG1 (Fig. 3A). For example, JG-294 had a Ki values of 0.35 ± 0.07 μM, while the control, JG-258, was inactive. To test whether these compounds could disrupt the PPI in cells, Hsp70 was immuno-precipitated from MCF-7 cells in the presence of JG-231 (10 μM) and immunoblotted for the representative family member, BAG3. As expected, JG-231 disrupted the Hsp70–BAG3 interaction (Fig. 3B). Thus, like JG-98, the analogs blocked the Hsp70-BAG interactions in vitro and in cells.
Figure 3:

70 with labelled Bag1 protein measured by flow cytometery. Results are the average of three independent experiments performed in triplicate. Error bars represent SD; (b) JG-231 inhibits the Hsp70-Bag3 interaction in treated MCF-7 cells as measured by immunoprecipitation. Hsp70 was immunoprecipitated and blotted for bound Bag3. The blot is representative of duplicate experiments
Hsp70 biomarkers suggest target engagement.
It is known that treatment of cancer cells with Hsp70 inhibitors leads to degradation of client proteins.33, 39 To confirm whether the JG-98 analogs share this activity, we treated MCF7 cells for 24 h and then performed western blots for Akt1, c-Raf-1, CDK4, IAP1 and HuR. We found that the levels of each biomarker were reduced after treatment with JG-194, JG-231, JG-294 or JG-345, but not JG-258 (Fig. 4). Consistent with previous reports,37 we found that treatment did not cause a stress response, as measured by the consistent levels of Hsp72 and Hsp90. Finally, Hsp70 inhibitors are known to initiate apoptosis in MCF7 cells.50 Consistent with this observation, each of the new compounds, but not the negative control, activated caspase3/7 in MCF7 cells (Suppl. Fig. 2).
Figure 4: Hsp70 inhibitors lead to degradation of oncoproteins without eliciting heat shock response.
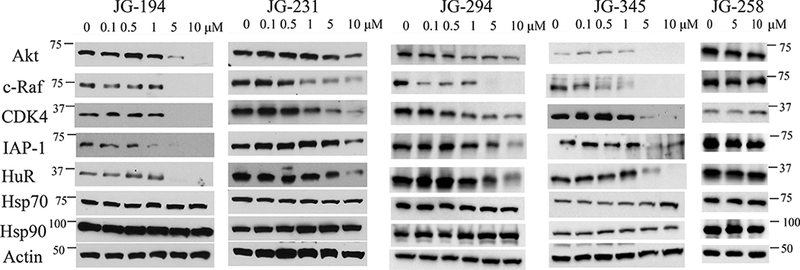
MCF-7 cells were treated with the indicated concentration of compound for 24 h, lysed and western blots performed. Results are representative of at least two independent experiments.
Genome wide CRISPRi screen supports Hsp70s as molecular targets.
Together, these biochemical and cell-based results were consistent with Hsp70 engagement, which might be expected from previous genetic and pulldown studies using other MKT-077 analogs.33, 35, 37, 43 However, to more rigorously explore this question, we performed a genome-wide CRISPRi functional genomics screen to identify genes that modulate the response to JG-294. This approach is emerging as a robust way to identify the target(s) of small molecules.51–53 Briefly, we infected a chronic myeloid leukemia (K562) cell line stably expressing dCas9-CRAB with a CRISPRi sgRNA library.54 Each sgRNA was expressed from a lentiviral vector and the infection ratio was selected such that cells would receive only one sgRNA. Moreover, the screening library contains ~1,000 negative control sgRNA sequences, which allows calculation of a p-value for the likelihood of a meaningful enrichment or depletion of the individual gene from the treated samples. A population of cells at the onset of the experiment (t0) was harvested (see below) and the remaining cells were split into two groups; the first was mock treated (1% DMSO) and the second treated with JG-294 at the EC80 value (0.25 μM). After 13 days of passaging, the genomic DNA was collected and the relative abundance of each sgRNA determined by next-generation deep sequencing (Fig. 5 and supplemental Table 1). The normalized difference in sgRNA abundance between the controls and JG-294 treated populations was then calculated for each gene.55 In this type of screen, knockdown is expected to either sensitize the cell to the chemical treatment or provide some measure of resistance. To visualize these hits, the strength of the enrichment is plotted against the p-value of its significance in a volcano plot (Fig 5). Those genes that provide sensitivity can include the molecular target, as partial reduction in the levels of the receptor often make the cell more sensitive to treatment. Conversely, resistance genes can indicate mechanisms for the cell to reduce compound efficacy. Strikingly, we identified the Hsp70 paralogs, mtHsp70/HSPA9 and BiP/HSPA5 as strong sensitivity genes (Fig. 5). Specifically, reducing the expression of these Hsp70s made the cells significantly more sensitive to JG-294. The identification of mtHsp70/HSPA9 was consistent with the observation that this paralog is an important target of the parent molecule, MKT-07735, while BiP/HSPA5 has also been reported as a good target in breast cancer46. The cytoplasmic orthologs, Hsc70/HSPA8 and Hsp72/HSPA1, were not identified by this approach, but it is known that both must be simultaneously knocked down to cause cell death,9 so they would be less likely to emerge from this study because the infection conditions were selected to favor only one knockdown per cell.
Figure 5:
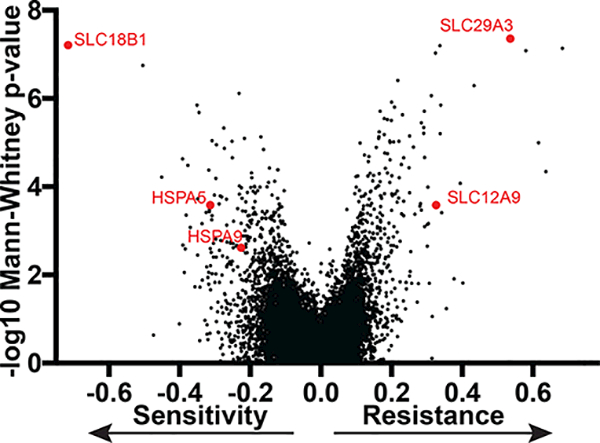
A genome-scale CRISPRi screen determines mechanisms of cellular response to JG-294 in K562 cells with stable expression of dCas9-CRAB. Gene knockdown either promoted or inhibited cell growth in the presence of JG-294 were identified. Each dot is the drug phenotype for a gene. The data is normalized and scaled as previously described.
In addition to the Hsp70s, this screen also revealed a number of other genes that impacted JG-294 activity. We were especially intrigued by the observation that repression of the transporters, SLC29A3 and SLC12A9, rendered cells resistant to JG-294, suggesting that the compound may require active transport. A number of the other top resistance genes were also transporters; for example, PQLC2 (glucose transporter) and ATP1A1 (cation transporter) (see Suppl. Table 1). Similarly, repression of the transporter, SLC18B1, increased sensitivity to JG-294, suggesting that it may be involved in efflux. Together, these studies support Hsp70s as important cellular targets and suggest possible mechanisms for inhibitor transport.
Animal pharmacokinetics and toxicity.
One of the key problems limiting use of JG-98 as a chemical probe in vivo is its relatively poor pharmacokinetics in animals.39 Accordingly, we wanted to explore the pharmacokinetics of JG-194, JG-231, JG-294 and JG-345 in mice. In the first experiments, we attempted oral administration in NSG mice, but failed to identify compound in plasma (data not shown). Moving to intraperitoneal (i.p.) injections, we performed single dose (5 mg/kg) injections, collected blood samples at 1, 2, 6 and 24 hours and measured the concentration in plasma by LC/MS/MS. This study suggested that the plasma levels were above EC50 values for ~1 or 2 hours after treatment (Table 5), while the half-life of JG-194, JG-231 and JG-294 were calculated to be approximately 20 hours or longer. Interestingly, although JG-345 was intended as an ester prodrug, the parent compound had a similar half-life to the other analogs. Although these exposures were modest, the AUC values were a significant improvement on JG-98.39 Next, we performed a maximum tolerated dose (MTD) study. In this experiment, we dosed NSG mice at 5 mg/kg, four times per week for 2 weeks. More frequent dosing was found to be toxic (data not shown), but, under these conditions, the compounds were tolerated, as judged by body weight (Suppl. Fig. 3). Although more work is needed to optimize solubility and formulation, we concluded that JG-231 and JG-345 could be used as proof-of-concept (POC) molecules in animals.
Table 5:
Pharmacokinetics of JG-98 analogs
| Pharmacokinetics Parameters | JG-98* | JG-194 | JG-231 | JG-294 | JG-345 |
|---|---|---|---|---|---|
| Cmax / nM | 73 | 64 | 159 | 58 | 87 |
| Tmax / h | 1 | 2 | 2 | 1 | 1 |
| t1/2 / h | 23 | >24 | 21.6 | >24 | 11.4 |
| AUC0 - ∞/ nM ×h | 1667 | 5074 | 3401 | 2429 | 1524 |
3mg/kg; from Li et al. 2015 Mol. Cancer Ther. 14:642–648.
JG-231 is active in breast cancer xenograft models.
To test whether JG-231 could engage with Hsp70 in vivo, it was administered i.p. for 3 consecutive days at 5 mg/kg in NSG mice bearing xenografted MCF7 cells. Three hours after the last dose, tumors were collected and homogenized. Western blots of these tumor lysates mirrored what we observed in cultured MCF7 cells; specifically, the levels of Akt and HuR decreased and the levels of Hsp70 and Hsp90 did not (Fig. 6), suggesting that Hsp70s were inhibited in vivo.
Figure 6:
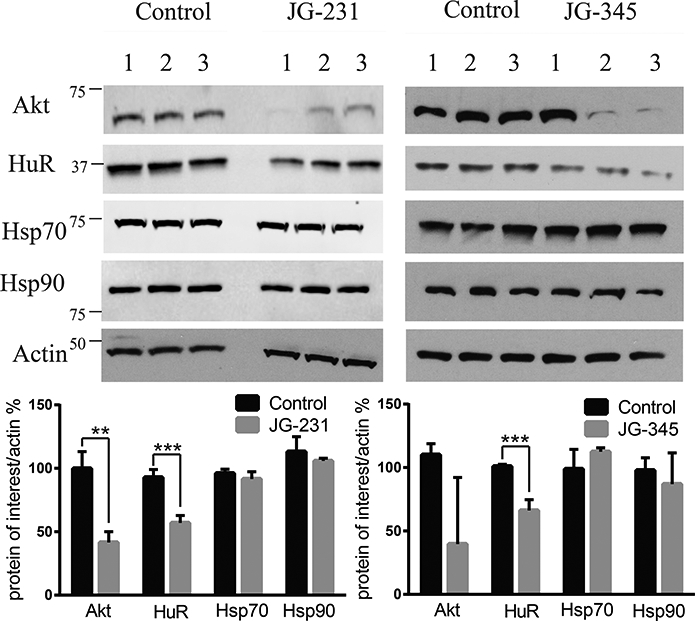
JG-231 and JG-345 show evidence of Hsp70 target engagement in vivo. MCF-7 xenografts were established in NSG mice treated for 3 days with JG-231, JG-345 or vehicle control. (See text). Tumors were harvested and blotted for Akt and HuR. Results are representative of at least two independent experiments.Quantification of band intensities was performed using ImageJ. ** indicates p < 0.05, *** p < 0.005.
Next, we turned to MDA-MB-231 xenograft models to begin probing the suitability of Hsp70 as a target in TNBC. Importantly, we considered this experiment to be a pilot study because the formulation and dosing was not yet optimized. Briefly, xenografts were first established in nude mice and, after tumors reached 100 mm3, JG-231 or vehicle was administered i.p. (4 mg/kg three days a week) for 4 weeks and then animals were followed for an additional week (arrows in Fig. 7a denote when animals were treated). The MDA-MB-231 tumors grew slowly in our hands, making effects of JG-231 on growth somewhat difficult to conclusively measure. However, relative to vehicle control animals, tumor growth was slowed in mice treated with JG-231 (Figs. 7a and 7b). Consistent with the MTD study in NSG mice, no overt changes in body weight were observed (Suppl. Fig 3b). These data provide proof of concept that Hsp70 may be a promising target for the treatment of TNBC (see below).
Figure 7.
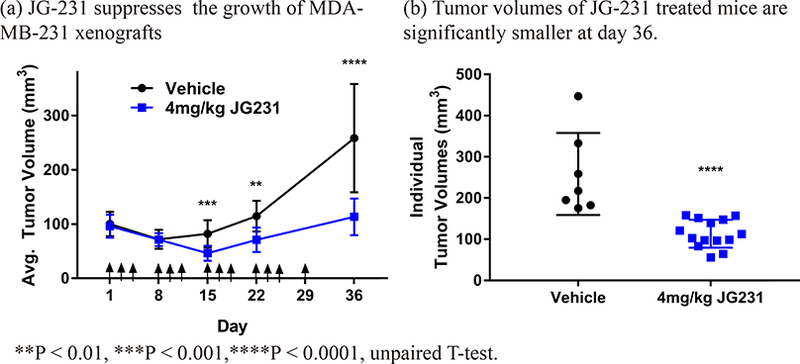
JG-231 is efficacious in triple negative breast cancer MDA-MB-231 xenografts. (a) JG-231 shows efficacy in MDA-MB-231 xenografted mice. Mice were treated with JG-231 for 4 weeks. Arrows indicate treatments. Vehicle group (n = 7), JG-231 treated group (n=14). (b) Tumor volume of individual mice treated with JG-231 was compared with vehicle group at day 36.
CONCLUSIONS
In this work, we advanced the allosteric Hsp70 inhibitor, JG-98, through structure-guided design. The best of the resulting compounds, such as JG-231 and JG-294, have mid-nanomolar potency and are relatively less toxic to non-transformed cells. Importantly, they seem to bind the expected allosteric site on Hsp70, interrupt the Hsp70-Bag3 interaction in vitro and in cells and destabilize known Hsp70 clients in cells and animals. This profile included reductions in IAP1, which has recently been shown to be responsive to inhibition of Hsp70 but not Hsp90.5, 47 Most strikingly, whole genome CRISPRi studies support the Hsp70 family members as putative targets. In that experiment, we also made the unexpected finding that SLC-family transporters seem to be important modulators of compound activity. Specifically, it seems that molecules of this series may utilize SLC29A3 and SLC12A9 for uptake and that SLC18B1 may be involved in its efflux.56 These particular solute carriers are not widely studied (SLC29A3 is annotated as a nucleotide transporter and SLC12A9 as a cation-chloride transporter), so additional studies will be required to verify these ideas. We consider it possible that the differential potency of the JG-98 analogs studied here might arise, in part, from their differential interactions with these transporters. In other words, compounds might be more/less active in cells due to their relative import/efflux kinetics,57 as well as their target engagement. More broadly, these results suggest that whole genome methods may be powerful ways of characterizing transport (and potential resistance) mechanisms. To our knowledge, CRISPRi has not yet been widely adopted as a method for unbiased target identification but our experience and recent literature reports53, 58 suggest that it could emerge as an unusually impactful platform to identify direct targets, putative off-targets and transport mechanisms51, 52. Together, these studies advance JG-231 and its analogs as POC chemical probes for studying Hsp70’s roles in cancer and other diseases.
While these studies represent an advance, it is important to be clear about the remaining hurdles. Firstly, although the benzothiazole-rhodacyanine pharmacophore is not regarded to be as problematic as the related rhodanines,59 it is still not preferred because it is planar, light sensitive and relatively difficult to formulate. Indeed, we found that solubility was a major issue for the in vivo experiments because the dose frequency seemed to be limited, in part, by precipitation. Optimization of the formulation is planned, which may improve the dosing of JG-231 and its analogs, allowing them to be used in a wider range of experiments. However, despite these challenges, we hoped that pursuing this series might produce chemical probes and that the SAR might inform future scaffold-hopping efforts by exploring the features of the allosteric binding pocket. For example, recent work has suggested alternative scaffolds that seem to bind the same site.60
Many studies have suggested that members of the Hsp70 family would be good drug targets for TNBC.61 Indeed, our results strongly support this idea. Why would breast cancer cells be more sensitive to inhibition of the Hsp70-BAG interaction? Recent studies have suggested that Hsp70 is selectively assembled into a larger chaperone complex in TNBC cells.10 This complex seems to be important for maintaining oncoproteins in an environment of high mutational burden, genomic instability and rapid growth rate, which likely puts stress on the protein homeostasis system.10 In addition, Hsp70-BAG3 complexes seem to play a role in tumor initiation and stromal cell interactions40, through remodeling a signaling pathway that is distinct from that in normal cells.33 Indeed, such observations have led to the hypothesis that Hsp70 and other chaperones represent a non-oncogene addiction.1 Thus, inhibitors that selectively target PPIs, rather than individual chaperones themselves, might represent an interesting way to target proteins that might otherwise be considered “housekeeping” factors.
EXPERIMENTAL SECTION
Chemistry.
Chemical reagents and solvents were obtained from commercial sources. When necessary, solvents were dried and/or purified by standard methods. 1H-NMR and 13C-NMR spectra were obtained using a Bruker 400 or 500 Ultrashield™ spectrometer. These were analyzed using the Bruker TOPSPIN program. Chemical shifts are reported in parts per million relative to the solvent chemical shift. Coupling constants (J) are quoted to the nearest 0.1 Hz. The following abbreviations are used: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; dd, doublet of doublets and td, triplet of doublets. Mass spectra were obtained using the Thermo Scientific™ MSQ Plus™ single quadrupole mass spectrometer. Purity for final compounds was greater than 95% and was measured using Agilent 1200 series high performance liquid chromatography with ZORBAX Eclipse XDB-C18 column (50 × 4.60 mm, product # 927975–902), UV detector at 214 and 254 nm, using system: 5 % ACN containing 0.1% TFA for 0.3 min, followed by linear gradient 5 – 95 % ACN over 3 min and 95% ACN over 1 min at a flow rate of 1.50 mL/min. Characterization by NMR and MS can be found in the Supporting Information.
Molecular modeling.
Compounds were docked to bovine HSPA8 NBD crystal structure (PDB accession code: 3HSC) using both InducedFit and Glide docking software (Software Suite 2017–3, Schrodinger Inc).62 The nucleotide binding domain of HSPA is 100% identical between bovine and human, so this model was considered to be informative of the human target. Prior to docking, we optimized the protein structure using Protein Preparation Wizard by removing water molecules, sodium and an inorganic phosphate ions, leaving the ADP and magnesium ion. In addition, hydrogen atoms were added and the protonation states of titratable residues were adjusted, allowing the overall structure to be energy minimized. In that process, heavy atoms were not allowed to move beyond 0.5 Å from their starting positions. Consistent with previous findings,36 the allosteric binding site was partially occluded in the crystal structure; requiring InducedFit docking. Briefly, the binding pocket was defined using residues: Thr13, Lys71, Tyr149, Glu175, Asp199 and Thr204, in addition to the position of ADP. This region was based on NMR chemical shift perturbations (CSPs) and contacts from NOE constraints.34, 36, 37After docking of JG-98, the remaining molecules were docked using Glide with extra-precision (XP). All inhibitors were built using Edit/Build panel of Maestro (Schrodinger Inc) and energy minimized using LigPrep software (v4.3016, Schrodinger Inc).
Solubility.
Aqueous solubility and mouse liver microsome stability measurements were performed as previously described.37, 63 Solubility assay: Eight two-fold serial dilutions of the test compound with an initial concentration at 10 mM were prepared in DMSO. 1 μL of the compound solution is added to 199 μL phosphate -buffered saline (pH 7.4) buffer and the final compound concentrations range between 3.90 μM and 500 μM. After 24 h incubation, the absorbance is measured at 620 nm and the kinetic solubility is estimated from the concentration of the test compound at which has a higher absorbance than the background.
Microsome stability.
4 μL of 100 μM compound was added to 10 μL mouse liver microsomes (20 mg/mL) in 366 μL 0.1 M phosphate buffer containing 3.3 mM MgCl2 and warmed at 37 °C for 3 min. Then 20 μL pre-warmed β-NADPH solution (20 mM in 0.1 M phosphate buffer with 3.3 mM MgCl2) was added to initiate the reaction at 37 °C. Aliquots of 40 μL of the mixture were collected at 0, 5, 10, 15, 30, 60 min, and then proteins were precipitated with 120 μL of ice-cold acetonitrile containing the internal standard YM-08 (100 ng/ml). The supernatants were collected after centrifugation at 13,000 rpm for 10 min and then subjected to LC-MS/MS analysis. The natural log of the amount of compound remaining was plotted against the time to determine the half-life of the tested compound.
Flow cytometry protein interaction assay (FCPIA).
Biotinylated Hsc70 on streptavidin coated beads (Spherotech) was incubated with compound and Alexa-Fluor® 488 labeled BAG1 (50 nM) in buffer A (25 mM HEPES, 5 mM MgCl2, 10 mM KCl, 0.3% Tween-20 pH 7.5). Plates were incubated for 15 minutes then analyzed using an Accuri® C6 Flow Cytometer. DMSO was used as a negative control and excess unlabeled Hsc70 (1 μM) was used as a positive control. Inhibition curves were calculated using Prism v.6.0c (GraphPad).
Co-immunoprecipitations.
MCF7 cell extracts were prepared in M-PER lysis buffer (Thermo Scientific) and adjusted to 5 mg of total protein in 1 mL of extract. Equal 500 μL samples were incubated with either a rabbit polyclonal for Hsp70 or Goat IgG. Samples received 5 μL of DMSO or JG-231 (10 μM). Samples were gently rotated overnight at 4 °C, followed by a 4 hr incubation with protein A/G-Sepharose Beads (Santa Cruz). The immune-complexes were then subjected to centrifugation at 1000 xg, washed with PBS pH 7.4, and eluted with SDS loading dye. Samples were separated on a 4–15% Tris-Tricine gel (Bio-Rad) and transferred to nitrocellulose membrane. The membranes were blocked in nonfat milk (5% milk in TBS, 0.1% Tween) for 1 hr, incubated with primary antibodies for Hsp70 and BAG3 overnight at 4 °C, washed, and then incubated with a horseradish peroxidase-conjugated secondary antibody (Anaspec) for 1 hr. Finally, membranes were developed using chemiluminescence (Thermo Scientific, Supersignal® West Pico).
Protein expression.
Hsp70 and its mutants were purified as previously described.64
Binding assay.
Hsc70 solutions (25 μL) in buffer (25 mM HEPES, 5 mM MgCl2, 10 mM KCl, pH 7.5) were added to Corning 384-well black, low volume plates. Then, 1 μL of test compound in DMSO was added and the solution incubated at room temperature for ~ 2 hours. Fluorescence readings of the compound’s intrinsic fluorescence were measured (Excitation 470 nM; Emission 580 nM) and quenching data were analyzed and fit using Prism v.6.0c (GraphPad).
Cell culture.
MCF-7, MDA-MB-231, MEF and cells were obtained from ATCC and cultured in DMEM (Corning) supplemented with 10% FBS, 100 units/mL streptomycin and 100 μg/mL penicillin. K562 cells were grown in RPMI-1640 with 25Mm HEPES and 2.0 g/L NaHCO3 in 10 % FBS, 2 mM glutamine, 100 units/mL streptomycin and 100 μg/mL penicillin. Cell authentication was performed by ATCC using short tandem repeat analysis. All cells were maintained in a 37 ºC and 5% CO2 humidified incubator, used for < 20 passages following thawing, and cultured for no longer than 3 months.
Cell viability assay.
MCF-7, MDA-MB-231 and MEF viabilities were determined by MTT cell proliferation assay kit from ATCC (ATCC number: 30–1010 K). EC50 values were derived from dose-response curves plotted and fitted using Prism v.6.0c (GraphPad).
High throughput pooled CRISPRi screening.
A chronic myeloid leukemia (K562) cell line stably expressing dCas9-KRAB was infected with a CRISPRi sgRNA library.54 These cells were used because they have been shown to be readily infected and high efficiency and they have stable expression of dCas9-KRAB. The genomic DNA from a population of these cells were harvested at the onset of the experiment (t0) and used as a comparison with the subsequent timepoint. Half the remaining cells were treated with JG-294 at the EC80 value (0.25 μM) and the two populations grown for thirteen days. At each passage, control and drug treated populations of cells were maintained such that there was >1000 cells per sgRNA. At the end of the seven days, genomic DNA was harvested. Then, the cassette encoding the sgRNA was amplified by PCR from the t0, t13 (untreated) and t13 (JG-294 treated) samples. The relative sgRNA abundance was determined by next generation sequencing as previously described.53, 55 Hit genes were ranked based on average phenotype of the 3 most extreme sgRNAs targeting them or by a Mann-Whitney test. Pathways and gene sets enriched among hit genes were identified using GSEA and DAVID software. A python script for processing the CRISPRi library sequencing data is available at https://github.com/mhorlbeck/ScreenProcessing.
Caspase-3/7 assay.
Activity of caspase 3/7 was measured in MCF-7 cells using the Apo-ONE® homogeneous caspase-3/7 assay kit (Promega, G7790). Method as described from the kit.
Western blots
Western blotting was performed as described previously.39 Antibodies used were as follows: Antibodies: The antibodies were purchased from Enzo: c-IAP1 (ALX-803– 335); Santa Cruz Biotechnology: Hsp90 (sc-13119), Hsp70/Hsc70 (sc-137239), anti-rat IgG-HRP (sc-2006); Cell Signaling: Akt-1 (9272), CDK4 (12790), c-Raf (9422), anti-mouse IgG-HRP (7076), anti-rabbit IgG-HRP (7074).
Animal toxicity study.
The animal experiments were carried out in accordance with guidelines of the UCSF Animal Care and Use Committee. NSG mice, which are often used for multiple myeloma studies, were dosed four times a week (Monday and Thursday) for 2 weeks with tested compounds intraperitoneally. Body weights were measured every 2 days and mice were closely monitored for any signs of toxicity or discomfort. The formulation is (10% DMSO, 18% Tween80 [Sigma], 3.6% dextrose, 68.4% 1 M HEPES).
Animal pharmacokinetics.
The animal experiments were carried out in accordance with guidelines of the UCSF Animal Care and Use Committee. NSG mice (three per group) were dosed with tested compounds or vehicle solution intraperitoneally. Blood samples were collected for plasma through tail vein at 1, 2, 6, and 24-hour time points. Compounds concentrations in plasma were determined by LC-MS-MS, using a published protocol.63
Xenografts.
The animal experiments were carried out in accordance with guidelines of the NIH Animal Care and Use Committee. Four-week-old female athymic mice (Charles River) were allowed to mature and acclimate for 2 weeks. To establish breast cancer xenografts, 2×106 low passage MDA-MB-231 cells in a 1:4 mixture of media/matrigel (v/v; BD Biosciences) were injected into the right flank of each mouse. Once tumors reached a volume of 100 mm3, mice were randomized into groups receiving either vehicle (n=7) or 4 mg/kg JG231 (n=14). Both groups were treated every other day by intraperitoneal (IP) injection for up to 4 weeks, followed by a 1 week recovery period. Tumor volumes were measured weekly and mouse body weights were measured every other day for accurate dosing.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by grants from the National Institutes of Health (R01NS059690) and the Tau Consortium.
ABBREVIATIONS USED
- AUC
area under the plasma concentration-time curve
- BAG
Bcl-2 associated anthanogene
- EC50
half maximal effective concentration
- Cmax
maximum observed plasma concentration
- Hsc70
heat shock cognate protein 70
- Hsp70
heat shock protein 70
- i.p
intraperitoneal
- Kd
dissociation constant
- MEF
mouse embryonic fibroblast
- MTD
maximum tolerated dose
- NEF
nucleotide exchange factor
- PK
pharmacokinetics
- PD
pharmacodynamics
- PPI
protein-protein interaction
- SAR
structure-activity relationship
- t1/2
elimination half-life
- TNBC
triple negative breast cancer
- CRISPR
clustered regularly interspaced short palindromic repeats
Footnotes
CONFLICTS
MPJ is a consultant to and shareholder of Schrodinger LLC, which licenses software used in this work.
SUPPORTING INFORMATION
The Supporting Information is available free of charge on the ACS Publications website. This material includes compound characterization (1H NMR, HPLC and ESI-MS), docking orientations, caspase activation results, body weight measurements in treated mice, HPLC traces for purity, table of top genes from the CRISPR screen and molecular strings formula.
REFERENCES
- 1.Luo J; Solimini NL; Elledge SJ Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 2009, 136, 823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murphy ME The HSP70 family and cancer. Carcinogenesis 2013, 34, 1181–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sherman MY; Gabai VL Hsp70 in cancer: back to the future. Oncogene 2015, 34, 4153–4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calderwood SK; Gong J Heat shock proteins promote cancer: It’s a protection racket. Trends Biochem Sci 2016, 41, 311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Srinivasan SR; Cesa LC; Li X; Julien O; Zhuang M; Shao H; Chung J; Maillard I; Wells JA; Duckett CS; Gestwicki JE Heat shock protein 70 (Hsp70) suppresses RIP1-dependent apoptotic and necroptotic cascades. Mol Cancer Res 2017, 16 58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Powers MV; Workman P Inhibitors of the heat shock response: biology and pharmacology. FEBS Lett 2007, 581, 3758–3769. [DOI] [PubMed] [Google Scholar]
- 7.Powers MV; Jones K; Barillari C; Westwood I; van Montfort RL; Workman P Targeting HSP70: the second potentially druggable heat shock protein and molecular chaperone? Cell Cycle 2010, 9, 1542–1550. [DOI] [PubMed] [Google Scholar]
- 8.Taldone T; Ochiana SO; Patel PD; Chiosis G Selective targeting of the stress chaperome as a therapeutic strategy. Trends Pharmacol Sci 2014, 35, 48–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Powers MV; Clarke PA; Workman P Dual targeting of HSC70 and HSP72 inhibits HSP90 function and induces tumor-specific apoptosis. Cancer Cell 2008, 14, 250–262. [DOI] [PubMed] [Google Scholar]
- 10.Rodina A; Wang T; Yan P; Gomes ED; Dunphy MPS; Pillarsetty N; Koren J; Gerecitano JF; Taldone T; Zong H; Caldas-Lopes E; Alpaugh M; Corben A; Riolo M; Beattie B; Pressl C; Peter RI; Xu C; Trondl R; Patel HJ; Shimizu F; Bolaender A; Yang C; Panchal P; Farooq MF; Kishinevsky S; Modi S; Lin O; Chu F; Patil S; Erdjument-Bromage H; Zanzonico P; Hudis C; Studer L; Roboz GJ; Cesarman E; Cerchietti L; Levine R; Melnick A; Larson SM; Lewis JS; Guzman ML; Chiosis G The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 2016, 538, 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Evans CG; Chang L; Gestwicki JE Heat Shock Protein 70 (Hsp70) as an emerging drug target. J Med Chem 2010, 53, 4585–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zuiderweg ER; Bertelsen EB; Rousaki A; Mayer MP; Gestwicki JE; Ahmad A Allostery in the Hsp70 chaperone proteins. Top Curr Chem 2013, 328, 99–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mayer MP Hsp70 chaperone dynamics and molecular mechanism. Trends Biochem Sci 2013, 38, 507–514. [DOI] [PubMed] [Google Scholar]
- 14.Zhuravleva A; Gierasch LM Substrate-binding domain conformational dynamics mediate Hsp70 allostery. Proc Natl Acad Sci U S A 2015, 112, E2865–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swain JF; Dinler G; Sivendran R; Montgomery DL; Stotz M; Gierasch LM Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Mol Cell 2007, 26, 27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vogel M; Mayer MP; Bukau B Allosteric regulation of Hsp70 chaperones involves a conserved interdomain linker. J Biol Chem 2006, 281, 38705–38711. [DOI] [PubMed] [Google Scholar]
- 17.English CA; Sherman W; Meng WL; Gierasch LM The Hsp70 interdomain linker is a dynamic switch that enables allosteric communication between two structured domains. J Biol Chem 2017, 292, 14765–14774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qi RF; Sarbeng EB; Liu Q; Le KQ; Xu XP; Xu HY; Yang J; Wong JL; Vorvis C; Hendrickson WA; Zhou L; Liu QL Allosteric opening of the polypeptide-binding site when an Hsp70 binds ATP. Nat Struct Mol Biol 2013, 20, 900–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Williamson DS; Borgognoni J; Clay A; Daniels Z; Dokurno P; Drysdale MJ; Foloppe N; Francis GL; Graham CJ; Howes R; Macias AT; Murray JB; Parsons R; Shaw T; Surgenor AE; Terry L; Wang YK; Wood M; Massey AJ Novel adenosine-derived Inhibitors of 70 kDa Heat Shock protein, discovered through structure-based design. J Med Chem 2009, 52, 1510–1513. [DOI] [PubMed] [Google Scholar]
- 20.Macias AT; Williamson DS; Allen N; Borgognoni J; Clay A; Daniels Z; Dokurno P; Drysdale MJ; Francis GL; Graham CJ; Howes R; Matassova N; Murray JB; Parsons R; Shaw T; Surgenor AE; Terry L; Wang Y; Wood M; Massey AJ Adenosine-derived inhibitors of 78 kDa glucose regulated protein (Grp78) ATPase: insights into isoform selectivity. J Med Chem 2011, 54, 4034–4041. [DOI] [PubMed] [Google Scholar]
- 21.Williams DR; Ko SK; Park S; Lee MR; Shin I An apoptosis-inducing small molecule that binds to heat shock protein 70. Angewandte Chemie-International Edition 2008, 47, 7466–7469. [DOI] [PubMed] [Google Scholar]
- 22. Cho HJ; Gee HY; Baek KH; Ko SK; Park JM; Lee H; Kim ND; Lee MG; Shin I A small molecule that binds to an ATPase domain of Hsc70 promotes membrane trafficking of mutant cystic fibrosis transmembrane conductance regulator. J Am Chem Soc 2011, 133, 20267–20276. [DOI] [PubMed] [Google Scholar]
- 23.Pettinger J; Le Bihan YV; Widya M; van Montfort RLM; Jones K; Cheeseman MD An irreversible inhibitor of HSP72 that unexpectedly targets lysine-56. Angewandte Chemie-International Edition 2017, 56, 3536–3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodina A; Patel PD; Kang YL; Patel Y; Baaklini I; Wong MJH; Taldone T; Yan PR; Yang CH; Maharaj R; Gozman A; Patel MR; Patel HJ; Chirico W; Erdjument-Bromage H; Talele TT; Young JC; Chiosis G Identification of an allosteric pocket on human Hsp70 reveals a mode of inhibition of this therapeutically important protein. Chem Biol 2013, 20, 1469–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taldone T; Kang YL; Patel HJ; Patel MR; Patel PD; Rodina A; Patel Y; Gozman A; Maharaj R; Clement CC; Lu A; Young JC; Chiosis G Heat shock protein 70 Inhibitors. 2. 2,5 ‘-thiodipyrimidines, 5-(phenylthio)pyrimidines, 2-(pyridin-3-ylthio)pyrimidines, and 3-(phenylthio)pyridines as reversible binders to an allosteric site on heat shock protein 70. J Med Chem 2014, 57, 1208–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang YL; Taldone T; Patel HJ; Patel PD; Rodina A; Gozman A; Maharaj R; Clement CC; Patel MR; Brodsky JL; Young JC; Chiosis G Heat shock protein 70 Inhibitors. 1. 2,5 ‘-thiodipyrimidine and 5-(phenylthio)pyrimidine acrylamides as irreversible binders to an allosteric site on heat shock protein 70. J Med Chem 2014, 57, 1188–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leu JI; Pimkina J; Frank A; Murphy ME; George DL A small molecule inhibitor of inducible heat shock protein 70. Mol Cell 2009, 36, 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hassan AQ; Kirby CA; Zhou W; Schuhmann T; Kityk R; Kipp DR; Baird J; Chen J; Chen Y; Chung F; Hoepfner D; Movva NR; Pagliarini R; Petersen F; Quinn C; Quinn D; Riedl R; Schmitt EK; Schitter A; Stams T; Studer C; Fortin PD; Mayer MP; Sadlish H The novolactone natural product disrupts the allosteric regulation of Hsp70. Chem Biol 2015, 22, 87–97. [DOI] [PubMed] [Google Scholar]
- 29.Leu JI; Zhang P; Murphy ME; Marmorstein R; George DL Structural basis for the inhibition of HSP70 and DnaK chaperones by small-molecule targeting of a C-terminal allosteric pocket. ACS Chem Biol 2014, 9, 2508–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Assimon VA; Gillies AT; Rauch JN; Gestwicki JE Hsp70 protein complexes as drug targets. Curr Pharm Design 2013, 19, 404–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bracher A; Verghese J The nucleotide exchange factors of Hsp70 molecular chaperones. Front Mol Biosci 2015, 2, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mayer MP; Bukau B Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci 2005, 62, 670–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Colvin TA; Gabai VL; Gong J; Calderwood SK; Li H; Gummuluru S; Matchuk ON; Smirnova SG; Orlova NV; Zamulaeva IA; Garcia-Marcos M; Li X; Young ZT; Rauch JN; Gestwicki JE; Takayama S; Sherman MY Hsp70-Bag3 interactions regulate cancer-related signaling networks. Cancer Res 2014, 74, 4731–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Young ZT; Rauch JN; Assimon VA; Jinwal UK; Ahn M; Li X; Dunyak BM; Ahmad A; Carlson GA; Srinivasan SR; Zuiderweg ER; Dickey CA; Gestwicki JE Stabilizing the Hsp70-tau complex promotes turnover in models of tauopathy. Cell Chem Biol 2016, 23, 992–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wadhwa R; Sugihara T; Yoshida A; Nomura H; Reddel RR; Simpson R; Maruta H; Kaul SC Selective toxicity of MKT-077 to cancer cells is mediated by its binding to the hsp70 family protein mot-2 and reactivation of p53 function. Cancer Research 2000, 60, 6818–6821. [PubMed] [Google Scholar]
- 36.Rousaki A; Miyata Y; Jinwal UK; Dickey CA; Gestwicki JE; Zuiderweg ERP Allosteric drugs: The interaction of antitumor compound MKT-077 with human Hsp70 chaperones. J Mol Biol 2011, 411, 614–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li XK; Srinivasan SR; Connarn J; Ahmad A; Young ZT; Kabza AM; Zuiderweg ERP; Sun DX; Gestwicki JE Analogues of the allosteric heat shock protein 70 (Hsp70) inhibitor, MKT-077, as anti-cancer agents. ACS Med Chem Lett 2013, 4, 1042–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Propper DJ; Braybrooke JP; Taylor DJ; Lodi R; Styles P; Cramer JA; Collins WC; Levitt NC; Talbot DC; Ganesan TS; Harris AL Phase I trial of the selective mitochondrial toxin MKT077 in chemo-resistant solid tumours. Ann Oncol 1999, 10, 923–927. [DOI] [PubMed] [Google Scholar]
- 39.Li XK; Colvin T; Rauch JN; Acosta-Alvear D; Kampmann M; Dunyak B; Hann B; Aftab BT; Murnane M; Cho M; Walter P; Weissman JS; Sherman MY; Gestwicki JE Validation of the Hsp70-Bag3 protein-protein Interaction as a potential therapeutic target in cancer. Molecular Cancer Therapeutics 2015, 14, 642–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gabai VL; Yaglom JA; Wang Y; Meng L; Shao H; Kim G; Colvin T; Gestwicki J; Sherman MY Anticancer Effects of Targeting Hsp70 in Tumor Stromal Cells. Cancer Res 2016, 76, 5926–5932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taguwa S; Maringer K; Li X; Bernal-Rubio D; Rauch JN; Gestwicki JE; Andino R; Fernandez-Sesma A; Frydman J Defining Hsp70 subnetworks in Dengue virus replication reveals key vulnerability in flavivirus infection. Cell 2015, 163, 1108–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khachatoorian R; Riahi R; Ganapathy E; Shao H; Wheatley NM; Sundberg C; Jung CL; Ruchala P; Dasgupta A; Arumugaswami V; Gestwicki JE; French SW Allosteric heat shock protein 70 inhibitors block hepatitis C virus assembly. Int J Antimicrob Agents 2016, 47, 289–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang AM; Miyata Y; Klinedinst S; Peng HM; Chua JP; Komiyama T; Li X; Morishima Y; Merry DE; Pratt WB; Osawa Y; Collins CA; Gestwicki JE; Lieberman AP Activation of Hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nat Chem Biol 2013, 9, 112–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fontaine SN; Rauch JN; Nordhues BA; Assimon VA; Stothert AR; Jinwal UK; Sabbagh JJ; Chang L; Stevens SM; Zuiderweg ERP; Gestwicki JE; Dickey CA Isoform-selective genetic inhibition of constitutive cytosolic Hsp70 activity promotes client tau degradation using an altered co-chaperone complement. J Biol Chem 2015, 290, 13115–13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yun CO; Bhargava P; Na Y; Lee JS; Ryu J; Kaul SC; Wadhwa R Relevance of mortalin to cancer cell stemness and cancer therapy. Sci Rep 2017, 7, 42016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sannino S; Brodsky JL Targeting protein quality control pathways in breast cancer. BMC Biol 2017, 15, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cesa LC; Shao H; Srinivasan SR; Tse E; Jain C; Zuiderweg ERP; Southworth DR; Mapp AK; Gestwicki JE X-linked inhibitor of apoptosis protein (XIAP) is a client of heat shock protein 70 (Hsp70) and a biomarker of its inhibition. J Biol Chem 2018, 293, 2370–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tatsuta N; Suzuki N; Mochizuki T; Koya K; Kawakami M; Shishido T; Motoji N; Kuroiwa H; Shigematsu A; Chen LB Pharmacokinetic analysis and antitumor efficacy of MKT-077, a novel antitumor agent. Cancer Chemother Pharmacol 1999, 43, 295–301. [DOI] [PubMed] [Google Scholar]
- 49.Rauch JN; Nie J; Buchholz TJ; Gestwicki JE; Kennedy RT Development of a capillary electrophoresis platform for identifying inhibitors of protein-protein interactions. Anal Chem 2013, 85, 9824–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Srinivasan SR; Cesa LC; Li X; Julien O; Zhuang M; Shao H; Chung J; Maillard I; Wells JA; Duckett CS; Gestwicki JE Heat shock protein 70 (Hsp70) suppresses RIP1-dependent apoptotic and necroptotic cascades. Mol Cancer Res 2018, 16, 58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jost M; Weissman JS CRISPR approaches to small molecule target identification. ACS Chem Biol 2018, 13, 366–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kampmann M Elucidating drug targets and mechanisms of action by genetic screens in mammalian cells. Chem Commun (Camb) 2017, 53, 7162–7167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jost M; Chen YW; Gilbert LA; Horlbeck MA; Krenning L; Menchon G; Rai A; Cho MY; Stern JJ; Prota AE; Kampmann M; Akhmanova A; Steinmetz MO; Tanenbaum ME; Weissman JS Combined CRISPRi/a-based chemical genetic screens reveal that rigosertib is a microtubule-destabilizing agent. Molecular Cell 2017, 68, 210–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gilbert LA; Horlbeck MA; Adamson B; Villalta JE; Chen Y; Whitehead EH; Guimaraes C; Panning B; Ploegh HL; Bassik MC; Qi LS; Kampmann M; Weissman JS Genome-scale CRISPR-mediated control of gene repression and activation. Cell 2014, 159, 647–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kampmann M; Bassik MC; Weissman JS Integrated platform for genome-wide screening and construction of high-density genetic interaction maps in mammalian cells. Proc Natl Acad Sci U S A 2013, 110, E2317–E2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.DeGorter MK; Xia CQ; Yang JJ; Kim RB Drug transporters in drug efficacy and toxicity. Annu Rev Pharmacol Toxicol 2012, 52, 249–73. [DOI] [PubMed] [Google Scholar]
- 57.Li Q; Shu Y Role of solute carriers in response to anticancer drugs. Mol Cell Ther 2014, 2, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.le Sage C; Lawo S; Panicker P; Scales TME; Rahman SA; Little AS; McCarthy NJ; Moore JD; Cross BCS Dual direction CRISPR transcriptional regulation screening uncovers gene networks driving drug resistance. Sci Rep 2017, 7, 17693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tomasic T; Masic LP Rhodanine as a scaffold in drug discovery: a critical review of its biological activities and mechanisms of target modulation. Expert Opinion on Drug Discovery 2012, 7, 549–560. [DOI] [PubMed] [Google Scholar]
- 60.Taylor IR; Dunyak BM; Komiyama T; Shao H; Ran X; Assimon VA; Kalyanaraman C; Rauch JN; Jacobson MP; Zuiderweg ERP; Gestwicki JE High throughput screen for inhibitors of protein-protein interactions in a reconstituted heat shock protein 70 (Hsp70) complex. J Biol Chem 2018, 293, 4014–4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Powers MV; Clarke PA; Workman P Death by chaperone: HSP90, HSP70 or both? Cell Cycle 2009, 8, 518–26. [DOI] [PubMed] [Google Scholar]
- 62.Sherman W; Day T; Jacobson MP; Friesner RA; Farid R Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem 2006, 49, 534–53. [DOI] [PubMed] [Google Scholar]
- 63.Miyata Y; Li XK; Lee HF; Jinwal UK; Srinivasan SR; Seguin SP; Young ZT; Brodsky JL; Dickey CA; Sun DX; Gestwicki JE Synthesis and initial evaluation of YM-08, a blood-brain barrier permeable derivative of the heat shock protein 70 (Hsp70) inhibitor MKT-077, which reduces tau levels. ACS Chem Neurosci 2013, 4, 930–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Assimon VA; Southworth DR; Gestwicki JE Specific binding of tetratricopeptide repeat proteins to heat shock protein 70 (Hsp70) and heat shock protein 90 (Hsp90) is regulated by affinity and phosphorylation. Biochemistry 2015, 54, 7120–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.