

ABSTRACT
Hedgehog (Hh) transduces signals by promoting cell surface accumulation and activation of the G-protein-coupled receptor (GPCR)-family protein Smoothened (Smo) in Drosophila, but the molecular mechanism underlying the regulation of Smo trafficking remains poorly understood. Here, we identified the Cul4–DDB1 E3 ubiquitin ligase complex as being essential for Smo ubiquitylation and cell surface clearance. We found that the C-terminal intracellular domain of Smo recruits Cul4–DDB1 through the β subunit of trimeric G protein (Gβ), and that Cul4–DDB1–Gβ promotes the ubiquitylation of both Smo and its binding partner G-protein-coupled-receptor kinase 2 (Gprk2) and induces the internalization and degradation of Smo. Hh dissociates Cul4–DDB1 from Smo by recruiting the catalytic subunit of protein kinase A (PKA) to phosphorylate DDB1, which disrupts its interaction with Gβ. Inactivation of the Cul4–DDB1 complex resulted in elevated Smo cell surface expression, whereas an excessive amount of Cul4–DDB1 blocked Smo accumulation and attenuated Hh pathway activation. Taken together, our study identifies an E3 ubiquitin ligase complex targeting Smo for ubiquitylation and provides new insight into how Hh signaling regulates Smo trafficking and cell surface expression.
KEY WORDS: Hedgehog, Hh, Smo, GPCR, Cul4, CRL4, DDB1, piccolo, Gβ, E3 ubiquitin ligase, PKA, GRK2, Ubiquitin, Trafficking, Signaling
Summary: The Cul4–DDB1–Gβ E3 ubiquitin ligase complex regulates Smoothened trafficking and Hedgehog signaling in Drosophila.
INTRODUCTION
Hedgehog (Hh) signaling plays essential roles in embryonic development and adult tissue homeostasis, and its deregulation has been implicated in a wide range of human disorders including congenital diseases and cancer (Briscoe and Thérond, 2013; Ingham and McMahon, 2001; Jiang and Hui, 2008; Taipale and Beachy, 2001; Villavicencio et al., 2000). Hh controls cell growth and pattern formation through a largely conserved signal transduction cascade that culminates in the activation of the latent transcription factors Cubitus interruptus (Ci) and Gli proteins (Briscoe and Thérond, 2013; Hui and Angers, 2011; Jiang and Hui, 2008; Wilson and Chuang, 2010). The Hh signal reception system contains two multi-span transmembrane proteins: the twelve-transmembrane protein Patched (Ptc) that functions as the Hh receptor, and the seven-transmembrane protein Smoothened (Smo) of the G-protein-coupled receptor (GPCR) family, which functions as the obligated signal transducer. In the absence of Hh ligand, Ptc inhibits Smo through an ill-defined mechanism. Hh binds to Ptc to release its inhibition of Smo, leading to Smo phosphorylation and activation (Chen and Jiang, 2013).
Hh regulates Smo by changing its subcellular localization and conformation. In vertebrates, Ptc restricts Smo ciliary localization and keeps Smo in an inactive conformation, whereas binding of Hh to Ptc promotes ciliary accumulation of phosphorylated Smo that adopts an active conformation (Chen et al., 2011; Corbit et al., 2005; Rohatgi et al., 2007). In Drosophila, Ptc inhibits, whereas Hh promotes, Smo cell surface accumulation and activation through regulating Smo phosphorylation (Denef et al., 2000; Jia et al., 2004; Li et al., 2014; Zhao et al., 2007). Although the mechanisms underlying the regulation of Smo trafficking and cell surface expression are still poorly understood, several recent studies revealed that ubiquitylation and subsequent degradation of Smo through both proteasome- and lysosome-dependent mechanisms are responsible to prevent Smo cell surface accumulation in the absence of Hh (Li et al., 2012; Xia et al., 2012; Yang et al., 2013). Upon stimulation, Hh inhibits Smo ubiquitylation and promotes its cell surface accumulation, at least in part, through protein kinase A (PKA)-mediated phosphorylation of Smo (Li et al., 2012; Xia et al., 2012). Furthermore, Hh induces sumoylation of Smo at amino acid (aa) residue K851 to recruit the hydrolase USP8 that antagonizes Smo ubiquitylation independent of PKA-mediated phosphorylation of Smo (Ma et al., 2016; Zhang et al., 2017). However, the E3 ubiquitin ligase(s) targeting Smo for ubiquitylation have not yet been identified. Hence, how PKA-mediated phosphorylation events promote Smo cell surface accumulation has so far remained obscure.
Previous genetic and RNA interference (RNAi) screens in Drosophila have not identified any E3 ubiquitin ligase that targets Smo for degradation owing to several reasons (Jiang and Struhl, 1995, 1998; Lum et al., 2003; Nybakken et al., 2005). First, multiple E3 ligases might be involved in targeting Smo for ubiquitylation; so, perturbation of any individual E3s might not result in a dramatic change in Smo activity. Second, even if Smo accumulated on the cell surface after inactivation of the E3 ligase(s) targeting Smo for ubiquitylation and degradation, it might not adopt the active conformation required for triggering activation of the Hh pathway. Third, Smo ubiquitylation might be catalyzed by E3 ligase(s) not dedicated for Smo, such that its inactivation would cause pleotropic defects that mask Hh-related phenotypes. Therefore, we decided to carry out an in vivo RNAi screen in wing imaginal discs, immunostaining for Smo as a readout. From this screen, we identified the cullin family member Cullin4 (Cul4) as a regulator of Smo cell surface expression. Cul4 forms multi-subunit E3 ubiquitin ligase complexes, in which DNA-damage-binding protein 1 (DDB1) bridges Cul4 to multiple DDB1-binding WD40 proteins (DWD) that recognize the substrates (Angers et al., 2006). We found that Smo recruited Cul4–DDB1 through the β subunit of trimeric G protein (Gβ) – a DWD protein – to ubiquitylate Smo and its binding partner G-protein-coupled-receptor-kinase 2 (Gprk2). Furthermore, we demonstrated that Hh inhibited Cul4-mediated ubiquitylation of Smo by recruiting PKA to phosphorylate DDB1, leading to the dissociation of Cul4–DDB1 from Smo.
RESULTS
Cul4 regulates Smo cell surface expression
During wing development, Hh is expressed in the posterior compartment, whereas Ci is expressed in the anterior compartment (Fig. 1A-A″) (Jiang and Hui, 2008). Smo is accumulated in posterior compartment cells as well as anterior (A) compartment cells adjacent to the anteroposterior (A/P) boundary in response to Hh stimulation (Fig. 1A-A″) (Denef et al., 2000). In an in vivo RNAi screen for novel regulators of Smo cell surface expression, we used the Cul4-targeting UAS-RNAi fly line VDRC #44829 crossed with the MS1096 Gal4 driver line (MS>Cul4-RNAi) to silence Cul4 expression, and found accumulation of Smo within A-compartment cells distant from the A/P boundary (Fig. 1B-B″; Fig. S1A-A″). We verified this phenotype by using two other independent Cul4 RNAi fly lines, BL #50614 and VDRC #105668 (Fig. 1C-C″; Fig. S1B-C″). In addition, knockdown of DDB1 (MS>DDB1-RNAi) also resulted in accumulation of Smo in A-compartment cells away from the A/P boundary (Fig. 1D-D″). Of note, depletion of Cul4 or DDB1 caused pleiotropic defects, such as morphological distortion of wing discs (Fig. 1 and Fig. S1A-C″) owing to the involvement of Cul4 family of E3 ligases in many cellular processes (Lee and Zhou, 2007). Although inactivation of Cul4 increased the abundancy of Smo in A-compartment cells away from the A/P boundary, examination of expression of the Hh target gene ptc indicated that Cul4 RNAi did not result in ectopic expression of ptc (Fig. S1D,E). We have previously shown that Hh not only increases Smo expression at the cell surface but also induces its active conformation to activate the downstream signaling events (Zhao et al., 2007). Lack of ectopic ptc expression in Cul4 RNAi wing discs suggested that the anteriorly accumulated Smo had adopted an inactive conformation.
Fig. 1.
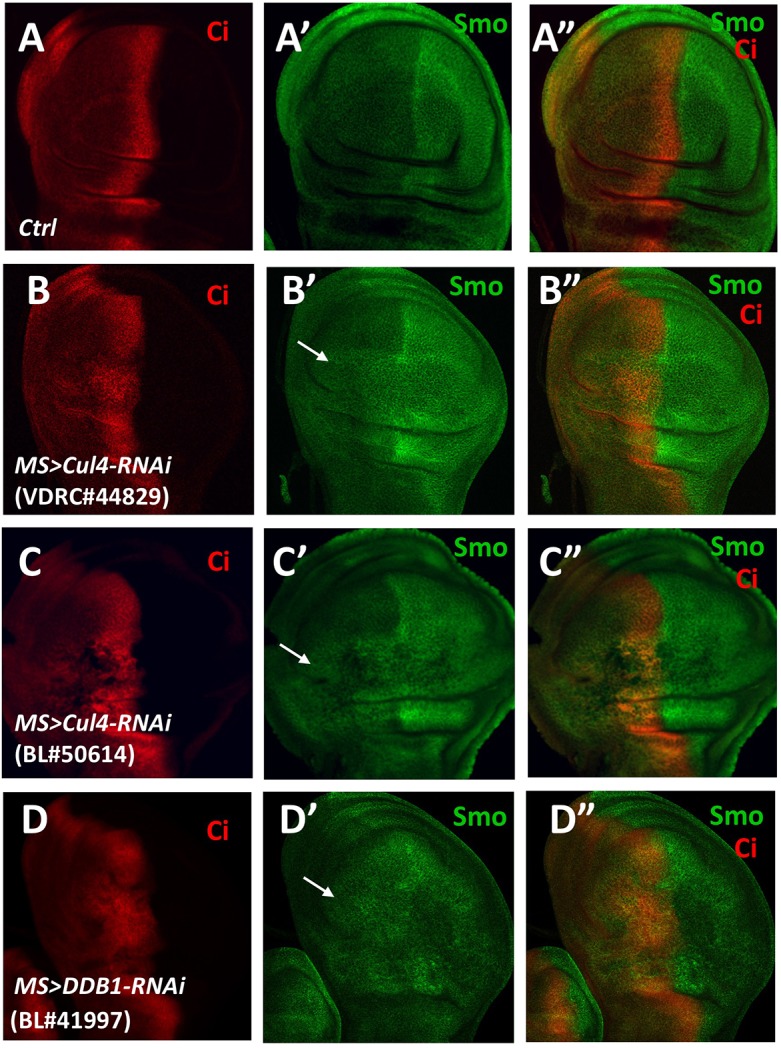
Cul4 or DDB1 knockdown causes Smo accumulation in A-compartment cells of developing wings. (A-D″) Late third control wing discs (A-A″) or wing discs expressing the UAS-Cul4-RNAi transgenes with MS1096 (B-C″) or UAS-DDB1-RNAi (D-D″) (as indicated) were immunostained for Smo (green) and Ci (red). Ci expression marks the anterior compartment. Smo ectopically accumulated in A-compartment cells distant from the A/P boundary (arrows) when Cul4 or DDB1 was knocked down.
We next examined the role of Cul4 in the regulation of Smo accumulation at the cell surface by using a cell-based assay (Jia et al., 2004; Zhao et al., 2007). For this, we generated a stable S2 cell line expressing an inducible Myc-tagged Smo transgene (Myc-Smo) under the control of the metallothionein (MT) promoter (pMT-Myc-Smo) (Li et al., 2012). Myc-Smo-expressing cells were treated with dsRNA targeting Cul4, DDB1 or both, and efficient knockdown of each gene was confirmed by quantitative RT-PCR measuring the mRNA abundance of Cul4 and DDB1 (Fig. S2). Although knockdown of Cul4 did not significantly change the surface abundance of Myc-Smo and knockdown of DDB1 only slightly increased Myc-Smo levels at the cell surface, simultaneous knockdown of Cul4 and DDB1 resulted in a significant increase of Myc-Smo at the cell surface (Fig. 2A), probably due a more-severe loss of the Cul4–DDB1 E3 ligase function.
Fig. 2.

Cul4–DDB1 binds to and ubiquitylates Smo. (A) Immunostaining with an anti-Myc antibody prior to membrane permeabilization (top) and quantification (bottom) of cell surface Myc signals in S2 cells stably expressing Myc-Smo and treated with control (Luc) or dsRNA as indicated. Data are mean±s.d. of three independent experiments. n=10 cells for each experimental condition. **P<0.01, ***P<0.001 (Student's t-test). (B) Immunoblot analysis of Myc-Smo ubiquitylation in S2 cells treated with dsRNA as indicated. (C) Immunoblot analysis of Myc-Smo ubiquitylation in S2 cells with or without co-transfection of Fg-Cul4. (D-E) Immunoblot analyses of CoIP experiments using S2 cells transfected with Smo and Fg-Cul4 (D) or Fg-DDB1 (E) constructs as indicated. Asterisks indicate monomeric forms of Myc-Smo and its C-terminal deletion mutant Myc-SmoΔC. (F) Immunoblot analysis, showing that SAID fused to Myc-tagged FZ2 (Myc-FS) enables binding of the chimera protein to Cul4–DDB1. (G) Immunoblot analysis of Myc-FZ2 and Myc-FS ubiquitylation in S2 cells with or without co-transfection of Fg-Cul4 and Fg-DDB1.
Cul4 binds to and ubiquitylates Smo
Next, we asked whether Cul4–DDB1 regulates Smo cell surface expression by targeting it for ubiquitylation. By using a cell-based ubiquitylation assay for Smo (Li et al., 2012), we found that the combined knockdown of Cul4 and DDB1 in cells expressing Myc-Smo diminished Smo ubiquitylation, whereas knockdown of either Cul4 or DDB1 alone had only a negligible effect (Fig. 2B). However, overexpression of Cul4 in S2 cells increased the ubiquitylation of coexpressed Myc-Smo (Fig. 2C). The effect that the loss of Cul4–DDB1 E3 ligase function has on Smo ubiquitylation is consistent with the effect on Smo cell surface expression (Fig. 2A), suggesting that Cul4-mediated Smo ubiquitylation promotes its downregulation. When coexpressed in S2 cells, Myc-Smo formed a complex with FLAG-tagged Cul4 and DDB1 (Fg-Cul4 and Fg-DDB1, respectively) as revealed by coimmunoprecipitation (CoIP) experiments (Fig. 2D,E). In addition, Myc-Smo interacted with endogenous Cul4 (Fig. S2). Deletion mapping revealed that Cul4–DDB1 interacted with the intracellular C-terminus of Smo (SmoC) but not with a truncated Smo version lacking the C-terminus (SmoΔC), suggesting that Cul4–DDB1 is recruited to Smo through its C-terminus.
The middle region (aa 661-818) of the Smo C-terminus contains the Smo auto-inhibitory domain (SAID), whose deletion resulted in constitutive Smo cell-surface expression and activation (Zhao et al., 2007). Deletion of the SAID from full-length Smo (SmoΔSAID) dramatically reduced, but did not completely block, the binding of Cul4 and DDB1 (Fig. 2D,E). Furthermore, fusion of SAID to the C-terminus of the Wg receptor Frizzled2 (FZ2) conferred binding of Cul4 and DDB1 to the chimeric FZ2-SAID protein (FS; Li et al., 2012) in S2 cells (Fig. 2F). Moreover, Cul4–DDB1 promoted ubiquitylation of coexpressed FS but not FZ2 (Fig. 2G). These results suggest that Cul4–DDB1 interacts with Smo mainly through the SAID in order to promote Smo ubiquitylation.
Hh inhibits Cul4-mediated Smo ubiquitylation through PKA
Hh promotes Smo cell surface accumulation mainly by inhibiting its ubiquitylation (Li et al., 2012; Xia et al., 2012). We found that the association between Myc-Smo and Fg-Cul4 or Fg-DDB1, as well as Cul4-mediated ubiquitylation of Myc-Smo, was diminished by treating transfected S2 cells with Hh-conditioned medium (Fig. 3A,B). In addition, Hh stimulation caused dissociation of endogenous Cul4 from Myc-Smo (Fig. S3).
Fig. 3.

Hh inhibits the binding of Cul4–DDB1 to Smo by recruiting PKAc. (A) Immunoblot analysis of CoIP experiments using S2 cells transfected with the indicated constructs and treated with or without Hh-conditioned medium in the absence or presence of the PKA inhibitor H89. (B) Immunoblot analysis of Myc-Smo ubiquitylation in S2 cells transfected with the indicated constructs and treated with or without Hh-conditioned medium, or H89. (C) Immunoblot analysis of CoIP experiments using S2 cells transfected with wild-type SmoWT, phosphorylation deficient (SmoSA) or phospho-mimetic Smo (SmoSD) together with Fg-Cul4 and Fg-DDB1. S2 cells were treated with the proteasome inhibitor MG132 to stabilize Smo. (D) Immunoblot analysis of CoIP experiments using S2 cells transfected with the indicated constructs and treated with or without Hh-conditioned medium. (E) Immunoblot analysis of ubiquitylation of the indicated Smo variants in S2 cells co-transfected with Cul4 and treated with or without Hh-conditioned medium. mC*-YFP, constitutively active PKA catalytic subunit tagged to YFP.
A previous study has revealed that PKA is required for Hh-induced Smo cell surface accumulation (Jia et al., 2004). Consistent with this, treatment of S2 cells with the pharmacological PKA inhibitor H89 prevented the dissociation of Fg-Cul4 and Fg-DDB1 from Myc-Smo stimulated by Hh (Fig. 3A). In addition, Hh failed to downregulate the Cul4-mediated Smo ubiquitylation in the presence of H89 (Fig. 3B). However, coexpression of a constitutively active PKA catalytic subunit (mC*) (Jiang and Struhl, 1995) resulted in the dissociation of Fg-Cul4 and Fg-DDB1 from Myc-Smo, and reduced Cul4-mediated ubiquitylation of Smo (Fig. 3A,B).
Previous studies have suggested that phosphorylation of the three clusters of PKA and CK1 phosphorylation sites within the Smo C-terminus promotes cell surface expression of Smo by inhibiting its ubiquitylation (Li et al., 2012; Xia et al., 2012). Therefore, we determined whether binding of Cul4–DDB1 to Smo is regulated by PKA- and CK1-mediated phosphorylation of the Smo C-terminus by carrying out CoIP experiments. S2 cells were transfected with Fg-Cul4 and Fg-DDB1 together with either wild-type Smo (Myc-SmoWT), phosphorylation-deficient Smo (Myc- SmoSA, comprising S667A, S687A and S740A mutation of the three PKA phosphorylation sites; Jia et al., 2004) or phospho-mimetic Smo (SmoSD, comprising mutations of the three PKA phosphorylation sites as well as mutation S670D, S673D, S690D, S693D, S743D and S746D of the six adjacent CK1 phosphorylation sites; Jia et al., 2004). To our surprise, Fg-Cul4 and Fg-DDB1 both bound to Myc-SmoWT, Myc-SmoSA and Myc-SmoSD with similar affinity (Fig. 3C). Moreover, binding of Fg-Cul4 to Myc-SmoSD, as well as Cul4-mediated ubiquitylation of Myc-SmoSD were still inhibited by Hh (Fig. 3D,E). These results suggest that Hh inhibits Cul4-mediated Smo ubiquitylation in a PKA activity-dependent manner but independently of PKA-mediated phosphorylation of the Smo C-terminus.
Our previous study has demonstrated that Hh stimulates the recruitment of the PKA catalytic subunit (PKAc) to the Smo C-terminus to promote phosphorylation of Smo and activation of the Hh pathway (Li et al., 2014). Binding of PKAc to Smo depends on three basic clusters within the membrane proximal region of the Smo C-terminus, as mutation of R565, K569, K570, K593, R594, K595, K645, R646 and R647 to A within these basic clusters (SmoRA1′2′3′) abolished PKAc binding, preventing, as a consequence, SmoRA1′2′3′ phosphorylation and activation in response to Hh (Li et al., 2014). To determine whether recruitment of PKAc to Smo is required for Hh-dependent inhibition of Cul4-mediated Smo ubiquitylation, S2 cells transfected with Myc-SmoRA1′2′3′ and Fg-Cul4 were treated with control or Hh-conditioned medium, followed by CoIP and ubiquitylation assays. We found that Hh stimulation failed to dissociate Fg-Cul4 from Myc-SmoRA1′2′3′ and no longer inhibited Cul4-mediated ubiquitylation of Myc-SmoRA1′2′3′ (Fig. 3D,E), suggesting that Hh-induced recruitment of PKAc to Smo is essential to inhibit Cul4-mediated ubiquitylation of Smo. Taken together, these observations suggest that PKA phosphorylates a component within the Smo–Cul4–DDB1 complex, leading to dissociation of Cul4–DDB1 from Smo and inhibition of Cul4-mediated Smo ubiquitylation.
Cul4 ubiquitylates Gprk2 in a manner facilitated by Smo but inhibited by Hh
Gprk2 forms a complex with Smo, and plays a dual role in the regulation of Smo activity and Hh signaling (Chen et al., 2010; Molnar et al., 2007). In the absence of Hh, Gprk2 inhibits Smo cell surface accumulation through an unknown mechanism (Chen et al., 2010; Molnar et al., 2007); however, in the presence of Hh, Gprk2 facilitates dimerization or oligomerization of the Smo C-terminus – in a manner that can be kinase activity dependent and independent – to increase Hh pathway activity (Chen et al., 2010; Li et al., 2016). Because ubiquitylation of receptor-binding proteins can also facilitate receptor internalization and degradation – as exemplified by GPCRs, whose endocytosis and desensitization involve the ubiquitylation of β-arrestin recruited to the receptors after ligand stimulation (Shenoy et al., 2001) – we speculated that Gprk2 itself can be ubiquitylated, which might contribute to its ability to downregulate Smo in the absence of Hh. Indeed, using a cell-based ubiquitylation assay, we observed that ubiquitylated forms of Gprk2 accumulated when S2 cells were treated with the proteasome inhibitor MG132 (Fig. 4A). Interestingly, Gprk2 ubiquitylation was inhibited upon Hh stimulation as well as by Smo RNAi (Fig. 4B), suggesting that Gprk2 was ubiquitylated in a manner that depending on Smo but inhibited by Hh.
Fig. 4.

Cul4–DDB1 binds to and ubiquitylates Gprk2 depending on Smo. (A) Immunoblot analysis of Myc-Gprk2 ubiquitylation in S2 cells treated with or without the proteasome inhibitor MG132. (B) Immunoblot analysis of Myc-Gprk2 ubiquitylation in S2 cells treated with or without Hh-conditioned medium or the indicated dsRNAs. (C) Immunoblot analysis of Myc-Gprk2 ubiquitylation in S2 cells treated with Luc dsRNA, Cul4 dsRNA, DDB1 dsRNA, or both Cul4 and DDB1 dsRNAs. (D) Immunoblot analysis of Myc-Gprk2 ubiquitylation in S2 transfected with Myc-Gprk2 and Fg-Cul4 and treated with or without Hh-conditioned medium or the indicated dsRNAs. (E,F) Immunoblot analyses of CoIP experiments using S2 cells expressing Myc-Gprk2 and Fg-Cul4 (E) or Fg-DDB1 (F) and treated with or without Hh-conditioned medium or the indicated dsRNAs.
We next determined whether Smo recruits Cul4–DDB1 to ubiquitylate Gprk2. Indeed, knockdown of either Cul4 or DDB1 reduced ubiquitylation of Gprk2, whereas their combined knockdown diminished Gprk2 ubiquitylation in S2 cells (Fig. 4C). Furthermore, overexpression of Cul4 increased Gprk2 ubiquitylation, which was inhibited by Hh stimulation or Smo RNAi (Fig. 4D). Co-immunoprecipitation experiments indicated that Gprk2 formed a complex with Cul4 and DDB1 in a manner that can be inhibited by Hh stimulation or Smo knockdown (Fig. 4E,F). Taken together, these results suggest that Smo-bound Cul4–DDB1 ubiquitylates not only Smo but also Gprk2, and that Hh inhibits ubiquitylation of both proteins by dissociating Cul4–DDB1 from the Smo–Gprk2 complex.
Cul4–DDB1 is recruited to Smo and Gprk2 by Gβ
Cul4–DDB is recruited to its substrates by a large family of DWD proteins that recognize the substrates (Angers et al., 2006). Interestingly, a recent study revealed that Gβ, which is also a DWD protein, formed a complex with GRK2 (the mammalian homolog of Gprk2) and targeted GRK2 for Cul4-mediated ubiquitylation and downregulation that is independent of its conventional role in the trimeric G protein complex (Zha et al., 2015). Indeed, all five Gβ proteins (Gβ1, Gβ2, Gβ3, Gβ4 and Gβ5) formed a complex with Cul4–DDB1 independently of Gγ (Zha et al., 2015). The Drosophila genome encodes three Gβ proteins: Gβ13F (CG10545), Gβ76C (CG8770) and Gβ5 (CG10763) (annotation symbol according to Flybase in parentheses). We found that knockdown of either Gβ13F or Gβ76C slightly reduced ubiquitylation of Gprk2, whereas their combined knockdown (for simplicity, hereafter referred to as Gβ RNAi) dramatically reduced it (Fig. 5A; Fig. S4). By contrast, knockdown of Gβ5 did not affect Gprk2 ubiquitylation in S2 cells, and combined knockdown of Gβ5 with either Gβ13F or Gβ76C did not have an additive effect on Gprk2 ubiquitylation (Fig. 5A; Fig. S4). Both Gβ13F and Gβ76C interacted with Gprk2 (Fig. 5B,C), and Gβ RNAi diminished the binding of Fg-Cul4 and Fg-DDB1 to Myc-Gprk2 (Fig. 5D), as well as the ability of Cul4 to promote Gprk2 ubiquitylation (Fig. 5E). Similarly, knockdown of either Gβ13F or Gβ76C slightly reduced Smo ubiquitylation whereas their combined knockdown reduced it more dramatically (Fig. 5F). In addition, Gβ RNAi inhibited the binding of Fg-Cul4 and Fg-DDB1 to Myc-Smo (Fig. 5G) and blocked Cul4-stimulated ubiquitylation of Smo (Fig. 5H). These results suggest that multiple Gβ proteins recruit Cul4–DDB1 to Smo and Gprk2, leading to their ubiquitylation.
Fig. 5.

Cul4–DDB1 binds to and ubiquitylates Smo and Gprk2 through Gβ. (A) Immunoblot analysis of Myc-Gprk2 ubiquitylation in S2 treated with the indicated Gβ dsRNAs individually or in different combinations. (B,C) Immunoblot analyses to show Myc-Gprk2 formed a complex with co-transfected Fg-Gβ13F (B) or Fg-Gβ76C (C) in S2 cells. (D) Immunoblot analysis of CoIP experiments using S2 cells transfected with Myc-Gprk2, Fg-Cul4, and Fg-DDB1 expressing constructs and treated with Luc dsRNA or Gβ13F and Gβ76C dsRNAs. (E) Immunoblot analysis of Myc-Gprk2 ubiquitylation in S2 cells transfected with the indicated constructs and treated with Luc dsRNA or Gβ13F and Gβ76C dsRNAs. (F) Immunoblot analysis of Myc-Smo ubiquitylation in S2 cells treated with control or the indicated Gβ dsRNAs. (G) Immunoblot analysis of CoIP experiments using S2 cells transfected with Myc-Smo, Fg-Cul4, and Fg-DDB1, and treated with control (luc) dsRNA or dsRNAs targeting both Gβ13F and Gβ76C. (H) Immunoblot analysis of Myc-Gprk2 ubiquitylation in S2 cells transfected with the indicated constructs and treated with Luc dsRNA or Gβ13F and Gβ76C dsRNAs. (I,J) Immunoblot analysis of CoIP experiments using S2 cells transfected with the indicated constructs and treated with the indicated dsRNA. The association of Myc-Smo with Fg-Gβ13F was not affected by Gprk2 RNAi (I) whereas the association of Myc-Gprk2 with Fg-Gβ13F was inhibited by Smo RNAi (J).
CoIP experiments showed that Gβ13F interaction with Smo was not affected by Gprk2 knockdown (Fig. 5I; Fig. S5). By contrast, Smo RNAi reduced the binding of Gβ13F to Gprk2 (Fig. 5J), which is consistent with the observation that Smo RNAi diminished the recruitment of Cul4–DDB1 to GPRK2 as well as the Cul4-mediated ubiquitylation of Gprk2. Hence, Cul4–DDB1 is recruited to the Smo–Gprk2 complex primarily through the interaction between Smo and Gβ, which leads to ubiquitylation of both Smo and Gprk2. Of note, the residual binding of Cul4–DDB1 to Smo in Gβ RNAi cells could be due to incomplete depletion of Gβ. Alternatively, Cul4–DDB1 could be recruited to the Smo–Gprk2 complex through an additional DWD.
Hh dissociates Cul4–DDB1 from Smo through PKA-mediated phosphorylation of DDB1
|Mutation of the PKAc binding sites but not of the PKA phosphorylation sites on Smo affected the ability of Hh to inhibit binding of Cul4–DDB1 to and Cul4-mediated ubiquitylation of Smo (Fig. 3D,E), which implies that PKA inhibits the recruitment of Cul4–DDB1 to Smo by phosphorylating a substrate different from Smo. It has been shown that PKA-mediated S645 phosphorylation of DDB1 enabled dissociation of Cul4–DDB1 from Gβ (Zha et al., 2015). Therefore, we hypothesized that Hh dissociates the Cul4–DDB1 E3 ligase complex from Smo–Gprk2 through PKA-mediated phosphorylation of DDB1 that disrupts its interaction with Gβ. Consistent with this, we found that Hh treatment dissociated Fg-Cul4 and Fg-DDB1 from Myc-Smo without affecting the binding of Fg-Gβ13F to Myc-Smo, and that the Hh-mediated dissociation of Cul4–DDB1 was inhibited by the PKA inhibitor H89 (Fig. 6A). In addition, association of endogenous Gβ13F to Myc-Smo was not affected by stimulation of Hh (Fig. S5). Importantly, mutation of the corresponding PKA phosphorylation site S644 in DDB1 (DDB1S644A) (S645 in mammalian DDB1) blocked dissociation of Cul4–DDB1S644A from Smo and Gprk2 in response to Hh (Hh-conditioned medium) (Fig. 6B; Fig. S6A). In addition, expression of DDB1S644A instead of DDB1 interfered with the ability of Hh to inhibit Cul4-mediated ubiquitylation of Smo and Gprk2 (Fig. 6C; Fig. S6B).
Fig. 6.

PKA-mediated phosphorylation of DDB1 dissociates Cul4–DDB1 from Smo. (A,B) Immunoblot analysis of CoIP experiments using S2 cells transfected with the indicated constructs and treated with or without Hh-conditioned medium or the PKA inhibitor H89. (C) Immunoblot analysis of Myc-Smo ubiquitylation in S2 cells transfected with the indicated constructs and treated with or without Hh-conditioned medium. (D-G′) Immunostaining of Myc epitope before cell permeabilization and of Flag epitope after cell permeabilization in Myc-Smo-expressing S2 cells transfected with Fg-DDB1WT (F,F′) or Fg-DDB1SA (G,G′) and treated without (D) or with Hh-conditioned medium (E-G′). (H) Quantification of Myc signals on the surface of the indicated S2 cells. Data are mean±s.d. from three independent experiments; n=10 cells for each experimental condition. **P<0.01, ***P<0.001 (Student's t-test). (I) ptc-luc reporter assay in cl8 cells transfected with the indicated constructs and treated with or without Hh-conditioned medium. Data are mean±s.d. from two independent experiments. n=10 cells for each experimental condition. **P<0.01, ***P<0.001 (Student's t-test).
To determine the consequences that failure to dissociate Cul4–DDB1 from Smo has on Hh signaling, Flag-tagged wild-type DDB1 (Fg-DDB1WT) or PKA phosphorylation-deficient DDB1 (Fg-DDB1S644A) was transfected into either S2 cells stably expressing Myc-Smo or cl-8 cells together with a ptc-luciferase (ptc-luc) reporter gene and with or without Fg-Cul4. The transfected cells were treated with either control or Hh-conditioned medium. We found that Hh stimulated cell surface accumulation of Myc-Smo (Fig. 6D,E), which was blocked by coexpression of Fg-DDB1S644A but not by Fg-DDB1WT (Fig. 6F-G′). Expression of Fg-DDB1S644A but not Fg-DDB1WT attenuated Hh-induced ptc-luc reporter gene expression in cl-8 cells (Fig. 6I). Coexpression of Fg-Cul4 with Fg-DDB1S644A increased the ability of Fg-DDB1S644A to inhibit the ptc-luc reporter gene expression in response to Hh, whereas coexpression of Fg-Cul4 with Fg-DDB1WT also reduced Hh-induced ptc-luc reporter gene expression, albeit to a lesser extent as coexpression of Fg-Cul4 with Fg-DDB1S644A (Fig. 6I). These results suggest that PKA-mediated phosphorylation of DDB1 is critical for Hh to be able to alleviate Cul4–DDB1-mediated downregulation of Smo expression at the cell surface and for Hh pathway activation.
Cul4–DDB1–Gβ acts in parallel with Smurf to regulate Smo cell surface expression
In a recently published study (Li et al., 2018), we have identified the Smurf family of E3 ubiquitin ligases – including Smurf, Nedd4 and Su(dx) – as Smo E3s in a cell-based RNAi screen. Whereas Smurf RNAi resulted in increased cell surface accumulation of Myc-Smo in S2 cells, combined knockdown of all three Smurf family members resulted in a more dramatic cell surface accumulation of Myc-Smo (Li et al., 2018). Interestingly, knockdown of Cul4 or Gβ (through treatment with dsRNA targeting Gβ13F and Gβ76C) also enhanced Smo cell surface accumulation caused by Smurf RNAi (Fig. S7) (Li et al., 2018), suggesting that the Cul4–DDB1–Gβ complex acts in parallel to the Smurf family of E3s in order to control Smo trafficking and cell surface expression.
DISCUSSION
Hedgehog (Hh) transduces signals by promoting Smo accumulation at the cell surface and Smo active conformation. It has been shown before that ubiquitylation of Smo regulates its trafficking and cell surface expression; however, the E3 ligase(s) that target Smo for ubiquitylation have remained unknown. In an in vivo RNAi screen for E3s that regulates Smo accumulation in wing imaginal discs, we identified Cul4–DDB1 as being essential to prevent Smo accumulation in A-compartment cells away from the A/P boundary. Our biochemical experiments demonstrated that Cul4 and DDB1 were recruited to Smo and Gprk2 through multiple Gβ subunits, and that the Cul4–DDB1–Gβ E3 ubiquitin ligase complex ubiquitylated both Smo and Gprk2, leading to reduced Smo expression at the cell surface. Furthermore, we found that Hh recruited PKAc to the Smo-bound Cul4–DDB1 to phosphorylate DDB1, causing dissociation of Cul4–DDB1 from the Smo–Gβ complex and inhibition of Cul4-mediated Smo ubiquitylation (Fig. 7). Hence, our study identified an E3 ubiquitin ligase complex that is targeting Smo, and shed light onto how Hh regulates Smo trafficking and cell surface expression.
Fig. 7.

Regulation of Smo cell surface expression by a Cul4–DDB1–Gβ E3 ubiquitin ligase complex. In the absence of Hh, Cul4–DDB1 is recruited to Smo through Gβ to promote ubiquitylation of both Smo and Gprk2, leading to Smo internalization and degradation. In the presence of Hh, recruitment of PKAc to Smo is stimulated, which phosphorylates DDB1 and dissociates Cul4–DDB1 from Smo. Consequently, Cul4-mediated Smo ubiquitylation is blocked, leading to increased Smo cell surface expression.
Previous studies have demonstrated that phosphorylation of the Smo C-terminus by PKA inhibits Smo ubiquitylation and promotes Smo cell surface accumulation (Jia et al., 2004; Li et al., 2012; Xia et al., 2012; Zhao et al., 2007). We were, therefore, surprised to observe that Cul4-mediated ubiquitylation of Smo was not regulated by the PKA-mediated phosphorylation of the Smo C-terminus. Nevertheless, we found that Cul4-mediated ubiquitylation of Smo was inhibited by PKA and identified DDB1 as the target for PKA-mediated regulation. We provided evidence that Hh-induced recruitment of PKAc to the membrane proximal region of the Smo C-terminus allows PKAc to phosphorylate DDB1 at S644, leading to the dissociation of Cul4–DDB1 from Smo–Gβ and the downregulation of Smo ubiquitylation (Fig. 7). Hence, Hh inhibits Cul4–DDB1–Gβ-mediated ubiquitylation of Smo through a PKA-dependent but Smo-phosphorylation-independent mechanism. But how does PKA-mediated phosphorylation of the Smo C-terminus inhibit Smo ubiquitylation? A likely explanation is that Cul4–DDB1–Gβ is not the only E3 ligase responsible for Smo and that Smo ubiquitylation can also be regulated by additional E3(s), whose activity or binding to Smo is inhibited by PKA-mediated phosphorylation of the Smo C-terminus. Indeed, in a parallel, recently published study (Li et al., 2018), we found that the Smurf family of E3 ubiquitin ligases promoted Smo ubiquitylation and that their association with Smo was inhibited by PKA-mediated phosphorylation of the Smo C-terminus. We provided further evidence that Cul4–DDB1–Gβ acts in parallel to Smurf in order to regulate Smo cell surface expression (Fig. S7) (Li et al., 2018). Hence, the Hh pathway employs multiple E3 ubiquitylation ligases to restrict Smo cell surface accumulation in the ‘signaling off’ state. Upon Hh stimulation, the catalytic subunit of PKA is recruited to Smo in order to inhibit the Smurf family of E3s and Cul4–DDB1 through phosphorylation of Smo and DDB1, respectively (Fig. 7) (Li et al., 2018). In addition, Hh stimulates sumoylation of Smo in a manner that is independent of PKA – which recruits the deubiquitylation enzyme USP8 to further antagonize Smo ubiquitylation (Ma et al., 2016; Zhang et al., 2017).
A previous study has shown that Gβ targets Cul4–DDB1 to ubiquitylate and downregulate GRK2, thereby modulating GRCP signaling (Zha et al., 2015). Here, we found that Cul4–DDB1–Gβ also interacted with and ubiquitylated Gprk2; however, Cul4-mediated ubiquitylation of Gprk2 requires Smo as Smo RNAi diminished Gprk2 ubiquitylation (Fig. 4). CoIP experiments indicated that Smo RNAi reduced the binding of Gβ to Gprk2, whereas Gprk2 RNAi did not affect binding of Gβ to Smo. These observations suggest that, compared with Smo, Gprk2 binds Gβ with lesser affinity, and that Smo functions as a scaffold to facilitate the interaction between Gprk2 and Cul4–DDB1–Gβ (Fig. 7). The dependence of Gprk2 ubiquitylation on Smo might allow Hh to selectively regulate a local pool of Gprk2 associated to Smo, thereby achieving pathway specificity. Because Gprk2 inhibits Smo accumulation at the cell surface in the absence of Hh (Chen et al., 2010; Cheng et al., 2010; Molnar et al., 2007), it is possible that Gprk2 ubiquitylation contributes to Smo internalization and degradation. Indeed, it has been shown several times that receptor endocytosis can be promoted by ubiquitylation of receptor-binding proteins (Bulut et al., 2013; Shenoy et al., 2001). Another possibility is that ubiquitylation of Gprk2 limits the amount of Gprk2 associated with Smo in the absence of Hh in order to reduce the basal activity of Smo, because Gprk2 can promote Smo dimerization and the active conformation of the Smo C-terminus (Chen et al., 2010). Further studies are needed to investigate the precise role of Gprk2 ubiquitylation in the regulation of Hh pathway activity. This is likely to be challenging because Gprk2 plays both positive and negative roles in Hh signaling.
The Cul4–DDB1–Gβ E3 ubiquitin ligase complexes might play broader roles in the regulation of receptor trafficking. Cul4–DDB1–Gβ can directly interact with receptors, such as GPCRs, to modulate their ubiquitylation and endocytosis – as we have shown here for Smo. Cul4–DDB1–Gβ complexes can also be brought to GPCRs through their association with GRK2. A traditional role of GRK2 in the regulation of GPCR signaling is its binding to and phosphorylation of ligand-occupied GPCRs, allowing β-arrestin to be recruited to internalize and desensitize GPCRs (Shenoy et al., 2001). However, emerging evidence has revealed that GRK2 regulates GPCR trafficking by using a kinase-independent mechanism (Evron et al., 2012). It is conceivable that GRK2 brings Cul4–DDB1–Gβ to its interacting GPCRs to facilitate their ubiquitylation and internalization. Therefore, it will be interesting to determine whether the Cul4–DDB1–Gβ E3 ubiquitin ligase complex plays a more general role in the regulation of receptor trafficking.
MATERIALS AND METHODS
Constructs and transgenes
Transgenic fly stocks were obtained from Vienna Drosophila Resource Center (VDRC) and Bloomington Drosophila Stock Center (BDSC). We used UAS-Cul4-RNAi (VDRC #44829, VDRC #105668 and BDSC line BL#50614; expressing dsRNA for RNAi of Cul4); UAS-DDB1-RNAi (BL#41997; expressing dsRNA for RNAi of Pic/DDB1) and MS1096-Gal4 (Capdevila and Guerrero, 1994; Wang et al., 1999). UAS-Smo constructs: Myc-Smo, Myc-SmoSA (SmoSA123), Myc-SmoSD (SmoSD123), Myc-SmoΔC, Myc-SmoΔSAID and Myc-SmoRA1′2′3′ have been previously described (Jia et al., 2003, 2004; Li et al., 2014; Zhao et al., 2007). Myc-tagged Fz2 and Myc-tagged Fz2-SAID fusion constructs (Myc-Fz and Myc-FS, respectively) have been described previously (Li et al., 2012). DNA constructs expressing Fg-Cul4, Fg-DDB1, Fg-Gβ13F and HA-Gβ76C were created by subcloning the corresponding cDNA coding sequence into the pUAST vector containing the FLAG (Fg) or hemagglutinin (HA) tag (Tong and Jiang, 2007). PCR-based site-directed mutagenesis was used to generate DDB1S644A.
Cell culture, transfection, immunoprecipitation, western blotting and immunostaining
Drosophila Schneider 2 (S2) cells were cultured in Drosophila serum-free medium (SFM, Invitrogen) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 mg/ml streptomycin at 23°C. cl-8 cells were cultured in Shields and Sang M3 Insect Medium (Sigma) with 2.5% FBS, 2.5% fly extract, insulin (0.125 IU/ml; 0.5 mg/ml) (Sigma), penicillin (100 U/ml), and streptomycin (100 mg/ml) at 24°C.
Transfection was carried out using the Calcium Phosphate Transfection Kit (Specialty Media) according to the manufacturer's instructions. Treatment with Hh-conditioned medium was carried out as described by Lum et al., 2003. Briefly, selection of S2 cells stably expressing the active Hh N-terminal fragment after culturing in Schneider medium (Gibco) supplemented with 200 µg/ml hygromycin. Hh-conditioned medium was obtained by culturing S2 cells in Schneider medium lacking hygromycin but supplemented with 0.7 mM CuSO4 for 1 day. The medium was harvested and sterilized by filtration. Unless mentioned otherwise, Hh-conditioned medium was diluted at a ratio of 6:4 with fresh Schneider medium.
Immunoprecipitation and western blot analyses were carried out using standard protocols as previously described (Zhang et al., 2005). For stabilization of Ptc, cells were treated with 50 μM of the proteasome inhibitor MG132 (Calbiochem) for 4 h or with 20 mM NH4Cl (Sigma) for 18 h prior to harvest and lysis. Cell surface staining of Smo prior to membrane permeabilization was carried out as previously described (Jia et al., 2003, 2004; Li et al., 2014; Zhao et al., 2007). Briefly, S2 cells stably expressing Myc-Smo were harvested and washed three times with PBS, fixed with 4% formaldehyde for 20 min at room temperature, and incubated with mouse anti-Myc antibody in PBS for 30 min at room temperature. Cells were washed 3× with PBS followed by secondary antibody staining. Quantification of surface Smo was performed using ImageJ software. Immunostaining of imaginal discs was carried out as described (Jiang and Struhl, 1995). Antibodies used in this study were: mouse anti-Smo (20C6, DSHB, 1:10) for immunostaining; mouse anti-Ci (2A1, DSHB, 1:50) for immunostaining; rabbit anti-FLAG (F7425, Sigma, 1:200) for immunostaining; mouse anti-FLAG (F3165, Sigma, 1:10,000) for western blotting; mouse anti-Myc (sc-40, Santa Cruz) at 1:200 for immunostaining and 1:10,000 for western blotting; mouse anti-HA (sc-7392, Santa Cruz, 1:10,000) for western blotting; rabbit anti-Cul4 (Lee et al., 2010; 1,1000) for western blotting; rabbit anti-Gβ13F (Hampoelz et al., 2005; 1,1000) for western blotting.
Quantitative RT-PCR
Total RNA was extracted from 1×106 cells using the RNeasy Plus mini kit (QIAGEN); cDNA was synthesized using the iScript cDNA synthesis kit (Bio-Rad Laboratories). RT-qPCR was performed using iQ SYBR Green System (Bio-Rad Laboratories). Reverse transcriptase (RT)-qPCR was performed in triplicate on each of three independent biological replicates; the following primer sequences were used: 5′-GATTAAGAACTTTAAGGATAA-3′ and 5′-TTGAGCTTAATATTGCGCTTT-3′ (for Cul4), 5′-ATGTCGCATCACTACGTGGTG-3′ and 5′-GAGCAAATCCTTGTTGCTGTC-3′ (for DDB1), 5′-ATGAATGAACTAGACAGTCTC-3′ and 5′-GGACACCCGGACGTTGCCCTC-3′ (for Gβ13F), 5′-ATGCCGAAAATTGACCCAGAA-3′ and 5′-AGGCATCGCGATTGTTCACAT-3′ (for Gβ76C), 5′-ATGTCGGAGGCGGCGGTGCCA-3′ and 5′-TTCGTGGTGAACGCATCCCAG-3′ (for Gβ5). Actin was used as a normalization control. Relative quantification of mRNA levels were calculated using the comparative CT method.
RNAi and dual-luciferase assay in Drosophila S2 cells
Double-stranded (ds) RNA was generated by using the MEGAscript High Yield Transcription Kit (Ambion: #AM1334) according to the manufacturer's instruction. The following primers were used to generate dsRNA, targeting:
Cul4 (5′-GAATTAATACGACTCACTATAGGGAGACGCGAATTCGCTGCAAAATTC-3′ and 5′-GAATTAATACGACTCACTATAGGGAGAGCGTCTAGAGCTCGTTCTCCT-3′), DDB1 (5′-GAATTAATACGACTCACTATAGGGAGAAATAATTCCCCGCTCCATTC-3′ and 5′-GAATTAATACGACTCACTATAGGGAGAAGAGGCACTATTGCGAAGT-3′), Smo (5′ UTR: 5′-GAATTAATACGACTCACTATAGGGAGAGTCGCACATTTGTTGCTTCAG-3′ and 5′-GAATTAATACGACTCACTATAGGGAGACCGCTTATAAAAATCATTAAA-3′). Gprk2 (5′-GAATTAATACGACTCACTATAGGGAGAGGGGGCGACGCCTTGGACGCC-3′ and 5′-GAATTAATACGACTCACTATAGGGAGAAAAATACATAGAGCTCTCAAA-3′), Gβ13F (5′-GAATTAATACGACTCACTATAGGGAGACATACCACGAACAAAGTCCA-3′ and 5′-GAATTAATACGACTCACTATAGGGAGAGATATCGAACAATCGACAGGT-3′), Gβ76C (5′-GAATTAATACGACTCACTATAGGGAGACGCCAACAAGGTGCAGAT-3′ and 5′-GAATTAATACGACTCACTATAGGGAGATCCATGTCGTGACCGAAG-3′). Gβ5 (5′-GAATTAATACGACTCACTATAGGGAGATGCGTTCACCACGAACAA-3′ and 5′-GAATTAATACGACTCACTATAGGGAGACTGCTGTCATCCGATCCA-3′). Smurf (5′-GAATTAATACGACTCACTATAGGGAGAAGCGGAGGAGGAAGTAGATCC-3′ and 5′-GAATTAATACGACTCACTATAGGGAGATCGAATCATTTGCAAAATACT-3′).
dsRNA targeting the firefly luciferase coding sequence was used as a control. For RNAi knockdown experiments, S2 cells were cultured in SFM medium containing the indicated dsRNA at 23°C for 8 h. After adding FBS to a final concentration of 10%, dsRNA-treated cells were cultured overnight before transfection with DNA constructs. After additional culturing for 2 days, cells were collected for analysis. Knockdown efficiency was measured by qRT-PCR. Dual-luciferase activity was measured with the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions. Briefly, Cl8 cells were transfected with 1 μg ptc-luc reporter construct and 50 ng RL-PolIII Renilla construct in 12-well plates together with 1 μg corresponding constructs. Cells were collected after 48 h incubation. For Hh treatment, two-thirds of the medium was replaced with Hh-conditioned medium, and cells were incubated for 24 h before harvest. Each sample was measured in triplicate by using a FLUOstar OPTIMA plate reader (BMG Labtech).
Ubiquitylation assay
Ubiquitylation assays were carried out according to the protocol described by Li et al., 2012. Briefly, cells were treated with 50 μM MG132 (Calbiochem) for 4 h to inhibit proteasome-mediated degradation prior to harvest. Cells were lysed in 100 µl of denaturing buffer (1% SDS/50 mM Tris pH 7.5/0.5 mM EDTA/1 mM DTT). After incubation for 5 min at 100°C, lysates were diluted 10-fold with lysis buffer and then subjected to immunoprecipitation and western blot analysis.
Supplementary Material
Acknowledgements
We thank Drs R. J. Duronio, C. T. Chien and J. A. Knoblich for providing reagents, Bloomington Drosophila Stock Center (BDSC) and Vienna Drosophila Resource Center (VDRC) for fly stocks, and the Developmental Studies Hybridoma Bank (DSHB) for antibodies.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: J.J.; Methodology: Shuang Li, Y.S.C., B.W., Shuangxi Li; Validation: Shuang Li; Formal analysis: Shuang Li, Y.b.C., Shuangxi Li, J.J.; Investigation: Shuang Li, Y.S.C., B.W., Shuangxi Li, J.J.; Data curation: Shuang Li, Y.S.C., Shuangxi Li, J.J.; Writing - original draft: J.J.; Writing - review & editing: J.J.; Visualization: Y.S.C.; Supervision: J.J.; Project administration: B.W., J.J.; Funding acquisition: J.J.
Funding
This work was supported by grants from the National Institutes of Health (NIH) [grant number: GM118063] and Welch Foundation [grant number: I-1603] to J.J., who is a Eugene McDermott Endowed Scholar in Biomedical Science at UTSW. Deposited in PMC for release after 12 months
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.218016.supplemental
References
- Angers S., Li T., Yi X., MacCoss M. J., Moon R. T. and Zheng N. (2006). Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 443, 590-593. 10.1038/nature05175 [DOI] [PubMed] [Google Scholar]
- Briscoe J. and Thérond P. P. (2013). The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 14, 418-431. 10.1038/nrm3598 [DOI] [PubMed] [Google Scholar]
- Bulut G. B., Sulahian R., Yao H. and Huang L. J. (2013). Cbl ubiquitination of p85 is essential for Epo-induced EpoR endocytosis. Blood 122, 3964-3972. 10.1182/blood-2013-05-506212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capdevila J. and Guerrero I. (1994). Targeted expression of the signalling molecule decapentaplegic induces pattern duplications and growth alterations in Drosophila wings. EMBO J. 13, 4459-4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. and Jiang J. (2013). Decoding the phosphorylation code in Hedgehog signal transduction. Cell Res. 23, 186-200. 10.1038/cr.2013.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Li S., Tong C., Zhao Y., Wang B., Liu Y., Jia J. and Jiang J. (2010). G protein-coupled receptor kinase 2 promotes high-level Hedgehog signaling by regulating the active state of Smo through kinase-dependent and kinase-independent mechanisms in Drosophila. Genes Dev. 24, 2054-2067. 10.1101/gad.1948710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Sasai N., Ma G., Yue T., Jia J., Briscoe J. and Jiang J. (2011). Sonic Hedgehog dependent phosphorylation by CK1alpha and GRK2 is required for ciliary accumulation and activation of smoothened. PLoS Biol. 9, e1001083 10.1371/journal.pbio.1001083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S., Maier D., Neubueser D. and Hipfner D. R. (2010). Regulation of smoothened by Drosophila G-protein-coupled receptor kinases. Dev. Biol. 337, 99-109. 10.1016/j.ydbio.2009.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbit K. C., Aanstad P., Singla V., Norman A. R., Stainier D. Y. R. and Reiter J. F. (2005). Vertebrate Smoothened functions at the primary cilium. Nature 437, 1018-1021. 10.1038/nature04117 [DOI] [PubMed] [Google Scholar]
- Denef N., Neubüser D., Perez L. and Cohen S. M. (2000). Hedgehog induces opposite changes in turnover and subcellular localization of patched and smoothened. Cell 102, 521-531. 10.1016/S0092-8674(00)00056-8 [DOI] [PubMed] [Google Scholar]
- Evron T., Daigle T. L. and Caron M. G. (2012). GRK2: multiple roles beyond G protein-coupled receptor desensitization. Trends Pharmacol. Sci. 33, 154-164. 10.1016/j.tips.2011.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampoelz B., Hoeller O., Bowman S. K., Dunican D. and Knoblich J. A. (2005). Drosophila Ric-8 is essential for plasma-membrane localization of heterotrimeric G proteins. Nat. Cell Biol. 7, 1099-1105. 10.1038/ncb1318 [DOI] [PubMed] [Google Scholar]
- Hui C.-C. and Angers S. (2011). Gli proteins in development and disease. Annu. Rev. Cell Dev. Biol. 27, 513-537. 10.1146/annurev-cellbio-092910-154048 [DOI] [PubMed] [Google Scholar]
- Ingham P. W. and McMahon A. P. (2001). Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 15, 3059-3087. 10.1101/gad.938601 [DOI] [PubMed] [Google Scholar]
- Jia J., Tong C. and Jiang J. (2003). Smoothened transduces Hedgehog signal by physically interacting with Costal2/Fused complex through its C-terminal tail. Genes Dev. 17, 2709-2720. 10.1101/gad.1136603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J., Tong C., Wang B., Luo L. and Jiang J. (2004). Hedgehog signalling activity of Smoothened requires phosphorylation by protein kinase A and casein kinase I. Nature 432, 1045-1050. 10.1038/nature03179 [DOI] [PubMed] [Google Scholar]
- Jiang J. and Hui C.-C. (2008). Hedgehog signaling in development and cancer. Dev. Cell 15, 801-812. 10.1016/j.devcel.2008.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J. and Struhl G. (1995). Protein kinase A and hedgehog signaling in Drosophila limb development. Cell 80, 563-572. 10.1016/0092-8674(95)90510-3 [DOI] [PubMed] [Google Scholar]
- Jiang J. and Struhl G. (1998). Regulation of the Hedgehog and Wingless signalling pathways by the F-box/WD40-repeat protein Slimb. Nature 391, 493-496. 10.1038/35154 [DOI] [PubMed] [Google Scholar]
- Lee J. and Zhou P. (2007). DCAFs, the missing link of the CUL4-DDB1 ubiquitin ligase. Mol. Cell 26, 775-780. 10.1016/j.molcel.2007.06.001 [DOI] [PubMed] [Google Scholar]
- Lee H. O., Zacharek S. J., Xiong Y. and Duronio R. J. (2010). Cell type-dependent requirement for PIP box-regulated Cdt1 destruction during S phase. Mol. Biol. Cell 21, 3639-3653. 10.1091/mbc.e10-02-0130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Chen Y., Shi Q., Yue T., Wang B. and Jiang J. (2012). Hedgehog-regulated ubiquitination controls smoothened trafficking and cell surface expression in Drosophila. PLoS Biol. 10, e1001239 10.1371/journal.pbio.1001239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Ma G., Wang B. and Jiang J. (2014). Hedgehog induces formation of PKA-Smoothened complexes to promote Smoothened phosphorylation and pathway activation. Sci. Signal. 7, ra62 10.1126/scisignal.2005414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Li S., Han Y., Tong C., Wang B., Chen Y. and Jiang J. (2016). Regulation of smoothened phosphorylation and high-level hedgehog signaling activity by a plasma membrane associated kinase. PLoS Biol. 14, e1002481 10.1371/journal.pbio.1002481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Li S., Wang B. and Jiang J. (2018). Hedgehog reciprocally controls trafficking of Smo and Ptc through the Smurf family of E3 ubiquitin ligases. Sci. Signal. 11, eaan8660 10.1126/scisignal.aan8660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum L., Yao S., Mozer B., Rovescalli A., Von Kessler D., Nirenberg M. and Beachy P. A. (2003). Identification of Hedgehog pathway components by RNAi in Drosophila cultured cells. Science 299, 2039-2045. 10.1126/science.1081403 [DOI] [PubMed] [Google Scholar]
- Ma G., Li S., Han Y., Li S., Yue T., Wang B. and Jiang J. (2016). Regulation of smoothened trafficking and Hedgehog signaling by the SUMO pathway. Dev. Cell 39, 438-451. 10.1016/j.devcel.2016.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnar C., Holguin H., Mayor F. Jr., Ruiz-Gomez A. and de Celis J. F. (2007). The G protein-coupled receptor regulatory kinase GPRK2 participates in Hedgehog signaling in Drosophila. Proc. Natl. Acad. Sci. USA 104, 7963-7968. 10.1073/pnas.0702374104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nybakken K., Vokes S. A., Lin T.-Y., McMahon A. P. and Perrimon N. (2005). A genome-wide RNA interference screen in Drosophila melanogaster cells for new components of the Hh signaling pathway. Nat. Genet. 37, 1323-1332. 10.1038/ng1682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R., Milenkovic L. and Scott M. P. (2007). Patched1 regulates hedgehog signaling at the primary cilium. Science 317, 372-376. 10.1126/science.1139740 [DOI] [PubMed] [Google Scholar]
- Shenoy S. K., McDonald P. H., Kohout T. A. and Lefkowitz R. J. (2001). Regulation of receptor fate by ubiquitination of activated beta 2-adrenergic receptor and beta-arrestin. Science 294, 1307-1313. 10.1126/science.1063866 [DOI] [PubMed] [Google Scholar]
- Taipale J. and Beachy P. A. (2001). The Hedgehog and Wnt signalling pathways in cancer. Nature 411, 349-354. 10.1038/35077219 [DOI] [PubMed] [Google Scholar]
- Tong C. and Jiang J. (2007). Using immunoprecipitation to study protein-protein interactions in the Hedgehog-signaling pathway. Methods Mol. Biol. 397, 215-229. 10.1007/978-1-59745-516-9_15 [DOI] [PubMed] [Google Scholar]
- Villavicencio E. H., Walterhouse D. O. and Iannaccone P. M. (2000). The sonic hedgehog-patched-gli pathway in human development and disease. Am. J. Hum. Genet. 67, 1047-1054. 10.1016/S0002-9297(07)62934-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G., Wang B. and Jiang J. (1999). Protein kinase A antagonizes Hedgehog signaling by regulating both the activator and repressor forms of Cubitus interruptus. Genes Dev. 13, 2828-2837. 10.1101/gad.13.21.2828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C. W. and Chuang P.-T. (2010). Mechanism and evolution of cytosolic Hedgehog signal transduction. Development 137, 2079-2094. 10.1242/dev.045021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia R., Jia H., Fan J., Liu Y. and Jia J. (2012). USP8 promotes smoothened signaling by preventing its ubiquitination and changing its subcellular localization. PLoS Biol. 10, e1001238 10.1371/journal.pbio.1001238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Mao F., Lv X., Zhang Z., Fu L., Lu Y., Wu W., Zhou Z., Zhang L. and Zhao Y. (2013). Drosophila Vps36 regulates Smo trafficking in Hedgehog signaling. J. Cell Sci. 126, 4230-4238. 10.1242/jcs.128603 [DOI] [PubMed] [Google Scholar]
- Zha Z., Han X., Smith M. D., Liu Y., Giguère P. M., Kopanja D., Raychaudhuri P., Siderovski D. P., Guan K.-L., Lei Q.-Y. et al. (2015). A non-canonical function of Gbeta as a subunit of E3 ligase in targeting GRK2 ubiquitylation. Mol. Cell 58, 794-803. 10.1016/j.molcel.2015.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Zhao Y., Tong C., Wang G., Wang B., Jia J. and Jiang J. (2005). Hedgehog-regulated costal2-kinase complexes control phosphorylation and proteolytic processing of cubitus interruptus. Dev. Cell 8, 267-278. 10.1016/j.devcel.2005.01.001 [DOI] [PubMed] [Google Scholar]
- Zhang J., Liu Y., Jiang K. and Jia J. (2017). SUMO regulates the activity of Smoothened and Costal-2 in Drosophila Hedgehog signaling. Sci. Rep. 7, 42749 10.1038/srep42749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Tong C. and Jiang J. (2007). Hedgehog regulates smoothened activity by inducing a conformational switch. Nature 450, 252-258. 10.1038/nature06225 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.