

ABSTRACT
Gap junctions (GJs) assembled from connexin (Cx) proteins allow direct cell–cell communication. While phosphorylation is known to regulate multiple GJ functions, much less is known about the role of ubiquitin in these processes. Using ubiquitylation-type-specific antibodies and Cx43 lysine-to-arginine mutants we show that ∼8% of a GJ, localized in central plaque domains, is K63-polyubiquitylated on K264 and K303. Levels and localization of ubiquitylation correlated well with: (1) the short turnover rate of Cxs and GJs; (2) removal of older channels from the plaque center; and (3) the fact that not all Cxs in an internalizing GJ channel need to be ubiquitylated. Connexins mutated at these two sites assembled significantly larger GJs, exhibited much longer protein half-lives and were internalization impaired. Interestingly, these ubiquitin-deficient Cx43 mutants accumulated as hyper-phosphorylated polypeptides in the plasma membrane, suggesting that K63-polyubiquitylation is triggered by phosphorylation. Phospho-specific anti-Cx43 antibodies revealed that upregulated phosphorylation affected serines 368, 279/282 and 255, which are well-known regulatory PKC and MAPK sites. Together, these novel findings suggest that the internalizing portion of channels in a GJ is K63-polyubiquitylated, ubiquitylation is critical for GJ internalization and that phosphorylation induces Cx K63-polyubiquitylation.
This article has an associated First Person interview with the first author of the paper.
KEY WORDS: Connexin 43, Endocytosis, Gap junctions, Phosphorylation, Phosphodegron, Ubiquitylation
Summary: Connexin 43 in gap junctions becomes K63-polyubiquitylated on lysines 264 and 303, which is required for gap junction endocytosis.
INTRODUCTION
Ubiquitylation, the addition of a small, 76 amino acid, 8.5 kDa protein to a target protein is known to play an important function in protein degradation (Komander and Rape, 2012; Nguyen et al., 2013; Ravid and Hochstrasser, 2008). Multiple types of ubiquitylation (mono- and several different types of poly-ubiquitylation) are known to execute these functions (Komander and Rape, 2012). Ubiquitin (Ub) is covalently attached to lysines (K) on target proteins by an enzyme cascade consisting of an E1 (Ub-activating), an E2 (Ub-conjugating) and an E3 (Ub-ligating) enzyme (Chen and Sun, 2009). Ub can also be removed from its target via proteases termed de-ubiquitylases (DUBs). Ub has seven internal lysines (K6, K11, K27, K29, K33, K48 and K63), all of which (and also the N-terminal methionine) are capable of forming linkages to the C-terminal glycine of subsequent Ub moieties, forming uniquely folded polyubiquitin (polyUb) chains. The two best-studied polyUb chains are K48-polyUb, which is known to lead to proteasomal degradation, and K63-polyUb, which – in addition to other functions – is known to signal endo- and phago-lysosomal degradation (Komander and Rape, 2012). Ubiquitylation of gap junction (GJ) proteins, which are termed connexins (Cxs), has also been reported (Girão et al., 2009; Laing and Beyer, 1995; Laing et al., 1997; Leithe and Rivedal, 2004; Ribeiro-Rodrigues et al., 2014); however, published results are inconsistent, suggesting that diverse types of ubiquitylation with potentially different functions may occur at individual stages of the Cx life-cycle. Whether Cx ubiquitylation plays a role in GJ internalization is particularly unclear.
Connexins are the four-pass transmembrane protein components of GJs that serve as a pathway for intercellular communication by physically coupling cells and allowing the passage of small metabolites, signaling molecules and ions. All Cxs have two extracellular loops, one intracellular loop and an intracellular N- and C-terminus. Six Cxs oligomerize to form a hemichannel or connexon, which is trafficked to the plasma membrane (PM) and docks with a hemichannel of an adjacent cell, forming the complete GJ channel. Accrual of multiple channels at the PM into clusters or two-dimensional arrays forms typical GJ plaques. Interestingly, once hemichannels dock at the PM, they can no longer be physiologically separated (Goodenough and Gilula, 1974). Therefore, in a process that requires the clathrin-mediated endocytic (CME) machinery, one of the two adjacent cells invaginates the GJ forming an annular gap junction (AGJ) vesicle or connexosome in the cytoplasm of one of the coupled cells (Falk et al., 2009; Gaietta et al., 2002; Jordan et al., 2001; Lauf et al., 2002; Piehl et al., 2007). Directionality may be achieved by phosphorylation-induced removal of a scaffolding protein, ZO-1, from the GJ plaque surface in the acceptor cell (Gilleron et al., 2009; Thévenin et al., 2017). The AGJ is then trafficked for degradation by autophago-lysosomal (under physiological, pathological and starvation conditions) (Bejarano et al., 2012; Fong et al., 2012; Hesketh et al., 2010; Lichtenstein et al., 2010) and possibly endo-lysosomal pathways upon treatment with the diacylglycerol analog TPA (12-O-tetradecanoylphorbol 13-acetate) (Fykerud et al., 2012; Leithe et al., 2009). In humans, there are 21 different Cx proteins, which are identified by molecular weight. Cx43 (also known as GJA1) is the best-studied Cx and is expressed in most tissues. Mutations in Cx43 have been linked to heart failure, ischemia and hypertrophy (Fontes et al., 2012), and the developmental bone malformation oculodentodigital dysplasia (ODDD) (Batra et al., 2012). Proper regulation of Cx43 trafficking and turnover is therefore imperative for human health and disease.
Cx43 ubiquitylation was initially discovered by Laing and Beyer in 1995 and characterized as a signal leading to Cx43 degradation by the proteasome (Laing and Beyer, 1995). Later, the same group published evidence suggesting that ubiquitylation plays a role in both proteasomal and lysosomal degradation (Laing et al., 1997). Afterwards, it was reported that Cx43 is likely to be modified by multiple mono-ubiquitylation events (Girão et al., 2009; Leithe and Rivedal, 2004), whereas more recent evidence suggests that Cx43 interacts with AMSH (associated molecule with the SH3 domain of STAM) (Ribeiro-Rodrigues et al., 2014). AMSH is a DUB with specificity toward K63-polyubiquitylated proteins (McCullough et al., 2004; Sato et al., 2008), suggesting that Cx43 may also become K63-polyubiquitylated (Ribeiro-Rodrigues et al., 2014). The different types of Cx43 ubiquitylation probably distinguish the degradation of single, misfolded Cx polypeptides (by the 26S proteasome) from the degradation of GJs and AGJs (by phago-/endo-lysosomal pathways). However, a rigorous analysis of where and when ubiquitylation occurs on Cxs and at which stages of its ‘life-cycle’ (Cx polypeptides, connexons, GJs, AGJs) has not been performed; nor is it known which signals trigger potential Cx43 ubiquitylation and whether Cx43 ubiquitylation has additional functions beyond its proteasomal degradation.
Here, we used Ub type-specific antibodies and generated Cx43 lysine (K) to arginine (R) mutations to investigate whether Cx43 ubiquitylation occurs in GJ plaques, what type of ubiquitylation occurs, and what its role might be for GJ function. We found that two juxtaposed lysine residues (K264, K303) in the C-terminal domain of Cx43 become K63-polyubiquitylated, and that ubiquitylation is required for constitutive GJ internalization. Interestingly, K63-polyUb-deficient Cx43 mutants accumulated as hyper-phosphorylated polypeptides in the PM, suggesting that phosphorylation triggers Cx43 ubiquitylation.
RESULTS
K63- but not K48-polyubiquitin-specific antibodies colocalize with plasma membrane and internalized gap junctions
To determine whether and what type of ubiquitylation occurs on Cx43 in GJs, we immunostained endogenously expressed Cx43 in primary porcine pulmonary artery endothelial cells (pPAECs) with antibodies directed against Cx43 and different types of ubiquitin and imaged these cells by acquiring image Z-stacks on a confocal microscope (Fig. 1). FK2, which recognizes both monoUb and polyUb chains, colocalized with Cx43 at GJ plaques (Fig. 1A, box 1) and internalized, annular GJs (AGJs) (Fig. 1A, box 2). Colocalization occurred over large areas of GJ plaques. Quantitative colocalization analyses revealed ∼16% colocalization by area (Fig. 1E). Next, we co-immunostained Cx43 with antibodies that detected only polyubiquitylated proteins (FK1). We found that colocalization of Cx43 GJ plaques and AGJs with FK1 was also evident. However, localization was much less pronounced and localized to distinct areas of GJs and small GJs only (Fig. 1B). Quantification revealed ∼8% colocalization (Fig. 1E). To test whether the detected polyubiquitin modification includes K63-based polyUb, we conducted co-immunostaining with an antibody that detects only K63-polyUb (HWA4C4). This antibody also revealed limited regions of colocalization (∼8%) (Fig. 1C). To investigate whether K48-polyubiqitylation (a known proteasomal degradation signal) also occurs in GJs, we co-immunostained PAECs with an antibody that detects only K48-polyUb chains. Although significant intracellular colocalization was evident, no convincing evidence of colocalization with K48-polyUb was found on Cx43 GJs and AGJs, suggesting that connexins and Cx-binding proteins in GJs are not K48-polyubiquitylated (Fig. 1D,E). Interestingly, while all three Ub-specific antibodies (FK1, FK2 and K63-polyUb-specific) localized to GJ plaques, localization of FK1 (polyUb-specific) and K63-polyUb-specific was much more distinct and localized to specific areas, suggesting that active, endocytic activity occurs specifically in these colocalization areas; a hypothesis that is further supported by the colocalization of these ubiquitin antibodies with internalized AGJ vesicles. Furthermore, areal colocalization of both, FK1 and K63-polyUb-specific antibodies was ∼8% (both probably detect the same Ub signal), whereas FK2 colocalization averaged ∼16%, suggesting that a second mono-ubiquitylation occurs on Cx43, or on a Cx43-binding protein in GJs. Taken together, our results indicate that Cx43 in GJs, or connexin-associated proteins in GJs and AGJs can become K63-polyubiquitylated. Our finding correlates with the enzyme specificity of the E3 ubiquitin ligase Nedd4-1 (neural precursor cell expressed developmentally downregulated protein 4-1), which preferentially K63-polyubiquitylates its substrates (Kim et al., 2007), and has been found to ubiquitylate Cx43 (Girão et al., 2009; Leykauf et al., 2006). Our data also support recent biochemical evidence suggesting that Cx43 interacts with the K63-polyUb-specific de-ubiquitylase AMSH (Ribeiro-Rodrigues et al., 2014).
Fig. 1.

Ubiquitin-specific antibodies colocalize with Cx43 in GJs and AGJs. Endogenously Cx43-expressing primary porcine pulmonary artery endothelial cells (pPAECs) were immunostained with antibodies directed against Cx43 (green) and ubiquitin-specific antibodies (red) and imaged by confocal microscopy. Below each panel, magnified insets highlight GJs (1) and AGJs (2). Cx43 antibodies robustly colocalize with monoUb- and polyUb-specific antibodies (FK2) (A), only polyUb chain-recognizing antibodies (FK1) (B), and with K63-polyUb-specific antibodies (HWA4C4) at distinct areas of GJs and with AGJs (C), but not with K48-polyUb-specific antibodies (Apu2) (D), suggesting that Cx43, or a Cx43-binding protein is K63-polyubiquitylated. (E) Average proportion of area per cell pair where Cx43- and ubiquitin-specific antibodies colocalize. Data are mean±s.e.m.; ns, not significant; ***P≤0.0005.
Cx43 in GJs can become K63-polyubiquitylated
To further investigate whether K63-polyubiquitylation in GJs occurs on Cx43 itself, or on a Cx43-associated protein, we performed Triton X-100 (TX-100) insolubility assays with pPAECs endogenously expressing Cx43, and MDCK cells exogenously expressing Cx43 (not expressing endogenous Cxs) (Dukes et al., 2011) to separate the TX-100-insoluble (GJs, AGJs) from the soluble (Cxs, connexons) fractions using a well-established method (Musil and Goodenough, 1991). MDCK cells were chosen in this and most other experiments as high transfection efficiencies (>75%) were achieved in this cell line. We also treated cells with 20 μM PR-619, a pan-deubiquitylase (DUB) inhibitor, for 1.5 h prior to lysis and fractionation. All fractions were analyzed via western blotting to detect Cx43 (Fig. 2A). Equal loading was verified by stripping the blots and assessing the amount of α-tubulin in the samples. In whole cell lysates analyzed in parallel and in all fractions, a well-characterized triplet of Cx43 bands was detected corresponding to the fastest migrating form of Cx43 (P0) and the slower-migrating phosphorylated forms of Cx43, commonly referred to as P1 and P2. Interestingly, the amount of Cx43 protein increased in the insoluble fraction of PR619-treated cells compared with untreated cells, consistent with an increase in protein ubiquitylation (as also seen in Fig. 2B). TX-100-insoluble fractions contained a number of additional, higher molecular weight bands migrating above Cx43 and appearing as a smear in some fractions (marked with brackets in Fig. 2A-C) that potentially corresponded to ubiquitylated Cx43 polypeptides. Two bands migrating around 70 kDa and 150 kDa were especially prominent in most fractions (Fig. 2A-C, asterisks). When TX-100-insoluble fractions of cells untreated or treated with 20 μM PR-619 were probed for monoUb/polyUb (FK2), polyUb (FK1) and K63-polyUb-specific antibodies (HWA4C4), dramatically increased levels of ubiquitylated proteins were detected in the fractions that migrated as a ladder or a smear of higher molecular weight proteins together with the bands migrating at around 70 and 150 kDa that also increased in the presence of PR-619 (Fig. 2B).
Fig. 2.

Both Cx43- and K63-polyUb-specific antibodies precipitate higher molecular weight Cx43 forms. Endogenously Cx43-expressing pPAECs (A,B), and exogenously Cx43-expressing MDCK cells (C) were lysed in buffer containing 1% TX-100, separated into TX-100-insoluble (GJ/AGJ) and TX-100-soluble (Cx, connexons) fractions, probed using Cx43- and ubiquitin-specific antibodies, and analyzed by western blotting. Total cell lysates, and cells treated with the de-ubiquitylase (DUB) inhibitor PR619 were also analyzed. (A) Mature forms of Cx43 (P0, P1, P2) migrating at the predicted molecular weight of ∼43 kDa were detected in total cell lysates and in both TX-100-soluble and -insoluble fractions. In addition, small amounts of higher molecular weight forms (bracket) including two species migrating at ∼70 and 150 kDa (asterisks) were detected in the insoluble fraction (that increased in PR619-treated cells) in this and the analyses shown in B and C, suggestive of potentially ubiquitylated Cx43. (B) PAECs were treated or not with PR-619, TX-100 extracted, and probed with mono/polyUb (FK2)-, polyUb (FK1)- and K63-polyUb (HWA4C4)-specific antibodies. In all instances, a smear of higher molecular weight bands (bracket) together with species migrating at ∼70 and 150 kDa (asterisks) were detected with all Ub-specific antibodies (and with K63-polyUb antibodies), especially in the lysates derived from cells in which protein deubiquitylation was blocked. (C) Cx43 and K63-polyubiquitylated proteins present in TX-100-insoluble fractions were precipitated from transfected MDCK cells and probed for K63-polyUb proteins and Cx43, respectively. Resulting blots again revealed a mixture of higher molecular weight bands (brackets) together with species migrating at ∼70 and 150 kDa (asterisks), further suggesting Cx43 K63-polyubiquitylation. Note that mature Cx43 migrating at ∼43 kDa was also detected when K63-polyubiquitylated proteins were precipitated and probed with Cx43-specific antibodies (right panel), but not in the inverse experiment (left panel), suggesting that only a portion of Cx43 subunits in connexons are ubiquitylated. Total cell lysates were analyzed in parallel.
To provide further evidence of Cx43 K63-polyubiquitylation, MDCK cells were transiently transfected with wild-type Cx43 cDNA, lysed and fractionated as described above for pPAECs 24 h post-transfection. The TX-100-insoluble pellet was then subjected to K63-polyUb and Cx43 immunoprecipitations. Total cell lysates were also probed with both antibodies. Cell lysates were analyzed on separate gels as exposure times for lysates and insoluble fractions differed vastly (8 s compared with 5 min). Higher molecular weight bands, including the 70 and 150 kDa bands were detected in the GJ-containing insoluble fraction when either Cx43 was precipitated and probed with K63-polyUb-specific antibodies (Fig. 2C, left panel) or K63-polyubiquitylated proteins were precipitated and probed with Cx43-specific antibodies (Fig. 2C, right panel), suggesting that Cx43 itself in GJs can become K63-polyubiquitylated. That the higher molecular weight signals corresponded to ubiquitylated Cx43-binding partner(s) is not likely as protein complexes containing Cx43 should have been dissociated in the SDS and β-mercaptoethanol-containing SDS-PAGE sample buffer. Protein-A Sepharose beads alone and immunoprecipitation reactions without antibodies analyzed in parallel as controls did not precipitate any proteins. It should be noted that the number of higher molecular weight bands detected in the insoluble fractions is small compared with the total level of non-ubiquitylated Cx43 (Fig. 2A, right panel), again suggesting that only a relatively small portion of Cx43 in a GJ plaque becomes K63-polyubiquitylated (the portion of the plaque that internalizes or triggers internalization), which is consistent with data presented in Fig. 1. Higher molecular weight, ubiquitylated Cx43 proteins were not detected in any of the analyzed total cell lysates, again consistent with the notion that only a very small portion of the total Cx43 polypeptides present within cells are K63-polyubiquitylated at any given time in steady-state cells. In addition, only some of the connexins in a connexon of an internalizing GJ channel appear to become ubiquitylated. This is indicated by the fact that significant amounts of unmodified Cx43 migrating at ∼40 kDa were detected along with ubiquitylated Cx43 when total K63-ubiquitylated proteins were precipitated and probed with Cx43-specific antibodies (Fig. 2C, right panel, band labeled Cx43); whereas unmodified Cx43 was not detected when Cx43 was precipitated and probed with K63-polyUb-specific antibodies (Fig. 2C, left panel). Together with our immuno-colocalization analyses shown in Fig. 1, these data suggest that internalizing Cx43 in GJs, in cells expressing Cx43 both endogenously and exogenously becomes K63-polyubiquitylated.
Cx43 GJs with critical lysine residues mutated to arginine accumulate in the plasma membrane
To determine which lysines in Cx43 GJs may become K63-polyubiquitylated, we transiently transfected HeLa cells with rat Cx43 constructs containing a set of C-terminal lysine (K) to arginine (R) mutations (K/R) in order to block ubiquitylation. We focused on mutating lysines in the C-terminus of Cx43 as this domain is highly post-translationally modified and is known to interact with numerous binding partners (reviewed in Thévenin et al., 2013). Mutant 6K/R has six lysines within the C-terminus (K258, K264, K287, K303, K345 and K346) mutated to arginines (Fig. 3A, boxed blue and green). Mutant 3K/R has the central lysines K264, K287 and K303 mutated to arginines (Fig. 3A, boxed green only). Twenty-four hours post-transfection, cells were fixed and immunostained using anti-Cx43 antibodies. Both mutants trafficked to the plasma membrane and assembled into GJ plaques with equivalent efficiency, and similarly to the wild-type protein. However, cells expressing either 6K/R or 3K/R mutants formed significantly more and significantly larger GJ plaques, both when measured individually or in average per cell pair (Fig. 3B, bottom; see Materials and Methods for how plaque size was measured). Representative images of wild-type and mutant GJ plaques are shown in Fig. 3B (top). Interestingly, 6K/R and 3K/R mutants gave similar results, implying that the additional peripheral lysine residues in the 6K/R mutant (K258, K345 and K346), or any other of the lysine residues that are present in the Cx43 polypeptide (Fig. 1A, intracellular located lysines labeled in red) did not become ubiquitylated. HeLa cells were used in this experiment, as they generally form larger, easier to quantify GJs due to the characteristic spreading morphology of HeLa cells compared with MDCK cells. Moreover, a comparable result was obtained when equal amounts of cell lysates were analyzed and probed for Cx43 protein content in Cx-deficient MDCK cells expressing wild-type and mutant Cx43 (Fig. 3C). Quantification of 6K/R and 3K/R mutants resulted in 8.88±2.70- and 7.71±2.95-fold increases in total amounts of Cx43 polypeptide compared with the wild-type, respectively (Fig. 3C). Taken together, these results indicate that mutating critical lysine residues in the Cx43 C-terminal domain causes accumulation of Cx polypeptides and GJs in the PM, which, based on the known role of K63-polyubiquitylation, suggests impairment of GJ internalization.
Fig. 3.

Cx43 mutants with lysine residues mutated form larger GJ plaques. (A) Amino acid sequence of human Cx43 with lysines (K) in the intracellular loop and the C-terminus potentially accessible to ubiquitylation in GJs highlighted in red. Lysines mutated to arginines (R) in the C-terminus of Cx43 are boxed in blue and green for mutant 6K/R, and green only for mutant 3K/R. (B) Cx-deficient HeLa cells (forming larger GJ plaques compared with PAEC and MDCK cells used in all other analyses) were transfected with wild-type or K/R mutants, immunostained with anti-Cx43 followed by Alexa Fluor 488-conjugated secondary antibodies, and GJ size and number determined. GJ size in 72, 81 and 94 cell pairs expressing Cx43 wild-type, 6K/R and 3K/R, respectively, were measured and plotted. Bar graphs depict the average plaque size per cell pair (the sum of individual plaque sizes for each cell pair), the average individual plaque size, and the average number of plaques/cell pair. Both K/R mutants showed significantly larger plaque sizes compared with those in cells expressing wild-type Cx43. (C) Total Cx43 protein levels of wild type, 6K/R and 3K/R mutants exogenously expressed in MDCK cells were analyzed by western blotting. In both mutants, ∼8 times the amount of Cx43, including a large amount of phosphorylated P1 and P2 variants, was detected. Lanes of blots are separated to indicate extraneous lanes between wild type and 6K/R, and wild type and 3K/R; however, lanes were derived from the same gels. Data are mean±s.e.m.; ns, not significant; *P<0.05, **P<0.005, ***P≤0.0005. Scale bar: 20 μm.
K63-polyubiquitin-deficient Cx43 mutants exhibit significantly longer protein half-lives
To further investigate the inhibitory effect of mutating lysine residues on GJ internalization, we analyzed the half-lives of K/R mutants expressed in Cx-deficient MDCK cells in which protein translation was blocked pharmacologically. Furthermore, we generated all possible double and single mutants of lysines 264, 287 and 303 (K264/303R, K264/287R, K287/303R, K264R, K287R and K303R) to determine if one or more lysine residue becomes ubiquitylated and how many residues needed to be mutated to inhibit GJ internalization. Twenty-four hours after transient transfection, protein translation was blocked by treatment with cycloheximide. Cells were lysed 0, 1, 2, 3, 4 and 6 h after cycloheximide addition and total wild-type and mutant Cx43 protein levels were analyzed using western blotting (Fig. 4). Un-cropped blots of all mutants are shown in Fig. S1. Consistent with Fig. 2, ubiquitylated Cx43 was again not detected in these whole-cell lysates. The half-life for wild-type Cx43 was determined to be ∼2.3 h, correlating with the known, short 1-5 h half-life described previously for Cx43 (Beardslee et al., 1998; Falk et al., 2009; Fallon and Goodenough, 1981) (Fig. 4A, top panel). Unlike the wild type, protein levels of Cx43 in 6K/R and 3K/R at 6 h post treatment had only decreased to 75% of starting Cx43 levels (Fig. 4A, middle and bottom panels). The observed 25% loss of Cx43 protein observed in these cells over the 6 h period is most likely due to a loss of cells caused by the cytotoxic effect of treating cells with cycloheximide, blocking protein biosynthesis in bulk, and associated negative metabolic side effects. Using a linear fit, the half-lives of these mutants were extrapolated as 15.7 and 13.1 h, respectively, making the 6K/R and 3K/R mutant half-lives 4- to 5-times longer than that of wild-type Cx43. 6K/R and 3K/R mutant half-lives were not significantly different, again suggesting that lysines 258, 345 and 346 are not targets for K63-polyubiquitylation, and apparently play no direct role in GJ internalization. Expression of double mutants (K287/303R, K264/303R and K264/287R) resulted in a similar significantly increased half-life of all three mutants (∼11.4, 6.6 and 7.7 h, respectively), suggesting that apparently at least two separate lysine residues become K63-polyubiquitylated (Fig. 4B). Finally, expressing the single lysine residue mutants also resulted in extended protein half-lives for K264 and K303 mutants (4.5 h for each), while the half-life of the K287 mutant corresponded to the protein half-life of wild-type Cx43 (2.5 h) (Fig. 4C). As already shown in Fig. 3C, relevant mutants (all except the K287R mutant) showed significantly increased Cx43 protein levels compared with wild-type Cx43 (compare Fig. 3C with Fig. 4). Taken together, these results identified two juxtaposed lysine residues in the Cx43 C-terminal domain (K264 and K303) as target sites for K63-polyubiquitylation and link K63-polyubiquitylation to GJ internalization.
Fig. 4.

Cx43 mutants with critical lysine residues mutated exhibit significantly extended protein half-lives. MDCK cells transfected with Cx43 wild-type or K/R mutant constructs were treated with cycloheximide for indicated times, lysed and analyzed by western blot. (A) Quantification of Cx43 protein levels revealed a half-life of 2.3 h for wild-type Cx43, with ∼20% of the starting Cx43 protein remaining after 6 h of treatment. Mutants 6K/R and 3K/R exhibited significantly increased half-lives extrapolated to 15.7 and 13.1 h, respectively. (B) All double K/R mutants also exhibited significantly extended half-lives (see table in C). (C) Mutating lysine 264 and 303, together or independently, also resulted in increased half-lives, whereas the half-life of the K287R mutant was unaffected and similar to that of the wild type (2.5 h). α-tubulin was probed as a loading control. Extrapolated half-lives were calculated using a linear fit curve. Data are mean±s.e.m.; ns, not significant; *P<0.05.
Immunoprecipitation of Cx43 mutants confirms K63-polyubiquitylation on lysines 264 and 303
To further investigate whether lysines 264 and 303 are indeed the residues that become K63-polyubiquitylated in Cx43, we again expressed Cx43 wild-type and relevant double and single K/R mutants (K264/303R, K264R, K287R and K303R) in MDCK cells. Cells were again fractionated into soluble (Cx, connexon) and insoluble pellet fractions (GJ, AGJ) using TX-100 extraction (Musil and Goodenough, 1991). Then, wild-type and mutant Cx43 polypeptides were immunoprecipitated and probed for the presence of K63-polyUb modification with K63-polyUb-specific antibodies (Fig. 5). Although faint, resulting western blots indicated that the wild type, as well as the Cx43 K287R mutant could become K63-polyubiquitylated (Fig. 5A) as indicated by the higher molecular weight band pattern that again included the weak, yet noticeable 70 and 150 kDa bands (marked with asterisks) in addition to a higher molecular weight smear (marked with bracket as detected previously under comparable conditions; compare Fig. 2C, left panel with Fig. 5A). In contrast, no higher molecular band patterns were detected in K264 and K303 double mutants or in single mutants, suggesting that these mutants did not become ubiquitylated and that mutating one lysine may be sufficient to also prevent ubiquitylation of the other remaining target lysine. Quantitative analysis of higher molecular weight Cx43 is shown in Fig. 5B. Control lanes consisting of lysate plus Protein-A Sepharose beads, beads alone, or beads plus Cx43 antibody, as expected, did not precipitate any K63-polyubiquitylated proteins. Amounts of total Cx43 and of K63-polyUb proteins (with α-tubulin as a loading control) present in the cell lysates were analyzed in control (Fig. 5A, bottom blots). Taken together, these results confirm that lysines 264 and 303 of Cx43 are target lysines for K63-polyubiquitylation in GJs. Our findings correlate with mass spectrometry evidence suggesting ubiquitylation of Cx43 on lysines 9 and 303 (Wagner et al., 2011).
Fig. 5.
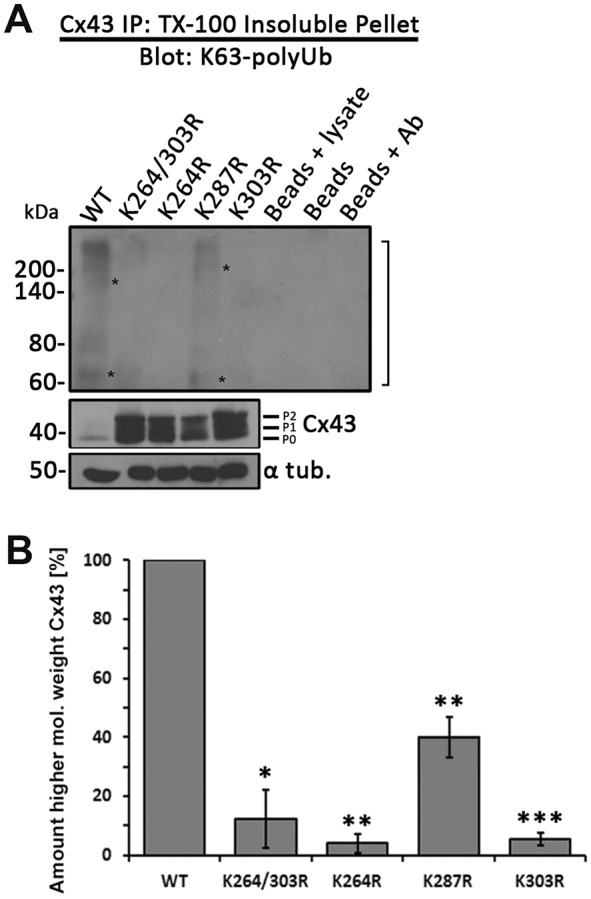
Cx43 K264R and K303R single and double mutants no longer become K63-polyubiquitylated. Cx43 was immunoprecipitated from MDCK cells transiently transfected with wild-type Cx43 or K264R, K287R, K303R and K264/303R mutants. Cells were lysed in 1% TX-100-containing buffer and separated into soluble and insoluble fractions. Cx43 was precipitated from insoluble fractions (GJs, AGJs) and probed for K63-polyUb by western blotting as in Fig. 2C. (A) Immunoprecipitation (top panel) and quantification (B) again revealed a higher-molecular weight smear (bracket) that includes bands migrating at ∼70 and 150 kDa (asterisks) typical of ubiquitylation in wild-type Cx43 (compare Fig. 2C, left panel) and in K287R mutant when probed with the K63-polyUb-specific antibodies. None of the other mutants revealed a similar higher molecular weight band pattern, further suggesting that K264 and K303, but not K287, in Cx43 can become K63-polyubiquitylated. Input cell lysates were probed in parallel for total K63-polyubiquitylated proteins (as in Fig. 2B, right panel), Cx43 and α-tubulin were detected as loading controls. Data are mean±s.e.m.; *P<0.05, **P<0.005, ***P≤0.0005.
Cx43 GJs with critical lysine residues mutated exhibit reduced K63-polyUb antibody colocalization
To further investigate K63-polyubiquitylation of GJs assembled from wild-type and K to R mutants, we expressed respective constructs in HeLa cells, fixed and stained Cx43 and K63poly-ubiquitin using specific antibodies and investigated the amount and pattern of colocalization using confocal imaging. The large size of GJs generally formed in HeLa cells allowed to perform these analyses. Representative confocal images are shown in Fig. 6A, and quantitative colocalization is shown in Fig. 6B. Significant co-localization was observed in GJs assembled from wild-type Cx43, consistent with observations in PAECs (Fig. 1). Interestingly, colocalization occurred predominantly in plaque centers and appeared speckled, consistent with a model of accrual of channels to plaque peripheries and simultaneous removal from plaque centers and ubiquitylation serving a signal for GJ channel internalization. Mutants in which lysines 264, 303, or both, were mutated exhibited significantly less colocalization, again suggesting that mutating one of two target lysines seems enough to prevent ubiquitylation of the other lysine. Interestingly, in mutants in which lysine 287 was mutated, ubiquitylation was increased, suggesting hyper-ubiquitylation similar to hyper-phosphorylation observed and described below (also see Discussion). Taken together, these results further support K63-polyubiquitylation on K264 and K303 in GJs.
Fig. 6.

Cx43 GJs with critical lysine residues mutated exhibit reduced K63-polyUb antibody colocalization. Wild-type Cx43 as well as single and double lysine mutants were transfected into HeLa cells, fixed and stained for Cx43 (green) and K63-polyUb (red) using respective antibodies. (A) Representative images depicting the distribution of K63 poly-ubiquitin in wild-type and mutant Cx43 GJ plaques. (B) Quantification of the ratio of mean fluorescence intensity in the red and green channels per cell pair. Asterisks indicate significance level compared to wild type. The K264R, K303R and K264/303R mutants each show a significant reduction in the ratio of K63 poly-ubiquitin to Cx43 fluorescence intensity consistent with Co-IP analyses presented in Fig. 5. Mutating K287 located in the main Cx43 endocytic domain (S3) may interfere with AP-2/clathrin binding and thus results in hyper-ubiquitylation, similarly to phosphorylation, as shown in Fig. 7. Data are mean±s.e.m.; ns, not significant; *P<0.05, ***P≤0.0005.
Preventing ubiquitylation on lysines 264 and 303 leads to hyper-phosphorylation of serines 368, 279/282, and 255
As described above, K63-polyubiquitylation-deficient Cx43 polypeptides and GJs were found to accumulate in the PM. Intriguingly, these Cx43 mutants accumulated as hyper-phosphorylated variants (compare the amount of Cx43 P1 and P2 forms in wild type and mutants in Figs 3C and 4; note that mutant K287R does not accumulate hyperphosphorylated), suggesting that phosphorylation at specific sites in Cx43 may precede and trigger Cx43 ubiquitylation. To further investigate which phosphorylation events preceded ubiquitylation, we analyzed and quantified phosphorylation levels of the Cx43 lysine mutants on serines 368, 279/282, 255 and 262 – modifications well known to down-regulate GJ-mediated cell-to-cell communication (reviewed in Thévenin et al., 2013). MDCK cells were lysed 24 h post transfection with wild-type or relevant K/R mutant constructs, and analyzed by western blotting using Cx43 phospho-specific antibodies (Fig. 7). Amounts of phosphorylated serine 368 (pS368), 279/282 (pS279/pS282), and 255 (pS255) Cx43 polypeptides were significantly increased in all double and single K/R mutants with the exception of the K287R mutant. Increased phosphorylation of serine 262 (pS262) was also observed; however, this occurred only in a few mutants (some mutants containing 287, and/or 303K/R exchanges), suggesting that mutating lysine 287 leads to side effects that impair GJ internalization (discussed below). Taken together, these results indicate that in all Cx43 mutants that harbor lysine to arginine exchanges on K264 and/or K303, serines 368, 279/282 and 255 become hyper-phosphorylated; suggesting that phosphorylation on one, or a combination of these serine residues may trigger K63-polyubiquitylation to induce GJ internalization.
Fig. 7.

K63-polyUb-deficient Cx43 K/R mutants become hyper-phosphorylated on S368, S279/S282 and S255. Wild-type Cx43 and single and double K/R mutants expressed in MDCK cells were analyzed by western blotting and probed using antibodies directed against total Cx43, and Cx43-phospho-specific antibodies directed against phosphorylated S368 (pS368), S279/S282 (pS279/pS282), S255 (pS255) and S262 (pS262) (specific Cx43 phosphorylation events known to downregulate GJIC). Blots were stripped and α-tubulin was analyzed as loading control. Densitometric analyses of Cx43 proteins normalized to α-tubulin. Dramatic increases in pS368, pS279/pS282 and pS255 were detected whenever K264 and/or K303 were mutated. Accumulation of Cx43 phosphorylated on serine 262 (pS262) was detected in only a few mutants (mutants containing K287R and/or K303R) and was attributed to potentially other, or non-specific mutagenesis-related side effects. Wild-type and mutant samples were analyzed on the same SDS-PAGE gels to allow direct comparison. Lanes of blots are shown separated when extraneous lanes were removed. Data are mean±s.e.m.; ns, not significant; **P<0.005, ***P≤0.0005.
DISCUSSION
Ubiquitylation is a post-translational modification that plays an important role in regulating protein degradation. In particular, the addition of K48- and K63-linked polyubiquitin chains act as well-known signals for targeting proteins and protein complexes to either proteasomal or endo-/phagolysosomal degradation (Komander and Rape, 2012). Here, we report for the first time the identification of two juxtaposed lysine residues in the C-terminus of Cx43, lysines 264 and 303, which can be K63-polyubiquitylated in GJs. Cx43 lysine 264 and 303 to arginine mutant proteins exhibited significantly longer protein half-lives and assembled significantly larger GJ plaques, suggesting that K63-polyubiquitylation of Cx43 is required for successful GJ internalization. Our additional discovery that Cx43 ubiquitin-deficient lysine mutants accumulate as hyper-phosphorylated proteins in the plasma membrane further suggests that K63-poly-ubiquitylation is triggered by Cx43 phosphorylation. Use of phospho-specific Cx43 antibodies revealed that hyper-phosphorylation occurred on serines 368, 279/282 and 255, which are well characterized PKC and MAPK target sites known to downregulate GJ-mediated intercellular communication (GJIC) (Fong et al., 2014; Kanemitsu et al., 1998; Lampe et al., 1998, 2000; Leykauf et al., 2003; Nimlamool et al., 2015; Petrich et al., 2002; Polontchouk et al., 2002; Sáez et al., 1997; Sirnes et al., 2009; Solan and Lampe, 2007, 2008) and GJ turnover (Fong et al., 2014; Nimlamool et al., 2015; Thévenin et al., 2017).
In our experiments, K63-polyubiquitylated Cx43 migrated as a smear of higher molecular weight forms (marked with brackets in Figs 2 and 5) including several distinct bands, e.g. 70 and ∼150 kDa (marked with asterisks in Figs 2 and 5) on SDS-PAGE gels as described by Ribeiro-Rodrigues and co-workers (Ribeiro-Rodrigues et al., 2014). Although ubiquitylated proteins are known to not necessarily migrate on SDS-PAGE gels according to their molecular weight (Hospenthal et al., 2015), polyubiquitin chains in general are believed to consist of at least four Ubs (Ravid and Hochstrasser, 2008). The molecular weight of four Ubs plus Cx43 would be ∼77 kDa, which is relatively close to the migration pattern of the prominently detected 70 kDa band, thus conforming to established criteria.
Ubiquitylation of Cx43 to facilitate its degradation has been known for quite some time (Girão et al., 2009; Laing and Beyer, 1995; Laing et al., 1997; Leithe and Rivedal, 2004). Ubiquitylation of Cx43 was discovered as a signal for proteasomal degradation (Laing and Beyer, 1995), thus regulating the degradation of mis-folded or aberrantly oligomerized Cx43 polypeptides shortly after their biosynthesis in the ER membrane. The later discovery that ubiquitylation may also play a role in the lysosomal degradation of Cx43 (Laing et al., 1997) suggested that ubiquitylation may also play a role in the degradation of oligomerized Cx43 complexes (connexons, GJ channels, and GJ plaques). However, the type of ubiquitylation, the lysine residues that become ubiquitylated and the signals that induce Cx43 ubiquitylation in GJs to aid in their internalization and degradation remained illusive. It is now well known that the type of Ub linkage conveys Ub signal specificity (Ikeda et al., 2010; Komander and Rape, 2012; MacGurn et al., 2012). Specifically, exposure of a hydrophobic patch on Ub and flexibility of the Ub C-terminus rely on the type of polyUb chain conformation (e.g. K48-linked versus K63-linked polyUb chains) and thus determine Ub signal specificity (Ye et al., 2012). Different E3 ligases add ubiquitins in a chain-specific manner. Nedd4-1, Wwp1 and Smurf2 (all HECT ligases) as well as Trim21 (a RING ligase) have been found to interact with Cx43 (Basheer et al., 2015; Chen et al., 2012; Fykerud et al., 2012; Leykauf et al., 2006). Of these, Nedd4-1 is known to preferentially interact with K63-polyubiquitylated proteins (Kim and Huibregtse, 2009) and to interact with 283PPGY286 in Cx43 via its WW2 domain (Leykauf et al., 2006) (see Fig. 8A). Moreover, phosphorylation of S279/S282 in vitro was recently found to increase the binding affinity of this E3 ligase for its Cx43 binding domain (Spagnol et al., 2016), further supporting the concept that Nedd4-1 K63-polyubiquitylates Cx43 in GJs to mediate their internalization and degradation.
Fig. 8.

Schematic of K63-polyUb-mediated internalization of Cx43 GJs. (A) C-terminal tail of Cx43 showing the relevant Nedd4-1 E3-Ub ligase binding site (brown circle), the AP-2/clathrin binding sites (purple boxes), the K63-polyubiquitylated lysine residues (green boxes) and the serine residues that are phosphorylated before Cx43 K63-polyubiquitylation (golden asterisks). Note that ubiquitylation occurs juxtaposed to the left and right of the Nedd4-1 and AP-2/clathrin binding sites. (B) Phosphorylation of Cx43 by PKC on S368, and MAPK on S279/S282, S255 and possibly on additional residues and kinases leads to Nedd4-1 recognition, K63-polyubiquitylation and AP-2/clathrin binding; resulting in GJ internalization. Mutating lysines 264 or 303 separately or together inhibits ubiquitylation, causes Cx43 hyper-phosphorylation on distinct residues and abolishes GJ internalization. Coupled S365 de-phosphorylation/S368 phosphorylation (a ‘gate-keeper’ event) may be critical, as it has been associated with a significant conformational re-arrangement of the Cx43 C-terminal domain that affects the upstream located, AP-2/clathrin binding region (Solan et al., 2007), and thus may allow MAPK, Nedd4-1 and AP-2/clathrin to access their respective Cx43-CT binding sites that otherwise may be sterically inaccessible in functional Cx43 GJ channels.
Cx43 domains exposed to the cytoplasm when oligomerized into GJ channels [intracellular (I)-loop and C-terminal (CT) domain] contain a large number of lysine residues (11 in the I-loop, 9 in the CT) that are all potential targets for ubiquitylation (labeled in red in Fig. 3A). However, as the majority of regulatory phosphorylation events occur in the Cx43-CT, we hypothesized that ubiquitylation – if occurring in GJs – would also occur in the Cx43-CT. In addition, the C-terminal region juxtaposed to the fourth transmembrane domain is known to harbor a microtubule binding site (Giepmans et al., 2001) and when mutated will likely interfere with Cx trafficking (Wayakanon et al., 2012). We thus left the lysines located within this region (K234, K237 and K241) untouched. Both mutants, with the six remaining C-terminal lysines (6K/R; K258, K264, K287, K303, K345, K346), and the mutant with the three central lysines mutated (3K/R; K264, K287, K303) behaved indistinguishably, resulting in significantly increased mutant protein half-lives, the accumulation of hyper-phosphorylated Cx43 polypeptides and larger GJ plaques in the PM (Figs 3 and 4); suggesting that one or more of these three (central) lysine residues become K63-polyubiquitylated. Generating and examining all possible double and single mutants then indicated that two of the three lysine residues, K264 and K303, can become K63-polyubiquitylated. Our finding that the protein half-lives of K264 and K303 single mutants were significantly longer compared with the wild type, although not as long as half-lives of the double lysine mutants (4.5 h and 6.6-11.4 h compared with 2.3 h for wild type, Fig. 4C) argues for a cumulative effect. However, we did not detect K63-polyubiquitylation of K264 and K303 single mutants (Figs 5 and 6), suggesting that mutating one lysine residue may inhibit K63-polyubiquitylation of Cx43 altogether. Of course, mutating lysines into arginines bears the risk of introducing conformational alterations of the Cx43-CT and this indirectly may inhibit GJ internalization; however, the fact that exactly two out of six mutated lysines inhibited GJ internalization, that K63-polyubiquitylation was detected on both locations (K264 and K303), and that the K287R mutant exhibited a half-life similar to that of the wild type and was not hyper-phosphorylated strongly argues against this possibility.
Phosphorylation of Cx43 is well established and has been shown to be a regulatory mechanism for Cx43 trafficking, GJ assembly, gating, plaque internalization and degradation (reviewed in Falk et al., 2016; Solan and Lampe, 2014; Thévenin et al., 2013). Cx43 phosphorylation by Akt (PKB), PKA (protein kinase A), and CK1 (casein kinase 1) is known to upregulate GJ intercellular communication (GJIC), whereas phosphorylation by PKC, CDC2 (cell division cycle protein 2), MAPKs and Src downregulate GJIC by closing GJ channels (Fong et al., 2014; Kanemitsu et al., 1998; Lampe et al., 1998, 2000; Leykauf et al., 2003; Nimlamool et al., 2015; Petrich et al., 2002; Polontchouk et al., 2002; Sáez et al., 1997; Sirnes et al., 2009; Solan and Lampe, 2007, 2008; van Zeijl et al., 2007). More recent findings from our lab also provided evidence for a link between Cx43 phosphorylation and GJ internalization. Specifically, phosphorylation of serines 368, 279/282, 255 (and in some cases 262) by PKC and MAPKs in response to epidermal growth factor (EGF) and vascular endothelial growth factor (VEGF) stimulation has been linked to acute GJ internalization in mouse embryonic stem cells and pPAECs (Fong et al., 2014; Nimlamool et al., 2015); and phosphorylation of serines 365, 368 and 373 regulates Cx43 and ZO-1 binding and release, an important early step in GJ turnover (Thévenin et al., 2017). Additionally, S279/S282 phosphorylation-deficient Cx43 mutants were shown to stabilize GJ plaques in human pancreatic tumor cells (Johnson et al., 2013), and upon EGF stimulation in HeLa cells (Schmitt et al., 2014). Recently, Solan and Lampe (Solan and Lampe, 2015) and we (Falk et al., 2016) suggested a kinase program consisting of PKC, MAPKs and Src that hierarchically phosphorylate Cx43 on serines 368, 279/282 (and potentially 255 and 262) and tyrosine 247, respectively, to spatiotemporally regulate GJ internalization. Of these events, phosphorylation of S368 is of particular interest. It has been found to preferentially localize to the center of GJ plaques (Cone et al., 2014), the region where GJ channel internalization occurs (Falk et al., 2009; Gaietta et al., 2002; Lauf et al., 2002). Furthermore, S368 phosphorylation is known to be dependent on S365 dephosphorylation (termed ‘gate keeper’), an event known to trigger a large conformational change of the Cx43-CT, which affects the upstream region that harbors the ubiquitylation sites described above (Solan et al., 2007). However, a link between phosphorylation and ubiquitylation had not been demonstrated.
Ub-mediated protein degradation is often regulated via protein phosphorylation (termed phosphodegron). Phosphorylation on one or more residues induces substrate changes (conformational or other) allowing the subsequent ubiquitylation of target lysines that then triggers the degradation of the phosphorylated and/or ubiquitylated protein (Nguyen et al., 2013; Ravid and Hochstrasser, 2008). As our data suggest that phosphorylation of S368, S279/S282 and S255 occurs prior to ubiquitylation, and preventing ubiquitylation on relevant lysine residues results in the build-up of hyper-phosphorylated mutant Cx43 protein and GJs in the plasma membrane, it is tempting to speculate that phosphorylation of Cx43 similarly induces K63-polyubiquitylation, which then triggers GJ internalization and degradation. We do not know yet which of the three phosphorylation events are required and in what order. However, as S365 dephosphorylation and S368 phosphorylation is linked to conformational Cx43-CT rearrangements (Solan et al., 2007), and S279/S282 phosphorylation is linked to enhancing Nedd4-1 binding (Spagnol et al., 2016), phosphorylation on at least these two sites and in this order is likely to precede Cx43 K63-polyubiquitylation.
How might K63-polyubiquitylation of Cx43 induce GJ internalization and degradation? It should be noted that at any given time we detected only a small portion of Cx43 in GJs to be K63-polyubiquitylated (∼8% of the GJ plaque area), a caveat that prevented us from detecting the K63-poly-ubiquitylated portion of Cx43 in unfractionated whole-cell lysates. Yet, ubiquitylation occurred in distinct GJ plaque domains in PAECs (Fig. 1), and in HeLa cells where GJ plaques usually become much larger, confocal analyses revealed a speckled, predominantly plaque-central localization (Fig. 6). Several published observations by us and others suggest that cells turn over GJs continuously by adding newly synthesized GJ channels to the plaque periphery while simultaneously internalizing small GJ plaque portions from central plaque areas (Gaietta et al., 2002; Lauf et al., 2002; Falk et al., 2009). The central location of ubiquitylation and its positioning within small domains is consistent with this model and the hypothesis that ubiquitylation signals GJ channel internalization. Earlier, we characterized two tyrosine-based sorting signals (S2, Y265AYF and S3, Y286KLV) (Fong et al., 2013) that are located in the vicinity of the K63 poly-ubiquitylation sites in the Cx43 C-terminal domain that recruit AP-2/clathrin to internalize GJs and GJ plaque portions (reviewed in Falk et al., 2016) (Fig. 8B). It is likely that ubiquitylation facilitates and/or regulates AP-2/clathrin access and thus controls the final step of GJ internalization. Interestingly, we found that mutating K287 increased Cx43 K63-polyubiquitylation in GJ plaques (Fig. 6B). Lysine 287 is located within the Cx43 S3-endocytic motif (see above) and mutating this lysine likely interferes with AP-2/clathrin recruitment and GJ internalization, and thus, similarly to Cx43 hyper-phosphorylation, leads to hyper-ubiquitylation, further supporting the role of K63-polyubiquitylation as a signal for GJ internalization.
Remarkably, we also gained evidence suggesting that only a portion of Cx subunits in a connexon of an internalizing GJ channel becomes ubiquitylated, as significant amounts of unmodified Cx43 (migrating at ∼40 kDa) were detected with ubiquitylated Cx43 (migrating at a higher molecular weight) when total K63 poly-ubiquitylated proteins were precipitated and probed with Cx43-specific antibodies (Fig. 2C). Of additional interest is that a second, mono-ubiquitylation event probably occurs on Cx43 or a Cx43-binding protein in GJ plaques, as areal colocalization of FK2 (recognizing both, mono and poly-Ub chains) was double (∼16%) that of FK1 (recognizing only poly-Ub chains) and K63-polyUb-specific antibodies. Taken together, the low percentage of colocalization and the localization characteristics (specific localization in central domains) correlate well with the short turnover rate of Cxs and GJs of only a few hours, suggesting that the ubiquitylated domains indeed corresponded to the plaque areas that were actively internalizing.
Alternatively, under certain conditions, clathrin may be recruited by another group of adaptor proteins, termed clathrin-associated sorting proteins (CLASPs), that specifically bind via a Ub-interacting motif (UIM) to a polypeptide sequence that is exposed in K63-polyubiquitin chains (Traub and Bonifacino, 2013). One such alternative clathrin adaptor, Eps15, was proposed to bind to Cx43 and facilitate GJ internalization (Catarino et al., 2011; Girão et al., 2009). Thus, K63-polyubiquitylation may allow Eps15 to bind to Cx43 and internalize GJs alternative to AP-2 (Gumpert et al., 2008; Piehl et al., 2007; Falk et al., 2016, 2014).
Previously, we and others also have demonstrated that Cx43 in GJs and internalized GJs interacts with a protein termed p62/SQSTM1, which sequesters AGJs to autophagosomal degradation (Bejarano et al., 2012; Fong et al., 2012; Lichtenstein et al., 2010). p62/SQSTM1 interacts specifically with K63-polyubiquitylated substrates via its UBA (ubiquitin-associated) domain (Seibenhener et al., 2004) to sequester targets for autophagosomal degradation (Bjørkøy et al., 2005; Pankiv et al., 2007). Depleting cells of autophagy-associated proteins such as LC3, Beclin1, Atg5/Atg12 and p62/SQSTM1 by RNAi significantly impaired GJ degradation (Bejarano et al., 2012; Fong et al., 2012; Lichtenstein et al., 2010). Thus, it is also likely that Cx43 K63-polyubiquitylation sequesters internalized GJs for autophagosomal degradation.
Taken together, our previous and current findings suggest that a series of post-translational modifications including ZO-1 binding and release, phosphorylation/de-phosphorylation and K63-polyubiquitylation transition functional into non-functional GJ channels that then are endocytosed (Fong et al., 2014; Nimlamool et al., 2015; Thévenin et al., 2017; reviewed in Falk et al., 2016). Blocking K63-polyubiquitylation apparently prevents GJ internalization and leads to the accumulation of hyper-phosphorylated Cx43 and GJs in the plasma membrane (scheme in Fig. 8B). GJ channels assembled from a Cx43 mutant with serine 368 exchanged to glutamic acid (E) (phospho-mimetic, simulating hyper-phosphorylation) are closed (did not transfer Lucifer Yellow) (Thévenin et al., 2017), suggesting that K63-polyUb-dead mutants are also closed. As E3-Ub-specific small molecule inhibitors are available, their clinical administration may prevent the acute loss of GJs from intercalated discs that is a typical culprit in a number of severe heart diseases (Fontes et al., 2012).
MATERIALS AND METHODS
cDNA constructs and mutagenesis
Full-length wild-type rat Cx43 cDNA was cloned into pEGFP-N1 vector (Clontech) as described (Falk, 2000). To generate full-length untagged wild-type Cx43, an authentic TAA stop codon was re-introduced (Fong et al., 2013). Untagged rat Cx43 mutant 3K/R and 9K/R cloned into pcDNA3.1 were generously provided by Vivian Su and Alan Lau (Natural Products and Cancer Biology Program, Cancer Research Center of Hawaii, Honolulu, USA) (Su et al., 2010). Untagged 6K/R Cx43 mutant was generated by restoring K234, K237 and K241 from 9K/R and adding a BglII restriction site for confirmation of mutagenesis. Double and single K/R mutants were generated by restoring one or two of the remaining lysines 264, 287 and 303 from the Cx43 mutant 3K/R using QuikChange Mutagenesis (Stratagene, Santa Clara, CA). Forward recovery mutagenesis primers were as follows (arginine recovered codons are underlined, BglII site in bold): R234_237_241K recovery, 5′-G CTC TTC TAC GTC TTC AAA GGC GTT AAG GAT CGC GTG AAG GGA AGA TCT GAT CC-3′; R264K recovery, 5′-CCA TCA AAA GAC TGC GGA TCT CCA AAA TAC GCC TAC TTC AAT GGC-3′; R287K recovery, 5′-CCT ATG TCT CCT GGG TAC AAG CTG GTT ACT GGT GAC AGA AAC AAT TCC-3′; and R303K recovery, 5′-CC TCG TGC CGC AAT TAC AAC AAG CAA GCT AGC GAG CAA AAC TGG-3′. PCR mutagenesis reactions were generated using proofreading Pfu Ultra II polymerase (Cat. No. 600670-51; Stratagene). Digestion of template cDNA with Dpn1 restriction endonuclease (Cat. No. R0176-S; New England Biolabs, Ipswich, MA) was followed by transformation into chemically competent DH5α E. coli cells (Cat. No. 18258-012; Invitrogen). Sequencing was performed on all cDNA constructs to confirm the presence of intended mutations and exclude the presence of additional unwanted mutations.
Cell culture and transient transfections
HeLa (gap junction deficient; Cat. No. CCL-2; American Type Culture Collection, Manassas, VA), Madine-Darby Canine Kidney (MDCK) (gap junction deficient; Cat. No. NBL-2; American Type Culture Collection) and pPAECs (expressing Cx43 and other Cxs, gap junction proficient; Cat. No. P302; Cell Applications, San Diego, CA) were maintained in low glucose Dulbecco's Modified Eagle Medium (DMEM) (Cat. No. SH30021.01; HyClone, Logan, UT) supplemented with 50 I U/ml penicillin and 50 μg/ml streptomycin (Cat. No.30-001-Cl; Corning, Manassas, VA), 2 mM L-glutamine (Cat. No. 25-005-C1; Mediatech, Manassas, VA) and 10% fetal bovine serum (Cat. No. S11150; Atlanta Biologicals, Flowery Branch, GA) at 37°C, 5% CO2 and 100% humidity. Cells were washed with 1× PBS and treated with 0.25% trypsin (Cat. No. 25-053-Cl; Corning) for passaging. 24-48 h after passaging, 60-80% confluent HeLa and MDCK cells were transiently transfected with wild-type and mutant constructs using Superfect (Cat. No. 301307; Qiagen, Hilden, Germany) or Lipofectamine2000 (Cat. No. 11668019; Invitrogen, Carlsbad, CA) reagents, respectively, according to the manufacturers’ recommendations.
Immunofluorescence staining and quantitative image analyses
pPAECs and HeLa cells were grown on pretreated poly-L-lysine (Cat. No. P8920; Sigma, St. Louis, MO)-coated coverslips in low glucose DMEM at 37°C, 5% CO2 and 100% humidity. Cells were fixed in 3.7% formaldehyde/PBS and permeabilized with 0.2% Triton X-100 (TX-100) (Cat. No. 3929-2; VWR, Radnor, PA) in PBS. Cells were blocked in 10% FBS/PBS for 30 min at room temperature (RT) and incubated with primary rabbit polyclonal anti-peptide Cx43 antibodies (Cat. No. 3512; Cell Signaling, Danvers, MA) or mouse monoclonal anti-Cx43 antibodies (clone 4E6.2) (Cat. No. MAB3067, Millipore, Billerica, MA) diluted 1:500 in 10% FBS/PBS at 4°C overnight. Additionally, pPAECs were incubated with primary mouse monoclonal anti-monoUb and polyUb (FK2) (Cat. No. PW8810; Enzo, Farmingdale, NY), polyUb (FK1) (Cat. No. PW8805; Enzo), K63-polyUb-specific (clone HWA4C4) (Cat. No. PW0600; Enzo) or primary monoclonal K48-polyUb-specific (clone Apu2) (Cat. No. 05-1307; Millipore) antibodies diluted 1:200 in 10% FBS/PBS at 4°C overnight. Similarly, transfected HeLa cells were incubated with mouse monoclonal anti-Cx43 antibodies (Cat. No. 138300, Invitrogen) diluted 1:200 in 10% FBS/PBS, then rabbit monoclonal anti-K63-polyUb-specific antibodies (Cat. No. 05-1308, clone Apu3; Millipore) diluted 1:200 in 10% FBS/PBS. Cells were incubated in secondary antibodies (goat anti-rabbit Alexa Fluor488, goat anti-mouse Alexa Fluor 568, goat anti-rabbit Alexa Fluor 568 and goat anti-mouse Alexa Fluor 488; Cat. No. A11008, A11031, A11011 and A11001, respectively; Molecular Probes/Invitrogen, Eugene, OR) diluted 1:200 or 1:500 in 10% FBS/PBS, respectively, for 1 h at RT. Cells were also stained with 1 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) (Cat. No. D1306; Molecular Probes). Coverslips were rinsed in distilled water and mounted using Fluoromount-G (Cat. No. 0100-01; Southern Biotechnology, Birmingham, AL). Endogenous GJ plaques in pPAECs and transiently expressed Cx43 wild-type and mutant GJs expressed in HeLa cells (presented in Figs 1 and 6) were imaged using a Zeiss 880 confocal microscope equipped with a 63× NA 1.4 Plan-Apochromat oil-immersion objective acquiring image z-stacks for each GJ plaque that extended well above and below the GJ plaque signal into the cell body. Exogenous GJ plaques in HeLa cells (presented in Fig. 3) were imaged using a Nikon Eclipse TE2000E wide-field inverted fluorescence microscope equipped with 40× NA 1.3 Plan Fluor and 60× NA 1.4 Plan-Apochromat oil-immersion objectives (Nikon Instruments, Melville, NY) and a forced-air cooled Photonics CoolSnap® HQ CCD camera (Roper Scientific, Duluth, GA). Images were acquired using MetaVue software version 6.1r5 (Molecular Devices, Sunnyvale, CA).
To quantify GJ plaque size and number (presented in Fig. 3), the longest distance along each plaque was measured using ImageJ (National Institutes of Health, Bethesda, MD) and was quantified for 251, 368 and 376 plaques across 72, 81 and 94 cell pairs expressing Cx43 wild type, 3K/R and 6K/R, respectively. Plaque number and total size per cell pair was calculated by summing the individual plaques/plaque lengths for each cell pair.
To quantify colocalization of ubiquitin with Cx43-specific antibodies in PAECs (Fig. 1), GJ plaques of entire lateral membranes were outlined using the freeform tool in the Fiji ImageJ distribution (Schindelin et al., 2012). This procedure was chosen as endogenous GJ plaques in these cells are numerous and too small to be outlined individually. The background fluorescence intensity for each antibody was calculated by averaging the mean image intensity of full field of view for the first and last z-stack sections, which contained cell bodies but extended beyond GJs. Thresholds for fluorescence signals for each antibody were chosen to be proportional to the background fluorescence intensity for each antibody. For each cell pair, the area where colocalization occurred was calculated using the ‘Colocalization’ plugin packaged with the Fiji distribution, which selected areas where both the Cx43 and Ub antibody fluorescence was above the threshold settings. This area was divided by the area where the Cx43 fluorescence was above the threshold to obtain the proportion of colocalization by area. This procedure was performed for each z-section, and sections were summed to produce the proportion of colocalization by area per GJ plaque. Finally, the proportion of colocalization by area for each Ub-specific antibody was normalized by setting the fluorescence in the K48-polyUb channel to zero. 127, 117, 92, and 79 cell pairs were quantified for the FK2, FK1, K63-polyUb- and K48-polyUb-specific antibodies, respectively.
To quantify relative colocalization of K63-polyUb antibodies with Cx43 antibodies in HeLa cells (Fig. 6), individual GJ plaques were outlined using the freeform tool in the Fiji ImageJ distribution. This was possible as exogenous GJ plaques in these cells are large enough to be recognized and outlined individually. The area and mean fluorescence intensity in the red (ubiquitin) and green (Cx43) channels were measured for each plaque. For each cell pair, the ratio of red/green intensity was calculated by summing the ratio of mean red/green intensity for each plaque in the cell pair while normalizing by the area of each plaque within the cell pair. 50, 63, 51, 80, 75, 71, and 57 cell pairs were quantified for the wt, K264R, K287R, K303, K264/303R, K264/287R and K287/303R conditions, respectively. To account for variations in staining intensity between independent experiments, the green fluorescence intensity of each cell pair was normalized between trials. This was done by multiplying the green fluorescence intensity in each trial by a single constant across all mutants such that the mean green fluorescence intensity in the wild-type Cx43 sample was the same between trials. The first and third statistical quartiles of each dataset were calculated using Excel. GJ plaques with abnormally high red fluorescence (cell pairs more than three interquartile distances above the third quartile, no more than 5 per Cx43 mutant) were defined as outliers and eliminated to produce the above-mentioned sets of cell pairs.
Triton X-100 solubility assays
To separate intracellular Cx proteins and connexons from plasma membrane GJs and likely also internalized AGJs, TX-100 solubility assays were performed based on the method described by Musil and Goodenough (Musil and Goodenough, 1991). MDCK cells transiently transfected with wild-type or Cx43K/R constructs or pPAECs were used. 24 h post transfection (MDCKs), or 48 h after seeding (pPAECs), cells were washed in ice-cold PBS. Prior to lysis, pPAECs were treated with 20 μM of the pan-DUB inhibitor, PR-619 (Cat. No. 662141; EMD Millipore) for 1.5 h (Fig. 2B). Cells were lysed on ice for 15 min in 400 μl pre-chilled lysis buffer containing 50 mM Tris-HCl, pH 7.4 (Cat. No. BP 153-1; Fisher Bioreagents, Fair Lawn, NJ), 150 mM NaCl (Cat. No. S671-3; Fisher Chemicals, Fair Lawn, NJ), 1 mM EDTA (Cat. No. 161-0729; Bio-Rad, Hercules, CA), 0.5% sodium deoxycholate (Cat. No. D6750; Sigma), 1% Triton X-100 (Cat. No. VW3929-2; VWR), and 1% Igepal (Cat. No. 18896; Sigma). The buffer was supplemented with the following protease and phosphatase inhibitors: 1 mM protease inhibitor cocktail (Cat. No. P8340; Sigma), 1 mM β-glycerol phosphate (Cat No. 157241; MP Biomedicals, LLC, Solon, OH), 1 mM sodium orthovanadate (Cat. No. 450234; Sigma), 20 mM N-ethylmalemide (Cat. No. E1271; Sigma), and 10 mM 1,10-phenanthroline monohydrate (Cat. No. AC130130050; ACROS Organics, NJ). Lysates were centrifuged at 10,000 g for 5 min at RT to pre-clear cell lysates. 180 μl of each resulting supernatant was centrifuged at 100,000 g in a Beckman Coulter Airfuge® ultracentrifuge for 10 min. For western blot analysis, 100,000 g supernatants (TX-100-soluble fractions) and pellets (TX-100-insoluble fractions) were re-suspended in SDS-PAGE sample buffer and boiled for 5 min.
SDS-PAGE and western blot analyses
Immunoprecipitation, protein half-life, and phosphorylation analyses samples were loaded onto 10% SDS-PAGE mini-gels (Bio-Rad). Proteins were transferred onto nitrocellulose membranes and blocked for 1 h at RT in 5% non-fat dry milk in TBST (TBS/0.1% Tween 20) or 5% bovine serum albumin (BSA) (Cat. No. A7906; Sigma) in TBST. Antibodies were diluted in 5% BSA/TBS as follows: rabbit anti-Cx43, rabbit anti-K63-polyUb (Cat. No. 05-1308; EMD Millipore), rabbit anti-Cx43 pS279/pS282, rabbit anti-Cx43 pS255, rabbit anti-Cx43 pS262 (Cat. No. sc-12900-R, sc-12899-R and sc-17219-R, respectively; Santa Cruz, Dallas, TX) and rabbit anti-Cx43 pS368 (Cat. No. 3511S; Cell Signaling) at 1:2000, and mouse anti-α-tubulin primary antibodies at 1:5000. Blots were incubated with primary antibodies at 4°C overnight, then washed three times with TBST. Secondary HRP-conjugated goat anti-rabbit or goat anti-mouse antibodies (Cat. No. 81-6520 or 81-6120, respectively; Zymed, San Francisco, CA) were diluted 1:5000 and secondary HRP-conjugated mouse anti-rabbit light chain-specific antibodies (Cat. No. 211-032-171; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) were diluted 1:15,000. Blots were incubated in secondary antibodies for 1 h at RT. Blots were stripped and re-probed with anti-α-tubulin-specific antibodies to verify equal loading. Protein bands were detected using Pierce® ECL2 Western Blotting Substrate (Cat. No. 80196; Thermo Scientific, Rockford, IL) and Kodak BioMax® Light Autoradiography Film (Cat. No. 1788207; Carestream Health, Rochester, NY). Autoradiographs were scanned and Cx43 wild-type and mutant protein were quantified using ImageJ software, normalized to α-tubulin, and protein band intensity plotted as bar graphs.
Cx43 protein half-life analyses
24 h post transfection, MDCK cells were treated with 50 μg/ml cycloheximide (Cat. No. C655; Sigma) for 0, 1, 2, 3, 4 and 6 h at 37°C, 5% CO2 and 100% humidity. Cells were lysed in SDS sample buffer at each time point and boiled for 5 min. Samples were separated on 10% SDS-PAGE gels using western blot protocols. Blots were probed with rabbit anti-Cx43 antibody, then stripped with stripping buffer and stringently washed in TBST (Cat. No. BP337; Fisher Bioreagents) before re-probing with mouse anti-α-tubulin antibodies (Cat. No. 9026; Sigma). Cx43 protein intensities were quantified using ImageJ and normalized to the corresponding α-tubulin intensities.
Immunoprecipitations
100,000 g pellets (TX-100-insoluble fraction) from MDCK cells transiently transfected with Cx43 wild-type or K/R mutants were resuspended in fresh lysis buffer without TX-100 and sonicated (six 1 s pulses), with 5 min intervals on ice between pulses. Immunoprecipitation samples were incubated with rabbit anti-Cx43 or mouse anti-K63-polyUb antibodies adsorbed to Protein-A Sepharose beads (Cat. No. 3391; Sigma) for 2 h at RT while rotating. After incubation, beads were washed in lysis buffer and immunoprecipitated protein was eluted from beads with SDS sample buffer and boiled for 5 min.
Statistical analyses
One-way ANOVA analyses with post-hoc Bonferroni corrections were performed on data sets including GJ plaque size measurements (Fig. 3B), Cx43 wild-type and mutant protein levels (Fig. 3C), and amounts of phosphorylated Cx43 in K/R mutants (Fig. 6) in SPSS software. Unpaired two-tailed Student's t-tests were performed in Excel to analyze Cx43 protein half-lives (Fig. 4) and amounts of higher molecular weight K63-polyUb immunoreactive Cx43 (Fig. 5C). All data are presented as mean±s.e.m. P-values of *P<0.05, **P<0.005 and ***P≤0.0005 were considered statistically significant.
Supplementary Material
Acknowledgements
We thank Vivian Su and Alan Lau (Natural Products and Cancer Biology Program, Cancer Research Center of Hawaii, Honolulu) for providing Cx43 3K/R and 9K/R constructs, Lynne Cassimeris (Department of Biological Sciences, Lehigh University, Bethlehem) for the use of specialized equipment (AirFuge), JoAnn Trejo and Neil Grimsey, and current and previous Falk lab members for constructive discussions.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: R.M.K., R.A.M., M.M.F.; Methodology: R.M.K., R.A.M., C.G.F., M.M.F.; Validation: R.M.K., R.A.M., M.M.F.; Formal analysis: R.M.K., R.A.M., C.G.F., M.M.F.; Investigation: R.M.K., R.A.M., C.G.F.; Data curation: C.G.F.; Writing - original draft: R.M.K.; Writing - review & editing: R.M.K., R.A.M., M.M.F.; Visualization: R.M.K., R.A.M.; Supervision: M.M.F.; Project administration: M.M.F.; Funding acquisition: M.M.F.
Funding
This work was supported by the National Institutes of Health NIGMS (GM55725) to M.M.F. and by Lehigh University. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.204321.supplemental
References
- Basheer W. A., Harris B. S., Mentrup H. L., Abreha M., Thames E. L., Lea J. B., Swing D. A., Copeland N. G., Jenkins N. A., Price R. L. et al. (2015). Cardiomyocyte-specific overexpression of the ubiquitin ligase Wwp1 contributes to reduction in Connexin 43 and arrhythmogenesis. J. Mol. Cell. Cardiol. 88, 1-13. 10.1016/j.yjmcc.2015.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batra N., Kar R. and Jiang J. X. (2012). Gap junctions and hemichannels in signal transmission, function and development of bone. Biochim. Biophys. Acta 1818, 1909-1918. 10.1016/j.bbamem.2011.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beardslee M. A., Laing J. G., Beyer E. C. and Saffitz J. E. (1998). Rapid turnover of connexin43 in the adult rat heart. Circ. Res. 83, 629-635. 10.1161/01.RES.83.6.629 [DOI] [PubMed] [Google Scholar]
- Bejarano E., Girão H., Yuste A., Patel B., Marques C., Spray D. C., Pereira P. and Cuervo A. M. (2012). Autophagy modulates dynamics of connexins at the plasma membrane in a ubiquitin-dependent manner. Mol. Biol. Cell 23, 2156-2169. 10.1091/mbc.e11-10-0844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørkøy G., Lamark T., Brech A., Outzen H., Perander M., Øvervatn A., Stenmark H. and Johansen T. (2005). p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171, 603-614. 10.1083/jcb.200507002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catarino S., Ramalho J. S., Marques C., Pereira P. and Girão H. (2011). Ubiquitin-mediated internalization of connexin43 is independent of the canonical endocytic tyrosine-sorting signal. Biochem. J. 437, 255-267. 10.1042/BJ20102059 [DOI] [PubMed] [Google Scholar]
- Chen Z. J. and Sun L. J. (2009). Nonproteolytic functions of ubiquitin in cell signaling. Mol. Cell 33, 275-286. 10.1016/j.molcel.2009.01.014 [DOI] [PubMed] [Google Scholar]
- Chen V. C., Kristensen A. R., Foster L. J. and Naus C. C. (2012). Association of connexin43 with E3 ubiquitin ligase TRIM21 reveals a mechanism for gap junction phosphodegron control. J. Proteome Res. 11, 6134-6146. 10.1021/pr300790h [DOI] [PubMed] [Google Scholar]
- Cone A. C., Cavin G., Ambrosi C., Hakozaki H., Wu-Zhang A. X., Kunkel M. T., Newton A. C. and Sosinsky G. E. (2014). Protein kinase Cdelta-mediated phosphorylation of Connexin43 gap junction channels causes movement within gap junctions followed by vesicle internalization and protein degradation. J. Biol. Chem. 289, 8781-8798. 10.1074/jbc.M113.533265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dukes J. D., Whitley P. and Chalmers A. D. (2011). The MDCK variety pack: choosing the right strain. BMC Cell Biol. 12, 43 10.1186/1471-2121-12-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk M. M. (2000). Connexin-specific distribution within gap junctions revealed in living cells. J. Cell Sci. 113, 4109-4120. [DOI] [PubMed] [Google Scholar]
- Falk M. M., Baker S. M., Gumpert A. M., Segretain D. and Buckheit R. W. III (2009). Gap junction turnover is achieved by the internalization of small endocytic double-membrane vesicles. Mol. Biol. Cell 20, 3342-3352. 10.1091/mbc.e09-04-0288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk M. M., Kells R. M. and Berthoud V. M. (2014). Degradation of connexins and gap junctions. FEBS Lett. 588, 1221-1229. 10.1016/j.febslet.2014.01.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk M. M., Bell C. L., Kells Andrews R. M. and Murray S. A. (2016). Molecular mechanisms regulating formation, trafficking and processing of annular gap junctions. BMC Cell Biol. 17 Suppl. 1, 22 10.1186/s12860-016-0087-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallon R. F. and Goodenough D. A. (1981). Five-hour half-life of mouse liver gap-junction protein. J. Cell Biol. 90, 521-526. 10.1083/jcb.90.2.521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong J. T., Kells R. M., Gumpert A. M., Marzillier J. Y., Davidson M. W. and Falk M. M. (2012). Internalized gap junctions are degraded by autophagy. Autophagy 8, 794-811. 10.4161/auto.19390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong J. T., Kells R. M. and Falk M. M. (2013). Two tyrosine-based sorting signals in the Cx43 C-terminus cooperate to mediate gap junction endocytosis. Mol. Biol. Cell 24, 2834-2848. 10.1091/mbc.e13-02-0111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong J. T., Nimlamool W. and Falk M. M. (2014). EGF induces efficient Cx43 gap junction endocytosis in mouse embryonic stem cell colonies via phosphorylation of Ser262, Ser279/282, and Ser368. FEBS Lett. 588, 836-844. 10.1016/j.febslet.2014.01.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontes M. S. C., van Veen T. A. B., de Bakker J. M. T. and van Rijen H. V. M. (2012). Functional consequences of abnormal Cx43 expression in the heart. Biochim. Biophys. Acta 1818, 2020-2029. 10.1016/j.bbamem.2011.07.039 [DOI] [PubMed] [Google Scholar]
- Fykerud T. A., Kjenseth A., Schink K. O., Sirnes S., Bruun J., Omori Y., Brech A., Rivedal E. and Leithe E. (2012). Smad ubiquitination regulatory factor-2 controls gap junction intercellular communication by modulating endocytosis and degradation of connexin43. J. Cell Sci. 125, 3966-3976. 10.1242/jcs.093500 [DOI] [PubMed] [Google Scholar]
- Gaietta G., Deerinck T. J., Adams S. R., Bouwer J., Tour O., Laird D. W., Sosinsky G. E., Tsien R. Y. and Ellisman M. H. (2002). Multicolor and electron microscopic imaging of connexin trafficking. Science 296, 503-507. 10.1126/science.1068793 [DOI] [PubMed] [Google Scholar]
- Giepmans B. N. G., Verlaan I., Hengeveld T., Janssen H., Calafat J., Falk M. M. and Moolenaar W. H. (2001). Gap junction protein connexin-43 interacts directly with microtubules. Curr. Biol. 11, 1364-1368. 10.1016/S0960-9822(01)00424-9 [DOI] [PubMed] [Google Scholar]
- Gilleron J., Carette D., Fiorini C., Benkdane M., Segretain D. and Pointis G. (2009). Connexin 43 gap junction plaque endocytosis implies molecular remodelling of ZO-1 and c-Src partners. Commun. Integr. Biol. 2, 104-106. 10.4161/cib.7626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girão H., Catarino S. and Pereira P. (2009). Eps15 interacts with ubiquitinated Cx43 and mediates its internalization. Exp. Cell Res. 315, 3587-3597. 10.1016/j.yexcr.2009.10.003 [DOI] [PubMed] [Google Scholar]
- Goodenough D. A. and Gilula N. B. (1974). The splitting of hepatocyte gap junctions and zonulae occludentes with hypertonic disaccharides. J. Cell Biol. 61, 575-590. 10.1083/jcb.61.3.575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumpert A. M., Varco J. S., Baker S. M., Piehl M. and Falk M. M. (2008). Double-membrane gap junction internalization requires the clathrin-mediated endocytic machinery. FEBS Lett. 582, 2887-2892. 10.1016/j.febslet.2008.07.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesketh G. G., Shah M. H., Halperin V. L., Cooke C. A., Akar F. G., Yen T. E., Kass D. A., Machamer C. E., Van Eyk J. E. and Tomaselli G. F. (2010). Ultrastructure and regulation of lateralized connexin43 in the failing heart. Circ. Res. 106, 1153-1163. 10.1161/CIRCRESAHA.108.182147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hospenthal M. K., Mevissen T. E. T. and Komander D. (2015). Deubiquitinase-based analysis of ubiquitin chain architecture using Ubiquitin Chain Restriction (UbiCRest). Nat. Protoc. 10, 349-361. 10.1038/nprot.2015.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda F., Crosetto N. and Dikic I. (2010). What determines the specificity and outcomes of ubiquitin signaling? Cell 143, 677-681. 10.1016/j.cell.2010.10.026 [DOI] [PubMed] [Google Scholar]
- Johnson K. E., Mitra S., Katoch P., Kelsey L. S., Johnson K. R. and Mehta P. P. (2013). Phosphorylation on Ser-279 and Ser-282 of connexin43 regulates endocytosis and gap junction assembly in pancreatic cancer cells. Mol. Biol. Cell 24, 715-733. 10.1091/mbc.e12-07-0537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan K., Chodock R., Hand A. R. and Laird D. W. (2001). The origin of annular junctions: a mechanism of gap junction internalization. J. Cell Sci. 114, 763-773. [DOI] [PubMed] [Google Scholar]
- Kanemitsu M. Y., Jiang W. and Eckhart W. (1998). Cdc2-mediated phosphorylation of the gap junction protein, connexin43, during mitosis. Cell Growth Differ. 9, 13-21. [PubMed] [Google Scholar]
- Kim H. C. and Huibregtse J. M. (2009). Polyubiquitination by HECT E3s and the determinants of chain type specificity. Mol. Cell. Biol. 29, 3307-3318. 10.1128/MCB.00240-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. T., Kim K. P., Lledias F., Kisselev A. F., Scaglione K. M., Skowyra D., Gygi S. P. and Goldberg A. L. (2007). Certain pairs of ubiquitin-conjugating enzymes (E2s) and ubiquitin-protein ligases (E3s) synthesize nondegradable forked ubiquitin chains containing all possible isopeptide linkages. J. Biol. Chem. 282, 17375-17386. 10.1074/jbc.M609659200 [DOI] [PubMed] [Google Scholar]
- Komander D. and Rape M. (2012). The ubiquitin code. Annu. Rev. Biochem. 81, 203-229. 10.1146/annurev-biochem-060310-170328 [DOI] [PubMed] [Google Scholar]
- Laing J. G. and Beyer E. C. (1995). The gap junction protein connexin43 is degraded via the ubiquitin proteasome pathway. J. Biol. Chem. 270, 26399-26403. 10.1074/jbc.270.44.26399 [DOI] [PubMed] [Google Scholar]
- Laing J. G., Tadros P. N., Westphale E. M. and Beyer E. C. (1997). Degradation of connexin43 gap junctions involves both the proteasome and the lysosome. Exp. Cell Res. 236, 482-492. 10.1006/excr.1997.3747 [DOI] [PubMed] [Google Scholar]
- Lampe P. D., Kurata W. E., Warn-Cramer B. J. and Lau A. F. (1998). Formation of a distinct connexin43 phosphoisoform in mitotic cells is dependent upon p34cdc2 kinase. J. Cell Sci. 111, 833-841. [DOI] [PubMed] [Google Scholar]
- Lampe P. D., TenBroek E. M., Burt J. M., Kurata W. E., Johnson R. G. and Lau A. F. (2000). Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J. Cell Biol. 149, 1503-1512. 10.1083/jcb.149.7.1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauf U., Giepmans B. N. G., Lopez P., Braconnot S., Chen S.-C. and Falk M. M. (2002). Dynamic trafficking and delivery of connexons to the plasma membrane and accretion to gap junctions in living cells. Proc. Natl. Acad. Sci. USA 99, 10446-10451. 10.1073/pnas.162055899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leithe E. and Rivedal E. (2004). Ubiquitination and down-regulation of gap junction protein connexin-43 in response to 12-O-tetradecanoylphorbol 13-acetate treatment. J. Biol. Chem. 279, 50089-50096. 10.1074/jbc.M402006200 [DOI] [PubMed] [Google Scholar]
- Leithe E., Kjenseth A., Sirnes S., Stenmark H., Brech A. and Rivedal E. (2009). Ubiquitylation of the gap junction protein connexin-43 signals its trafficking from early endosomes to lysosomes in a process mediated by Hrs and Tsg101. J. Cell Sci. 122, 3883-3893. 10.1242/jcs.053801 [DOI] [PubMed] [Google Scholar]
- Leykauf K., Dürst M. and Alonso A. (2003). Phosphorylation and subcellular distribution of connexin43 in normal and stressed cells. Cell Tissue Res. 311, 23-30. 10.1007/s00441-002-0645-5 [DOI] [PubMed] [Google Scholar]
- Leykauf K., Salek M., Bomke J., Frech M., Lehmann W.-D., Durst M. and Alonso A. (2006). Ubiquitin protein ligase Nedd4 binds to connexin43 by a phosphorylation-modulated process. J. Cell Sci. 119, 3634-3642. 10.1242/jcs.03149 [DOI] [PubMed] [Google Scholar]
- Lichtenstein A., Minogue P. J., Beyer E. C. and Berthoud V. M. (2010). Autophagy: a pathway that contributes to connexin degradation. J. Cell Sci. 124, 910-920. 10.1242/jcs.073072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGurn J. A., Hsu P.-C. and Emr S. D. (2012). Ubiquitin and membrane protein turnover: from cradle to grave. Annu. Rev. Biochem. 81, 231-259. 10.1146/annurev-biochem-060210-093619 [DOI] [PubMed] [Google Scholar]
- McCullough J., Clague M. J. and Urbé S. (2004). AMSH is an endosome-associated ubiquitin isopeptidase. J. Cell Biol. 166, 487-492. 10.1083/jcb.200401141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musil L. S. and Goodenough D. A. (1991). Biochemical analysis of connexin43 intracellular transport, phosphorylation, and assembly into gap junctional plaques. J. Cell Biol. 115, 1357-1374. 10.1083/jcb.115.5.1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L. K., Kolch W. and Kholodenko B. N. (2013). When ubiquitination meets phosphorylation: a systems biology perspective of EGFR/MAPK signalling. Cell Commun. Signal 11, 52 10.1186/1478-811X-11-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimlamool W., Andrews R. M. K. and Falk M. M. (2015). Connexin43 phosphorylation by PKC and MAPK signals VEGF-mediated gap junction internalization. Mol. Biol. Cell 26, 2755-2768. 10.1091/mbc.e14-06-1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankiv S., Clausen T. H., Lamark T., Brech A., Bruun J.-A., Outzen H., Øvervatn A., Bjørkøy G. and Johansen T. (2007). p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131-24145. 10.1074/jbc.M702824200 [DOI] [PubMed] [Google Scholar]
- Petrich B. G., Gong X., Lerner D. L., Wang X., Brown J. H., Saffitz J. E. and Wang Y. (2002). c-Jun N-terminal kinase activation mediates downregulation of connexin43 in cardiomyocytes. Circ. Res. 91, 640-647. 10.1161/01.RES.0000035854.11082.01 [DOI] [PubMed] [Google Scholar]
- Piehl M., Lehmann C., Gumpert A., Denizot J.-P., Segretain D. and Falk M. M. (2007). Internalization of large double-membrane intercellular vesicles by a clathrin-dependent endocytic process. Mol. Biol. Cell 18, 337-347. 10.1091/mbc.e06-06-0487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polontchouk L., Ebelt B., Jackels M. and Dhein S. (2002). Chronic effects of endothelin 1 and angiotensin II on gap junctions and intercellular communication in cardiac cells. FASEB J. 16, 87-89. 10.1096/fj.01-0381fje [DOI] [PubMed] [Google Scholar]
- Ravid T. and Hochstrasser M. (2008). Diversity of degradation signals in the ubiquitin-proteasome system. Nat. Rev. Mol. Cell Biol. 9, 679-690. 10.1038/nrm2468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro-Rodrigues T. M., Catarino S., Marques C., Ferreira J. V., Martins-Marques T., Pereira P. and Girão H. (2014). AMSH-mediated deubiquitination of Cx43 regulates internalization and degradation of gap junctions. FASEB J. 28, 4629-4641. 10.1096/fj.13-248963 [DOI] [PubMed] [Google Scholar]
- Sáez J. C., Nairn A. C., Czernik A. J., Fishman G. I., Spray D. C. and Hertzberg E. L. (1997). Phosphorylation of connexin43 and the regulation of neonatal rat cardiac myocyte gap junctions. J. Mol. Cell. Cardiol. 29, 2131-2145. 10.1006/jmcc.1997.0447 [DOI] [PubMed] [Google Scholar]
- Sato Y., Yoshikawa A., Yamagata A., Mimura H., Yamashita M., Ookata K., Nureki O., Iwai K., Komada M. and Fukai S. (2008). Structural basis for specific cleavage of Lys 63-linked polyubiquitin chains. Nature 455, 358-362. 10.1038/nature07254 [DOI] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676-682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt M., Leykauf K., Reinz E., Cheng H., Alonso A. and Schenkel J. (2014). Mutation of human connexin43 amino acids s279/s282 increases protein stability upon treatment with epidermal growth factor. Cell Biochem. Biophys. 69, 379-384. 10.1007/s12013-013-9811-5 [DOI] [PubMed] [Google Scholar]
- Seibenhener M. L., Babu J. R., Geetha T., Wong H. C., Krishna N. R. and Wooten M. W. (2004). Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol. Cell. Biol. 24, 8055-8068. 10.1128/MCB.24.18.8055-8068.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirnes S., Kjenseth A., Leithe E. and Rivedal E. (2009). Interplay between PKC and the MAP kinase pathway in Connexin43 phosphorylation and inhibition of gap junction intercellular communication. Biochem. Biophys. Res. Commun. 382, 41-45. 10.1016/j.bbrc.2009.02.141 [DOI] [PubMed] [Google Scholar]
- Solan J. L. and Lampe P. D. (2007). Key connexin 43 phosphorylation events regulate the gap junction life cycle. J. Membr. Biol. 217, 35-41. 10.1007/s00232-007-9035-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan J. L. and Lampe P. D. (2008). Connexin 43 in LA-25 cells with active v-src is phosphorylated on Y247, Y265, S262, S279/282, and S368 via multiple signaling pathways. Cell Commun. Adhes. 15, 75-84. 10.1080/15419060802014016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan J. L. and Lampe P. D. (2014). Specific Cx43 phosphorylation events regulate gap junction turnover in vivo. FEBS Lett. 588, 1423-1429. 10.1016/j.febslet.2014.01.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan J. L. and Lampe P. D. (2015). Kinase programs spatiotemporally regulate gap junction assembly and disassembly: Effects on wound repair. Semin. Cell Dev. Biol. 50, 40-48. 10.1016/j.semcdb.2015.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solan J. L., Marquez-Rosado L., Sorgen P. L., Thornton P. J., Gafken P. R. and Lampe P. D. (2007). Phosphorylation at S365 is a gatekeeper event that changes the structure of Cx43 and prevents down-regulation by PKC. J. Cell Biol. 179, 1301-1309. 10.1083/jcb.200707060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spagnol G., Al-Mugotir M., Kopanic J. L., Zach S., Li H., Trease A. J., Stauch K. L., Grosely R., Cervantes M. and Sorgen P. L. (2016). Secondary structural analysis of the carboxyl-terminal domain from different connexin isoforms. Biopolymers 105, 143-162. 10.1002/bip.22762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su V., Nakagawa R., Koval M. and Lau A. F. (2010). Ubiquitin-independent proteasomal degradation of endoplasmic reticulum-localized connexin43 mediated by CIP75. J. Biol. Chem. 285, 40979-40990. 10.1074/jbc.M110.170753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thévenin A. F., Kowal T. J., Fong J. T., Kells R. M., Fisher C. G. and Falk M. M. (2013). Proteins and mechanisms regulating gap-junction assembly, internalization, and degradation. Physiology 28, 93-116. 10.1152/physiol.00038.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thévenin A. F., Margraf R. A., Fisher C. G., Kells-Andrews R. M. and Falk M. M. (2017). Phosphorylation regulates connexin43/ZO-1 binding and release, an important step in gap junction turnover. Mol. Biol. Cell 28, 3595-3608. 10.1091/mbc.e16-07-0496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub L. M. and Bonifacino J. S. (2013). Cargo recognition in clathrin-mediated endocytosis. Cold Spring Harbor Perspect. Biol. 5, a016790 10.1101/cshperspect.a016790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zeijl L., Ponsioen B., Giepmans B. N. G., Ariaens A., Postma F. R., Várnai P., Balla T., Divecha N., Jalink K. and Moolenaar W. H. (2007). Regulation of connexin43 gap junctional communication by phosphatidylinositol 4,5-bisphosphate. J. Cell Biol. 177, 881-891. 10.1083/jcb.200610144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S. A., Beli P., Weinert B. T., Nielsen M. L., Cox J., Mann M. and Choudhary C. (2011). A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol. Cell. Proteomics 10, M111.013284 10.1074/mcp.M111.013284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayakanon P., Bhattacharjee R., Nakahama K. and Morita I. (2012). The role of the Cx43 C-terminus in GJ plaque formation and internalization. Biochem. Biophys. Res. Commun. 420, 456-461. 10.1016/j.bbrc.2012.03.018 [DOI] [PubMed] [Google Scholar]
- Ye Y., Blaser G., Horrocks M. H., Ruedas-Rama M. J., Ibrahim S., Zhukov A. A., Orte A., Klenerman D., Jackson S. E. and Komander D. (2012). Ubiquitin chain conformation regulates recognition and activity of interacting proteins. Nature 492, 266-270. 10.1038/nature11722 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.