

Abstract
The spread of antibiotic resistance is a major challenge for treatment of Mycobacterium tuberculosis infection. In addition, efficacy of drugs is often limited by the restricted permeability of the mycomembrane. Frontline antibiotics inhibit mycomembrane biosynthesis leading to rapid cell death. Inspired by this mechanism we exploit β-lactones as putative mycolic acid mimics to block serine hydrolases involved in their biosynthesis. Among a collection of β-lactones we found one hit with potent anti-mycobacterial and bactericidal activity. Chemical proteomics using an alkynylated probe identified Pks13 and Ag85 serine hydrolases as major targets. Validation via enzyme assays and customized 13C metabolite profiling showed that both targets are functionally impaired by the β-lactone. Co-administration with front-line antibiotics enhanced the potency against M. tuberculosis by more than 100-fold demonstrating a therapeutic potential of targeting mycomembrane biosynthesis serine hydrolases.
Keywords: activity-based protein profiling, antibacterials, Mycobacterium tuberculosis, proteomics
Trick and treat
A β-lactone resembling an electrophilic mimic of mycolic acid blocks serine hydrolases essential for mycomembrane biosynthesis. Activity-based protein profiling paired with metabolic labeling studies confirmed the mechanism of action responsible for the potent antibiotic activity against Mycobacterium tuberculosis.
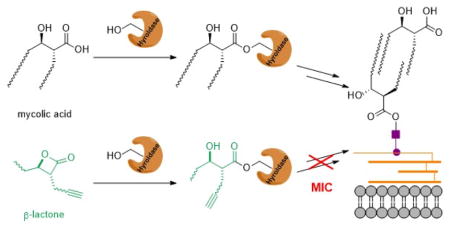
Tuberculosis is caused by the intracellular pathogen Mycobacterium tuberculosis (Mtb) and represents one of the most prevalent deadly infectious diseases worldwide.[1] Treatment of tuberculosis is extremely difficult, in part because of the mycomembrane, a lipid bilayer forming the outer bacterial barrier.[2] Mycolic acids (MAs) are hallmarks of the mycobacterial cell envelope and essential for growth.[3] These branched-chain β-hydroxylated fatty acids consist of an α-chain (C22–C26) and a mero-chain (up to C60).[4] The two chains are assembled by fatty acid synthases[5] from acetyl-CoA and linked by polyketide synthase 13 (Pks13) via a Claisen-type condensation (Figure 1A).[6] Several studies have shown the importance of Pks13 for mycobacterial viability[7] and successful inhibition of an individual domain has been reported.[8] For maturation of MAs the product of Pks13, β-keto mycolate, is transferred onto trehalose and reduced to yield trehalose monomycolate (TMM).[9] For incorporation into the outer cell membrane TMMs are transported into the periplasm and either linked onto arabinogalactan (AG) or TMM forming trehalose dimycolates (TDM).[10] Subsequently, free MAs, TMM and TDM are non-covalently associated to the mycomembrane. A group of orthologous serine hydrolases, antigen 85A/B/C (Ag85), catalyzes these essential transacylation reactions.[11] Although individual Ag85 genes are dispensable, loss of at least two is lethal.[12] Crystal structures of the orthologous proteins exhibit conserved active sites[13], and treatment with inhibitors, such as ebselen, resulted in decreasing TDM levels and accumulation of TMM.[14]
Figure 1.

(A) Schematic overview of mycolic acid biosynthesis by assembly of fatty acid chains (FAS I & II), condensation (Pks13) and maturation (CmrA) to TMM. Transfer across the inner cell membrane is receptor guided (MmpL3). Incorporation of mycolates onto the outer cell membrane is enzyme assisted (Ag85). (B) Formation of mycolic acid-hydrolase complex by acylation of active site serine. (C) Enzymatic opening of β-lactone EZ120 and acylation by active site serine. FAS: fatty acid synthase, Pks: Polyketide synthase, Ag85: Antigen 85, TMM: trehalose monomycolate, TDM: trehalose dimycolate, MA: mycolic acid, CoA: coenzyme A, AMP: adenosine monophosphate.
Given the crucial role of diverse serine hydrolases in mycomembrane biosynthesis and their binding of mycolates as β-keto or -hydroxy esters (Figure 1B), we rationalized that long-chain aliphatic compounds equipped with an electrophilic group could act as covalent inhibitors of several of these key steps.[15] β-Lactones are attractive molecules for this endeavor as their substitution pattern is reminiscent of the signature mycolic acid β-hydroxy motif, formed by ring opening and covalent active site acylation (Figure 1C). Previous studies with a small number of β-lactones in mycobacteria revealed targeting of diverse lipases[16] as well as two caseinolytic proteases termed ClpP1 and ClpP2.[17] Here we profile a panel of β-lactones for anti-mycobacterial activity and identify one hit compound with bactericidal activity. In-depth mode of action analysis reveals Pks13 and Ag85 enzymes as predominant targets.
Previously, several β-lactone inhibitors of ClpP were explored for their anti-virulence properties in S. aureus.[18] In the search for mimics of fatty acid substrates blocking essential serine hydrolases a collection of β-lactones was thus tested for antimicrobial activity against a panel of Gram positive and Gram negative bacteria. Importantly, one compound, EZ120, exhibited a minimal inhibitory concentration (MIC) of 1.6 μM (0.68 μg/mL) against Mtb H37Rv while all other lactone compounds including previously established ClpP inhibitors were less active (Table S1). Moreover, among all bacterial strains tested, EZ120 was selective for mycobacteria. Furthermore, it exhibited a minimal bactericidal concentration (MBC) of 6 μM (2.5 μg/mL) (Figure S1). Cytotoxicity against mouse macrophages was more than 200 fold higher compared to the MIC value indicating a suitable therapeutic window (Figure S2).
To identify molecular targets of EZ120, we utilized activity-based protein profiling (ABPP)[19] by synthesizing an alkyne-modified probe version (EZ120P) (Scheme S1) which retained initial bioactivity (Table S1).
As insoluble fractions of Mtb H37Rv could not be handled outside the biosafety level 3 laboratory for proteomic studies, we included Mycobacterium smegmatis mc2 155 (Msmeg) as a substitute. Following standard gel-free ABPP workflow and label-free protein quantification (see SI for details) we obtained mycobacterial targets of EZ120P displayed as volcano plots (Figure 2). For analysis of soluble and insoluble protein fractions enrichment exceeding a factor of 4 (Mtb) and 8 (Msmeg), as well as a p-value ≤ 0.05 were selected. In agreement with previous results a variety of hydrolases across mycobacterial strains was detected (Table S2).[15, 17, 20] To determine potential targets responsible for the observed antibacterial effect only proteins described as essential for growth were considered as hits.[21] Interestingly, only Pks13 and AroG were identified as essential proteins in the soluble fraction of Mtb. Ag85A, Ag85B, ClpP2 and a secreted serine protease were found as hits in the insoluble fraction of Msmeg. Respective orthologue genes in Mtb have been annotated as essential for growth. Although there are certainly proteomic differences between Msmeg and Mtb (Figure S3), major pathways such as MA biosynthesis are highly conserved. We also performed SDS-PAGE of the insoluble fraction in Msmeg to ensure solubilization of a wide range of proteins using click chemistry[22] with a linker consisting of a rhodamine and a biotin moiety. Fluorescent scanning showed a distinct set of protein bands (Figure S4) that were analyzed by LC-MS/MS. In accordance with the gel-free results, Ag85 enzymes were most prominently enriched compared to control (Table S3). In line with the hypothesis of a mycolate mimic these results indeed highlight a preference of EZ120P for targeting conserved cell wall hydroalses Pks13 and Ag85 enzymes in mycobacteria which is the focus of this work. Of note, other protein hits such as AroG, involved in chorismate biosynthesis and ClpP, important for cell homeostasis, could contribute to the EZ120 mode of action as well.
Figure 2.

Gel-free proteomic analysis of mycobacterial targets of EZ120P by volcano plot presentation of (A) Mtb H37Rv soluble fraction, and (B) Msmeg mc2 155 insoluble fraction. Cut-off values indicated by dotted lines (log2-fold enrichment ≥ 2 for soluble and ≥ 3 for insoluble targets; p-value ≤ 0.05). Protein hits are highlighted in green (non-essential) or red (essential). Protein accession numbers are given in case of non-specifically identifying protein names. Data results from nine different experiments. For details of enriched proteins please see Supplementary table 2.
In fact, ClpP has been previously reported to be an anti-mycobacterial target of β-lactones.[17] The clpP1 and clpP2 genes are essential for survival of mycobacteria. We thus tested the initial set of β-lactones for their effect on purified Mtb ClpP1/P2 peptidase activity using an in-vitro assay (Figure S5). While positive control compound P1 inhibited the protease at low μM concentration, EZ120 was a very weak inhibitor suggesting that the antibacterial effect largely results from other targets.
Pks13 is a cytoplasmic polyketide synthase assembling fatty acid chains during mycolic acid synthesis and consists of 5 domains. The C-terminal thioesterase (TE) domain is a serine hydrolase and thus a candidate binding site for EZ120. The TE domain is responsible for product release and its turnover can be monitored using a fluorescent substrate.[23] Incubation of Pks13-TE with EZ120 resulted in inhibition suggesting active site acylation (Figure S6). To confirm this mechanism, we performed gel-based ABPP experiments with wildtype (wt) enzyme and an active site mutant (S1533A) with EZ120P. Contrary to the wt TE domain, which yielded a strong fluorescent signal upon expression, the mutant did not show pronounced labeling (Figure 3A, S7). In addition, intact-protein MS on wt (Figure 3B) and mutant enzyme (Figure 3C) with the inhibitor only showed a mass increase in the presence of the active site serine. Remarkably, covalent binding could neither be detected for the initially tested β-lactone D3 nor the more promiscuous antimycobacterial hydrolase inhibitor lalistat (Figure S8) demonstrating high selectivity for EZ120.
Figure 3.

(A) In-situ gel-based ABPP fluorescent scan of Mtb Pks13TE wt and S1533A mutant in E. coli with EZ120P. (B, C) Intact-protein MS of purified PKS13 wt and S1533A of Mtb with EZ120. (D) Immunoblot analysis of Ag85A and (E) loading control (background protein band) in wt, knock-out and complement Msmeg strain. (F) In situ gel-based ABPP fluorescent scan of Msmeg wt and ΔAg85A insoluble protein fractions with EZ120P. IPTG: isopropyl β-D-1-thiogalactopyranoside; MW: molecular weight.
Ag85 enzymes represent a group of homologous serine hydrolases with a conserved catalytic triad (Figure S9) suggesting functional redundancy.[24] They act in the periplasm to transfer acyl groups of donor molecules (i.e. TMM) onto acceptor substrates (i.e. TMM, AG) (Figure 1A).[25] To verify that Ag85 is a target for EZ120 in situ we generated an Msmeg ΔAg85A strain and validated the deletion by immunoblot analysis (Figure 3D, E, S10). Remarkably, gel-based ABPP experiments showed selective labeling of proteins at the 30 kDa range and comparison between wt and ΔAg85A revealed the disappearance of a fluorescent band (Figure 3F, S11). Given the high homology of Ag85A-C and their binding to EZ120 as obtained by ABPP studies, a consolidated inhibition of more than one enzyme likely impairs transacylation of mycolates for cell wall incorporation as previously reported.[12]
Since Pks13 and Ag85 enzymes catalyze essential steps in MA synthesis, we determined the impact of drug treatment on steady state MA biosynthesis. Isoniazid (INH) and other drugs that reduce MA biosynthesis typically cause drastic reductions on newly synthesized MAs but only partially deplete whole cell MA pools before causing bacterial death. To avoid 14C labeling in the biosafety level 3 laboratory, we developed a new MS-based 13C labeling method. We biosynthetically labeled Mtb lipids by supplementing [1,2-13C] acetate in media for 48 hours. MAs were analyzed after saponification by negative mode HPLC-MS and showed a complex profile of 13C labeling compared to the unlabeled spectra (Figure 4A). This complexity stems from MA molecules comprising different functional groups throughout the molecule as well as overlapping isotope peaks derived from endogenous and supplemented 13C (see SI for details). The contribution from natural isotopes versus label-induced isotopes in fatty acids could be accurately assessed by collision-induced dissociation (CID). Mycolate α-chains exclusively comprise unbranched, saturated C24 and C26 fatty acids, yielding signature fragment ions of 367.355 and 395.390, respectively (Figure 4B–D, S12). By taking the intensity ratio of the sum of all label-derived ions to that from m/z 367.36 and 395.39, we can compare the degree of isotope labeling between different culture conditions. Using this approach, we set up an experiment by treating Mtb with DMSO, isoniazid (INH), and different doses of EZ120 ranging from 1× to 10× MIC for 20 hours under labeling conditions (Figure 4D). As expected, INH almost completely abolished 13C incorporation. EZ120 treatment reduced 13C incorporation in a dose-dependent manner and not as strong as INH (Figure 4E). Nevertheless, the results indicate that the inhibition of MA synthesis was the result of the treatment. To determine whether the drug effect was due to the specific target of MAs or the nonspecific toxicity, we inspected the free fatty acids in the extracted bacterial total lipids. Isotope enriched fatty acids were observed under all conditions suggesting that bacteria were indeed able to actively ingest 13C acetate (Figure S13). Taken together, these results support that EZ120 inhibits MA biosynthesis.
Figure 4.

Detection of EZ120’s impact on newly synthesized MAs by 13C labeling. (A) Negative mode MS spectra of saponified cell wall-bound MAs of Mtb harvested from media supplemented without (top) or with (bottom) 3 mg/ml of [1, 2-13C] acetate for 48 hours. (B) CID-MS spectra of m/z 1192.23 generated fragment ions of m/z 367.355 and m/z 395.390, corresponding to C24 and C26 fatty acid fragments, respectively. (C) Structures of CID fragmentation of MAs as described in (B). Arrows indicate cleavage sites. (D) CID-MS spectra at m/z 1308.35. Mtb was treated with DMSO (−), INH (MIC: 5x, 6.5 μM), EZ120 (MIC: 10x, 16 μM; 5x, 8 μM; 1x, 1.6 μM) for 4 hours before adding [1, 2-13C] acetate (3 mg/ml). The experiment was performed in triplicate and one representative set of data is shown here. ‘×5’ in (B) and ‘×10’ in (C) indicate the enlarged factors. (E) Ratio of the total intensity of 13C incorporation peaks to the intensity of unlabeled fatty acids (m/z 367 and 395). Data were presented as mean ± SD and using a t-test for statistical analysis. ***, p< 0.001; **, p< 0.01; *, p< 0.05; ns, not significant.
Previous studies on mycobacterial strains with reduced Ag85 genes showed an increased susceptibility to front-line antibiotics.[26] We therefore tested MICs for antibiotics with various cellular targets on Msmeg ΔAg85A. These included rifampicin (targeting RNA polymerase), vancomycin (cell wall assembly) and INH (MA synthesis). Interestingly, ΔAg85A cells were 8- and 16-fold more susceptible to rifampicin and vancomycin, respectively (Figure S14, 15). Application of MA targeting compounds INH and EZ120 showed little to no effect on MIC. We concluded synergistic effects on growth inhibition for drugs targeting different molecular pathways and tested our hypothesis via checkerboard resazurin assay on wt Msmeg applying EZ120 with each of the above drugs individually (Figure S16–18). Rifampicin and INH showed no significant effect. However, the combined application of EZ120 and vancomycin shows growth inhibition at 100-fold and 4-fold reduced concentration, respectively, compared to individual treatment. Utilizing the fractional inhibitory concentration index clearly confirms a synergistic effect and leads to the conclusion that mycobacteria are more susceptiple to cell wall targeting drugs when maintainance of the cellular envelope is already impaired.
In summary, EZ120 represents an activated β-hydroxy acid and structural homologue of mycolates, binding to hydrolase sites crucial for mycobacterial cell wall biosynthesis. The potent MIC and MBC, the low toxicity against human cells, the excellent penetration of the mycomembrane and the simultaneous targeting of essential enzymes for cell wall biosynthesis make EZ120 an important tool compound for studying MA biosynthesis hydrolases. Further chemical optimization of the core scaffold could become a testing groud for new inhibitors against intracellular Mtb as well as novel combinations of drugs in medical application.
Supplementary Material
Acknowledgments
We gratefully thank the Deutsche Forschungsgemeinschaft SFB749. J.L. was supported by the Studienstiftung des deutschen Volkes. We thank Mathias Hackl, Matthias Stahl, Katja Bäuml and Liam Guthrie for support and discussion on mass-spectrometry experiments. We thank Shoko Minami for assistance with Mtb experiments. This work was supported by the National Institute of Health AI U19 111224.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/
References
- 1.WHO. Antimicrobial resistnace: global report on surveillance. 2014. [Google Scholar]
- 2.Koul A, Arnoult E, Lounis N, Guillemont J, Andries K. Nature. 2011;469:483–490. doi: 10.1038/nature09657. [DOI] [PubMed] [Google Scholar]
- 3.Lederer E, Adam A, Ciorbaru R, Petit JF, Wietzerbin J. Mol Cell Biochem. 1975;7:87–104. doi: 10.1007/BF01792076. [DOI] [PubMed] [Google Scholar]
- 4.a) Marrakchi H, Laneelle MA, Daffe M. Chem Biol. 2014;21:67–85. doi: 10.1016/j.chembiol.2013.11.011. [DOI] [PubMed] [Google Scholar]; b) Takayama K, Wang C, Besra GS. Clin Microbiol Rev. 2005;18:81–101. doi: 10.1128/CMR.18.1.81-101.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhatt A, Molle V, Besra GS, Jacobs WR, Jr, Kremer L. Mol Microbiol. 2007;64:1442–1454. doi: 10.1111/j.1365-2958.2007.05761.x. [DOI] [PubMed] [Google Scholar]
- 6.a) Gavalda S, Bardou F, Laval F, Bon C, Malaga W, Chalut C, Guilhot C, Mourey L, Daffe M, Quemard A. Chem Biol. 2014;21:1660–1669. doi: 10.1016/j.chembiol.2014.10.011. [DOI] [PubMed] [Google Scholar]; b) Portevin D, De Sousa-D’Auria C, Houssin C, Grimaldi C, Chami M, Daffe M, Guilhot C. Proc Natl Acad Sci. 2004;101:314–319. doi: 10.1073/pnas.0305439101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gande R, Gibson KJ, Brown AK, Krumbach K, Dover LG, Sahm H, Shioyama S, Oikawa T, Besra GS, Eggeling L. The J Biol Chem. 2004;279:44847–44857. doi: 10.1074/jbc.M408648200. [DOI] [PubMed] [Google Scholar]
- 8.a) Wilson R, Kumar P, Parashar V, Vilcheze C, Veyron-Churlet R, Freundlich JS, Barnes SW, Walker JR, Szymonifka MJ, Marchiano E, Shenai S, Colangeli R, Jacobs WR, Jr, Neiditch MB, Kremer L, Alland D. Nat Chem Bio. 2013;9:499–506. doi: 10.1038/nchembio.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Thanna S, Knudson SE, Grzegorzewicz A, Kapil S, Goins CM, Ronning DR, Jackson M, Slayden RA, Sucheck SJ. Org Biomol Chem. 2016;14:6119–6133. doi: 10.1039/c6ob00821f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Ahibo-Coffy A, Aurelle H, Lacave C, Prome JC, Puzo G, Savagnac A. Chem Phys Lipids. 1978;22:185–195. doi: 10.1016/0009-3084(78)90025-7. [DOI] [PubMed] [Google Scholar]; b) Lea-Smith DJ, Pyke JS, Tull D, McConville MJ, Coppel RL, Crellin PK. J Biol Chem. 2007;282:11000–11008. doi: 10.1074/jbc.M608686200. [DOI] [PubMed] [Google Scholar]
- 10.a) Tahlan K, Wilson R, Kastrinsky DB, Arora K, Nair V, Fischer E, Barnes SW, Walker JR, Alland D, Barry CE, 3rd, Boshoff HI. Antimicrob Agents Chemother. 2012;56:1797–1809. doi: 10.1128/AAC.05708-11. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) La Rosa V, Poce G, Canseco JO, Buroni S, Pasca MR, Biava M, Raju RM, Porretta GC, Alfonso S, Battilocchio C, Javid B, Sorrentino F, Ioerger TR, Sacchettini JC, Manetti F, Botta M, De Logu A, Rubin EJ, De Rossi E. Antimicrob Agents Chemother. 2012;56:324–331. doi: 10.1128/AAC.05270-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Belisle JT, Vissa VD, Sievert T, Takayama K, Brennan PJ, Besra GS. Science. 1997;276:1420–1422. doi: 10.1126/science.276.5317.1420. [DOI] [PubMed] [Google Scholar]; b) Warrier T, Tropis M, Werngren J, Diehl A, Gengenbacher M, Schlegel B, Schade M, Oschkinat H, Daffe M, Hoffner S, Eddine AN, Kaufmann SHE. Antimicrob Agents Chemother. 2012;56:1735–1743. doi: 10.1128/AAC.05742-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Armitige LY, Jagannath C, Wanger AR, Norris SJ. Infect Immun. 2000;68:767–778. doi: 10.1128/iai.68.2.767-778.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Favrot L, Lajiness DH, Ronning DR. J Biol Chem. 2014;289:25031–25040. doi: 10.1074/jbc.M114.582445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Favrot L, Grzegorzewicz AE, Lajiness DH, Marvin RK, Boucau J, Isailovic D, Jackson M, Ronning DR. Nat Comm. 2013;4:2748. doi: 10.1038/ncomms3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bachovchin DA, Ji T, Li W, Simon GM, Blankman JL, Adibekian A, Hoover H, Niessen S, Cravatt BF. Proc Natl Acad Sci. 2010;107:20941–20946. doi: 10.1073/pnas.1011663107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ravindran MS, Rao SP, Cheng X, Shukla A, Cazenave-Gassiot A, Yao SQ, Wenk MR. Mol Cell Proteomics. 2014;13:435–448. doi: 10.1074/mcp.M113.029942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Compton CL, Schmitz KR, Sauer RT, Sello JK. ACS Chem Biol. 2013;8:2669–2677. doi: 10.1021/cb400577b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Böttcher T, Sieber SA. J Am Chem Soc. 2008;130:14400–14401. doi: 10.1021/ja8051365. [DOI] [PubMed] [Google Scholar]; b) Zeiler E, Korotkov VS, Lorenz-Baath K, Böttcher T, Sieber SA. Bioorg Med Chem. 2011;20:583–591. doi: 10.1016/j.bmc.2011.07.047. [DOI] [PubMed] [Google Scholar]
- 19.a) Barglow KT, Cravatt BF. Nature Methods. 2007;4:822–827. doi: 10.1038/nmeth1092. [DOI] [PubMed] [Google Scholar]; b) Fonovic M, Bogyo M. Expert Rev Proteomics. 2008;5:721–730. doi: 10.1586/14789450.5.5.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Böttcher T, Sieber SA. Angew Chem Int Ed Engl. 2008;47:4600–4603. doi: 10.1002/anie.200705768. [DOI] [PubMed] [Google Scholar]
- 21.Sassetti CM, Boyd DH, Rubin EJ. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 22.a) Rostovtsev VV, Green JG, Fokin VV, Sharpless KB. Angew Chem Int Ed Engl. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; b) Tornøe CW, Christensen C, Meldal M. J Org Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 23.Aggarwal A, Parai MK, Shetty N, Wallis D, Woolhiser L, Hastings C, Dutta NK, Galaviz S, Dhakal RC, Shrestha R, Wakabayashi S, Walpole C, Matthews D, Floyd D, Scullion P, Riley J, Epemolu O, Norval S, Snavely T, Robertson GT, Rubin EJ, Ioerger TR, Sirgel FA, van der Merwe R, van Helden PD, Keller P, Bottger EC, Karakousis PC, Lenaerts AJ, Sacchettini JC. Cell. 2017;170:249–259.e225. doi: 10.1016/j.cell.2017.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ronning DR, Vissa V, Besra GS, Belisle JT, Sacchettini JC. J Biol Chem. 2004;279:36771–36777. doi: 10.1074/jbc.M400811200. [DOI] [PubMed] [Google Scholar]
- 25.Matsunaga I, Naka T, Talekar RS, McConnell MJ, Katoh K, Nakao H, Otsuka A, Behar SM, Yano I, Moody DB, Sugita M. J Biol Chem. 2008;283:28835–28841. doi: 10.1074/jbc.M805776200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.a) Nguyen L, Chinnapapagari S, Thompson CJ. J Bacteriol. 2005;187:6603–6611. doi: 10.1128/JB.187.19.6603-6611.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Favrot L, Ronning DR. Expert Rev Anti Infect Ther. 2012;10:1023–1036. doi: 10.1586/eri.12.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.