

Abstract
To investigate large library docking’s ability to find molecules with joint activity against on-targets and selectivity versus antitargets, the dopamine D2 and serotonin 5-HT2A receptors were targeted, seeking selectivity against the histamine H1 receptor. In a second campaign, κ-opioid receptor ligands were sought with selectivity versus the μ-opioid receptor. While hit rates ranged from 40% to 63% against the on-targets, they were just as good against the antitargets, even though the molecules were selected for their putative lack of binding to the off-targets. Affinities, too, were often as good or better for the off-targets. Even though it was occasionally possible to find selective molecules, such as a mid-nanomolar D2/5-HT2A ligand with 21-fold selectivity versus the H1 receptor, this was the exception. Whereas false-negatives are tolerable in docking screens against on-targets, they are intolerable against antitargets; addressing this problem may demand new strategies in the field.
Introduction
The efficacy of many drugs and reagents depends on activities on multiple targets,1−7 and this is particularly true of molecules active against G protein-coupled receptors (GPCRs) for psychiatric diseases. Conversely, the unwanted activity of drugs on related GPCR antitargets can cause adverse reactions. Accordingly, there has been much interest in the design of drugs with focused polypharmacology and specificity.8−10 With the surge of GPCR structures determined to atomic resolution11−16 and their exploitation for ligand discovery,17−24 there is an opportunity to adopt a structure-based approach for focused polypharmacology and antitarget selectivity. Using structural models of the on- and off-targets, libraries may be docked for those that complement the on-targets well and fit the off-targets poorly.
Structure-based screens for focused polypharmacology face at least three technical challenges in addition to the common liabilities of docking.25 First, one must select molecules that complement the sites of two or more targets; many molecules optimal for one target will fit subsequent targets poorly, reducing ligand possibilities. Second, one must often use homology models to address disease-relevant polypharmacology,26 as the structures of many targets remain experimentally undetermined. Third, while false-negatives are typically acceptable in a docking screen against a single target, they are much less tolerable when seeking molecules that are selective against an antitarget. The use of one or a small number of receptor conformations, which is common when only looking for true positives, may be a dubious proposition when selecting against a flexible receptor.
We thought to explore these questions in two docking campaigns: one for molecules that antagonized both the serotonin 5-HT2A (HTR2A) and the dopamine D2 (DRD2) receptors and, at the same time, that did not antagonize the histamine H1 (HRH1) receptor antitarget, and a second campaign for molecules that bound to the κ-opioid receptor (KOR) without affinity for the μ-opioid receptor (MOR). Coantagonists of HTR2A and DRD2 would speak to the first two questions, that of finding molecules able to modulate two different targets at the same time and of using homology models, as the structures of these two receptors had not been determined at the time of the study (we note that we do use crystal structures for MOR, KOR, and the antitarget HRH1). Nevertheless, the two structures were readily modeled based on the DRD3 crystal structure, which was available and which shares 78% and 40% transmembrane sequence identity to DRD2 and HTR2A, respectively. This is considered well within the range of sequence identity to serve as a template for a GPCR docking screen.24,26,27 The insistence on not antagonizing HRH1 speaks to the third challenge, that of false-negatives against an antitarget. The triplet of receptors is therapeutically relevant, as coantagonism of DRD2 and HRT2A is crucial to the efficacy of atypical antipsychotics like clozapine, while antagonism of HRH1 by many antipsychotics and antidepressants, such as clozapine and olanzapine, is thought to lead to the weight gain typical of these molecules.28,29 Indeed, clozapine has a higher affinity for the HRH1 antitarget than for the therapeutic targets HTR2A and DRD2 (1.2 nM versus 5.4 nM and 256 nM, respectively),28 while even a much newer and selective drug like ziprasidone has affinities 0.3 nM for HTR2A, 9.7 nM for DRD2, and 43 nM for HRH1.28,30
The second docking campaign, against the two opioid targets, allows us to consider simple selectivity, with only one on- and one off-target and without resorting to homology models, as crystal structures in the inactive state were available for both KOR and MOR. Here, too, selectivity is therapeutically relevant: KOR-selective antagonists have been mooted as potential antidepressants without the unwanted effects of MOR antagonists like naloxone, while peripheral KOR-selective agonists could confer analgesia without activating the reward pathways associated with MOR.31,32
In both campaigns, the docking screens found new chemotypes with the desired mechanism, either joint antagonism of DRD2/HTR2A or modulation of KOR, with high hit rates, and in both campaigns, compounds selective against the antitargets were found. However, even in the simpler case of KOR vs MOR, most of the new molecules were unselective against the antitarget, and indeed, hit rates against an antitarget, either HRH1 or MOR, were as high or higher as for the on-targets. Efforts to overcome this problem through flexible receptor docking, again tested prospectively with new ligands, will be considered, as will the challenge of docking against promiscuous antitargets.
Results
Receptor Modeling and Retrospective Docking
Whereas the structures of many pharmacological target-pairs are only acessible via homology modeling, not all may be modeled with sufficient reliability to support library docking screens. To ensure that homology models of the DRD2 and the HTR2A could do so, we investigated retrospective enrichment of known ligands against decoy molecules by the models.21,24,27 We began with homology models of the two targets based on the DRD3 template, the most homologous template available to us at the time (PDB code 3PBL),33 using MODELLER v9.8 to generate 400 models for each receptor.34,35 To investigate the models’ prioritization of known ligands, we selected a set of 68 and 85 diverse HTR2A and DRD2 ligands, respectively, from the ChEMBL10 database.36 Only ligands with lead-like properties37 (molecular weights between 250 and 350, log P less than 3.5, and 7 or less rotatable bonds) and with affinity better than 100 nM were chosen. Enrichment was measured against over 2500 property-matched decoys21 and experimentally confirmed nonbinders from ChEMBL10 for all homology models of both receptors. To measure enrichment, we used the metric of adjusted log AUC, which compares the prioritization of known ligands over generated decoy molecules versus what would be expected at random (an adjusted log AUC of 0 represents random, with a maximum of 85.5). The log-weighted enrichment, where enrichment, for instance, in the top 0.1–1% of the ranked library counts as much as enrichment in the top 1–10%, emphasizes the performance of the highest-ranking molecules. One homology model was chosen for DRD2 and one for HTR2A, with log AUC values of 15 and 11, respectively, of the known ligands to the on-targets (Figure S1). Fortunately, the two on-target models also favorably enriched the other on-target’s ligands. The DRD2 model showed an adjusted log AUC of 5.7 for HTR2A ligands, and similarly, the HTR2A model showed an adjusted log AUC of 12.3 for DRD2 ligands.
We note that recently, after the completion of this study, a crystal structure of the DRD2 bound to risperidone appeared.38 The docking-prioritized DRD2 homology model superposes to the DRD2 crystal structure (PDB code 6CM4) with an all-atom binding site root-mean-square deviation (rmsd) of 0.9 Å (Figure 1A, superposition based on all-receptor atom overlay). The model has a remarkably similar binding site shape in the upper portion of the orthosteric site, though it does not predict the risperidone-induced opening of the binding pocket in the transmembrane core. Similarly, superposition of the docking-prioritized HTR2A homology model to the HTR2B crystal structure (PDB code 4IB4(39)) gives a binding site all-atom rmsd of 1.0 Å, again with good agreement (Figure 1B). Reassuringly, the new ligands discovered from docking against the DRD2 homology model (see below) also had high docking scores against the DRD2 crystal structure, with scores ranging from −67 to −37 kcal/mol, and docked with reasonable poses.
Figure 1.

Docking-prioritized homology models superpose well on subsequently determined crystal structures (0.9–1 Å all atom binding site rmsd). (A) Binding site of the DRD2-risperidone cocrystal structure (PDB code 6MC4, green) superposition based on all receptor atom overlay on the docking-prioritized homology model used in the docking screens (magenta). (B) Binding site of HTR2B ergotamine cocrystal structure (PDB code 4IB4, orange) superposition based on all receptor atoms overlaid on the docking-prioritized homology model used in the docking screen (cyan).
To minimize the likelihood of finding docking hits for the antitarget, HRH1, we docked to the DRD2 and HTR2A homology models an additional set of 50 diverse HRH1 ligands from ChEMBL10 against a corresponding set of over 1600 property-matched decoys. Each model showed little or no enrichment of HRH1 ligands, with an adjusted log AUC of −2.44 of known HRH1 ligands by the DRD2 model and 0.47 of known HRH1 ligands by the HT2RA model. The antitarget HRH1 crystal structure (PDB code 3RZE(40)) had a high retrospective enrichment, with an adjusted log AUC of 41 for its 50 ligands over property-matched decoys and annotated nonbinders (Figure S2). The utility of this structure has previously been shown in prospective virtual screening, giving a 73% hit rate in a screen of novel, chemically diverse fragments.20
Prospective Docking for New Ligands Binding to DRD2/HTR2A but Not HRH1
Satisfied with the high retrospective enrichments of both the on-target and antitarget, we launched a docking campaign with the 3 million lead-like subset of ZINC (http://zinc15.docking.org)41,42 for molecules that complemented HTR2A and DRD2 well and that fit HRH1 poorly, using DOCK3.6.43 The 5862 molecules that ranked in the top 1% to both HTR2A and DRD2 were selected for detailed evaluation (insisting on this union of high ranking molecules dropped the number of candidates by 80% over either target considered alone). Any of these DRD2/HTR2A high-ranked molecules with an ECFP4-based Tanimoto coefficient (Tc) of 0.5 or greater to any of the top 50 000 ranked molecules docked to HRH1 were discarded, increasing the likelihood that we would find molecules that do not complement HRH1. We further insisted that no DRD2/HTR2A high-ranking molecule had Tc > 0.35 to any known HRH1 binder in the ChEMBL10 database, further increasing dissimilarity of the docked hit list to known HRH1 ligands. This left 354 docking hits, from which we eliminated molecules with ECFP4-based Tc values of >0.7 to known DRD2 or HT2RA ligands in ChEMBL10. With ECFP4 fingerprints, molecules with this level of similarity are either identical or very close analogs. Ultimately 28 top-ranking molecules were selected by visual inspection for experimental testing. Molecules were prioritized based on the formation of the key salt-bridge to D3.32 (Ballesteros–Weinstein numbering44) in the docked pose to both HTR2A and DRD2, a complementary fit to the HTR2A and DRD2 binding sites and a poor fit to the HRH1 binding site, pragmatic availability from the vendor, and diversity among the candidate ligands (Table S1). While a basic center was not an a priori filtering criterion, one was present in all 354 top scoring molecules.
Five New Ligands with Dual DRD2/HTR2A Binding and with HRH1 Specificity
From the initial 28 molecules, 17 displaced [3H]ketanserin from HTR2A (60% hit rate, Table S1), 10 displaced [3H]N-methylspiperone from DRD2 (35% hit rate, Table S1), and 8 molecules bound to both (29%, Table 1, compounds 5, 9, 12, 14, 19, 21, 23, 25), all with Ki values of <10 μM. None of these were previously annotated in the public databases to bind to either target, and all had Tc values of <0.65 (ECFP4 fingerprints) to any known ligand for these receptors when compared to the most recent list of ChEMBL annotated ligands (September 2017) and so likely represent new chemotypes (Table 1). The high hit rate and novelty are balanced by the relatively weak affinities, compared to hits previously identified in docking campaigns against aminergic GPCRs; only three molecules had mid-nanomolar Ki values against HTR2A, and only compound 21 had mid-nanomolar Ki values against both targets. Conversely, five of the eight dual ligands showed substantial specificity vs HRH1 when tested for binding by displacement of radiolabeled [3H]pyrilamine: compounds 5, 14, 19, 21, and 25. Four of these (5, 14, 19, and 25) had HRH1 Ki values worse than 100 μM and corresponding selectivity values of better than 19-fold to better than 130-fold for HTR2A vs HRH1 and from 3.4-fold to 31-fold for DRD2 vs HRH1. However, these molecules had only modest affinity for the on-targets, and they did show signs of weak affinity for HRH1. The molecule with the best dual affinity for HTR2A/DRD2 and with appreciable selectivity against HRH1 was 21, with binding affinities of 55 nM, 334 nM, and 1144 nM for HTR2A, DRD2, and HRH1, respectively (Table 1, docked poses shown in Figure 2 and binding curves shown in Figure 3, top row). This represents a 21- and 3-fold selectivity, respectively, for the therapeutic targets over the antitarget and is comparable to that of the most selective antipsychotic currently on the market, ziprasidone (discussed above). Although on-target affinities are about 100-fold worse than those of ziprasidone, they are within the range of known antipsychotics (Psychoactive Drug Screening Program (PDSP) Ki database8).
Table 1. Hit Compounds at HTR2A, DRD2, and HRH1 with Their Respective Docking Ranks, Binding Constants, and Tanimoto Similarity Coefficients (Tc)a.

Footnotes: *Weak binding detected at high concentration but Ki could not be calculated. aNo agonist activity measured in calcium flux assay at concentrations up to 100 μM.
Figure 2.

Docking can predict dual-binders for on-targets but cannot reliably predict nonbinders for an antitarget. (A–C) Cut-away view of the orthosteric binding sites for HTR2A model with LSD bound; DRD2 model with eticlopride bound; HRH1 cocrystal structure with doxepin. (D–F) Docked pose of the most selective compound, compound 21, to HTR2A; DRD2; HRH1. Clashes with the HRH1 crystal structure are shown as red circles. (H–J) Docked pose of the least selective compound, compound 6, docked to HTR2A; DRD2; HRH1. Clashes with the HRH1 crystal structure are shown as red circles.
Figure 3.

Radioligand displacement binding affinities for HTR2A, DRD2, and HRH1. Reference ligands are [3H]ketanserin, [3H]N-methylspiperone, and [3H]pyrilamine, respectively. (Top) Specific binding of the 20-fold selective docking hit, compound 21. (Bottom) Compound 6, a molecule that was both a docking false-positive (it does not bind to the on-targets HTR2A and DRD2) and a false-negative (it does bind to the off-target HRH1 with subnanomolar affinity).
Prospective Docking of Hit Analogs Fails To Improve Selectivity
To improve the moderate affinities and selectivities of the initial hits, we investigated commercially available analogs of compounds 5, 19, and 21. On the basis of docking fits to the receptors, we selected seven analogs of compound 5 (5a–g in Table 2), three analogs of compound 19 (19a–c in Table 2), and five analogs of compound 21 for experimental testing (21a–e in Table 2), using the same target and antitarget criteria as in our initial docking screen. Despite the strict filtering and improved starting molecules, analogs either improved HRH1 affinity or reduced HTR2A and/or DRD2 affinity. No analog improved selectivity over the antitarget.
Table 2. Analogs of Hit Compounds 5, 19, and 21 with Their Respective HTR2A, DRD2, and HRH1 Docking Ranks, Binding Constants, and Selectivity Ratiosa.


Footnotes: *Weak binding detected at high concentration but Ki could not be calculated. aNo agonist activity measured in calcium flux assay at concentrations up to 100 μM.
A Problematic False Negative Rate against HRH1
While docking had a substantial true positive rate for dual HTR2A/DRD2 ligands, the false-negative rate against HRH1 was disappointingly high, with 16/28 molecules (57%) displacing [3H]pyrilamine. Not only was this hit rate for the antitarget similar to that for HTR2A and higher than that for DRD2, several of the HRH1 hits bound with high affinity. For instance, compound 6, in spite of a relatively poor rank of 326 040 out of 3 million docked, had a Ki of 0.8 nM (Table 1), among the tightest binding compounds found in a GPCR structure-based screen45,46 (docked pose shown in Figure 2 and binding curves in Figure 3, bottom row). Worse still, compound 6 had no measurable affinity for the on-targets HTR2A/DRD2, in the teeth of much better ranks. Indeed, median affinities were higher for the discovered HRH1 antitarget ligands, despite the emphasis on low docking rank and poor predicted HRH1 binding site fit (median Ki = 3430 nM, 4276 nM, and 1042 nM, with median ranks of 5784, 2097, and 1 030 000, for DRD2, HTR2A, and HRH1, respectively).
Modeling HRH1 Receptor Flexibility To Reduce Docking False Negatives
Inspection of the HRH1 false-negatives suggested that receptor flexibility might play a role in the unwanted activity of these molecules. For instance, compound 6, a subnanomolar HRH1 binder, ranks poorly in the HRH1 docking screen (though still in the top 10%, rank 326 040). The van der Waals score of −5.8 DOCK score units (putatively kcal/mol) reflects poor steric fit; top-ranked molecules in this receptor typically score over −30 DOCK units. The molecule docks with the key salt-bridge to D3.32 intact but clashes with the backbone carbonyl of D178 on the extracellular loop; this clash may likely be relieved by modest receptor relaxation (Figure 2J).
To test whether receptor flexibility contributed to the high false-negative rate, we first tested induced fit docking (IFD) to model local protein rearrangement upon ligand binding.47 Flexibility modeled through IFD rescued 5 out of the 16 false-negatives, with high scores and well docked poses. However, a high scoring pose was not found for the remaining 9 HRH1 binders, representing 70% of the false-negatives, including compound 6, suggesting that the IFD protocol was insufficient to fully model receptor flexibility. We also worried that the IFD procedure would be too expensive computationally to be practical for high-throughput de novo screening.
Instead, we turned to elastic normal modes to model the conformational changes that might occur upon ligand binding in a more computationally efficient way. We worried that many annotated HRH1 ligands may be too bulky to dock into the HRH1 crystal structure, with its small and compact orthosteric site (Figure 2C), indeed one well-suited to fragment-discovery.20 We used the program 3K-ENM48 to pregenerate 3700 expanded orthosteric site models. We selected seven large, topologically diverse ligands from the annotated HRH1 ligands in ChEMBL10. While five of these docked into the crystallographic orthosteric site with sterics, two did not. Docking these seven large ligands into the 3700 ENM models, we selected the seven models that best ranked them. Adding the seven enlarged-site models to the crystal structure docking did not affect enrichment of known ligands (50 diverse molecules from ChEMBL over a background of property-matched decoys and experimental nonbinders, log AUC = 35 for the crystal structure only, log AUC = 32 for the combined docking, where for each molecule the docking score was selected as the highest score in the crystal structure and model docking). While the crystal structure alone did not enrich the false-negatives in our prospective screen, the combined docking did (log AUC of −1.7 vs 14, respectively, over the same background of property-matched decoys and experimental nonbinders).
Ultimately, we found that using all seven expanded models was not necessary; a single expanded-site model sufficed to enrich what were formerly the docking false-negatives. Accordingly, we selected the model that prioritized the ligand with the largest solvent accessible surface area (Figure S5). This model was reasonable on visual inspection, and known ligands docked with believable poses. Docking to this model combined with the crystal structure enriched known binders (log AUC = 31 for the 50 diverse ChEMBL ligands over a background of property-matched decoys and experimental nonbinders). Importantly, this model was not selected using any knowledge of those specific false-negatives but rather on the ability to recognize large, already annotated HRH1 ligands. It was however able to rescue many of the false-negatives from our previous docking screen (log AUC = 14 over the same background of property-matched decoys and experimental nonbinders).
Prospective Screening with an Expanded HRH1 Model
Prospective docking screens were repeated against the HTR2A, DRD2, the new model of the HRH1, and its crystal structure, again with the ZINC lead-like database. To further increase the chances of finding specific molecules, the top ranked molecules were filtered more stringently. Again, molecules that ranked in the top 1% docked to both HTR2A and DRD2 were selected and molecules having a Tc > 0.5 to the top 50 000 HRH1 docked models were excluded. All DRD2/HTR2A docking hits with Tc > 0.8 to the top 500 000 docking-ranked molecules to HRH1 were also excluded. In this way, we ensured that all molecules we selected and their close analogs were even more poorly ranked against HRH1 (below 500 000 in the crystal structure and expanded site). Twenty new molecules, none previously known to bind to the HTR2A or the DRD2, were selected for testing against the three receptors (Table S1, second screening round, compounds 29–48).
Testing the New Molecules for Binding to DRD2/HTR2A and Selectivity against HRH1
Despite the higher stringency of this second screen, the on-target hit rates remained high, with 13/20 and 9/20 (65% and 45%) molecules binding to HTR2A and DRD2, respectively, and 6 (30%) binding to both with Kd < 10 μM (Table 1, compounds 31, 35, 38, 39, 42, 47). Unfortunately, the false-negative rate actually rose, with 15/20 (75%) of the molecules binding to HRH1 (Table 1). Affinities were comparable to the first screen, with median affinities of 1970 nM, 6442 nM, and 1271 nM and median ranking of 14 466, 15 595, and 726 846 to the HTR2A, DRD2, and HRH1, respectively. HRH1 hits again generally had higher affinity, with five submicromolar compounds (compared to three for HTR2A and none for DRD2).
Impact of Chemical Similarity Filters and Scoring Functions
In the initial docking screen, we filtered molecules by dissimilarity to any known HRH1 binder, insisting on Tc < 0.35 using radial ECFP4 fingerprints. To investigate whether a different fingerprint could have better prefiltered these HRH1 false-negatives, we used MACCS structural keys49 and dendritic and linear fingerprints50 to recalculate the similarity of the docking hits to known HRH1 ligands in ChEMBL. While MACCS similarity was high for all the false-negatives, suggesting that it may have been able to remove the HRH1 false-negatives, it was also high for all the true negatives, suggesting that this fingerprint had low discriminatory power overall. Meanwhile, similarity to the false-negatives was low for both the dendritic and linear fingerprints, with a similar distribution of Tc similarities for true and false-negatives for all three fingerprints (Figure S3). At a perhaps more fundamental level, we would note that while similarity filtering is certainly useful and pragmatic, it does not address the problem of false-negatives from structure-based docking.
Arguably, this could be addressed by improved scoring functions. Accordingly, we docked all experimentally tested compounds with GLIDE and rescored with molecular mechanics/generalized Born surface area (MM-GBSA) (Schrodinger, Inc.51). While a tight correlation between MM-GBSA scores and experimental binding affinities is not expected, this is often a suitable docking and rescoring protocol to discriminate actives from inactive compounds.52 However, we found no discrimination using the MM-GBSA scores (Figure S4). In fact, our most potent false-negative, compound 6 (HRH1 Ki = 0.8 nM), was given a poor MM-GBSA score of −6 (putative units of kcal/mol) against HRH1 whereas the relatively selective compound 21 scored −31 kcal/mol, and compounds that had no detectable HRH1 activity scored as high as −67 kcal/mol. It is likely that there are scoring functions that could better discriminate than those we have investigated here; our own view is that scoring functions alone cannot fully address the docking “false-negative” problem on which this study has foundered (see below).
Prospective Chemoinformatic Screening for Selective Compounds
To deconvolute the influence of 2D chemical similarity on aminergic selectivity, we screened the same ZINC lead-like library with the similarity ensemble approach (SEA), a statistical model that ranks the significance of chemical similarity of a query molecule to a set of ligands for a target.53 We insisted on compounds having an expectation value (E value) cutoff of less than 10–10 to both HTR2A and DRD2 but one greater than 1 to HRH1. Further, molecules had to have Tc < 0.4 to any known HRH1 ligand. This filtering led to seven purchasable molecules that were experimentally tested for HTR2A, DRD2, and HRH1 affinities (compounds 49–55 in Table 3). While no molecules showed appreciable affinity for HRH1, only compound 49 showed midmicromolar affinity for DRD2 and compound 50 for HTR2A. No tested molecules had the desired selectivity profile.
Table 3. Compounds Predicted To Be HRH1-Selective by the Similarity Ensemble Approach (SEA) with Their Respective E Values, Binding Constants, and Selectivity Ratios.

Docking for Compounds Selective for KOR vs MOR
A possible problem with the HTR2A/DRD2/HRH1 campaign was the reliance on homology models for the structures of the on-targets; a second concern was that HRH1 might be unusually promiscuous. To partly control for these concerns, we turned to docking for selective ligands of the κ-opioid receptor (KOR) over the μ-opioid receptor (MOR). The structures of both receptors had been determined by crystallography, and in an earlier study we had been able to find agonists functionally selective for MOR versus KOR and versus the δ-opioid and nociception opioid receptors54 (i.e., the reverse selectivity). Here again, a set of 60 diverse antagonists were used to retrospectively measure enrichment against several thousand property-matched decoys. Unlike docking to the aminergic receptors, the best adjusted log AUC achieved was 6.5, reflecting the rich diversity of ligands annotated to these targets and their large, solvent-exposed binding sites. While this retrospective adjusted log AUC is lower than what we have observed with small neurotransmitter GPCRs, it is similar to that seen in the successful prospective screen against MOR,54 and so we proceeded. Three million lead-like molecules from ZINC were docked into the KOR orthosteric site using DOCK3.6, and the top 1% of the ranked molecules were more closely examined. We calculated the ratio of each compound’s rank in the KOR screen to the compound’s rank in the MOR screen, and the binding poses of the molecules with the biggest rank ratio were visually inspected. Molecules were selected on the basis of key salt-bridges to D3.32 in the KOR binding pose, a complementary fit to the orthosteric site, and interactions with residues that are either specific to KOR or exist in a pose-conflicting and different rotamer in the MOR crystal structure (Glu209 in the second extracellular loop, Glu2976.58, Tyr3127.35, and Thr1112.56, Gln1152.60, Tyr3207.43; Figure 4; residue numbering from KOR), thus making a similar binding pose in MOR unlikely.
Figure 4.

KOR and MOR binding sites. (A) Orthosteric sites for KOR (left) and MOR (right), with their respective cocrystallized ligand. The residues shown as sticks were those used to discriminate predicted selective compounds. (B) A selective compound, 2, is shown in the docked pose to KOR (left) and MOR (right).
Testing for Binding to KOR and Selectivity against MOR
Of the 22 molecules tested, nine specifically displaced radiolabeled [3H]U69593 from KOR with Ki values between 1.8 and 14 μM (41% hit rate; Table 4; Table S2). Here again, hit rates were high, and while affinities were worse than have been observed against several small neurotransmitter GPCRs,10,19,20,27 they were comparable to affinities observed against other peptide and protein receptors.54,55 As previous docking screens at the inactive opioid receptor structures had found agonists,55,54 we tested the active compounds in Gi functional assays. While most compounds were, in fact, KOR antagonists, compounds 106 and 114 were agonists (Table 4). Of the nine active compounds, two, 103 and 122, had better than 18-fold specificity for KOR over MOR.
Table 4. Hit Compounds at KOR and MOR with Their Respective Docking Ranks, Binding Constants, and Selectivity Ratios.


A Problematic False Negative Rate against MOR
Correspondingly, and like the HTR2A/DRD2 screen, the selectivity that was a key goal for the KOR vs MOR campaign was low: seven of nine KOR actives were also active against MOR, displacing radiolabeled [3H]DAMGO. Indeed, three of the 22 molecules tested were actually specific to MOR vs KOR (the opposite of the intended selectivity). Median affinities were similar for each receptor: 4.7 μM for KOR vs 4.4 μM for MOR, despite a median docking rank of 6842 to KOR but of 251 804 to MOR, each out of the same 3 million compound library. Here too, success in finding novel compounds for the on-target was often belied by the inability to select against the antitarget.
Discussion and Conclusions
Ligands with focused polypharmacology have attracted much recent interest.1,5,7,8,15,56−59 In principle, these are accessible from docking screens against on- and off-targets. Returning to the challenges that motivated this study, docking must find molecules that jointly complement two or more targets, reducing the number of candidates. For biologically relevant polypharmacology, the screens must often use homology models, adding to uncertainty. Lastly, for selectivity, the docking screens cannot afford the false-negatives tolerated for on-targets; rather, molecules that might bind to the off-target must be stringently identified and discarded. Three major observations emerge from this study. First, docking can find molecules that modulate a pair of modeled targets with a high hit rate, with mid-nanomolar (occasionally) to the low micromolar (more typically) binding affinities. This is despite the inevitable reduction in chemical space due to the requirement for joint complementarity. Second, while a handful of these molecules were, in fact, selective for the on-targets vs the off-targets, most compounds chosen for their supposed inability to fit the off-targets in fact bound them well, sometimes with high affinity. Third, flexible treatment of the HRH1 antitarget, in an effort to find a receptor conformation that could identify the false-negatives from the initial screen, was unsuccessful, as was an effort to exclude the false-negatives by 2D ligand similarity.
If the discovery of novel GPCR ligands by library docking has been established by studies in the past decade,17,19−21,54,55,60 including for modeled structures,24,26,27,45,61 the ability to do so for two targets, much less two modeled targets, simultaneously, has received less attention. In this limited sense, the results of this study are encouraging. Even with the chemotype restrictions implicit fitting to two targets, hit rates for the on-targets were as high or higher than typical for an unbiased library docking screen, at 63%, 40%, and 41% for the HTR2A, DRD2, and KOR. Admittedly, many of the new compounds had only low micromolar affinity, and this does perhaps reflect the restraints of dual inhibition. Four compounds with micromolar binding against HTR2A and DRD2 had 29- to 120-fold selectivity vs HRH1 (Figure 5). However, one compound, 21, had mid-nanomolar affinity for HTR2A and DRD2 and 21-fold selectivity vs the HRH1 (Table 1). This potency and selectivity of 21 place it among the few serotonin/dopamine receptor antagonists with substantial HRH1 selectivity.
Figure 5.

Visual summary of the initial screen of 28 molecules tested against the HT2RA, DRD2, and HRH1 receptors.
Still, the larger story is perhaps the remarkably high hit-rates for the antitargets. Against the HRH1, the first-round hit rate was 57%. This grew to a daunting 75% in the second-round screen. This second round conformation had a larger orthosteric site that identified many of the false-negatives from the first screen; this, however, did not prevent it from missing new false-negatives in the next prospective screen. For the MOR antitarget, false-negative rates were about as bad, despite selecting docked molecules interacting with residues specific to KOR or in pose that conflicted with the rotamers adopted in MOR.
Discounting the slender successes of selective compounds such as 5, 14, 19, 21, 25, 103, and 122, three broad explanations may be considered for the failure to select against HRH1 and MOR. First, we did not properly model low-energy receptor conformations that could accommodate the high-scoring molecules from the on-targets. Second, the antitargets might be so permissive that they will always be a selectivity challenge, at least without a much larger and more diverse docking library. Finally, it could be that our docking scoring function is approximate and inaccurate enough to prevent the distinction among what are, after all, related aminergic targets recognizing related primary neurotransmitters (serotonin, dopamine, and histamine or, in the case of KOR and MOR, opioid peptides). We can perhaps discount this third possibility; as approximate and inaccurate as our scoring function remains, the false-negatives come from their ability to fit targets that sterics alone should have largely excluded. Thus, we focus on the first two possibilities here.
Crystal structures are typically snapshots of a receptor in one conformation, but biophysical studies and simulations suggest that GPCRs exist in an ensemble of low energy, distinguishable states.62−65 In docking for new ligands for an on-target, any one of these low-energy conformations, certainly including the crystal structure, will do. As long as the docked ligand fits the selected low-energy conformation well, mass balance will make up for the ignored accessible states, typically with only a minor energy penalty. Said another way, only representing one good low energy conformation of the on-target will certainly miss plausible ligands (false-negatives), but since the goal is simply to find some new ligands, this is tolerable. For selectivity against antitarget, however, the molecule must not fit any accessible conformation of the receptor. This is not only a substantial sampling problem but also one of weighting the receptor states in the available ensemble against each other.66
After the problems selecting against HRH1 in the first docking screen, we modeled an alternative HRH1 conformation with an expanded binding site that, together with the HRH1 crystal structure, could accommodate even the largest known HRH1 ligands. Had we used both structures in the initial docking screen, the first-round false-negatives would have scored well against HRH1 and so not been selected. Unfortunately, this had little predictive value in the second prospective screen, which had an even higher hit rate against the antitarget. We considered whether an alternative method, induced-fit docking, which optimizes orthosteric site residues around a docked pose and then re-docks the molecule, would have found acceptable poses for these false-negatives. Unfortunately, only five (30%) of the false-negatives would have been rescued and correctly predicted to score well in the HRH1 site; among the nine (70%) false-negatives that continued not to fit, for instance, was the 0.8 nM HRH1 ligand, which was still predicted as a nonbinder. This suggests that induced-fit docking alone could not have solved our selectivity problem; to do so would have likely required the sampling of more conformations of the overall receptor structure, not just local accommodations around two particular states.
A second explanation for the antitarget false-negatives might be that HRH1 and MOR are unusually promiscuous, and selective chemotypes may simply be absent from the docking library. The inability to improve any hit compound’s selectivity through docking may support this idea. While only a small percentage of ligands in ChEMBL23 are annotated to bind to all three of DRD2, HTR2A, and HRH1, 18.2% and 21.6% of the HRH1 compounds are annotated for HTR2A or DRD2, respectively (note that many more ligands are annotated for DRD2 and HTR2A vs HRH1, which may reflect the historical interests of medicinal chemistry campaigns, Figure 6). More broadly, 40.1% of the HRH1 annotated ligands are active against at least two other aminergic receptors, 21.2% are active against three other aminergic receptors in ChEMBL23, and chemoinformatics of the overall ligand-based similarity among receptors suggests that HRH1 ligands are not only among the most diverse of the aminergic GPCRs (suggesting a broad recognition of chemotypes associated with different classes of receptors) but also among those with the greatest ligand overlap with other receptors.53,67 Similarly, while 71.3% of KOR ligands also bind to MOR, 72.6% of MOR ligands bind to KOR with affinity better than 10 μM. In short, the antitargets chosen here, though clinically relevant, might be so similar in their structure and share so many ligands and chemotypes with the on-targets that finding just the right selective molecules will be difficult with available libraries. Consistent with this view, tightening of our original topological similarity or docking rank filters would have removed essentially all the aminergic molecules in the library, removing not only false-negatives but also true-positives. If this argument were true (and we moot it only tentatively), then an expansion of our libraries to sample new chemotypes may help in finding selective and still potent molecules, direct from a structure-based screen, against highly related targets such as those sampled here.
Figure 6.
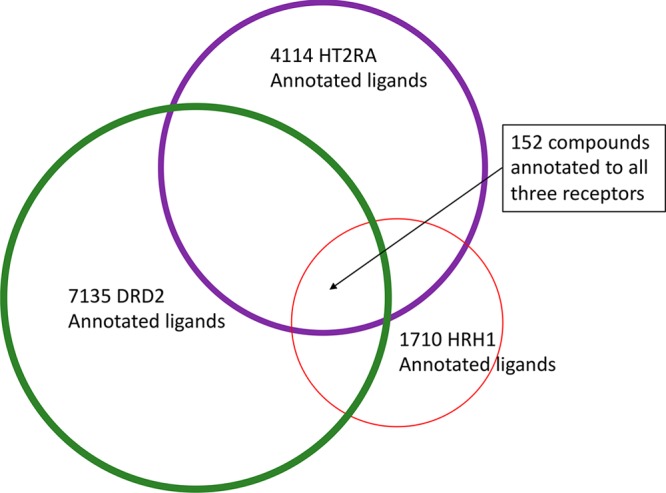
Venn diagram of annotated ChEMBL ligands for the three targets.
Certain gaps in this study temper its conclusions. While it may be that HRH1 is too similar in structure and ligand recognition to HTR2A and DRD2 to reliably dock for selective molecules, several such molecules were in fact found, and others have been optimized from medicinal chemistry campaigns. Beginning with a compound with nascent potency and selectivity, new compounds have been synthesized with focused polypharmacology and selectivity, even against highly related targets.68 This partly reflects the synthesis of chemotypes not present even among the 3 million commercially available molecules. We also note that we largely ignored functional effects of docked molecules; for most compounds, we did not consider whether they were agonists or inverse agonists. Arguably, this was not crucial for this proof-of-concept study, though it would be important in a true effort toward focused polypharmacology. Throughout, we insisted on aminergic molecules that made well-precedented interactions in the heart of the orthosteric sites of each of the D2, 5HT2a, and KOR receptors. Such cationic molecules are almost canonical for these on-targets, but this is a feature shared with H1 ligands. Allowing for nonaminergic ligands, which are very rare for this family of receptor but not entirely unheard of,69 or expanding our search to putative allosteric sites where sequence diversity is higher59 may have improved our chances of finding selective ligands, though it would likely have reduced hit-rates against the on-targets themselves. Finally, our failure to reliably select against the antitargets at least partly reflects well-known weaknesses in our methods. Among the most important of these was the failure to consider multiple low-energy receptor states in modeling our off-targets, although more rigorous treatment will be challenging in high-throughput.
These caveats should not obscure the main observations from this study. Structure-based docking may be pragmatic for focused polypharmacology, here finding multiple molecules that can jointly modulate the HTR2A and DRD2 GPCRs. Homology models of these targets can support these efforts, at least when they are sufficiently similar to structural templates and are vetted in control experiments (e.g., retrospective benchmarking). Crucially, however, we struggled to find molecules selective against the off-targets, with hit rates against HRH1 and MOR as high or higher than for the on-targets. Overcoming this problem may demand expanding chemotypes in dockable libraries,70−72 so that just the right molecule might be found, and certainly better modeling of the accessible conformations of the antitargets.73−80
Experimental Section
Homology Modeling and Docking
The alignments for the construction of the DRD2 and HTR2A models were generated using PROMALS3D.81 Homology models of the DRD2 and HTR2A receptors were built with MODELLER 9v8 using the crystal structure of the dopamine D3 receptor (PDB code 3PBL) as the template. Elastic network models for the expanded HRH1 binding site were produced by the program 3K-ENM.48 We used DOCK3.643 to screen the ZINC database (Results). In DOCK3.6, ligand conformational ensembles are precalculated in a frame of reference defined by their rigid fragments, and fragment atoms are fit onto binding site matching spheres, which represent favorable positions for individual ligand atoms. Once a sphere-atom superposition is defined for a rigid-fragment, the conformational ensemble may be oriented in the site using the defined rotation–translation matrix of that rigid fragment.27,43 Each ligand pose is scored as the sum of the receptor–ligand electrostatic and van der Waals interaction energies and corrected for context-dependent ligand desolvation.82 Partial charges from the united-atom AMBER force field were used for all receptor atoms except for, in the KOR, Asp1383.32, Glu209 in ECL2, and Glu2976.58, for which the magnitude of the partial atomic charges in the carboxylate was increased, as previously described;18 the net charges were not changed. When selecting molecules from the KOR screen, those with high internal-energy interactions that do not appear in the Cambridge Structural Database were manually discarded, as is common as a last step in the docking-aided selection of new molecules.25
Induced Fit Docking
The induced-fit docking used a three-step protocol: (1) each molecule was docked into the receptor; (2) for each pose, the receptor side chains and backbone were minimized around the posed ligand using PLOP;83 (3) the compound was re-docked into the optimized receptor binding site.
Binding Affinity and Functional Assays
Radioligand binding and functional (GloSensor, Tango, and FLIPR) assays at the DRD2, HTR2A, HRH1, KOR, and MOR were carried out at the National Institute of Mental Health Psychoactive Drug Screening Program, as described.84
Compound Sources
Compounds were obtained from commercial suppliers and used without further purification (a full list is provided in the Supporting Information as a tab-delimited files with vendors and SMILES strings). All active compounds reported for the serotonin, dopamine, and histamine receptors were tested for purity by liquid chromatography/mass spectrometry and were at least 95% homogeneous by peak height and identity. Compounds were counterscreened for aggregation using detergent-dependent inhibition of AmpC β-lactamase as a proxy, as has been widely done,85 and no substantial inhibition was observed. Also, the dose–response curves in the GPCR assays themselves were well-behaved with Hill coefficients close to 1.
Acknowledgments
We thank Alison Doak for testing compounds for aggregation, and we thank Anat Levitt and Josh Pottel for critical reading of this manuscript. This work was supported by U.S. National Institutes of Health Grant R35 GM122481 (to B.K.S.), by Grant F32 GM093580 (to D.R.W.), and by the NIMH Psychoactive Drug Screening Program (NIMH PDSP, to B.L.R.).
Glossary
Abbreviations Used
- GPCR
G protein-coupled receptor
- DRD2
dopamine D2 receptor
- HTR2A
serotonin 5-HT2A receptor
- HRH1
histamine H1 receptor
- MOR
μ-opioid receptor
- KOR
κ-opioid receptor
- log AUC
logarithm of area under the curve
- rmsd
root-mean-square deviation
- ECFP4
extended connectivity fingerprint of diameter 4
- Tc
Tanimoto coefficient
- IFD
induced-fit docking
- SEA
similarity ensemble approach
- E value
expectation value
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b00718.
Additional figures illustrating enrichment plots and model binding site comparisons (PDF)
SMILES strings and list of vendors for compounds (CSV)
Coordinates information for structure representation (PDB)
Coordinates information for structure representation (PDB)
Coordinates information for structure representation (PDB)
Author Contributions
§ D.R.W., J.K., and X.-P.H. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Frantz S. Drug Discovery: Playing Dirty. Nature 2005, 437 (7061), 942–943. 10.1038/437942a. [DOI] [PubMed] [Google Scholar]
- Roth B. L.; Sheffler D. J.; Kroeze W. K. Magic Shotguns Versus Magic Bullets: Selectively Non-Selective Drugs for Mood Disorders and Schizophrenia. Nat. Rev. Drug Discovery 2004, 3 (4), 353–359. 10.1038/nrd1346. [DOI] [PubMed] [Google Scholar]
- Fishman M. C.; Porter J. A. Pharmaceuticals: A New Grammar for Drug Discovery. Nature 2005, 437 (7058), 491–493. 10.1038/437491a. [DOI] [PubMed] [Google Scholar]
- Urban J. D.; Clarke W. P.; von Zastrow M.; Nichols D. E.; Kobilka B.; Weinstein H.; Javitch J. A.; Roth B. L.; Christopoulos A.; Sexton P. M.; Miller K. J.; Spedding M.; Mailman R. B. Functional Selectivity and Classical Concepts of Quantitative Pharmacology. J. Pharmacol. Exp. Ther. 2007, 320 (1), 1–13. 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L. Network Pharmacology: The Next Paradigm in Drug Discovery. Nat. Chem. Biol. 2008, 4 (11), 682–690. 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- Metz J. T.; Hajduk P. J. Rational Approaches to Targeted Polypharmacology: Creating and Navigating Protein-Ligand Interaction Networks. Curr. Opin. Chem. Biol. 2010, 14 (4), 498–504. 10.1016/j.cbpa.2010.06.166. [DOI] [PubMed] [Google Scholar]
- Dar A. C.; Das T. K.; Shokat K. M.; Cagan R. L. Chemical Genetic Discovery of Targets and Anti-Targets for Cancer Polypharmacology. Nature 2012, 486 (7401), 80–84. 10.1038/nature11127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besnard J.; Ruda G. F.; Setola V.; Abecassis K.; Rodriguiz R. M.; Huang X. P.; Norval S.; Sassano M. F.; Shin A. I.; Webster L. A.; Simeons F. R.; Stojanovski L.; Prat A.; Seidah N. G.; Constam D. B.; Bickerton G. R.; Read K. D.; Wetsel W. C.; Gilbert I. H.; Roth B. L.; Hopkins A. L. Automated Design of Ligands to Polypharmacological Profiles. Nature 2012, 492 (7428), 215–220. 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D.; Bernat V.; Brox R.; Tschammer N.; Kolb P. Identifying Modulators of Cxc Receptors 3 and 4 with Tailored Selectivity Using Multi-Target Docking. ACS Chem. Biol. 2015, 10 (3), 715–724. 10.1021/cb500577j. [DOI] [PubMed] [Google Scholar]
- Kaczor A. A.; Silva A. G.; Loza M. I.; Kolb P.; Castro M.; Poso A. Structure-Based Virtual Screening for Dopamine D2 Receptor Ligands as Potential Antipsychotics. ChemMedChem 2016, 11 (7), 718–729. 10.1002/cmdc.201500599. [DOI] [PubMed] [Google Scholar]
- Jazayeri A.; Rappas M.; Brown A. J. H.; Kean J.; Errey J. C.; Robertson N. J.; Fiez-Vandal C.; Andrews S. P.; Congreve M.; Bortolato A.; Mason J. S.; Baig A. H.; Teobald I.; Dore A. S.; Weir M.; Cooke R. M.; Marshall F. H. Crystal Structure of the Glp-1 Receptor Bound to a Peptide Agonist. Nature 2017, 546 (7657), 254–258. 10.1038/nature22800. [DOI] [PubMed] [Google Scholar]
- Ma Y.; Yue Y.; Ma Y.; Zhang Q.; Zhou Q.; Song Y.; Shen Y.; Li X.; Ma X.; Li C.; Hanson M. A.; Han G. W.; Sickmier E. A.; Swaminath G.; Zhao S.; Stevens R. C.; Hu L. A.; Zhong W.; Zhang M.; Xu F. Structural Basis for Apelin Control of the Human Apelin Receptor. Structure 2017, 25 (6), 858–866. 10.1016/j.str.2017.04.008. [DOI] [PubMed] [Google Scholar]
- Congreve M.; Oswald C.; Marshall F. H. Applying Structure-Based Drug Design Approaches to Allosteric Modulators of Gpcrs. Trends Pharmacol. Sci. 2017, 38 (9), 837–847. 10.1016/j.tips.2017.05.010. [DOI] [PubMed] [Google Scholar]
- Robertson N.; Rappas M.; Dore A. S.; Brown J.; Bottegoni G.; Koglin M.; Cansfield J.; Jazayeri A.; Cooke R. M.; Marshall F. H. Structure of the Complement C5a Receptor Bound to the Extra-Helical Antagonist Ndt9513727. Nature 2018, 553 (7686), 111–114. 10.1038/nature25025. [DOI] [PubMed] [Google Scholar]
- Peng Y.; McCorvy J. D.; Harpsoe K.; Lansu K.; Yuan S.; Popov P.; Qu L.; Pu M.; Che T.; Nikolajsen L. F.; Huang X. P.; Wu Y.; Shen L.; Bjorn-Yoshimoto W. E.; Ding K.; Wacker D.; Han G. W.; Cheng J.; Katritch V.; Jensen A. A.; Hanson M. A.; Zhao S.; Gloriam D. E.; Roth B. L.; Stevens R. C.; Liu Z. J. 5-Ht2c Receptor Structures Reveal the Structural Basis of GPCR Polypharmacology. Cell 2018, 172 (4), 719–730. 10.1016/j.cell.2018.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Qiao A.; Yang L.; Van Eps N.; Frederiksen K. S.; Yang D.; Dai A.; Cai X.; Zhang H.; Yi C.; Cao C.; He L.; Yang H.; Lau J.; Ernst O. P.; Hanson M. A.; Stevens R. C.; Wang M. W.; Reedtz-Runge S.; Jiang H.; Zhao Q.; Wu B. Structure of the Glucagon Receptor in Complex with a Glucagon Analogue. Nature 2018, 553 (7686), 106–110. 10.1038/nature25153. [DOI] [PubMed] [Google Scholar]
- Kolb P.; Rosenbaum D. M.; Irwin J. J.; Fung J. J.; Kobilka B. K.; Shoichet B. K. Structure-Based Discovery of Beta2-Adrenergic Receptor Ligands. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (16), 6843–6848. 10.1073/pnas.0812657106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson J.; Yoo L.; Gao Z. G.; Irwin J. J.; Shoichet B. K.; Jacobson K. A. Structure-Based Discovery of A2a Adenosine Receptor Ligands. J. Med. Chem. 2010, 53 (9), 3748–3755. 10.1021/jm100240h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V.; Jaakola V. P.; Lane J. R.; Lin J.; Ijzerman A. P.; Yeager M.; Kufareva I.; Stevens R. C.; Abagyan R. Structure-Based Discovery of Novel Chemotypes for Adenosine a(2a) Receptor Antagonists. J. Med. Chem. 2010, 53 (4), 1799–1809. 10.1021/jm901647p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf C.; Kooistra A. J.; Vischer H. F.; Katritch V.; Kuijer M.; Shiroishi M.; Iwata S.; Shimamura T.; Stevens R. C.; de Esch I. J.; Leurs R. Crystal Structure-Based Virtual Screening for Fragment-Like Ligands of the Human Histamine H(1) Receptor. J. Med. Chem. 2011, 54 (23), 8195–8206. 10.1021/jm2011589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysinger M. M.; Weiss D. R.; Ziarek J. J.; Gravel S.; Doak A. K.; Karpiak J.; Heveker N.; Shoichet B. K.; Volkman B. F. Structure-Based Ligand Discovery for the Protein-Protein Interface of Chemokine Receptor Cxcr4. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (14), 5517–5522. 10.1073/pnas.1120431109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazayeri A.; Dias J. M.; Marshall F. H. From G Protein-Coupled Receptor Structure Resolution to Rational Drug Design. J. Biol. Chem. 2015, 290 (32), 19489–19495. 10.1074/jbc.R115.668251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez D.; Ranganathan A.; Carlsson J. Discovery of Gpcr Ligands by Molecular Docking Screening: Novel Opportunities Provided by Crystal Structures. Curr. Top. Med. Chem. 2015, 15 (24), 2484–2503. 10.2174/1568026615666150701112853. [DOI] [PubMed] [Google Scholar]
- Lansu K.; Karpiak J.; Liu J.; Huang X. P.; McCorvy J. D.; Kroeze W. K.; Che T.; Nagase H.; Carroll F. I.; Jin J.; Shoichet B. K.; Roth B. L. In Silico Design of Novel Probes for the Atypical Opioid Receptor Mrgprx2. Nat. Chem. Biol. 2017, 13 (5), 529–536. 10.1038/nchembio.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin J. J.; Shoichet B. K. Docking Screens for Novel Ligands Conferring New Biology. J. Med. Chem. 2016, 59 (9), 4103–4120. 10.1021/acs.jmedchem.5b02008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X. P.; Karpiak J.; Kroeze W. K.; Zhu H.; Chen X.; Moy S. S.; Saddoris K. A.; Nikolova V. D.; Farrell M. S.; Wang S.; Mangano T. J.; Deshpande D. A.; Jiang A.; Penn R. B.; Jin J.; Koller B. H.; Kenakin T.; Shoichet B. K.; Roth B. L. Allosteric Ligands for the Pharmacologically Dark Receptors Gpr68 and Gpr65. Nature 2015, 527 (7579), 477–483. 10.1038/nature15699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson J.; Coleman R. G.; Setola V.; Irwin J. J.; Fan H.; Schlessinger A.; Sali A.; Roth B. L.; Shoichet B. K. Ligand Discovery from a Dopamine D3 Receptor Homology Model and Crystal Structure. Nat. Chem. Biol. 2011, 7 (11), 769–778. 10.1038/nchembio.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeze W. K.; Hufeisen S. J.; Popadak B. A.; Renock S. M.; Steinberg S.; Ernsberger P.; Jayathilake K.; Meltzer H. Y.; Roth B. L. H1-Histamine Receptor Affinity Predicts Short-Term Weight Gain for Typical and Atypical Antipsychotic Drugs. Neuropsychopharmacology 2003, 28 (3), 519–526. 10.1038/sj.npp.1300027. [DOI] [PubMed] [Google Scholar]
- Kim S. F.; Huang A. S.; Snowman A. M.; Teuscher C.; Snyder S. H. From the Cover: Antipsychotic Drug-Induced Weight Gain Mediated by Histamine H1 Receptor-Linked Activation of Hypothalamic Amp-Kinase. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (9), 3456–3459. 10.1073/pnas.0611417104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi T.; Horisawa T.; Tokuda K.; Ishiyama T.; Ogasa M.; Tagashira R.; Matsumoto K.; Nishikawa H.; Ueda Y.; Toma S.; Oki H.; Tanno N.; Saji I.; Ito A.; Ohno Y.; Nakamura M. Pharmacological Profile of Lurasidone, a Novel Antipsychotic Agent with Potent 5-Hydroxytryptamine 7 (5-Ht7) and 5-Ht1a Receptor Activity. J. Pharmacol. Exp. Ther. 2010, 334 (1), 171–181. 10.1124/jpet.110.167346. [DOI] [PubMed] [Google Scholar]
- Negri A.; Rives M. L.; Caspers M. J.; Prisinzano T. E.; Javitch J. A.; Filizola M. Discovery of a Novel Selective Kappa-Opioid Receptor Agonist Using Crystal Structure-Based Virtual Screening. J. Chem. Inf. Model. 2013, 53 (3), 521–526. 10.1021/ci400019t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spetea M.; Asim M. F.; Noha S.; Wolber G.; Schmidhammer H. Current Kappa Opioid Receptor Ligands and Discovery of a New Molecular Scaffold as a Kappa Opioid Receptor Antagonist Using Pharmacophore-Based Virtual Screening. Curr. Pharm. Des. 2013, 19 (42), 7362–7372. 10.2174/138161281942140105162601. [DOI] [PubMed] [Google Scholar]
- Chien E. Y.; Liu W.; Zhao Q.; Katritch V.; Han G. W.; Hanson M. A.; Shi L.; Newman A. H.; Javitch J. A.; Cherezov V.; Stevens R. C. Structure of the Human Dopamine D3 Receptor in Complex with a D2/D3 Selective Antagonist. Science 2010, 330 (6007), 1091–1095. 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N.; Webb B.; Marti-Renom M. A.; Madhusudhan M. S.; Eramian D.; Shen M. Y.; Pieper U.; Sali A., Comparative Protein Structure Modeling Using Modeller. Current Protocols in Bioinformatics; Wiley, 2007. l Chapter 2, Unit 2 9, 10.1002/0471250953.bi0506s15. [DOI] [PubMed] [Google Scholar]
- Webb B.; Sali A. Comparative Protein Structure Modeling Using Modeller. Curr. Protoc. Protein Sci. 2016, 86, 2.9.1–2.9.37. 10.1002/cpps.20. [DOI] [PubMed] [Google Scholar]
- Gaulton A.; Bellis L. J.; Bento A. P.; Chambers J.; Davies M.; Hersey A.; Light Y.; McGlinchey S.; Michalovich D.; Al-Lazikani B.; Overington J. P. Chembl: A Large-Scale Bioactivity Database for Drug Discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprea T. I.; Davis A. M.; Teague S. J.; Leeson P. D. Is There a Difference between Leads and Drugs? A Historical Perspective. J. Chem. Inf. Comput. Sci. 2001, 41 (5), 1308–1315. 10.1021/ci010366a. [DOI] [PubMed] [Google Scholar]
- Wang S.; Che T.; Levit A.; Shoichet B. K.; Wacker D.; Roth B. L. Structure of the D2 Dopamine Receptor Bound to the Atypical Antipsychotic Drug Risperidone. Nature 2018, 555 (7695), 269–273. 10.1038/nature25758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D.; Wang C.; Katritch V.; Han G. W.; Huang X. P.; Vardy E.; McCorvy J. D.; Jiang Y.; Chu M.; Siu F. Y.; Liu W.; Xu H. E.; Cherezov V.; Roth B. L.; Stevens R. C. Structural Features for Functional Selectivity at Serotonin Receptors. Science 2013, 340 (6132), 615–619. 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura T.; Shiroishi M.; Weyand S.; Tsujimoto H.; Winter G.; Katritch V.; Abagyan R.; Cherezov V.; Liu W.; Han G. W.; Kobayashi T.; Stevens R. C.; Iwata S. Structure of the Human Histamine H1 Receptor Complex with Doxepin. Nature 2011, 475 (7354), 65–70. 10.1038/nature10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin J. J.; Shoichet B. K. Zinc–a Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45 (1), 177–182. 10.1021/ci049714+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin J. J.; Sterling T.; Mysinger M. M.; Bolstad E. S.; Coleman R. G. Zinc: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model. 2012, 52 (7), 1757–1768. 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorber D. M.; Shoichet B. K. Hierarchical Docking of Databases of Multiple Ligand Conformations. Curr. Top. Med. Chem. 2005, 5 (8), 739–749. 10.2174/1568026054637683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros J.; Weinstein H. Integrated Methods for Modeling G-Protein Coupled Receptors. Methods Neurosci. 1995, 25, 366–428. 10.1016/S1043-9471(05)80049-7. [DOI] [Google Scholar]
- Evers A.; Klabunde T. Structure-Based Drug Discovery Using Gpcr Homology Modeling: Successful Virtual Screening for Antagonists of the Alpha1a Adrenergic Receptor. J. Med. Chem. 2005, 48 (4), 1088–1097. 10.1021/jm0491804. [DOI] [PubMed] [Google Scholar]
- Becker O. M.; Dhanoa D. S.; Marantz Y.; Chen D.; Shacham S.; Cheruku S.; Heifetz A.; Mohanty P.; Fichman M.; Sharadendu A.; Nudelman R.; Kauffman M.; Noiman S. An Integrated in Silico 3d Model-Driven Discovery of a Novel, Potent, and Selective Amidosulfonamide 5-Ht1a Agonist (Prx-00023) for the Treatment of Anxiety and Depression. J. Med. Chem. 2006, 49 (11), 3116–3135. 10.1021/jm0508641. [DOI] [PubMed] [Google Scholar]
- Sherman W.; Day T.; Jacobson M. P.; Friesner R. A.; Farid R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49 (2), 534–553. 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- Yang Q.; Sharp K. A. Building Alternate Protein Structures Using the Elastic Network Model. Proteins: Struct., Funct., Genet. 2009, 74 (3), 682–700. 10.1002/prot.22184. [DOI] [PubMed] [Google Scholar]
- Durant J. L.; Leland B. A.; Henry D. R.; Nourse J. G. Reoptimization of Mdl Keys for Use in Drug Discovery. J. Chem. Inf. Comput. Sci. 2002, 42 (6), 1273–1280. 10.1021/ci010132r. [DOI] [PubMed] [Google Scholar]
- Sastry M.; Lowrie J. F.; Dixon S. L.; Sherman W. Large-Scale Systematic Analysis of 2d Fingerprint Methods and Parameters to Improve Virtual Screening Enrichments. J. Chem. Inf. Model. 2010, 50 (5), 771–784. 10.1021/ci100062n. [DOI] [PubMed] [Google Scholar]
- Halgren T. A.; Murphy R. B.; Friesner R. A.; Beard H. S.; Frye L. L.; Pollard W. T.; Banks J. L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47 (7), 1750–1759. 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- Greenidge P. A.; Kramer C.; Mozziconacci J. C.; Sherman W. Improving Docking Results Via Reranking of Ensembles of Ligand Poses in Multiple X-Ray Protein Conformations with Mm-Gbsa. J. Chem. Inf. Model. 2014, 54 (10), 2697–2717. 10.1021/ci5003735. [DOI] [PubMed] [Google Scholar]
- Keiser M. J.; Roth B. L.; Armbruster B. N.; Ernsberger P.; Irwin J. J.; Shoichet B. K. Relating Protein Pharmacology by Ligand Chemistry. Nat. Biotechnol. 2007, 25 (2), 197–206. 10.1038/nbt1284. [DOI] [PubMed] [Google Scholar]
- Manglik A.; Lin H.; Aryal D. K.; McCorvy J. D.; Dengler D.; Corder G.; Levit A.; Kling R. C.; Bernat V.; Hubner H.; Huang X. P.; Sassano M. F.; Giguere P. M.; Lober S.; Duan D.; Scherrer G.; Kobilka B. K.; Gmeiner P.; Roth B. L.; Shoichet B. K. Structure-Based Discovery of Opioid Analgesics with Reduced Side Effects. Nature 2016, 537 (7619), 185–190. 10.1038/nature19112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negri A.; Rives M. L.; Caspers M. J.; Prisinzano T. E.; Javitch J. A.; Filizola M. Discovery of a Novel Selective Kappa-Opioid Receptor Agonist Using Crystal Structure-Based Virtual Screening. J. Chem. Inf. Model. 2013, 53, 521–526. 10.1021/ci400019t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anighoro A.; Bajorath J.; Rastelli G. Polypharmacology: Challenges and Opportunities in Drug Discovery. J. Med. Chem. 2014, 57 (19), 7874–7887. 10.1021/jm5006463. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Li Q.; Wu M.; Chen C.; Cen S. Progress in the Rational Design for Polypharmacology Drug. Curr. Pharm. Des. 2016, 22 (21), 3182–3189. 10.2174/1381612822666160224125121. [DOI] [PubMed] [Google Scholar]
- Wang S.; Wacker D.; Levit A.; Che T.; Betz R. M.; McCorvy J. D.; Venkatakrishnan A. J.; Huang X. P.; Dror R. O.; Shoichet B. K.; Roth B. L. D4 Dopamine Receptor High-Resolution Structures Enable the Discovery of Selective Agonists. Science 2017, 358 (6361), 381–386. 10.1126/science.aan5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korczynska M.; Clark M. J.; Valant C.; Xu J.; Moo E. V.; Albold S.; Weiss D. R.; Torosyan H.; Huang W.; Kruse A. C.; Lyda B. R.; May L. T.; Baltos J. A.; Sexton P. M.; Kobilka B. K.; Christopoulos A.; Shoichet B. K.; Sunahara R. K. Structure-Based Discovery of Selective Positive Allosteric Modulators of Antagonists for the M2 Muscarinic Acetylcholine Receptor. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (10), E2419–E2428. 10.1073/pnas.1718037115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabio M.; Jones K.; Topiol S. Use of the X-Ray Structure of the Beta2-Adrenergic Receptor for Drug Discovery. Part 2: Identification of Active Compounds. Bioorg. Med. Chem. Lett. 2008, 18 (20), 5391–5395. 10.1016/j.bmcl.2008.09.046. [DOI] [PubMed] [Google Scholar]
- Evers A.; Klebe G. Ligand-Supported Homology Modeling of G-Protein-Coupled Receptor Sites: Models Sufficient for Successful Virtual Screening. Angew. Chem., Int. Ed. 2004, 43 (2), 248–251. 10.1002/anie.200352776. [DOI] [PubMed] [Google Scholar]
- Provasi D.; Bortolato A.; Filizola M. Exploring Molecular Mechanisms of Ligand Recognition by Opioid Receptors with Metadynamics. Biochemistry 2009, 48 (42), 10020–10029. 10.1021/bi901494n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygaard R.; Zou Y.; Dror R. O.; Mildorf T. J.; Arlow D. H.; Manglik A.; Pan A. C.; Liu C. W.; Fung J. J.; Bokoch M. P.; Thian F. S.; Kobilka T. S.; Shaw D. E.; Mueller L.; Prosser R. S.; Kobilka B. K. The Dynamic Process of Beta(2)-Adrenergic Receptor Activation. Cell 2013, 152 (3), 532–542. 10.1016/j.cell.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A.; Kobilka B. The Role of Protein Dynamics in Gpcr Function: Insights from the Beta2ar and Rhodopsin. Curr. Opin. Cell Biol. 2014, 27, 136–143. 10.1016/j.ceb.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isogai S.; Deupi X.; Opitz C.; Heydenreich F. M.; Tsai C. J.; Brueckner F.; Schertler G. F.; Veprintsev D. B.; Grzesiek S. Backbone Nmr Reveals Allosteric Signal Transduction Networks in the Beta1-Adrenergic Receptor. Nature 2016, 530 (7589), 237–241. 10.1038/nature16577. [DOI] [PubMed] [Google Scholar]
- Fischer M.; Coleman R. G.; Fraser J. S.; Shoichet B. K. Incorporation of Protein Flexibility and Conformational Energy Penalties in Docking Screens to Improve Ligand Discovery. Nat. Chem. 2014, 6 (7), 575–583. 10.1038/nchem.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H.; Sassano M. F.; Roth B. L.; Shoichet B. K. A Pharmacological Organization of G Protein-Coupled Receptors. Nat. Methods 2013, 10 (2), 140–146. 10.1038/nmeth.2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besnard J.; Hopkins A. L.. De Novo Design of Ligands against Multitarget Profiles. In De Novo Molecular Design; Schneider G., Ed.; John Wiley and Sons: New York, NY, U.S., 2013. [Google Scholar]
- Gray D. L.; Allen J. A.; Mente S.; O’Connor R. E.; DeMarco G. J.; Efremov I.; Tierney P.; Volfson D.; Davoren J.; Guilmette E.; Salafia M.; Kozak R.; Ehlers M. D. Impaired Beta-Arrestin Recruitment and Reduced Desensitization by Non-Catechol Agonists of the D1 Dopamine Receptor. Nat. Commun. 2018, 9 (1), 674. 10.1038/s41467-017-02776-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevillard F.; Kolb P. Scubidoo: A Large yet Screenable and Easily Searchable Database of Computationally Created Chemical Compounds Optimized toward High Likelihood of Synthetic Tractability. J. Chem. Inf. Model. 2015, 55 (9), 1824–1835. 10.1021/acs.jcim.5b00203. [DOI] [PubMed] [Google Scholar]
- Visini R.; Awale M.; Reymond J. L. Fragment Database Fdb-17. J. Chem. Inf. Model. 2017, 57 (4), 700–709. 10.1021/acs.jcim.7b00020. [DOI] [PubMed] [Google Scholar]
- Taylor R. D.; MacCoss M.; Lawson A. D. Combining Molecular Scaffolds from Fda Approved Drugs: Application to Drug Discovery. J. Med. Chem. 2017, 60 (5), 1638–1647. 10.1021/acs.jmedchem.6b01367. [DOI] [PubMed] [Google Scholar]
- Spyrakis F.; Benedetti P.; Decherchi S.; Rocchia W.; Cavalli A.; Alcaro S.; Ortuso F.; Baroni M.; Cruciani G. A Pipeline to Enhance Ligand Virtual Screening: Integrating Molecular Dynamics and Fingerprints for Ligand and Proteins. J. Chem. Inf. Model. 2015, 55 (10), 2256–2274. 10.1021/acs.jcim.5b00169. [DOI] [PubMed] [Google Scholar]
- Spyrakis F.; Cavasotto C. N. Open Challenges in Structure-Based Virtual Screening: Receptor Modeling, Target Flexibility Consideration and Active Site Water Molecules Description. Arch. Biochem. Biophys. 2015, 583, 105–119. 10.1016/j.abb.2015.08.002. [DOI] [PubMed] [Google Scholar]
- Miao Y.; Goldfeld D. A.; Moo E. V.; Sexton P. M.; Christopoulos A.; McCammon J. A.; Valant C. Accelerated Structure-Based Design of Chemically Diverse Allosteric Modulators of a Muscarinic G Protein-Coupled Receptor. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (38), E5675–5684. 10.1073/pnas.1612353113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Ehmann U.; Zacharias M. Monte Carlo Replica-Exchange Based Ensemble Docking of Protein Conformations. Proteins: Struct., Funct., Genet. 2017, 85 (5), 924–937. 10.1002/prot.25262. [DOI] [PubMed] [Google Scholar]
- Wieder M.; Garon A.; Perricone U.; Boresch S.; Seidel T.; Almerico A. M.; Langer T. Common Hits Approach: Combining Pharmacophore Modeling and Molecular Dynamics Simulations. J. Chem. Inf. Model. 2017, 57 (2), 365–385. 10.1021/acs.jcim.6b00674. [DOI] [PubMed] [Google Scholar]
- Feng X.; Ambia J.; Chen K. M.; Young M.; Barth P. Computational Design of Ligand-Binding Membrane Receptors with High Selectivity. Nat. Chem. Biol. 2017, 13 (7), 715–723. 10.1038/nchembio.2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertazzo M.; Bernetti M.; Recanatini M.; Masetti M.; Cavalli A. Fully Flexible Docking Via Reaction-Coordinate-Independent Molecular Dynamics Simulations. J. Chem. Inf. Model. 2018, 58 (2), 490–500. 10.1021/acs.jcim.7b00674. [DOI] [PubMed] [Google Scholar]
- Martinez-Rosell G.; Harvey M. J.; De Fabritiis G. Molecular-Simulation-Driven Fragment Screening for the Discovery of New Cxcl12 Inhibitors. J. Chem. Inf. Model. 2018, 58, 683–691. 10.1021/acs.jcim.7b00625. [DOI] [PubMed] [Google Scholar]
- Pei J.; Kim B. H.; Grishin N. V. Promals3d: A Tool for Multiple Protein Sequence and Structure Alignments. Nucleic Acids Res. 2008, 36 (7), 2295–2300. 10.1093/nar/gkn072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysinger M. M.; Shoichet B. K. Rapid Context-Dependent Ligand Desolvation in Molecular Docking. J. Chem. Inf. Model. 2010, 50 (9), 1561–1573. 10.1021/ci100214a. [DOI] [PubMed] [Google Scholar]
- Jacobson M. P.; Friesner R. A.; Xiang Z.; Honig B. On the Role of the Crystal Environment in Determining Protein Side-Chain Conformations. J. Mol. Biol. 2002, 320 (3), 597–608. 10.1016/S0022-2836(02)00470-9. [DOI] [PubMed] [Google Scholar]
- Allen J. A.; Yost J. M.; Setola V.; Chen X.; Sassano M. F.; Chen M.; Peterson S.; Yadav P. N.; Huang X. P.; Feng B.; Jensen N. H.; Che X.; Bai X.; Frye S. V.; Wetsel W. C.; Caron M. G.; Javitch J. A.; Roth B. L.; Jin J. Discovery of Beta-Arrestin-Biased Dopamine D2 Ligands for Probing Signal Transduction Pathways Essential for Antipsychotic Efficacy. Proc. Natl. Acad. Sci. U. S. A. 2011, 108 (45), 18488–18493. 10.1073/pnas.1104807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassano M. F.; Doak A. K.; Roth B. L.; Shoichet B. K. Colloidal Aggregation Causes Inhibition of G Protein-Coupled Receptors. J. Med. Chem. 2013, 56 (6), 2406–2414. 10.1021/jm301749y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.