

Abstract
Objective
To explore valproic acid (VPA) as a potentially beneficial drug in cellular and worm models of oculopharyngeal muscular dystrophy (OPMD).
Methods
Using a combination of live cell imaging and biochemical measures, we evaluated the potential protective effect of VPA in a stable C2C12 muscle cell model of OPMD, in lymphoblastoid cell lines derived from patients with OPMD and in a transgenic Caenorhabditis elegans OPMD model expressing human mutant PABPN1.
Results
We demonstrated that VPA protects against the toxicity of mutant PABPN1. Of note, we found that VPA confers its long-term protective effects on C2C12 cell survival, proliferation, and differentiation by increasing the acetylated level of histones. Furthermore, VPA enhances the level of histone acetylation in lymphoblastoid cell lines derived from patients with OPMD. Moreover, treatment of nematodes with moderate concentrations of VPA significantly improved the motility of the PABPN-13 Alanines worms.
Conclusions
Our results suggest that VPA helps to counteract OPMD-related phenotypes in the cellular and C elegans disease models.
Oculopharyngeal muscular dystrophy (OPMD) (MIM #164300) is an adult-onset disorder characterized by progressive eyelid drooping, swallowing difficulties, and proximal limb weakness.1 Currently, no effective treatment exists for OPMD. In 1990, our group began collecting samples from affected families1 and, in 1998, we identified the poly(A) binding protein nuclear 1 gene (PABPN1) as causative.2 Normal PABPN1 gene (NG_008239) contains 10 (GCN) repeats of a polyalanine (polyAla) tract, and mutated (expPABPN1) has 11–18.2
Our group previously observed a reduced level of histone acetylation in both mammalian and Caenorhabditis elegans OPMD models.3 We sought to test whether a Food and Drug Administration–approved compound could restore the level of histone acetylation and be protective for OPMD. Valproic acid (VPA) is known to be a direct inhibitor of histone deacetylase (HDAC) classes I and II.4,5 In order to study the effect of VPA over a long period and test whether it could protect muscle cell death at later stages of OPMD, we conducted a longitudinal study using C2C12 myoblasts that stably expressed human PABPN1 encoding a polyAla (17 Ala) expPABPN1. We observed that VPA reduced cell death in this model, and this protection appears to be mediated via an increase of histone acetylation levels. We further validated the protective effect of VPA in another OPMD model in which C elegans also expressed human PABPN1 encoding a polyAla (13 Ala). VPA was shown to effectively ameliorate the worms' motility. Our results confirm the perturbation of histone acetylation deemed to be at play in OPMD and support further testing of VPA as a therapeutic avenue for OPMD.
Methods
Plasmid constructs
The complementary DNAs encoding wild-type (wt) PABPN1 with 10 Ala and expPABPN1-17Ala (the longest Ala repeat mutation seen in patients) were cloned into the pEGFP-C2 vector to create N-terminal green fluorescent protein (GFP) fusion of PABPN1 proteins.6–8 Site mutagenesis was performed on wtPABPN1-10Ala to delete the Ala tract and create GFP-PABPN1-0Ala. The DNA sequence of every construct was verified using Sanger sequencing.
Cell culture and differentiation
C2C12 were maintained in Dulbecco's modified Eagle medium (DMEM) containing 20% fetal calf serum. When cultivated in growth media, which is DMEM containing 10% fetal bovine serum, proliferating C2C12 cells grow as mononucleated flattened cells in a monolayer. When confluent cells were incubated in differentiation DMEM media (DM), DMEM contained 2% horse serum. The majority of C2C12 cells assumed elongated morphology and fused to become multinucleated myotubes.7
Establishment of a stable C2C12 muscle cell model for OPMD
To establish stable clones, C2C12 cells were transfected with 1 μg of a plasmid (GFP-wtPABPN1-10Ala, GFP-expPABPN1-17Ala, GFP-PABPN1-0Ala, and GFP) containing a neomycin resistance marker, using Jet prime reagent (Polyplus-transfection Inc., Illkirch, France), and we named the PABPN1 cell lines as C2C12-10Ala, C2C12-17Ala, and C2C12-0Ala. Forty-eight hours after transfection, cells were transferred to media containing 0.4 mg/mL G418 (Invitrogen, Carlsbad, CA) to select for stable integration of the plasmid. After 2 weeks of G418 selection, multiple resistant colonies were isolated from each transfection. Clones that maintained stable expression of GFP fluorescence over several passages were used for further analysis. Stable cell lines were identified and confirmed by immunofluorescence, reverse transcriptase PCR, and Western blot.
Standard protocol approvals, registrations, and patient consents
We obtained ethics approval on using human samples. Informed consent was obtained from all patients.
Human lymphoblastoid cell line cultures
We used the same methodology as the one used in a previous study.7 Briefly, lymphoblastoid cell lines (LCLs) were established from peripheral blood samples. Cells were maintained in RPMI 1640 with 2 mM l-glutamine and 10% fetal bovine serum in a 37°C incubator (5% CO2). Control LCL used in this study is 34299. Patient OPMD LCLs used in this study are 34260 and 34262.
Immunocytochemistry and measurement of myogenic fusion index
The cells were incubated with antibodies directed against anti-myosin MF-20 = 1:200 (Developmental Studies Hybridoma Bank, University of Iowa) in 2% normal goat serum/phosphate buffered saline, or desmin = 1:100 (Cedarlane, Hornby, Canada). Cells were visualized using a confocal microscope (Leica TCS SP5, DMI6000, program Leica Application Suite; Leica Microsystems, Mannheim, Germany) equipped with a standard camera. The myogenic index was determined as the ratio of the nuclei number in the cells containing 2 or more nuclei to the total nuclei number in desmin-stained myotubes of C2C12 cells. The number of nuclei within each individual myotube was counted for 100 myotubes per well. At least 3 different fields were counted. Data are mean ± SE of 3 independent experiments.
VPA treatment
VPA 0.5 mM was the most appropriate dose, because it did not cause cell toxicity and therefore was used throughout the cell culture experiments. VPA was applied 2 days after growing stable cell lines in culture and continued to be administered to the cells every 48 hours. Basically, every 2 days, we carefully removed the old medium and replaced it with a fresh G418-containing medium and then added 0.5 mM VPA. Controls in which no drug was applied to cells were also included. The cells were monitored daily under the live-stage microscope for 24 consecutive days.
Western blotting
Proteins were blotted into nitrocellulose membrane and probed with specific antibodies against GFP (1:2,000) (Clontech, Mountain View, CA), PABPN1 (1:2,000) (Abcam, Cambridge, UK), desmin (1:100), and myosin (1:200). Actin was used to confirm equal loading of protein. Data are available from Dryad (Methods, doi.org/10.5061/dryad.37628q6). Quantification was performed using ImageJ.
Histone acetylation analysis
We used the same methodology as the one used in a previous study.3 Briefly, histones were extracted under acidic conditions as described.3 The histones were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis and blotted with rabbit anti-Histone H3 acetylated and H4 acetylated antibodies (1:1,000; Calbiochem, Billerica, MA), and mouse anti-Histone H1 for equal loading (1:1,000; Chemicon, Temecula, California).
Quantification of cell survival
We used an automated fluorescent microscope and fluorescence-based flow cytometry (FFC) as previously descibed7 and calculated the percentage of cell survival.
C elegans strains, maintenance, drug treatment, and measurements of motility
Standard methods for culturing and handling the worms were used.9 The strains used in this study were obtained from our collaborator Dr. Christian Neri.3 For motility of the nematode, a synchronized worm population was obtained using hypochlorite extraction. Worms were grown on solid media up to day 1 of adulthood. At day 1, 30 worms per well were placed in S basal with OP-50 Escherichia coli (optical density 0.5) in a flat-bottom 96-well plate; at least 3 wells were done per condition. PABPN1-13Ala worms treated with 0.25 mM VPA or with vehicle were let to rest in the dark for 1 hour before recording their motility. We then used the 96-well–based infrared locomotor tracking system, known as the Microtracker (Phylumtech, Santa Fe, Argentina) method to detect the motion of the worms on a continuous basis. The system detects animal movement through infrared microbeam light scattering.10 In brief, each microtiter well is crossed by at least one infrared microbeam, scanned more than 10 times per second. The detected signal is then digitally processed to register the amount of animal movement in a fixed period of time. For any plate, the system allowed us to set groups of replicate wells; in this way, the software will inform the activity of each well and the average activity of the group. At the end of each run, the system generated reports with the activity over time of each replicate group, well, and channel. The activity is informed as the accumulated movement counts of fixed periods of time.
Statistical analysis
All values represent the mean of at least 5 independent experiments (mean ± SE). The percentage of cell survival generated from the live-stage microscope and FFC as well as motility of C elegans were analyzed with single-factor analysis of variance. In every analysis, values of p < 0.05 compared with any other groups (analysis of variance) were designated as statistically significant.
Data availability
The supplementary data can be accessed via the Dryad Digital Repository (doi.org/10.5061/dryad.37628q6). The authors will share the data of this study by request from any qualified investigator. Any data not published within the article is available in a public repository and will be shared by request from any qualified investigator.
Results
Development of a stable C2C12 model
Here, we developed a stable skeletal myoblast OPMD model by expressing expPABPN1-17Ala in C2C12 murine cells. Protein expression could be assessed by immunofluorescence using a live-stage microscope (figure 1A), by Western blot (data available from Dryad: figure e-1, doi.org/10.5061/dryad.37628q6), and by reverse transcriptase PCR (data available from Dryad: figure e-2, doi.org/10.5061/dryad.37628q6).
Figure 1. Development of a stable C2C12 muscle model for OPMD.

(A) Representative images of C2C12-10Ala, C2C12-17Ala, C2C12-0Ala, and GFP. Cells were monitored using a live-stage microscope every day and up to 21 days in culture. Images were captured at the specified days: 3, 10, and 21 post culture. All cell lines were able to exhibit myotube formation at day 10. Scale bar 100 μM. (B) Muscle fusion was detected in all cells stably expressing different PABPN1 proteins including C2C12-17Ala at the earlier stages of disease progression. Fluorescent images were captured using the live-stage microscope at day 10 post culture showing that C2C12-17Ala as well as C2C12-0Ala are able to differentiate into myotubes, similar to the C2C12-10Ala. (C) VPA is protective for OPMD. C2C12-17Ala die at later stages of culture (C.a), while VPA is protective at all time points (C.b). At later stages of disease, when the cells are lost and more cell death observed (day 14, day 21), VPA continues to show a remarkable recovery of cell survival. Cells were visualized daily under the live-stage microscope for morphology and viability. Representative images were captured on day 7, day 14, and day 21 posttreatment with the drug. Phase contrast images show the viability and myotube formation after VPA treatment. GFP = green fluorescent protein; OPMD = oculopharyngeal muscular dystrophy; VPA = valproic acid.
We examined different C2C12-PABPN1 stable lines for the presence of PABPN1-positive aggregates. All PABPN1 proteins led to the formation of aggregates containing PABPN1 (within a diffuse background) at different time points (figure 1A). We observed the presence of GFP-positive aggregates in C2C12-10Ala, C2C12-17Ala, and C2C12-0Ala myoblast lines. We also observed the movement and fusion of these aggregates in real time and the division of cells containing them (data available from Dryad: figure e-3, doi.org/10.5061/dryad.37628q6).
We were interested to explore whether any of the PABPN1 proteins might interfere with the process of myogenic differentiation. The differentiation of myoblasts into myotubes was therefore monitored on a daily basis using live-stage microscopy. It is of interest that our analysis of muscle differentiation showed that none of the PABPN1 proteins blocked the differentiation of C2C12 myoblasts at an early stage of culture (figure 1A, all proteins on day 10, and figure 1B). At day 3, all cells were mononucleic (figure 1A), and by day 5, they fused to form small myotubes, which then formed large myotubes and sheets of large multinucleated myotubes by day 10 (figure 1A).
Although C2C12-17Ala myoblasts showed a pattern of differentiation that was similar to the one observed in the control C2C12 lines at days 5 to 10 (figure 1B; and data available from Dryad: figure e-4, doi.org/10.5061/dryad.37628q6), the cells did not exhibit myotubes after day 14 in culture. This is because cells began to shrink and die at later stages of culture (day 14, day 24) (figure 1A, second column; figure 1C, left panels; and data available from Dryad: figure e-5, doi.org/10.5061/dryad.37628q6).
A striking observation from our stable myoblast model of OPMD is the progressive cell death that occurs in C2C12-17Ala lines by comparison to what is seen in control. In C2C12-17Ala, the cell death started at day 10 of culture; this became hastened over time and reached its highest point after day 21 of culture (figure 1A, second column, day 21). The myotubes of C2C12-17Ala showed an increased number of apoptotic cells over time (figure 1C, left panels; and data available from Dryad: figure e-5, doi.org/10.5061/dryad.37628q6) and by comparison to control myoblasts, they also lost their GFP signals and detached from the plates before they eventually died after 2 weeks in culture (data available from Dryad: figure e-6, doi.org/10.5061/dryad.37628q6).
VPA rescues cell death in C2C12-17Ala
We have previously reported a decrease in histone acetylation level in C elegans and cell culture OPMD models.3 If OPMD pathology involves suppression of histone acetylation, then one would predict that inhibiting the histone hypoacetylation process via a pharmacologic intervention could rescue the cell death associated with it. To test this, we used VPA in our stable C2C12 model of OPMD. First, we monitored the viability of C2C12 myoblasts treated with 0.5 mM of VPA using an automated live-stage fluorescent microscope7 (figure 1C; and data available from Dryad: figure e-7, doi.org/10.5061/dryad.37628q6). Second, we validated these visual observations of cell survival using a quantitative method of FFC assay.7
C2C12-17Ala myoblasts that were not treated with VPA presented more cell death that was characterized by the presence of fragmented cells and condensed nuclei (figure 1C, left panels). C2C12-17Ala also exhibited a progressive loss of GFP signal over the course of the experiments (figure 1C, left panels). However, VPA-treated cells continued to proliferate with a growing number of viable green cells for up to 21 days posttreatment (figure 1C, right panels; and data available from Dryad: figure e-7, doi.org/10.5061/dryad.37628q6). Percentages of surviving cells at all time points showed that VPA rescued cells expressing C2C12-17Ala from cell death (p < 0.001 vs nontreated samples) (figure 2A). It is important to mention that the degree of VPA protective activity is clearly higher in C2C12-17Ala than in C2C12-10Ala (figure 2A). In addition to enhancing cell survival, VPA improved the cell morphology; a good example of this effect can be seen in VPA-treated C2C12-17Ala myoblasts at day 18 (figure 2B; and data available from Dryad: figure e-7, doi.org/10.5061/dryad.37628q6). At day 18 of VPA treatment, myoblasts were more proliferative, differentiated, and appeared healthier than nontreated myoblasts (figure 2B, bottom panel). Of interest, VPA could also protect from the cell death associated with C2C12-17Ala expression when it was introduced at late stage (18–22 days) (figure 1C; and data available from Dryad: figure e-8, doi.org/10.5061/dryad.37628q6). It is also noteworthy that myoblast cells containing GFP-positive aggregates were those predominantly surviving (data available from Dryad: figure e-8, doi.org/10.5061/dryad.37628q6).
Figure 2. VPA is protective for OPMD.

(A) Percentage of cell survival was determined by live-stage microscopy at different days. VPA-treated C2C12-17Ala shows a striking increased in the percentage of cell survival compared to nontreated C2C12-17Ala at different time points (mean ± SE, *p < 0.001 vs nontreated samples) (analysis of variance). This increase seems to be specific to C2C12-17Ala as C2C12-10Ala did not exhibit any change in cell survival after VPA treatment. After they were seeded, cells were counted every 24 hours for 21 consecutive days using the live-stage microscope. Cell survival was quantified using Volocity software (PerkinElmer, Waltham, MA). The percentage of transfected living cells represents the variation of the amount of transfected living cells at different time points compared with the number of transfected cells obtained on day 1. (B) A positive effect of VPA on the morphology of C2C12-17Ala at later stage of treatment. Cells treated with 0.5 mM VPA are more proliferative, differentiated, and look healthier than the nontreated samples. Both fluorescent and phase contrast microscopy images were captured on day 18 of treatment. (C) A 2-color FACS dotplot from a representative experiment in which the C2C12-17Ala were treated with 0.5 mM VPA compared with nontreated cells, stained with 7-AAD to label dead cells, and cosorted for GFP (green) and 7-AAD (red) fluorescence. Grid lines were positioned after calibrating the flow cytometry. Upper right quadrants signify 7-AAD–labeled dead or dying transfected cells (GFP+, 7-AAD+), i.e., Q2-2. The experiment was performed on day 18 post drug treatment and repeated 3 times. (D) VPA significantly rescues C2C12-17Ala–associated cell death as assessed by FFC. Quantification of cell death measured by FFC. Percentage of cell death on day 18 was calculated by dividing the number of 7-AAD–stained transfected GFP C2C12 cells over the total number of transfected cells. The experiment was performed on day 18 post drug treatment and repeated 3 times (*p < 0.001 versus nontreated cells). 7-AAD = 7-amino actinomycin D; FACS = fluorescence-activated cell sorting; FFC = fluorescence flow cytometry; FITC = fluorescein isothiocyanate; GFP = green fluorescent protein; OPMD = oculopharyngeal muscular dystrophy; VPA = valproic acid.
When FFC was used to assess cell survival in 0.5 mM VPA-treated myoblasts, C2C12-17Ala myoblasts were observed to survive for a longer period of time than untreated cells (figure 2, C and D) (p < 0.001 vs nontreated cells). Overall, the 2 survival approaches led to the conclusion that VPA conferred protection against the pathologic effects of polyAla repeat expansion in the OPMD stable cell model.
VPA treatment enhances myogenic differentiation in C2C12-17Ala
To monitor the effect of VPA on the myogenic differentiation of C2C12, the cells were grown in differentiating media (DM) with and without VPA. After 5 days in DM, the mononucleated myoblasts that were dividing became postmitotic and fused to form multinucleated myotubes (Figure 1A, middle panels). From this point (day 5) to day 21 in DM, it was possible to calculate the myogenic fusion index (data available from Dryad: figure e-9, doi.org/10.5061/dryad.37628q6). In the early days of differentiation, we observed that C2C12-17Ala myoblasts exhibited a similarly positive muscle differentiation index whether or not their DM contained VPA (figure 3A). However, after 15 days in DM, the fusion index appeared to be sharply decreased in C2C12-17Ala myoblasts (figure 3A), and after 18 days, the myoblasts appeared to stop fusing. In contrast, C2C12-17Ala maintained in DM with VPA at day 18 showed a remarkably higher fusion index (p < 0.001 vs nontreated cells) (figure 3A; and data available from Dryad: figure e-10, doi.org/10.5061/dryad.37628q6). Even at later days, VPA stimulated the ability of C2C12-17Ala myoblasts to fuse into multinucleated myotubes (figure 3A) (p < 0.001 vs nontreated cells). It is important to mention that the degree of VPA differentiation in C2C12-17Ala is clearly higher than in C2C12-10Ala (figure 3A).
Figure 3. VPA increases muscle differentiation of C2C12-17Ala.

(A) VPA increases the differentiation index of C2C12-17Ala. This increase seems to be specific to C2C12-17Ala as C2C12-10Ala did not exhibit any change in differentiation after VPA treatment. The histogram represents the fusion index calculated at the indicated days after the addition of differentiation medium to cells that have been treated with 0.5 mM VPA. Myogenic index was determined as the ratio of the nuclei number of cells containing 2 or more nucleic to the total nuclei number of desmin C2C12-stained myotubes. At least 3 different fields containing 100 myotubes cells were counted. Data are the mean ± SE of 3 independent experiments. *p < 0.001 vs nontreated samples (analysis of variance). (B) Confocal images of immunocytochemistry detection made at day 14 showing an increased number of cells staining desmin after VPA treatment. Desmin-positive cells (red signal) in C2C12-17Ala (green signal) treated with VPA were higher than nontreated cells. (C) Western blots showing the effect of VPA treatment at day 14 on the myosin’s level. VPA increases the level of myosin protein in cells treated with VPA. Actin antibody was used to confirm equal loading. Graph shows quantification of the ratio of myosin to β-actin. Bars represent mean ± SE obtained from 3 independent experiments. VPA = valproic acid.
The differentiated nature of these cells was confirmed by performing immunodetection for 2 myogenic markers (myosin and desmin) (figure 3B; and data available from Dryad: figure e-11, doi.org/10.5061/dryad.37628q6). In C2C12-17Ala myoblasts maintained in DM with VPA, the myosin- and desmin-positive myoblast population exceeded 80% (figure 3B; and data available from Dryad: figure e-11, doi.org/10.5061/dryad.37628q6). Western blot immunodetections (figure 3C; and data available from Dryad: figure e-12, doi.org/10.5061/dryad.37628q6) confirmed the increased expression of both myosin and desmin in VPA-treated C2C12-17Ala. However, this increase was not obvious in VPA-treated C2C12-10Ala. Altogether, it appears that VPA can improve the differentiation index of C2C12-17Ala myoblasts at the later stage of our assay and this likely occurs through an increased expression of muscle-specific proteins (data available from Dryad: figure e-13, doi.org/10.5061/dryad.37628q6).
VPA restores the histone acetylation level in C2C12-17Ala and in LCLs from patients with OPMD
First, we analyzed acetylation levels of histones H3 in C2C12 cells transiently transfected with PABPN1 constructs and confirmed that the expression of expPABPN1-17Ala reduced acetylation of H3, compared with wtPABPN1-10Ala (figure 4A).3 When the C2C12-17Ala myoblasts were treated with VPA, the level of H3 acetylation was clearly increased (figure 4B). No change in PABPN1 protein level was detected in cells treated with VPA (figure 4C), indicating that the increased cell survival is attributable to increase of histone acetylation and not to a reduced expression of the PABPN1 transgenes.
Figure 4. VPA reverses the histone acetylation deficit in a stable muscle cell model of OPMD and in the OPMD patient LCLs.

(A) Western blot shows a clear reduction of the histone acetylation level in cells transfected with GFP-expPABPN1-17Ala compared to the control samples. H1 was used as loading control for total histones. (B) VPA increases the histone level in a stable C2C12 model of OPMD. Representative images of Western blot results showing the increased level of H3 acetylation after 0.5 mM VPA treatment in C2C12-17Ala. VPA treatment led to increased level of histone acetylation in C2C12-17Ala, while leaving the other samples unaffected. Histones were extracted on day 5 posttreatment. H1 was used as loading control for total histones. (C) Western blot, using PABPN1 antibody, showing no change in PABPN1 protein level in cells treated with VPA. (D) OPMD LCLs are responsive to VPA treatment. Western blot analysis of histones extracts from OPMD LCLs, untreated or VPA-treated for 72 hours (0.5 mM), showing an increase in histone acetylation using acetylated-H3 antibody. H1 was used as loading control for total histones. expPABPN1 = expanded PABPN1; GFP = green fluorescent protein; LCL = lymphoblastoid cell line; OPMD = oculopharyngeal muscular dystrophy; VPA = valproic acid; wtPABPN1-10Ala = wild-type PABPN1-10Ala.
We next sought to determine the effect of VPA on LCLs from patients with OPMD. One advantage of patient LCLs is that expression of expPABPN1 is likely to be closer to the endogenous levels seen in patients with OPMD. As shown in figure 4D, VPA treatment led to an increase in the level of histone acetylation in OPMD LCLs when compared to nontreated samples. Therefore, OPMD LCLs appear to respond to the VPA treatment (figure 4D).
VPA improves the motility of worms expressing a human 13 Ala expPABPN1
Finally, we sought to test the effect of VPA in an in vivo model of OPMD. We therefore tested VPA in our established C elegans model of OPMD that we previously used.3 The OPMD phenotype of these transgenic nematodes includes adult-stage abnormalities, notably reduced motility and muscle degeneration.3 These phenotypes were only observed in transgenic C elegans strains that expressed the human 13 Ala expPABPN1 protein and not in strains that expressed normal PABPN1 (10 Ala) expression.3 We applied VPA and observed that it has a striking effect (p < 0.05) on suppressing the paralysis caused by PABPN1-13Ala at a concentration of 0.25 mM (figure 5). As detected with an automated motility tracking system, VPA-treated PABPN-13Ala worms showed a strikingly improved motility compared to controls throughout all time points (figure 5). Altogether, this suggests that VPA conferred protection in both in vivo and in vitro OPMD models.
Figure 5. VPA improves the motility of Caenorhabditis elegans transgenics expressing human PABPN1 with 13 Ala.
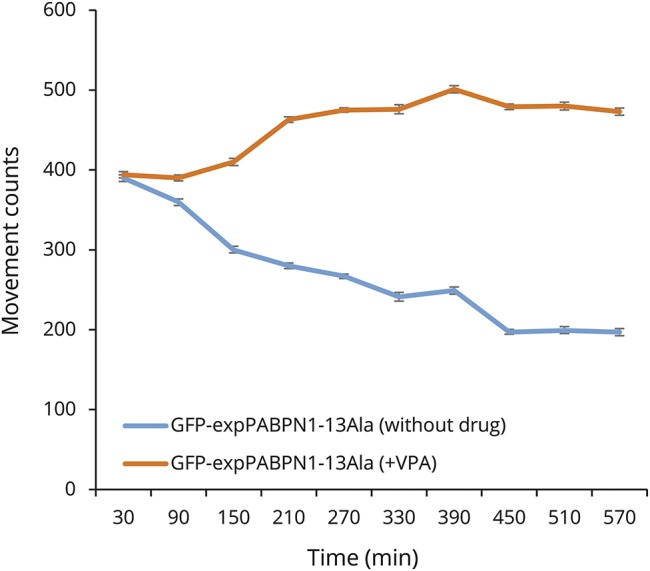
A striking improvement of motility of VPA-treated PABPN1-13 Ala worms. Abnormal motility induced by mutant PABPN1 in C elegans transgenics expressing human PABPN1 with 13 Ala was significantly reduced after VPA treatment starting at 90 minutes post recording. Animals grown on media supplemented with 0.25 mM VPA have a faster motility than the ones grown on normal media (no drug) (*p < 0.0001). Motility recordings were generated using a Microtracker device, an automated tracking system that detects the animal movement through infrared microbeam light scattering. Simply, each microtiter well is crossed by at least one infrared microbeam, scanned more than 10 times per second. The detected signal is then digitally processed to register the amount of animal movement in a fixed period of time. At the indicated time points, the number of movements was scored at 22°C, for 10 consecutive hours. expPABPN1 = expanded PABPN1; GFP = green fluorescent protein; VPA = valproic acid.
Discussion
Our stable C2C12 model recapitulates the major pathogenic events of OPMD, including the formation of protein aggregation and muscle cell death. By opposition to cells transiently expressing the expanded form of PABPN1, myoblast lines stably expressed the GFP-expPABPN1-17Ala fusion proteins in a more uniform manner and, as such, they present a more reliable model to allow observation over time.
Our observations on PABPN1 aggregates in living cells confirm our previous results of these structures to be dynamic as the cells containing them are able to divide and their aggregates do reappear in daughter cells.8,11,12 The presence of GFP-positive aggregates in C2C12-10Ala, C2C12-17Ala, and C2C12-0Ala myoblast lines reinforces the notion that the sole presence of aggregates containing polyAla expPABPN1 is not sufficient to induce cell death. The role of aggregates in OPMD is being vigorously disputed, as strategies targeting protein aggregates (e.g., doxycycline and HSP70) diminish their formation and cell death in parallel.6,13 Such effects on cell survival may be explained by the role of these strategies in preventing apoptosis.14 Toxicity may not result from the aggregates, but rather from the soluble form of expPABPN1.8 Nuclear aggregates that have proven to be protective delay cell death most likely by depleting the misfolded expPABPN1 from the soluble pool.8 However, aggregate formation may prove to be insufficient to halt the disease, as the accumulation of the soluble toxic form of expPABPN1 overwhelms the cellular defense mechanisms (proteasome/ubiquitin and heat shock response) with aging leading eventually to muscle cell degeneration. The soluble expPABPN1 may interfere with the RNA processing proteins15 and impede their functions. It has been reported that normal PABPN1 interacts with RNA polymerase II during transcription.16 Also, normal PABPN1 binds to the histone deacetylase component SKIP (SKI-interacting protein) and MyoD to regulate muscle cell differentiation.17 Inappropriate interactions between the soluble form of expPABPN1 and components of transcription complex16,17/the histone acetylation17,18 machinery may compromise the expression of genes essential for muscle cell survival, proliferation, and differentiation and lead to toxicity.
The fact that PABPN1-0Ala did not prevent the differentiation of C2C12 myoblasts into myotubes suggests that the Ala tract of PABPN1 is not required for muscle cell differentiation. The observed impairment of differentiation in C2C12-17Ala myoblasts suggests that polyAla expansion of PABPN1 would not impair myogenesis early in the disease process but might rather interfere with this process at later stages. One could hypothesize that polyAla expansion of PABPN1 begins to further reduce the fusion of myoblasts into myotubes at this point in the life of a patient with OPMD. In agreement with our results, an earlier study of cricopharyngeal muscles obtained from patients with OPMD has revealed a reduced myogenicity, as well as a rapid decrease in the proliferation of myoblasts.19
The VPA effect is specific for OPMD pathology as the cell survival and differentiation were significantly enhanced in C2C12-17Ala compared to C2C12-10Ala. Hence, we could conclude that the VPA effect is specific for OPMD pathology. Our current study showed that both the histone acetylation disturbance and decreased cell viability associated with the expression of mutant PABPN1 was mitigated by the addition of VPA at a concentration of 0.25–0.5 mM, well within the therapeutic range. In a normal cell, protein acetylation reflects a balance between the rate of acetylation by coactivators or other histone acetyltransferases (HATs) and the rate of histone deacetylation by HDACs.20 It is possible that the HAT activity of specific proteins is reduced (or the activity of HDAC proteins is increased) as they become entrapped in the protein aggregates containing polyAla expPABPN.18 Alternatively, interactions between the soluble polyAla expPABPN1 and these proteins could directly affect their HAT/HDAC enzymatic activities. It is intriguing that our previous study on a transcriptomic analysis derived from a mouse model of OPMD has revealed a list of genes related to progressive muscle atrophy, 4 of which (F-box protein 32, ATF2, BAZ1A, and HDAC5) are known to be implicated in the HAT/HDAC pathway and interact with the myocyte enhancer factor-2.21 Modulators of HAT and HDAC have been shown to have important roles in the myogenesis process.22 Deacetylase inhibitors were shown to increase muscle cell size by promoting myoblast recruitment and fusion through induction of follistatin.22 HDAC members are found in different regulatory complexes and were previously reported to associate with muscle regulatory factors in myoblasts.23,24
In a study conducted on mice with spinal muscular atrophy (SMA), the muscle-specific E3 ligases atrogin-1 and MuRF1 were shown to be upregulated.25 The study provided evidence that the HDAC inhibitor trichostatin A mitigates SMA disease manifestations by inhibition of atrogene upregulation.25 It is also noteworthy that in a previously reported OPMD mouse model, an increased expression of MuRF-1 was on the top list of dysregulated genes.21 It is important to mention that SMA is a motor neuron disorder while OPMD is a primary muscle disease. HDAC inhibitors might work on different mechanisms in these diseases. Trichostatin A, unlike VPA, works through a myogenin-dependent pathway in SMA by modulating the upstream regulators of myogenin expression, HDAC4, and Dach2.25 Of note, this treatment has no effect when atrogene induction occurs independently of myogenin.25
A 2006 study demonstrated that HDAC inhibitors enhanced skeletal myogenesis and promoted muscle regeneration.22 The same report also found that HDAC inhibitors increased muscle cell size by promoting myoblast recruitment and fusion through induction of follistatin.22 Prior to that report, an independent study had shown that treating dystrophic mouse muscle with HDAC inhibitors led to both a functional and morphologic recovery.26 Moreover, VPA was shown to improve pathologic endpoints in a Duchenne muscular dystrophy mouse model through inducing muscle hypertrophy and inhibiting apoptosis in myotubes through an activation of the PI3K/Akt/mTOR pathway.27
Our results indicate that VPA (a Food and Drug Administration–approved compound) may be worthy of further testing in a clinical trial of patients with OPMD.
Acknowledgment
The authors thank the patients for providing blood samples. They also thank Dr. Helene Catoire and Dr. Martine Therrien for their technical support on C elegans.
Glossary
- DM
differentiation medium
- DMEM
Dulbecco's modified Eagle medium
- expPABPN1
expanded PABPN1
- FFC
fluorescence flow cytometry
- GFP
green fluorescent protein
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- LCL
lymphoblastoid cell line
- OPMD
oculopharyngeal muscular dystrophy
- polyAla
polyalanine
- RPMI
Roswell Park Memorial Institute medium
- SMA
spinal muscular atrophy
- VPA
valproic acid
- wt
wild-type
Author contributions
Aida Abu-Baker conceived, designed the study and performed all experiments and analyzed the data. Alex Parker helped in C elegans work and revised the manuscript. Siriram Ramalingam genotyped the C elegans worms and confirmed Western blot of lymphoblastoid cell lines derived from patients with OPMD. Janet Laganiere helped in microscopy and immunocytochemistry experiments. Bernard Brais provided all PABPN1 complementary DNAs. Christian Neri provided the C elegans strains of OPMD. Patrick Dion revised the first and final draft of the manuscript. Guy Rouleau supervised the study and revised the manuscript.
Study funding
Supported by the Muscular Dystrophy Association, the Canadian Institutes of Health Research, and the Federation Foundation of Greater Philadelphia.
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Brais B, Rouleau GA, Bouchard JP, Fardeau M, Tome FM. Oculopharyngeal muscular dystrophy. Semin Neurol 1999;19:59–66. [DOI] [PubMed] [Google Scholar]
- 2.Brais B, Xie YG, Sanson M, et al. The oculopharyngeal muscular dystrophy locus maps to the region of the cardiac alpha and beta myosin heavy chain genes on chromosome 14q11.2-q13. Hum Mol Genet 1995;4:429–434. [DOI] [PubMed] [Google Scholar]
- 3.Catoire H, Pasco MY, Abu-Baker A, et al. Sirtuin inhibition protects from the polyalanine muscular dystrophy protein PABPN1. Hum Mol Genet 2008;17:2108–2117. [DOI] [PubMed] [Google Scholar]
- 4.Gottlicher M, Minucci S, Zhu P, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J 2001;20:6969–6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kramer OH, Zhu P, Ostendorff HP, et al. The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J 2003;22:3411–3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abu-Baker A, Messaed C, Laganiere J, Gaspar C, Brais B, Rouleau GA. Involvement of the ubiquitin-proteasome pathway and molecular chaperones in oculopharyngeal muscular dystrophy. Hum Mol Genet 2003;12:2609–2623. [DOI] [PubMed] [Google Scholar]
- 7.Abu-Baker A, Laganiere J, Gaudet R, et al. Lithium chloride attenuates cell death in oculopharyngeal muscular dystrophy by perturbing Wnt/β-catenin pathway. Cell Death Dis 2013;4:e821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Messaed C, Dion PA, Abu-Baker A, et al. Soluble expanded PABPN1 promotes cell death in oculopharyngeal muscular dystrophy. Neurobiol Dis 2007;26:546–557. [DOI] [PubMed] [Google Scholar]
- 9.Stiernagle T. Maintenance of C. elegans. In: WormBook: The Online Review of C elegans Biology. The C. elegans Research Community, WormBook; 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simonetta SH, Golombek DA. An automated tracking system for Caenorhabditis elegans locomotor behavior and circadian studies application. J Neurosci Methods 2007;161:273–280. [DOI] [PubMed] [Google Scholar]
- 11.Tavanez JP, Calado P, Braga J, Lafarga M, Carmo-Fonseca M. In vivo aggregation properties of the nuclear poly(A)-binding protein PABPN1. RNA 2005;11:752–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marie-Josée Sasseville A, Caron AW, Bourget L, et al. The dynamism of PABPN1 nuclear inclusions during the cell cycle. Neurobiol Dis 2006;23:621–629. [DOI] [PubMed] [Google Scholar]
- 13.Davies JE, Wang L, Garcia-Oroz L, et al. Doxycycline attenuates and delays toxicity of the oculopharyngeal muscular dystrophy mutation in transgenic mice. Nat Med 2005;11:672–677. [DOI] [PubMed] [Google Scholar]
- 14.Yew EH, Cheung NS, Choy MS, et al. Proteasome inhibition by lactacystin in primary neuronal cells induces both potentially neuroprotective and pro-apoptotic transcriptional responses: a microarray analysis. J Neurochem 2005;94:943–956. [DOI] [PubMed] [Google Scholar]
- 15.Fan X, Messaed C, Dion P, et al. HnRNP A1 and A/B interaction with PABPN1 in oculopharyngeal muscular dystrophy. Can J Neurol Sci 2003;30:244–251. [DOI] [PubMed] [Google Scholar]
- 16.Bear DG, Fomproix N, Soop T, Bjorkroth B, Masich S, Daneholt B. Nuclear poly(A)-binding protein PABPN1 is associated with RNA polymerase II during transcription and accompanies the released transcript to the nuclear pore. Exp Cell Res 2003;286:332–344. [DOI] [PubMed] [Google Scholar]
- 17.Kim YJ, Noguchi S, Hayashi YK, Tsukahara T, Shimizu T, Arahata K. The product of an oculopharyngeal muscular dystrophy gene, poly(A)-binding protein 2, interacts with SKIP and stimulates muscle-specific gene expression. Hum Mol Genet 2001;10:1129–1139. [DOI] [PubMed] [Google Scholar]
- 18.Abu-Baker A, Rouleau GA. Oculopharyngeal muscular dystrophy: recent advances in the understanding of the molecular pathogenic mechanisms and treatment strategies. Biochim Biophys Acta 2007;1772:173–185. [DOI] [PubMed] [Google Scholar]
- 19.Perie S, Trollet C, Mouly V, et al. Autologous myoblast transplantation for oculopharyngeal muscular dystrophy: a phase I/IIa clinical study. Mol Ther 2014;22:219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strahl BD, Allis CD. The language of covalent histone modifications. Nature 2000;403:41–45. [DOI] [PubMed] [Google Scholar]
- 21.Trollet C, Anvar SY, Venema A, et al. Molecular and phenotypic characterization of a mouse model of oculopharyngeal muscular dystrophy reveals severe muscular atrophy restricted to fast glycolytic fibres. Hum Mol Genet 2010;19:2191–2207. [DOI] [PubMed] [Google Scholar]
- 22.Iezzi S, Di Padova M, Serra C, et al. Deacetylase inhibitors increase muscle cell size by promoting myoblast recruitment and fusion through induction of follistatin. Dev Cell 2004;6:673–684. [DOI] [PubMed] [Google Scholar]
- 23.McKinsey TA, Zhang CL, Olson EN. Control of muscle development by dueling HATs and HDACs. Curr Opin Genet Dev 2001;11:497–504. [DOI] [PubMed] [Google Scholar]
- 24.Puri PL, Iezzi S, Stiegler P, et al. Class I histone deacetylases sequentially interact with MyoD and pRb during skeletal myogenesis. Mol Cell 2001;8:885–897. [DOI] [PubMed] [Google Scholar]
- 25.Bricceno KV, Sampognaro PJ, Van Meerbeke JP, Sumner CJ, Fischbeck KH, Burnett BG. Histone deacetylase inhibition suppresses myogenin-dependent atrogene activation in spinal muscular atrophy mice. Hum Mol Genet 2012;21:4448–4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Minetti GC, Colussi C, Adami R, et al. Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat Med 2006;12:1147–1150. [DOI] [PubMed] [Google Scholar]
- 27.Gurpur PB, Liu J, Burkin DJ, Kaufman SJ. Valproic acid activates the PI3K/Akt/mTOR pathway in muscle and ameliorates pathology in a mouse model of Duchenne muscular dystrophy. Am J Pathol 2009;174:999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The supplementary data can be accessed via the Dryad Digital Repository (doi.org/10.5061/dryad.37628q6). The authors will share the data of this study by request from any qualified investigator. Any data not published within the article is available in a public repository and will be shared by request from any qualified investigator.