

Abstract
Chromosome translocations can be detected by cytogenetic analysis, but it is hard to characterize the breakpoints at the sequence level. Chromosome sorting by flow cytometry produces flow karyotypes that enable the isolation of abnormal chromosomes and the generation of chromosome-specific DNA. In this study, a derivative chromosome t(9; 14) and its homologous normal chromosomes 9 and 14 from the Ishikawa 3-H-12 cell line were sorted to collect homologue-specific samples. Chromosome sequencing identified the breakpoint junction in the der(9) at 9p24.3 and 14q13.1 and uncovered the formation of a fusion gene, WASH1–NPAS3. Amplicon sequencing targeted for neighbouring genes at the fusion breakpoint revealed that the variant frequencies correlate with the allelic copy number. Sequencing of sorted chromosomes permits the assignment of allelic variants and can lead to the characterization of abnormal chromosomes. We show that allele-specific chromosome sequencing of homologues is a robust technique for distinguishing alleles and this provides an efficient approach for the comprehensive analysis of genomic changes.
Keywords: chromosome sorting, allelic profile, fusion gene, breakpoint junctions, variant frequency
1. Introduction
Next-generation sequencing provides whole genome analysis of structural variants as well as sequence variants.1,2 The technique has made a significant contribution to cancer genomics and to the analysis of human cancer cell lines that have been accumulated in the Cancer Cell Line Encyclopedia3 and the Catalogue of Somatic Mutations in Cancer.4 It is noted that clinical tumour samples are often contaminated with normal cells and show heterogeneity due to clonal evolution5,6 which is also observed in tumour cell lines.7 These two features present problems in sequencing tumour samples. Although recent advances in single cell analysis can distinguish differences between cells and help to uncover genetic heterogeneity in tumour cells,8 the investigation of allelic differences remains challenging.
Chromosomes can be distinguished in fluid suspensions using a bivariate fluorescence-activated cell sorter which separates them by size and base pair ratio and displays the results in the form of a flow karyotype,9 allowing accurate estimation of chromosome size and GC content.10,11 The procedure enables the isolation and collection not only of aberrant chromosomes but sometimes also of normal homologues that contain measurable differences in repetitive DNA.12 Analysis of sorted chromosomes can characterize cryptic allelic variants occurring in a reciprocal chromosome translocation.13 Sorted chromosome samples have been extensively exploited in diagnostic cytogenetics and in the production of chromosome-specific DNA for chromosome painting14,15 and chromosome sequencing.16,17 The latter serves to focus on regions of interest and has application in refining genome analysis and assisting genome assembly.
In this study, chromosome-specific sequencing reveals the breakpoint junction and identifies a novel fusion gene in a translocation between chromosomes 9 and 14 observed in a human cancer cell line. The sequence analysis of homologous regions on chromosomes separated by flow sorting has enabled alleles at several loci to be distinguished by their nucleotide differences.
2. Materials and methods
2.1. Cell lines and DNA preparation
The Ishikawa 3-H-12 cell line registered as JCRB1505 was established from an endometrial cancer.18 Cells were cultured using MEM with 15% non-heat-inactivated foetal bovine serum. Genomic DNA was extracted using the AllPrep DNA/RNA Mini Kit (Qiagen). The karyotypes and SNP array profiles are available from our previous study (Supplementary Fig. S1A).7 Because t(9; 14) was detected in all the metaphases examined and therefore expected to be a reference marker for this cell line, this study was focused on der(9).
2.2. Chromosome preparation and staining
Mitotic cells were collected by shake-off from three 175-cm2 flasks at passage 25* after colcemid treatment (KaryoMAX Colcemid, Gibco) at a final concentration of 0.1 μg/ml for 5 h. Cells were centrifuged at 250 g for 8 min in 50 ml tubes. The supernatant was discarded and replaced by 10 ml of hypotonic solution (75 mM KCl, 0.2 mM spermine, 0.5 mM spermidine, 10 mM MgSO4·7H2O, pH 8.0) and incubated at RT for 15 min. After centrifugation, the cell pellet was resuspended in 1 ml of ice-cold polyamine buffer (2 mM EDTA, 0.5 mM EGTA, 80 mM KCl, 0.25% Triton X-100, 0.2 mM spermine, 0.5 mM spermidine, 20 mM NaCl, pH 7.2) and incubated on ice for 10 min. The suspension was then vortexed vigorously for 12 s and the chromosomes were stained by 5 μg/ml of Hoechst 33258 (Sigma-Aldrich, B2883), 40 μg/ml of Chromomycin A3 (Sigma-Aldrich, C2659), and 10 mM MgSO4. 10 mM sodium citrate and 25 mM sodium sulphite were added 1 h before flow sorting.
2.3. Chromosome sorting
Flow cytometric analysis was carried out at the Cambridge Resource Centre for Comparative Genomics. Stained chromosome suspensions were analysed on a flow cytometer (MoFlo, Beckman Coulter, Fullerton, CA, USA) equipped with two water-cooled lasers (Spectra-Physics Model 2020) spatially separated at the flow chamber. The first laser was tuned to emit multiline UV (330–360 nm) which excites Hoechst 33258. The second laser was tuned to emit light at 457 nm to excite Chromomycin A3. The power of both lasers was set to 300 mW and kept constant using light control feedback. Fluorescence emitted from Hoechst 33258 was collected using a 400 nm long pass filter combined with a 480 nm short pass filter. Chromomycin A3 fluorescence was collected using a 490 nm long pass filter. The instrument was configured for two-way sorting with high speed sorting at a data rate of 8,000–12,000 events per second with the optimal setting of the sheath pressure to ∼ 60 psi and the drop drive frequency to ∼ 95 kHz using a 70 μm cytonozzle tip. The flow karyotype was displayed by Hoechst 33258 versus Chromomycin A3 fluorescence and acquired by a total of 50,000–100,000 events at a rate of 1,000 events per second. Data collected from the experiments were analysed using Summit Software Version 4.1 (Beckman Coulter). Approximately 2,000 chromosomes were flow sorted into a 0.5-ml UV-treated PCR tube from each chromosome peak.
2.4. Chromosome sequencing
For chromosome-specific sequencing, chromosomes were amplified by the REPLI-g single cell kit (Qiagen) and a genomic DNA fragment library was prepared using the Ion Xpress Plus Fragment Library kit (Life Technologies). Emulsion PCR of the library was performed by the Ion One Touch 2 system using the Ion PGM Template OT2 400 kit (Life Technologies). Sequencing was carried out by the Ion PGM sequencer using the Ion PGM Sequencing 400 kit and the Ion 318 Chip (Life Technologies). All protocols followed the manufacturer’s instructions. Alignment of sequencing reads with the human genome reference (GRCh37/hg19) was performed using the Torrent Suite v4.1.2 software. The bam file was mapped to the hg19 reference using the TMAP software v5.0.13 with standard options (Supplementary Table S1). This was visualized in the Integrative Genomics Viewer (IGV) using the mapping quality value as 1. BLAT search in the UCSC Genome tools was used to find similarity of sequence reads in the genome database.
2.5. Fusion point analysis
The breakpoint junction was amplified by PCR at an annealing temperature of 65°C using the following primer pairs designed using the Primer3 software; der9-BPf: GGAAGGCTCAGACAGAGAGG and der9-BPr: CTCTCCCTTGCACTCAGCTC. Total RNA was prepared using the AllPrep DNA/RNA Mini Kit (Qiagen) and treated with DNaseI. Reverse transcription of RNA into cDNA was performed using the PrimeScript RT-PCR Kit (Takara) with random hexamers. Amplification of the WASH1–NPAS3 fusion transcript was performed with annealing temperature at 63°C using the PCR primer pair of WASH1f: GGAAGGCTCAGACAGAGAGG and NPAS3r: AGCATCTCGGGATTTCTCCT.
2.6. Variant analysis
Multiplex primer pools for eight genes at 9p24.3, 10 genes at 14q13 and the breakpoint boundary region at 14q13.1 were obtained using the Ion AmpliSeq Designer and are listed in Supplementary Table S2. The Ampliseq custom panels for 9p24.3 and 14q13 consist of 118 and 120 amplicons, covering 33,607 and 33,275 bp in total size, respectively. Sequence library and template preparations were performed using the Ion AmpliSeq Kit for Chef DL8 and Ion PGM Hi-Q Chef Kit (Life Technologies), respectively. Sequencing was run on the Ion PGM using the Ion 316 chip. The analysis was carried out using the Variant Caller plug-in in the Torrent Suit and the parameters are given in Supplementary Table S3.
3. Results
3.1. Flow karyotype
The flow karyotype of the Ishikawa 3-H-12 cell line at passage 25* is shown in Fig. 1, indicating the peaks resulting from chromosome 9, one of two chromosomes 14 (14S) and der(9). Based on array data, the chromosome size of der(9) is estimated to be ∼ 215 Mb. It is contained in an abnormal peak, close to the normal chromosome 3 peak, and weaker than the other normal peaks because it contains only half the number of chromosomes, i.e. only the derivative chromosome rather than the pairs of homologues present in other peaks. Chromosome 14 is heteromorphic and this allows the separation of one of the two chromosome 14 homologues by sorting. In this study, the sorted chromosome 14 is referred to as 14S, and the other unsorted homologue is referred to as 14U.
Figure 1.

Flow karyotype of the Ishikawa 3-H-12 cell line at P25* without removing the background manually by image processing was obtained after sorting a larger number of chromosomes than usual to show clearly the der(9) peak close to the normal chromosome 3 peak. The der(9) peak is weaker than other peaks that have pairs of normal chromosomes as it exists in only one copy in each cell. Normal chromosome 9 and one of the chromosome 14 homologues, 14S, are also selected for sorting.
3.2. Sequence analysis of der(9) and characterization of the fusion breakpoint
In order to identify the genomic breakpoint junction in der(9), two sequence libraries were prepared from amplification of the sorted der(9) chromosomes. The sequence data from these two runs show 1.7 and 1.0Gb consisting of 6.4 and 4.0 M reads with the read length modes of 372 and 416 bp, respectively (Supplementary Table S4). Coverage analysis based on the human reference genome revealed that most sequence reads map to chromosomes 9 and 14, with some on chromosome 3 (Fig. 2) corresponding to a small amount of contamination during chromosome sorting due to the close proximity of the der(9) and chromosome 3 peaks. Although a small number of reads were not aligned to the reference, more than 99% of the reads were aligned with 95 or 96% identities of base pairs (Supplementary Table S4). This indicates that some reads are partially aligned with soft-clipped sequences, corresponding to breakpoint regions. Mapping data were displayed in the IGV software focusing on the distal end of the 9p region (Supplementary Fig. S2). Three reads measuring 242, 356 and 400 bp including unmapped sequences prove to be fragments that cover the fusion point. These three sequences were subjected to a BLAT search, which show homologies to both chromosomes 9 and 14 (Supplementary Fig. S2). The fragments localize to the second introns of both the WASH1 (9p24.3) and NPAS3 (14q13.1) genes (Fig. 3A). Absence of sequence homology between chromosomes 9 and 14 at the breakpoints indicates non-homologous end joining. The junction was amplified by PCR on genomic DNA using primers from chromosomes 9 and 14. A fusion transcript is formed as expected based on the orientation of two genes. RT-PCR followed by sequencing confirms the in-frame fusion of WASH1 exon 2 and NPAS3 exon 3 (Fig. 3B). WASH1 is located at the distal end of the short arm of chromosome 9, ∼ 22 kb from the telomere according to the genome database. As the SNP microarray reported in our previous study7 does not have markers located at the distal end, this rearrangement could not be characterized. The chromosome sequence data and the breakpoint sequence are deposited in DRA/DDBJ (accession number: DRA006485 and LC331302).
Figure 2.

Reference coverage charts from chromosome sequencing of der(9) are shown based on average base read depth. (A) Coverage on each chromosome indicates its contents aligned to chromosomes 9 and 14, with some sequences mapped to chromosome 3 because of contamination from the closely located peak in the flow karyotype. (B) Sequence reads are detected along chromosome 9, with a large gap at the region of the centromere and pericentromeric heterochromatin. (C) The majority of reads appear in the 14q13-qter region, which indicates the breakpoint at 14q13.1 shown by an arrowhead.
Figure 3.

(A) Breakpoint junction sequence of the der(9) chromosome. Upper, middle and lower sequences indicate reference sequence of chromosome 14, derivative chromosome and reference sequence of chromosome 9, respectively. Breakpoint positions are marked with arrowheads. Numbers are based on the nucleotide position in the UCSC genome browser (GRCh37/hg19). Cen, centromere; tel, telomere. (B) The nucleotide and amino acid sequences of the fusion gene. The chimeric WASH1–NPAS3 gene shows in-frame fusion of exon 2 of WASH1 to exon 3 of NPAS3.
3.3. Variant profiles in the neighbourhood genes of the breakpoint
Amplicon sequencing on total genomic DNA reveals that 117 and 118 target regions in 9p24 and 14q13, respectively, close to the breakpoint are amplified with at least 200 reads. As three amplicons in the 14q13 region have less than 30 reads, these targets are excluded from the data analysis. The average number of reads per amplicon is calculated to be 2,198 and 1,982 for 9p24 and 14q13 regions, respectively.
Nucleotide variants in the total genomic DNA sample are detected at 37 positions in the 9p24.3 region, but five positions were of low quality. The remaining 32 positions which did not include missense mutations serve as informative markers for distinguishing between alleles at variant loci. According to their frequency in the sequence reads, each variant allele can be classified into two types, around 50 or nearly 100% of reads. This suggests that two alleles are present in this region, supported by our previous data based on the SNP microarray and M-FISH which showed two chromosome copies (Supplementary Fig. S3A).7 Of the 32 sequence variants, 16 were detected at 100% frequency, reflecting their presence in both chromosomes (Table 1). The other 16 variants with a frequency of around 50% indicate their presence in one of the two chromosomes, but these could not be localized to a chromosome in the genomic DNA sample. Chromosome-specific sequencing on der(9) and normal chromosome 9 reveal that allelic variants were detected at 23 and 25 positions, respectively, all of which showed nearly 100% frequency. This demonstrates that seven and nine variants are specific for der(9) and the normal chromosome 9, respectively, i.e., one allele at each locus. Because the other nine positions are not amplified in sorted chromosome samples, the alleles at these positions could not be distinguished.
Table 1.
Variant frequency for the 9p24.3 region
| Gene ID | Position | Ref | Variant | Total | der(9) | chr 9 |
|---|---|---|---|---|---|---|
| WASH1 | 14665 | G | A | 61.2 | 100 | |
| FOXD4 | 116800 | C | G | 48.0 | 100 | |
| FOXD4 | 116832 | T | C | 47.5 | 100 | |
| FOXD4 | 118173 | C | G | 100 | 100 | 100 |
| CBWD1 | 154795 | T | C | 49.9 | 96.1 | |
| DOCK8 | 286593 | C | A | 49.9 | 99.3 | |
| DOCK8 | 327938 | G | C | 53.6 | 100 | |
| DOCK8 | 327957 | T | G | 53.4 | 100 | |
| DOCK8 | 328006 | T | C | 53.3 | 99.1 | |
| DOCK8 | 334524 | G | A | 42.6 | 100 | |
| DOCK8 | 417990 | T | C | 100 | 100 | 98.9 |
| DOCK8 | 418003 | T | C | 100 | 100 | 100 |
| DOCK8 | 422157 | T | C | 100 | 100 | 100 |
| DOCK8 | 432330 | C | G | 100 | 100 | 100 |
| DOCK8 | 433978 | A | G | 100 | 100 | 100 |
| DOCK8 | 439453 | G | C | 100 | 100 | 100 |
| DOCK8 | 439467 | A | C | 100 | 100 | 100 |
| DOCK8 | 441827 | C | T | 100 | 100 | 100 |
| DOCK8 | 441952 | G | A | 100 | 100 | 100 |
| DOCK8 | 442030 | C | T | 100 | 100 | 100 |
| DOCK8 | 446352 | G | C | 100 | 100 | 100 |
| DOCK8 | 449700 | G | T | 100 | 100 | 100 |
| KANK1 | 730054 | A | G | 47.7 | 100 | |
| KANK1 | 738434 | C | T | 55.1 | 100 | |
| KANK1 | 740901 | C | T | 51.3 | 100 | |
| KANK1 | 741084 | C | T | 52.2 | 100 | |
| KANK1 | 742342 | A | G | 100 | 100 | 100 |
| DMRT1 | 841825 | C | T | 53.2 | 89.0 | |
| DMRT1 | 841971 | T | A | 52.6 | 89.1 | |
| DMRT1 | 967981 | G | C | 100 | 100 | 100 |
| DMRT3 | 990840 | C | T | 100 | 100 | 100 |
| DMRT2 | 1057318 | A | G | 46.5 | 100 |
Color table is available in online version.
Variant analysis of 10 gene loci at 14q13 in the total genomic DNA reveals 36 informative positions (Table 2, Supplementary Table S5). The allele frequencies at these positions show three classes; either around 33 or 66, or nearly 100% of reads, corresponding to the presence of variants in one of three, two of three or all three alleles at these positions, respectively. This suggests that this region in 14q13 is present in three copies, which agrees with the SNP microarray profile and the karyotype analysis that reveal three copies of chromosome 14 (Supplementary Fig. S1).7 The common variants present in all three alleles with 100% frequency are detected at six positions. The remaining 23 and 7 variants could be detected by frequencies of 33 and 66%, corresponding to their presence in one and two chromosomes, respectively. Chromosome sequencing was run twice in each sample and showed that variant frequencies were all nearly 100% (Supplementary Table S5). By sorting 14S separately from the unsorted chromosome 14 (14U), variant frequencies in 14S are either 0 or 100% (Table 2), indicating successful chromosome sorting without contamination between homologues. This demonstrates that der(9) and 14S samples have 13 and 29 variants, respectively (Table 2). Of these, 23 variants are specific for 14S. The total DNA sample indicates the presence of variants in 7 loci in two of three chromosomes, but they are only detected in der(9) and absent in 14S (Table 2). This implies that these variants must be shared between the chromosome 14 homologous region on der(9) and 14U, indicating that the chromosome 14 region in der(9) represents a duplication of 14U. Among the variants in the 10 genes, five missense mutations are identified (Supplementary Table S5). One of them is known to be a polymorphism, localized to der(9) and 14U. The other four mutations involving three different gene loci are included in 14S (Supplementary Table S5).
Table 2.
Variant frequency for the 14q13 region
| Gene ID | Position | Ref | Variant | Total | der(9) | 14S | 14U |
|---|---|---|---|---|---|---|---|
| EGLN3 | 34395149 | A | – | 100 | 100 | 100 | |
| EAPP | 34985645 | G | A | 34.1 | 100 | ||
| EAPP | 34998705 | T | C | 33.1 | 100 | ||
| SNX6 | 35062166 | T | C | 100 | 100 | 100 | |
| SNX6 | 35062312 | C | T | 100 | 100 | 99.4 | |
| SNX6 | 35075022 | G | T | 100 | 100 | 100 | |
| BAZ1A | 35222627 | – | TTGT | 32.3 | 90.0 | ||
| BAZ1A | 35223956 | A | G | 35.0 | 91.6 | ||
| BAZ1A | 35223971 | A | G | 34.9 | 92.5 | ||
| BAZ1A | 35227880 | G | T | 33.1 | 89.9 | ||
| BAZ1A | 35228090 | G | A | 30.2 | 89.2 | ||
| BAZ1A | 35230906 | G | A | 32.7 | 91.6 | ||
| BAZ1A | 35233740 | T | C | 34.5 | 93.1 | ||
| BAZ1A | 35245748 | A | G | 29.5 | 87.3 | ||
| SRP54 | 35483882 | A | C | 36.4 | 100 | ||
| SRP54 | 35488309 | A | G | 96.9 | 100 | 100 | |
| SRP54 | 35492128 | C | T | 60.9 | 100 | ||
| SRP54 | 35498443 | G | A | 35.3 | 98.9 | ||
| PPP2R3C | 35565856 | A | T | 32.3 | 95.8 | ||
| PPP2R3C | 35568756 | – | AA | 100 | 100 | 100 | |
| PPP2R3C | 35576458 | A | C | 32.4 | 100 | ||
| PPP2R3C | 35578797 | C | T | 71.0 | 98.1 | ||
| PPP2R3C | 35579927 | G | A | 30.7 | 100 | ||
| PPP2R3C | 35591174 | T | C | 61.2 | 98.6 | ||
| PPP2R3C | 35591316 | G | A | 62.3 | 98.2 | ||
| KIAA0391 | 35592491 | C | A | 34.2 | 100 | ||
| KIAA0391 | 35592538 | C | G | 66.3 | 100 | ||
| KIAA0391 | 35593599 | C | G | 67.3 | 98.2 | ||
| KIAA0391 | 35596092 | C | A | 32.9 | 100 | ||
| KIAA0391 | 35736075 | A | G | 33.2 | 100 | ||
| PSMA6 | 35761573 | C | T | 31.9 | 100 | ||
| PSMA6 | 35761675 | C | G | 68.5 | 96.8 | ||
| PSMA6 | 35777232 | C | T | 33.4 | 100 | ||
| PSMA6 | 35777243 | A | G | 33.8 | 100 | ||
| PSMA6 | 35777270 | A | C | 33.4 | 100 | ||
| PSMA6 | 35777278 | C | G | 33.5 | 100 |
Color table is available in online version.
3.4. Copy number changes at the 14q13 region during passage
The SNP microarray performed in our previous study shows differences of the Ishikawa 3-H-12 genome profiles between low and high passages.7 Although the der(9) has already been found in all cells at low passage, the enlarged profiles focused on the breakpoint region at 14q13.1 indicate copy number changes occurring in one locus at P3 and three loci at P20 (Supplementary Fig. S3B). The change observed at both P3 and P20 corresponds to the breakpoint that forms der(9). The additional changes at P20 between the other two loci involve breakpoints that cause a decrease in copy number. Because this is not detected at P3, the cryptic deletion must have arisen in the cell population after long-term culture. Amplicon sequencing targeted for the 10 kb region around the breakpoint revealed that the whole region is amplified in 14S (Supplementary Fig. S4). As the region upstream of the der(9) breakpoint is absent in der(9), only the downstream region is amplified. Similar profiles of the downstream region shared by 14S and der(9) suggest that no additional chromosome rearrangement has occurred in the region. The patterns of variant frequencies across this region in the total genomic DNA sample demonstrates the change from 50 to 33/66/100% at P3 and from 100 to 50/100% at P20 (Supplementary Table S6), corresponding to the chromosome copy number changes from 2 to 3 and from 1 to 2 that have occurred in this region (Fig. 4). Variants detected in the der(9) would be expected to be present at P3, indicating that three of them, 66% at P3, are shared with 14U. However, these variants are not detected in total genomic DNA at P20. This implies that two additional breaks have occurred in 14U and led to a cryptic deletion. Because the other variants detected in one of three loci at P3 are specific for der(9), these changes can be related to the breakpoint event that occurred following duplication of chromosome 14.
Figure 4.

Genome structure at the 14q13.1 region is illustrated from the array profiles and combined with variant frequencies obtained from amplicon sequencing, with comparisons between passages, P3 (A) and P20 (B). The breakpoint in the der(9) is located at the position 33,583,729 bp. Variants are assigned to der(9) and 14S by amplicon sequencing in the chromosome-specific DNA. The frequencies are distinguished by four different symbols. Variant assignments on chromosome homologues at P3 are estimated from the P20 profile. A series of 100, 50 and 33/66% in total genomic DNA correspond to 1, 2 and 3 copies, respectively. Frequencies at three positions with 66% at low passage change to 50% at high passage, suggesting the presence of these variants in the 14U deletion indicated by a dashed line. gDNA: total genomic DNA.
4. Discussion
Chromosome translocation is a common feature of chromosome rearrangements in tumour cells and can lead to the formation of a fusion gene.19,20 Although fusion genes are found by RNA-seq, it is hard to identify the genomic breakpoint because normal and abnormal alleles usually coexist in total genomic DNA samples and cannot be distinguished by conventional analysis.21 The human genome size is 3,099 Mbp according to the golden path length of the GRCh37.p13 assembly. As the majority of the Ishikawa karyotypes are 45, X, -X and have some gains,7 the size of each Ishikawa nuclear DNA is estimated to be ∼ 6,200 Mbp, including one copy of the der(9). When the breakpoint junction is identified by conventional whole genome sequencing using total genomic DNA, theoretically it can appear in 1 per 15,500,000 fragments based on a 400 bp sequencing sample. However, it can be increased to 1 per 500,000 fragments when the sample is from the sorted der(9) chromosome, that is more efficient than whole genome sequencing. It is reported that alignment of single-end long reads is straightforward to identify fusion junctions.22 We demonstrate that identification of genomic breakpoints detected by conventional karyotypes can be achieved by mapping analysis of single end reads on 400 bp scale without de novo assembly. Although considerable efforts involving conventional cloning techniques have been devoted to uncover breakpoint junctions in tumour genomes,23, chromosome sequencing helps the detection and analysis of the genomic breakpoint junction.
Fusion genes serve as genetic markers and can be associated with disease phenotypes.24 Sequence analysis of the der(9) chromosome-specific DNA isolated by flow sorting in our study reveals a WASH1–NPAS3 fusion gene at the translocation breakpoint. This chimeric gene is formed mostly by the NPAS3 component, suggesting that altered NPAS3 expression may be oncogenic. Although no report has shown fusion genes between these two genes, NPAS3 is a transcription factor of a basic helix–loop–helix Per-ARNT-Sim domain family associated with tumour development in multiple forms of cancer.25 Also, it is reported that NPAS3 acts as a tumour suppressor in human malignant astrocytomas.26 The fusion detected by the genomic breakpoint junction and confirmed by the transcript can be a definitive marker for the Ishikawa cell line and used, possibly, for future studies on endometrial carcinoma.
Amplicon sequencing of the genes neighbouring the breakpoint demonstrates that variant frequencies in the total genomic DNA are linked to chromosome copy number. Variant frequencies of 50% of reads detected in chromosome 9 indicate the presence of the variant in one of two homologues. The variant frequencies of 33 or 66% of reads in chromosome 14 reflect one or two of three homologues, respectively (Fig. 4). This suggests that a series of patterns consisting of frequencies of approximate 50/100 or 33/66/100% are strongly correlated to the presence of copy numbers of two or three chromosomes, respectively. While the fusion can be clearly distinguished at the chromosome level, sequence variants cannot be regarded as allelic unless the chromosomes are sequenced separately. When amplicon sequencing is performed on chromosome-specific DNA, the variant frequencies of 0 or 100% of reads indicate that the variant is either absent or present in an amplicon. This permits chromosome assignment of each variant to a specific chromosome and enables distinction between homologues. Two chromosomes sharing the same allelic profile suggest that they have a common origin and are formed by duplication. Allele frequencies of ∼ 66% of reads reflects the presence of the same variant in two of the three alleles in three chromosomes corresponding to one in der(9) and one in 14U (Table 2, Fig. 5). Further analysis of the breakpoint by amplicon sequencing of the 10 kb region that encompasses the junction in the second intron of NPAS3 identified a cryptic deletion occurring in the chromosome 14 allele shared with der(9). These detailed variant analyses elucidate the process of chromosome rearrangements involving chromosomes 9 and 14. SNP allele peaks show two different allelic profiles of chromosome 14 in Ishikawa cells,7 indicating that the duplication of 14q13–ter has occurred prior to the formation of der(9). Although the variant frequency at 14q13 reveals that the cryptic deletion and the breakpoint in the der(9) occur in different chromosome 14 homologues, these two chromosomes must have been formed through duplication because of their otherwise identical allelic profiles. As the deletion cannot be identified in the array profile at low passage, it must have occurred after the formation of der(9) and during cell culture. These two rearrangements have occurred independently, possibly suggesting a tendency to breakage in the region.
Figure 5.
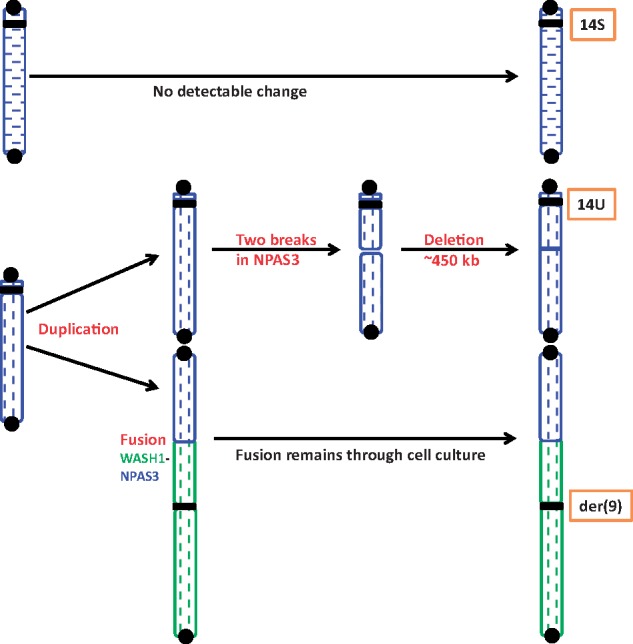
The process of chromosome rearrangement involved in chromosome 14. Chromosomes 9 and 14 regions are marked by green and blue, respectively, and the two homologues of chromosome 14 are distinguished by vertical and horizontal patterns. Because chromosome 14 consists of two different allele contents in the SNP array, it must have originated from a normal pair with different allele profiles. Variant analysis in the sorted chromosome samples show that 14S and der(9) have different chromosome 14 allelic profiles, but der(9) and 14U have the same chromosome 14 allelic profiles. This indicates that the origin of der(9) was associated with duplication of 14U. One of the homologues, 14S, does not have detectable changes at chromosomal level and der(9) remains unchanged during cell culture. However, duplication of the long arm of 14U is fused with 9p to form der(9). The 14U chromosome at passage P20 has a cryptic deletion in the same region as the breakpoint in der(9). Black circles and squares show telomere and centromere, respectively. Color figure is available in online version.
Our chromosome sorting using a dual laser flow cytometer was first introduced in 1992 to generate chromosome-specific DNA for diagnostic cytogenetics.14 This led to the development of chromosome painting probes for phylogenetic studies in a wide variety of species including human that has revealed homologous syntenic blocks conserved between species not only in mammals but also among birds and reptiles.15,27,28 These studies provide many examples of chromosome-specific mapping and sequencing. In one study in dogs (2n = 78), although there are many acrocentric chromosomes of similar size and seven peaks in the flow karyotype that contain 2 chromosome pairs,29 each dog chromosome was successfully isolated, some by single chromosome sorting; a complete set of painting probes was produced with each probe specific for a single type of dog chromosome.30 Another study demonstrated that chicken microchromosomes can be characterized by single chromosome sorting.31 Single chromosome sorting has been applied more recently to haplotyping normal human chromosome 19, by demonstrating allele heterozygosity.32 When derivative chromosomes are different in size from the normal homologues, they form a unique peak in the flow karyotype and can be resolved easily and collected by flow sorting.33 We show here the isolation of chromosome homologues in a tumour cell line by chromosome sorting that allows homologue-specific sequencing, chromosome assignment of allelic variants and improved characterization of abnormal chromosomes.
Supplementary Material
Acknowledgements
Authors would like to thank Dr. Ryoka Miki (Thermo Fisher Scientific, Japan) and Mr. Takashi Ishikura (Thermo Fisher Scientific, Japan) for their technical comments on data analysis.
Conflict of interest
None declared.
References
- 1. Henson J., Tischler G., Ning Z.. 2012, Next-generation sequencing and large genome assemblies, Pharmacogenomics, 13, 901–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pabinger S., Dander A., Fischer M., et al. 2014, A survey of tools for variant analysis of next-generation genome sequencing data, Brief. Bioinformatics, 15, 256–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barretina J., Caponigro G., Stransky N.. 2012, The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity, Nature, 483, 603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Forbes S. A., Beare D., Boutselakis H., et al. 2017, COSMIC: somatic cancer genetics at high-resolution, Nucleic Acids Res., 45, D777–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marusyk A., Polyak K.. 2010, Tumor heterog eneity: causes and consequences, Biochim. Biophys. Acta, 1805, 105–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Greaves M., Maley C.C.. 2012, Clonal evolution in cancer, Nature, 481, 306–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kasai F., Hirayama N., Ozawa M., Iemura M., Kohara A.. 2016, Changes of heterogeneous cell population in the Ishikawa cell line during long-term culture; proposal of an in vitro clonal evolution model of tumor cells, Genomics, 107, 259–66. [DOI] [PubMed] [Google Scholar]
- 8. Potter N.E., Ermini L., Papaemmanuil E., et al. 2013, Single-cell mutational profiling and clonal phylogeny in cancer, Genome Res., 23, 2115–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carter N.P., Ferguson-Smith M.E., Affara N.A., Briggs H., Ferguson-Smith M.A.. 1990, Study of X chromosome abnormality in XX males using bivariate flow karyotype analysis and flow sorted dot blots, Cytometry, 11, 202–7. [DOI] [PubMed] [Google Scholar]
- 10. Kasai F., O'Brien P.C.M., Ferguson-Smith M.A.. 2012, Reassessment of genome size in turtle and crocodile based on chromosome measurement by flow karyotyping: close similarity to chicken, Biol. Lett., 8, 631–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kasai F., O'Brien P.C.M., Ferguson-Smith M.A.. 2013, Afrotheria genome; overestimation of genome size and distinct chromosome GC content revealed by flow karyotyping, Genomics, 102, 468–71. [DOI] [PubMed] [Google Scholar]
- 12. Ferguson-Smith M.A. 1997, Genetic analysis by chromosome sorting and painting: phylogenetic and diagnostic applications, Eur. J. Hum. Genet., 5, 253–65. [PubMed] [Google Scholar]
- 13. Kasai F., Yoshihara M., Matsukuma S., O'Brien P., Ferguson-Smith M.A.. 2007, Emergence of complex rearrangements at translocation breakpoints in a transgenic mouse; implications for mechanisms involved in the formation of chromosome rearrangements, Cytogenet. Genome Res., 119, 83–90. [DOI] [PubMed] [Google Scholar]
- 14. Carter N.P., Ferguson-Smith M.A., Perryman M.T., et al. 1992, Reverse chromosome painting: a method for the rapid analysis of aberrant chromosomes in clinical cytogenetics, J. Med. Genet., 29, 299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ferguson-Smith M.A., Yang F., O'Brien P.C.. 1998, Comparative mapping using chromosome sorting and painting, ILAR J., 39, 68–76. [DOI] [PubMed] [Google Scholar]
- 16. Edwards A.M., Cameron E.Z., Pereira J.C., Wapstra E., Ferguson-Smith M.A., Horton S.R., Thomasson K.. 2016, Gestational experience alters sex allocation in the subsequent generation, R. Soc. Open Sci., 3, 160210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gomes A.J., Nagamachi C.Y., Rodrigues L.R., et al. 2016, Chromosomal phylogeny of Vampyressine bats (Chiroptera, Phyllostomidae) with description of two new sex chromosome systems, BMC Evol. Biol., 16, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nishida M. 1985, Establishment of a new human endometrial adenocarcinoma cell line, Ishikawa cells, containing estrogen and progesterone receptors, Nihon Sanka Fujinka Gakkai Zasshi, 37, 1103–11. [PubMed] [Google Scholar]
- 19. Aplan P. D. 2006, Causes of oncogenic chromosomal translocation, Trends Genet., 22, 46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Roukos V., Misteli T.. 2014, The biogenesis of chromosome translocations, Nat. Cell Biol., 16, 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Suzuki T., Tsurusaki Y., Nakashima M., Miyake N., Saitsu H., Takeda S., Matsumoto N.. 2014, Precise detection of chromosomal translocation or inversion breakpoints by whole-genome sequencing, J. Hum. Genet., 59, 649–54. [DOI] [PubMed] [Google Scholar]
- 22. Ge H., Liu K., Juan T., Fang F., Newman M., Hoeck W.. 2011, FusionMap: detecting fusion genes from next-generation sequencing data at base-pair resolution, Bioinformatics, 27, 1922–8. [DOI] [PubMed] [Google Scholar]
- 23. Ford A.M., Mansur M.B., Furness C.L., et al. 2015, Protracted dormancy of pre-leukemic stem cells, Leukemia, 29, 2202–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kawamura M., Taki T., Kaku H., Ohki K., Hayashi Y.. 2015, Identification of SPAG9 as a novel JAK2 fusion partner gene in pediatric acute lymphoblastic leukemia with t(9; 17)(p24; q21), Genes Chromosomes Cancer, 54, 401–8. [DOI] [PubMed] [Google Scholar]
- 25. Bersten D.C., Sullivan A.E., Peet D.J., Whitelaw M.L.. 2013, bHLH-PAS proteins in cancer, Nat. Rev. Cancer, 13, 827–41. [DOI] [PubMed] [Google Scholar]
- 26. Moreira F., Kiehl T.R., So K., et al. 2011, NPAS3 demonstrates features of a tumor suppressive role in driving the progression of Astrocytomas, Am. J. Pathol., 179, 462–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ferguson-Smith M.A., Yang F., Rens W., O'Brien P.C.. 2005, The impact of chromosome sorting and painting on the comparative analysis of primate genomes, Cytogenet. Genome Res., 108, 112–21. [DOI] [PubMed] [Google Scholar]
- 28. Kasai F., O'Brien P.C., Martin S., Ferguson-Smith M.A.. 2012, Extensive homology of chicken macrochromosomes in the karyotypes of Trachemys scripta elegans and Crocodylus niloticus revealed by chromosome painting despite long divergence times, Cytogenet. Genome Res., 136, 303–7. [DOI] [PubMed] [Google Scholar]
- 29. Yang F., O'Brien P.C., Milne B.S., et al. 1999, A complete comparative chromosome map for the dog, red fox, and human and its integration with canine genetic maps, Genomics, 62, 189–202. [DOI] [PubMed] [Google Scholar]
- 30. Graphodatsky A.S., Yang F., Serdukova N., Perelman P., Zhdanova N.S., Ferguson-Smith M.A.. 2000, Dog chromosome-specific paints reveal evolutionary inter- and intrachromosomal rearrangements in the American mink and human, Cytogenet. Cell Genet., 90, 275–8. [DOI] [PubMed] [Google Scholar]
- 31. Masabanda J.S., Burt D.W., O'Brien P.C., et al. 2004, Molecular cytogenetic definition of the chicken genome: the first complete avian karyotype, Genetics, 166, 1367–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang H., Chen X., Wong W.H.. 2011, Completely phased genome sequencing through chromosome sorting, Proc. Natl Acad. Sci. U S A., 108, 12–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gribble S.M., Ng B.L., Prigmore E., Fitzgerald T., Carter N.P.. 2009, Array painting: a protocol for the rapid analysis of aberrant chromosomes using DNA microarrays, Nat. Protoc., 4, 1722–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.