

Abstract
Teak (Tectona grandis L. f.) is one of the precious bench mark tropical hardwood having qualities of durability, strength and visual pleasantries. Natural teak populations harbour a variety of characteristics that determine their economic, ecological and environmental importance. Sequencing of whole nuclear genome of teak provides a platform for functional analyses and development of genomic tools in applied tree improvement. A draft genome of 317 Mb was assembled at 151× coverage and annotated 36, 172 protein-coding genes. Approximately about 11.18% of the genome was repetitive. Microsatellites or simple sequence repeats (SSRs) are undoubtedly the most informative markers in genotyping, genetics and applied breeding applications. We generated 182,712 SSRs at the whole genome level, of which, 170,574 perfect SSRs were found; 16,252 perfect SSRs showed in silico polymorphisms across six genotypes suggesting their promising use in genetic conservation and tree improvement programmes. Genomic SSR markers developed in this study have high potential in advancing conservation and management of teak genetic resources. Phylogenetic studies confirmed the taxonomic position of the genus Tectona within the family Lamiaceae. Interestingly, estimation of divergence time inferred that the Miocene origin of the Tectona genus to be around 21.4508 million years ago.
Keywords: teak, genome sequencing, SSRs, phylogeny, divergence
1. Introduction
Teak (Tectona grandis L. f.; 2n = 2x = 36) belonging to the mint family Lamiaceae is one of the world’s highly valued tropical timber species that occurs naturally in India, Laos, Myanmar and Thailand.1–4 The timber is highly valued because of its extreme durability, strength, stability as well as resistance to pests, chemicals and water. Quinones and other extractives found abundant in the teakwood are responsible for its anti-termite and anti-fungal properties conferring the longevity of its timber. Thus, the wood is used in building ships, railway carriages, sleepers, construction, furniture, veneer and carving. Owing to its admirable timber qualities and aesthetical properties (Fig. 1), teak has been successfully established as pure plantations in India and elsewhere since 1850.5 The recent log export ban imposed by Myanmar has resulted steep rise in international prices of plantation grown teak from Latin America and Africa leading to expansion of plantation area. The estimated planted area of teak is about 4.25–6.89 million ha with over 1.7 million ha in India.5 Though the share of teak is <2% of tropical round wood production,6 its high value continuously attracts new planters. At the same time, natural populations are continuously diminishing due to illegal logging, anthropogenic pressures and climate change. A recent study on the effect of climate change in teak expresses the risks of biological invasion into teak habitats and recommends conservation of crucial teak growing areas and suitable management planning.7 Population structure of teak across natural and introduced locations reveal that the landraces in introduced locations have comparatively narrow genetic diversity,8 thus demanding exploration of genetic diversity of natural provenances and their conservation.
Figure 1.
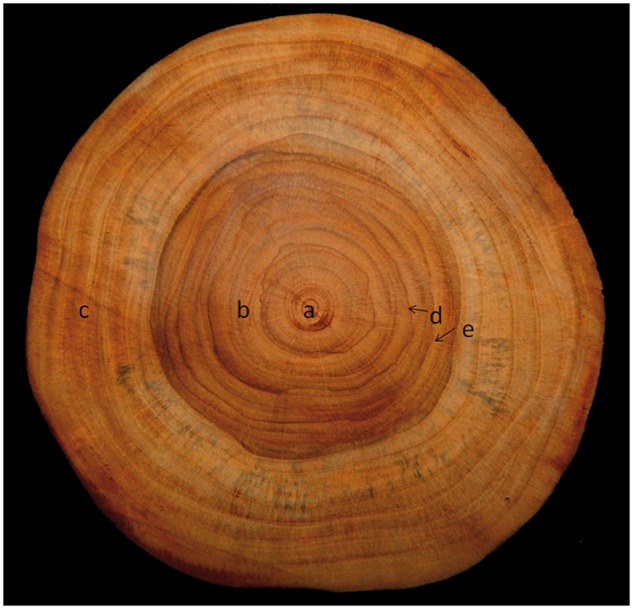
Cross section of teak wood showing major features (a) pith; (b) heart wood; (c) sap wood; (d) growth ring; and (e) medullary rays.
Teak has several intrinsic genetic qualities that allow its genetic improvement for timber production. Wide and discontinuous natural distribution across varying edaphic and climatic conditions in India offers enormous potential for capturing adaptive genetic variation for genetic improvement. As a first step in the genetic improvement programme at a global level, a series of seventy five international provenance trials co-ordinated by Danish International Development Agency were established during 1973–76 across sixteen countries. Evaluation of the provenances indicated wide variation for survival, growth rate, stem form, flowering, fruit yield and wood characteristics.9,10 The results of genetic improvement in teak showed an overall positive trend, however, possible existence of non-additive genetic control for economically important traits in seed progeny generated high genetic variability even within a family.11,12 Further, it was suggested that selection of teak stem size can be carried out at the age of 3 years, wherein indirect selection on flowering age will improve forking height.13 Clonal propagation through budding, rooting of cuttings and in vitro propagation has facilitated tree improvement and deployment of superior performers for commercial cultivation towards increasing timber yield.14 Globally, although clonal seed orchards (CSOs) were established for production of quality seed stock, reproductive fitness and success of CSO was a chronic problem largely pivoted to asynchronous flowering.15
Economic importance, concerns for conservation of natural populations and increase in plantation area, demand understanding the genetic basis of the economic traits in teak. Hence, like many other forest plantation species (e.g. Pinus, Populus and Eucalyptus), genetic and genomic resources in teak needs to be comprehended. Genetic diversity in natural and introduced populations of teak has been assessed with markers such as random amplified polymorphic DNA,16 amplified fragment length polymorphism17,18 and simple sequence repeat markers (SSRs) or Microsatellites.19,20 These studies generated information on population genetic structure of natural teak populations. Indian teak is genetically very distinct from Thai and Indonesian provenances and African landraces.17,19 Knowledge generated in these studies is highly useful to implement conservation programmes in teak to improve sustainable management of teak forests.20 Recently, transcriptomes of secondary wood21 and vegetative to flowering transition stage22 were developed, leading to identification of genes involved in lignification, secondary metabolite production and flower formation. Although numerous studies focused on various life history traits exist in teak, comprehensive understanding on the complete genome information remains unexplored. Next generation sequencing-based whole genome sequencing (NGS-WGS) yields more information on genomic scans of polymorphism to precisely estimate various population genetic parameters including demographic history. In this context, to enrich genomic resources in teak, the present study reports whole genome of teak through Illumina Hiseq 2000 NGS platform followed by de novo contig assembly, gene annotation and subsequent discovery of SSR polymorphism.
2. Materials and methods
2.1. Plant material and genomic DNA sample preparation
Open-pollinated seeds were collected from six dominant trees, one each from a provenance covering the entire latitudinal range of natural distribution of teak in India (Table 1). Seedlings were raised with family identity and one vigorously growing seedling randomly chosen from each family was used in this study. Genomic DNA was extracted from fresh and young leaves using standard CTAB method23 and was purified using DNAeasy Plant Mini kit (Qiagen, USA). The quantity and quality of the genomic DNA were assessed using Nanodrop2000 (Thermo Fisher Scientific, USA), Qubit (Thermo Fisher Scientific, USA) and agarose gel electrophoresis.
Table 1.
Details of plant materials used in this study
| Accession ID | Sample code | Name of the provenance | State | Latitude | Longitude | Altitude (m) | Rainfall (mm) |
|---|---|---|---|---|---|---|---|
| 1 | NR | Nilambur | Kerala | 11° 17' N | 76° 19' E | 49 | 2,600 |
| 2 | AU | Arienkavu | Kerala | 8° 96’ N | 77° 14’ E | 240 | 2,600 |
| 3 | WR | Walayar | Kerala | 10° 52' N | 76° 46' E | 216 | 1,500 |
| 4 | DI | Dandeli | Karnataka | 15° 07' N | 74° 35' E | 510 | 2,200 |
| 5 | HI | Hojai | Assam | 26° 39’ N | 92° 36’ E | 69 | 1,750 |
| 6 | TP | Topslip | Tamilnadu | 10° 26' N | 76° 50' E | 640 | 1,350 |
2.2. Library preparation and genome sequencing
WGS was performed using Illumina HiSeq 2000 platform and Oxford Nanopore Technologies MinION device by the Genotypic Technology, Bengaluru, India in accordance to standard protocols. Accession 2 was selected for the generation of high quality reference genome assembly. Accessions 1, 3, 4, 5 and 6 were subjected to low coverage genome sequencing to identify polymorphic SSRs. In the case of accession 2, one paired end (PE) (150 bp × 2) library of size 300–700 bp, two mate pair (MP) libraries (2–4 and 4–6 kb fragments) and one nanopore library with genomic DNA (2 μg) were prepared for sequencing. In Illumina HiSeq 2000 platform, one lane of the flow cell was used for each sequencing library. Nanopore sequencing was performed using R9.4 flow cells on a MinION Mk 1B device (Oxford Nanopore) with the MinKNOW software (versions 1.0.5–1.5.12) and base calling was performed using Albacore 1.1.0 (Oxford Nanopore). Template reads were exported as FASTA using poretools version 0.6. In the case of other five accessions (1, 3, 4, 5 and 6) one PE library for each with the size of 300–700 bp was sequenced at ∼15× coverage through Illumina Hiseq 2000 platform. The sequence data is uploaded in genome database of GenBank (Project id: PRJNA374940). The assembled genome, protein sequences and its annotation, GO and pathway information are available in the web link https://biit.cs.ut.ee/supplementary/WGSteak/.
2.3. De novo genome assembly
The Illumina PE raw reads were filtered using FastQC and the raw reads were processed by in-house (Genotypic Technology, Bangaluru, India) ABLT script for low-quality bases and adapters removal. The MP reads were processed using Platanus24 internal trimmer for adapters and low-quality regions towards 3’-end. The processed PE reads along with MP and nanopore reads were used for contig generation using MaSuRCA v 3.2.2 de novo assembler.25 To assemble the genome following command was used in MaSuRCA assembler: GRAPH_KMER_SIZE = auto, LIMIT_JUMP_COVERAGE = 300, JF_SIZE = 38000000000, DO_HOMOPOLYMER_TRIM = 1. Scaffolding of the assembled contigs was performed using SSPACE v 2.0.526 with processed PE and MP reads followed by gap filling using Gap Closer v 1.12.27 The genome size was estimated automatically during read computing stage which utilized both the Illuimna and Nanopore reads. Similarly, the low depth Illumina reads generated for five accessions of teak were assembled using accession 2 as reference. The sequenced data was uploaded to the Genome database of GenBank (Project id: PRJNA421422).
2.4. Genome annotation
For a functional overview of draft genome, assembled scaffolds were converted to FASTA formatted sequences, hard masked by RepeatMasker tool (RepeatMasker Open-3.0; www.repeatmasker.org (10 November 2017, date last accessed)). Repeats of Arabidopsis thaliana were used as reference for genome masking. Gene prediction was carried out using Augustus 3.0.228 programme and predicted proteins were searched against the Uniprot non-redundant plant protein database (Taxonomy = Viridiplante) with BlastX algorithm with an e-value (e-10) for gene ontology and annotation. Pathway annotation was performed by mapping the sequences obtained from Blast2GO to the contents of the KEGG Automatic Annotation Server (http://www.genome.jp/kegg/kaas/ (10 November 2017, date last accessed)). List of eudicot plants used as reference organism for pathway identification in KASS server is given in Supplementary Table S1.
2.5. Identification of SSRs and detection of polymorphism
FASTA formatted scaffolds of teak were analysed for frequency and density of SSRs using the Perl script MIcroSAtelitte (MISA; http://pgrc.ipk-gatersleben.de/misa/ (20 November 2017, date last accessed)). Initially SSRs of 1–6 nucleotides motifs were identified with the minimum repeat unit defined as 10 for mononucleotides, 6 for dinucleotides, 5 for trinucleotides, 4 for tetra-nucleotides, and 3 each for penta and hexa-nucleotides. Compound SSRs were defined as ≥2 SSRs interrupted by ≤100 bases. To design primers flanking the microsatellite loci, two interface Perl script modules were used to interchange data between MISA and the primer designing software Primer 3. The SSR containing scaffolds were used to design the primers with the following parameters. Primer length 18–25 bp, with 20 bp as optimum; primer GC content = 30–0%, with the optimum value of 50%; primer Tm 57–63°C, and product size ranged 100–300 bp.
Polymorphic SSRs across five samples were analysed using accession 2 as reference. Polymorphic SSR retrieval tool (PSR)29 comprising two modules (PSR_read_retrieval and PSR_poly_finder) were deployed to detect SSR length polymorphisms of perfect repeats from NGS data. It is to be noted that PSR tool identifies length polymorphisms in perfect microsatellites only. Also, it filters out all the reads that match twice or more on the reference sequence as well as non-overlapping paired-end reads that are aligned on the same microsatellite locus. Minimum number of supporting reads and read depth was fixed to 10 and 30, respectively. This process detects polymorphic SSRs based on the availability of left and right border unique sequences based on the complete coverage of the SSR region in the sequence data.
The polymorphic SSR data generated from PSR software was validated through gel electrophoresis. Totally 10 SSRs representing di, tri and tetra-nucleotide motifs were randomly chosen and amplified with 10 randomly selected teak trees from Topslip (Latitude: 10°29’09.5’N; Longitude: 75°50’03.8’E Altitude: 736m) provenance (Supplementary Table S2). PCR amplification were set to 10 µl volume containing 5 ng of template DNA, 2.5 µM MgCl2, 2.5 µl 10 × PCR buffer, 0.5 µM of primer, 0.5 U Taq DNA polymerase, and 2.5 mM of dNTPs. The PCR cycling profile was programmed to 94°C for 5 min, 35 cycles at 94°C for 45 s, 58°C (annealing temperature varied for each locus) for 45 s (Table 2), 72°C for 45 s, and a final extension at 72°C for 10 min. Banding pattern was visualised by silver staining after denaturing polyacrylamide gel (5%) electrophoresis.
Table 2.
Details on 10 SSR markers used for amplification of teak germplasm
| SSR code | SSR Motif | Primers (5’-3’) | Annealing temperature (oC) | Number of alleles | Product size (bp) |
|---|---|---|---|---|---|
| IFT83 | (AG)21 | F5’AATTGGCATAAAGCGTGCTACTR5’CGCACGTCCTATTTTGGTTTAT | 54 | 5 | 312-393 |
| IFT821 | (AC)24 | F5’CCCCAATTATGTCAACCGACT R5’GGCATTATCTAAGATCGCAAGG | 53.9 | 3 | 331-350 |
| IFT63 | (ATG)12 | F5’CCCAAAGCGAATAATATCCTAC R5’CATGACTTGTTCGATGGGCTAAT | 54 | 3 | 250-275 |
| IFT168 | (TCT)12 | F5’ATCTTCAGCAGAGGAGGCTATG R5’GTGCCCTTTTCTCTCTTCTTCA | 55 | 4 | 287-306 |
| IFT479b | (GGA)11 | F5’GTGAAGATTCGGGTATGGAGAG R5’TACTCCCAGATTTCCCAATCAC | 55 | 3 | 331-343 |
| IFT382 | (TATG)7 | F5’TACTCATCACTGTCCCCAGTTG R5’GAACGGGAATCTAGAGTTGTGG | 56 | 3 | 337-350 |
| IFT 28 | (AAAG)6 | F5’CAGCCTCTGCATGTCAAATAAA R5’TTAGAGCTGGATATGCCATTGA | 53.6 | 3 | 381-393 |
| IFT14 | (TTCT)9 | F5’TGTGGTATTGGACCATCTGAAA R5’GGTAACCCACCAACAAATATGC | 54 | 5 | 265-278 |
| IFT3 | (GAAAG)5 | F5’TTCCACCTACTGGTTAAGGAAC R5’ATGGCTTACCAATTTACCAAACC | 54 | 1 | 330 |
| IFT777 | (TCAGG)6 | F5’TACTAACCGGAAGAGGGAAACC R5’TGTCGCTATGGACAGTTCATCT | 56 | 4 | 312-343 |
2.6. Phylogenetic tree construction
Plastid gene sequences of psbB, rbcL, psaA and ycf2 from 16 species of the family Lamiaceae were accessed from the Genbank public domain (https://www.ncbi.nlm.nih.gov/genbank (13 February 2018, date last accessed)) for constructing the phylogenetic tree with Olea europaea (Oleaceae) as the out group (Supplementary Table S3). Sequences were aligned using multiple sequence alignment tool implemented in ClustalX ver. 2.0.30 The sequences were manually refined in BioEdit ver. 7.0.9.31 Phylogenetic analyses were performed for concatenated sequences of plastid gene regions. JModeltest ver. 2.1.732 was used to choose the appropriate model of sequence evolution according to the Akaike information criterion (AIC).33 Bayesian interference analysis was performed in MrBayes ver. 3.2.6.34 The Markov chain Monte Carlo algorithm was run for ten million generations, over four chains each, sampled every 1000 generations. The estimated sample size was checked using Tracer ver. 1.4 (http://beast.bio.ed.ac.uk/Tracer (22 February 2018, date last accessed)). The first 25% of the sampled trees was discarded as burn-in. The phylogenetic tree out of MrBayes ver.3.2.6 was visualized in FigTree ver.1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/ (22 February 2018, date last accessed)).
2.7. Divergence time estimation
The estimation of divergence time was performed using Bayesian approach implemented in BEAST ver. 2.4.4 programme.35 Bayesian approach was deployed to estimate divergence time of the genus Tectona with respect to the other subfamilies and genera within the family Lamiaceae. The HKY model was used based on the result of AIC from JModeltest under an uncorrelated lognormal relaxed clock model. Yule speciation model was used as tree prior. Two calibration points (57.6 million years ago, Mya) for Nepetoideae and 23.9 Mya for Lamioideae) were used based on the previous reports to determine specific nodes prior and lognormal distributions.36 Markov Chain was run for 109 generations, while every 1,000 generations were sampled. The chronograms shown were calculated using the median clade credibility tree plus 95% confidence intervals. Results were analysed using Tracer ver. 1.4 to assess the convergence statistics of the sequences. The effective sample sizes for all parameters and the tree files from the four runs of BEAST were combined using LogCombiner ver. 2.4.4.35 Twenty percent of the trees were removed as burn-in and the resulting trees were summarized with Treeannotator ver. 2.4.4.37 Finally, the summarized single trees were visualized in FigTree ver. 1.4.2.
3. Results and discussion
Several members of the order Lamiales are well known for their secondary metabolites of medicinal value38 and 17 species of the family were sequenced for the whole genome (https://www.ncbi.nlm.nih.gov/genome/? term=Lamiales (13 February 2018, date last accessed)) due to their economic importance. However, the woody species teak, one of the world’s premier timber species cultivated across 65 countries has only 3,269 nucleotide accessions including 6 ESTs available so far in the public domain (7 November 2017, date last accessed). Owing to the commercial importance, this study was undertaken to unravel the genome structure to facilitate conservation and improvement of teak genetic resources. The only available genomic resource in teak is the de novo assembly with transcriptome in 12- and 60-year-old trees to generate unigenes related to lignin biosynthesis.21 All the earlier gene assemblies were based on short-read technology. This work on draft genome assembly using long read technology like MP and MinION nanopore sequencing would provide an excellent resource to comprehend genome structure, genetic variation and conservation.
3.1. De novo assembly and characterization of genomic sequences
The study has generated a high quality reference genome for the accession 2 which was assembled from PE, MP and nanopore library sequences. The numbers of raw and processed reads are summarized in Table 3. High depth (109×) PE sequencing provided a global overview of teak genome with over 137.2 million PE sequences. After suitable filtration, a total of 128.2 million sequences, representing 93.43% of the raw reads were obtained. Two different MP libraries with 2–4 and 4–6 Kb generated raw reads of about 7,681 (20×) and 5,819 Mbp (15×) of which processed read length was 2,408 and 1,898 Mbp, respectively.
Table 3.
Raw data statistics of Illumina PE, MP and Nanopore reads of teak genome
| Platform | Chemistry | Number of raw reads | Total bases of raw reads (bp) | No of processed reads (bp) | Total bases after processing (bp) | Coverage (×) |
|---|---|---|---|---|---|---|
| Illumina HiSeq | PE (150× 2) | 137,231,716 | 41,443,978,232 | 128,115,515 | 37,507,019,850 | 109 |
| Illumina HiSeq (2-4 Kb) | MP (150× 2) | 25,436,869 | 7,681,934,438 | 10,776,772 | 2,408,197,618 | 20 |
| Illumina HiSeq (4-6Kb) | MP (150× 2) | 19,268,378 | 5,819,050,156 | 8,470,072 | 1,898,380,817 | 15 |
| Nanopore | Long read (5-1,345,484) | 782,591 | 2,685,280,348 | 782,591 | 2,685,280,348 | 7.06 |
| Total coverage | 151× | |||||
Nanopore library generated 782,591 reads with read length of 2,685, 280,348 bp, corresponding to 7.06× coverage of the genome. Longest read was 1,345,484 bp and the average read length was 3,431 bp. All these sequences with 151× coverage were included in whole genome assembly. Application of nanopore sequencing was challenging mainly due to large size and repetitive nature of the plant genome. However, high yield of nanopore using 9.4 chemistry has resolved this issue. Genomic applications to genetic resource conservation and breeding in forest trees is expected to harness much benefits due to the long read sequencing technologies.39 The processed PE reads along with MP and nanopore reads were assembled using MaSuRCA de novo assembler. It used all the Illumina and Nanopore reads choosing a kmer size of 105 and estimated the teak genome size of 371,016,305 bp. Genome size of teak was estimated 465 Mbp through flow cytometry (1C = 0.48 pg).40 Typically, data obtained from the WGS approach in plants with genome size exceeding a few hundred mega bases are difficult to assemble satisfactorily due to highly repetitive DNA.41 The final draft genome used for downstream analysis had 2,993 filtered contigs (>1 Kbp) with maximum, minimum and average contig length of 1,718,606, 1,100 and 106,098 bp, respectively (Table 4). The N50 value of the assembly was 357,576 bp. Comparative WGS analysis including estimated genome size, assembly statistics and annotation details of Lamiaceae members is provided in Supplementary Table S4.
Table 4.
Draft genome assembly statistics of teak
| Parameters | Contig | Scaffold | Gap closer | Draft genome |
|---|---|---|---|---|
| Contigs generated | 3,500 | 3,004 | 3,004 | 2,993 |
| Maximum contig length (bp) | 1,718,119 | 1,718,322 | 1,718,606 | 1,718,606 |
| Minimum contig length (bp) | 332 | 445 | 445 | 1,100 |
| Average contig length (bp) | 90,394 | 105,594 | 105,712 | 106,098 |
| Total contigs length (bp) | 316,377,938 | 317,203,315 | 317,558,121 | 317,551,182 |
| Total number of non-ATGC characters | 2,084,563 | 2,374,171 | 808,446 | 808,446 |
| Percentage of non-ATGC characters | 0.659 | 0.748 | 0.255 | 0.255 |
| Contigs ≥ 500 bp | 3,484 | 3,003 | 3,003 | 2,993 |
| Contigs ≥ 1 Kbp | 3,478 | 2,993 | 2,993 | 2,993 |
| Contigs ≥ 10 Kbp | 2,431 | 2,069 | 2,069 | 2,069 |
| Contigs ≥ 1 Mbp | 8 | 18 | 18 | 18 |
| N50 value (bp) | 277,872 | 357,576 | 357,576 | 357,576 |
3.2. Repetitive genome elements
Repetitive DNAs and transposable elements are ubiquitously present in eukaryotic genomes. They provide wide variety of variations across plant species. Repetitive elements are fast evolving components of nuclear genome that play an important role in evolution of the species42 and interspecific divergence.43 The internal sequence variability of various repeat elements depends on the ratio between mutation and homogenization/fixation rates within a species.44 Repeat masking of the reference genome of teak with A. thaliana showed that a total of 19,046,577 bp (6% of the genome) had repeat elements which was very low when compared to Salvia repeat elements.45 The classification of repetitive elements of teak genome is provided in Table 5. The simple repeats were dominant occupying about 3.36% of the total genome that can aid in molecular marker development. A total of 6,976 (1.62%) retroelements, 6,518 (1.58%) long terminal repeats (LTRs), 4,431 (0.24%) DNA transposons, 2,687 (0.70%) Copia-type, and 2,499 (0.81%) Gypsy-type sequences were predicted in teak. Number of repeat elements in teak differed from other Lamiaceae members, Mentha longifolia46 and Ocimum sanctum.47,48 In M. longifolia46 LTR elements were predicted as 3,866, whereas in teak it was 6,518. In Ocimum tenuiflorum48, the percent distribution of long interspersed nuclear elements (LINEs), LTR, unclassified repeat elements and total interspersed repeats was 0.3, 11.07, 27.99, and 40.71, respectively, but in teak the distribution was 0.04, 1.58, 0.02, and 1.88 respectively. In contrast, small RNA repeats were not recorded in O. tenuiflorum47 but 0.03% observed in teak. Presence of higher number of simple repeats, retroelements and interspersed repeats are common in plants,49 which was reflected in teak genome as well. Overall, the percent distribution of repeat elements in teak genome seemed to be very low compared to pine genome, where 82% of genome is repetitive in nature.50 The sequencing methods, per cent coverage and regions covered in the genome influence the representation of repeat elements.51 The teak genome is estimated to be 317 Mb in length, and at least 11% of its sequence is observed to be made of repeat elements. Variations in repeat elements among the members of Lamiaceae could be due to the variations in chromosome number and ploidy level among the species which may reflect on evolutionary distances between genomes.43 Further, in this study, only 6% of the genome was used for masking and WGS assembly may have missed out some regions that are rich in repeated sequences.
Table 5.
Overview of repeat elements in teak genome
| Type | Number of elements | Length occupied (bp) | Percentage in genome |
|---|---|---|---|
| Retroelements | 6,976 | 5,153,927 | 1.62 |
| SINEs | 1 | 46 | 0.00 |
| LINEs | 457 | 122,681 | 0.04 |
| L1/CIN4 | 457 | 122,681 | 0.04 |
| LTR elements | 6,518 | 5,031,200 | 1.58 |
| Ty1/Copia | 2,687 | 2,212,758 | 0.70 |
| Gypsy/DIRS1 | 2,499 | 2,564,424 | 0.81 |
| DNA transposons | 4,431 | 749,766 | 0.24 |
| hobo-Activator | 647 | 139,917 | 0.04 |
| Tc1-IS630-Pogo | 1,298 | 216,636 | 0.07 |
| Tourist/Harbinger | 285 | 70,800 | 0.02 |
| Unclassified | 242 | 63,153 | 0.02 |
| Total interspersed repeats | − | 5,966,846 | 1.88 |
| Small RNA | 158 | 104,298 | 0.03 |
| Satellites | 1 | 54 | 0.00 |
| Simple repeats | 253,260 | 10,655,131 | 3.36 |
| Low complexity | 46,994 | 2,328,770 | 0.73 |
| Total bases masked | 19,046,577 | 6.00 | |
3.3. Annotation and gene prediction
Functional annotation, process of identifying sequence similarity to other known genes or proteins, in teak was carried out using the hard masked draft genome for gene prediction. A total of 36,172 proteins were predicted of which 31,126 (86%) proteins were annotated against Viridiplantae (Supplementary Table S5) and 5,046 proteins were unannotated. In this study, length of the longest and shortest annotated protein sequence was 5,648 and 32 amino acids, respectively. The number of predicted genes is in accordance with Lamiaceae members, Mentha and Ocimum, except for one cultivar of O. tenuiflorum, where 53,480 genes were predicted.46–48 List of species considered for pathway analysis by homology based alignment of the T. grandis draft genome is given in the Supplementary Table S6. The proteins with >30% identity cut off were taken for pathway analysis. Overall, 344 taxa had similarity hits when searched against Uniprot Viridiplantae protein database for similarity using BLASTP programme with an e-value of e-10. Fifteen plant species showed high homology is listed out in Supplementary Table S7. Among the 31,126 predicted protein-coding genes, 17,353 genes (55%) showed high similarity to Erythranthe guttata 2,478 to Coffea canephora, 1,922 to Solanum tuberosum and 1,049 to Vitis vinifera. Highest per cent of similarity in the predicted coding genes with E. guttata (17,353 genes) could reflect genetic relationship of this genus with teak as both these belonging to the class Lamioideae.52
Gene ontology analysis revealed that 48.08% genes related to molecular functions (Fig. 2a), 34.35% genes related to cellular components (Fig. 2b) and 12.56% involved in biological processes (Fig. 2c). In terms of biological processes, the major categories were transcription, regulation of transcription and metabolic and defense processes. Cellular component consisted of a major portion of integral component of membrane, followed by nucleus and cytoplasm components. In terms of molecular function, the top three GO terms were ATP binding, DNA binding and metal ion binding activities. Classification of all the protein sequences grouped under five categories such as metabolism, cellular processes, environmental information processing, genetic information processing and organismal systems. Metabolism related sequences were represented in the highest number, in which genes were representing carbohydrate metabolism (1,100) ranked first, followed by amino acid metabolism (643) (Supplementary Table S8). The top five pathways were involved in plant-pathogen interaction, plant hormone signal transduction, carbon metabolism, ribosomes, and protein processing (Supplementary Table S9).
Figure 2.

Characterization of teak genome sequence by gene ontology categories: (a) Biological process; (b) Molecular function; (c) Cellular component.
Teak is known for its natural resistance against various decaying agents and is highly durable. Many biochemical studies on teak wood indicated the role of several secondary metabolites and phenolic compounds including flavanoids, alkaloids, terpenoids, quinines and tannins that play a major role for its durability.53,54 This study has identified a total of 615 gene sequences that directly code for enzymes involved in the synthesis of specialized secondary metabolites (363 genes) and biosynthesis of terpenoids and polyketides (252 genes). Lipid metabolism related genes were also represented in higher number (542) (Supplementary Table S8). The colour of wood is associated with extractive content, and is a useful parameter to estimate the durability of heartwood. Identification of several genes responsible for the production of above compounds in the teak genome would pave way for understanding the basis of natural resistance of teak timber. Recently, it was shown that heartwood specific transcriptome signatures were responsible for the presence of particular secondary metabolites through functional genomics studies in Santalum album.55 Similar approaches would provide further insights into secondary wood formation in teak.
3.4. The frequency and distribution of SSR types in teak genome
In this study, totally 2,993 scaffolds amounting to 317.5 Mbp were examined for SSRs, of which 2,938 sequences were harbouring SSRs. Further, 2,846 sequences had more than one SSR and 11,255 SSRs were in compound form. Different types of SSR recorded in the teak genome are shown in Table 6. A total of 182,712 SSR motifs were identified, where perfect SSRs were represented in maximum numbers (170,574) with an overall frequency of 537.15 loci/Mbp accounting for 93% of SSRs. Compound, complex and interrupted types constituted 7% of the total SSRs, where interrupted complex type was the least in number. Among the pure repeat motifs, mononucleotide repeats were represented in maximum counts (88,766) followed by di (81,215), tri (14,654), tetra (8,086), penta (1,967) and hexanucleotides (1,161) (Table 7). Predominant (>1,000) repeat times were 12–22 for mononucleotides, 7–16 for dinucleotides, 5–8 for trinucleotides, 4–7 for tetranucleotides and 4 for pentanucleotides. The major repeat motifs with over 5000 loci were (A)n, (T)n, (C)n, (G)n, (AC)n, (AT)n, (AG)n, (GT)n and (CT)n. Nine trinucleotide, four tetranucleotide and two pentanucleotide motifs were predominant (Supplementary Table S10). (AT)n repeat motif with frequency of 131.2 loci/Mb was the most predominant dinucleotide SSRs, accounting for over 51.3% of the total dinucleotide SSRs. Primers were designed for 86,854 SSRs which had sufficient left and right sequences (Supplementary Table S11). Presence of large number of short repeat type SSR loci in the teak genome may be due to the higher genomic mutation rate and long evolutionary history of the genus.56
Table 6.
Characteristics of six types of SSRs in teak genome
| SSR type | Total counts | Total length (bp) | Average length (bp) | Frequency (loci/Mb) | Density (bp/Mb) |
|---|---|---|---|---|---|
| cd | 6,309 | 248,207 | 39.34 | 19.87 | 781.63 |
| cx | 227 | 13,835 | 60.95 | 0.71 | 43.57 |
| icd | 4,049 | 170,803 | 42.18 | 12.75 | 537.88 |
| icx | 670 | 43,668 | 65.18 | 2.11 | 137.51 |
| ip | 883 | 34,519 | 39.09 | 2.78 | 108.7 |
| p | 170,574 | 3,006,200 | 17.62 | 537.15 | 9,466.82 |
Table 7.
The number, length, frequency and density of six different types of SSRs
| Nucleotide | Total counts | Total length (bp) | Average length (bp) | Frequency (loci/Mb) | Density (bp/Mb) | SSRs in the whole genome (%) |
|---|---|---|---|---|---|---|
| Mononucleotide | 88,766 | 1,321,753 | 14.89 | 279.53 | 4,162.3 | 45.32 |
| Dinucleotide | 81,215 | 1,664,278 | 20.49 | 255.75 | 5,241 | 41.4 |
| Trinucleotide | 14,654 | 286,074 | 19.52 | 46.15 | 900.88 | 7.48 |
| Tetranucleotide | 8,086 | 146,728 | 18.15 | 25.46 | 462.06 | 4.13 |
| Pentanucleotide | 1,967 | 42,960 | 21.84 | 6.19 | 135.29 | 1 |
| Hexanucleotide | 1,161 | 29,724 | 25.6 | 3.66 | 93.604 | 0.59 |
Cd, compound; cx, complex; icd, interrupted compound; icx, interrupted complex; ip, imperfect; p, perfect.
3.5. Selection of polymorphic SSRs and validation
SSRs have become powerful markers for population genetic analysis, QTL mapping and other related genetic and genomic studies.57 The conventional methods for SSR genotyping are labour intensive, time consuming and costly, especially for tree species that lack DNA sequence information in the public databases. The recent advances in NGS methods offer rapid identification of repeat size variations by sequencing. Five teak accessions were re-sequenced with coverage level of 7.3x–11.6x (Table 8). The PSR tool was used to compare sequence variants among the five assemblies against the SSR sequences of reference genome. Among the 170,574 perfect SSRs found in teak genome, 16,252 showed polymorphisms across these genotypes and primer pairs were developed for 13,007 SSRs (Supplementary Table S12). Heterozygous and homozygous conditions at each microsatellite locus across all genotypes were detected by the comparative module (PSR poly finder).
Table 8.
Read Statistics of the teak samples sequenced at low depth coverage for identification of polymorphic SSRs
| Accession ID | Sample code | Total raw reads (bp) | Total processed reads (bp) | Total reference covered (%) | Coverage (×) |
|---|---|---|---|---|---|
| 1 | NR | 22,503,220 | 21,315,714 | 90.1 | 9.6 |
| 3 | WR | 28,219,627 | 26,462,008 | 90.6 | 11.6 |
| 4 | DI | 19,670,649 | 18,335,390 | 87.6 | 7.8 |
| 5 | HI | 17,029,396 | 16,043,025 | 85.8 | 7.3 |
| 6 | TP | 18,838,068 | 18,451,073 | 94.3 | 8.2 |
| Average | 8.9 | ||||
Gel electrophoresis of all the 10 primer pairs developed for polymorphic SSRs generated by the PSR software produced perfect banding pattern and no optimization of primer annealing temperature were required. All the amplified loci generated polymorphism except one locus as in the PSR results.29 Validation of more number of primer pairs in efficient allele separation systems like capillary electrophoresis would strengthen the SSR marker development. These results showed that identification of polymorphic SSRs by sequencing is highly cost efficient and rapid compared to conventional methods SSR identification.
3.6. Phylogeny and divergence time estimation
The Lamiaceae (Labiatae) family contains 236 genera and over 7,000 species, and is one of the largest families of seed plants.3 The family Verbenaceae is closely related to Lamiaceae and differentiated mainly on the basis of terminal (Verbenceae)/gynobasis (Lamiaceae) style with difficulties in separating members of one family from the other.58 Previously, several genera from Verbenaceae were transferred to Lamiaceae including Tectona2,59 but the systematic position of genus Tectona still lacks clarity. Out of 236 genera of Lamiaceae, 226 were placed under seven subfamilies (Ajugoideae, Lamioideae, Nepetoideae, Prostantheroideae, Scutellarioideae, Symphorematoideae and Viticoideae) and 10 genera were listed as Incertae sedis3 (of ‘uncertain placement’) by considering morphology, secondary metabolites and molecular phylogeny.60–64 However, a recent study on chloroplast phylogeny proposed additional three subfamilies in Lamiaceae to encompass eight genera of Incertae sedis, leaving Tectona and Callicarpa unassigned.4 In the present study, potential phylogenetic plastid marker sequences, ycf2 and psbB were used to disentangle the taxonomic position of the genus Tectona. Although several different genes were used for phylogeny analysis of teak, the ycf2 and psbB genes were not reported so far. The plastid gene psbB which codes for the core protein of Photosystem II, has the highest level of translation among the chloroplast genes and provides an excellent opportunity to investigate an unusual evolutionary situation.65 Phylogenetic utility of psbB gene sequences has been tested in many plant families under the Order Lamiales such as Lamium,66Chelonopsis,67Salvia68 and Prosanthera69 among others. Similar to psbB, studies on molecular systematics have undoubtedly proved that ycf gene is more variable than matK in many taxa to resolve the phylogeny issues.70,71 This gene harbours highest level nucleotide genetic diversity among the angiosperm plastid genomes.72 As the number of entries of Lamiaceae members in the public domain is very less, combination of two genes, ycf2 and psbB sequences generated phylogeny tree with 16 Lamiaceae species. Inclusion of other genes (rbcL and psaA) resulted in phylogenetic tree with lesser number (10–14) of Lamiaceae species (data not shown). The resulted phylogenetic tree retained the previously reported uncertain taxonomic status of the genus Tectona, which formed a separate sister clade to the clades comprising of Laminoideae, Ajugoideae, Nepetoideae, Scutellarioideae and Premonoideae (Fig. 3). Monophyly of all the sub families under Lamiaceae were also reported.4
Figure 3.

Bayesian tree generated by analysis of two plastid sequences psb and ycf2. Posterior probabilities are shown above the branches. Scale bar specifies mean branch length. Six major clades representing subfamilies of the Lamiaceae family are indicated.
Earlier classifications on morphology considered Tectona under tribe Tectoneae in subfamily Viticoideae.73–76 Molecular phylogeny analysis and novel combination of morphological features delineated Tectona to be an earliest diverging lineage in Lamiaceae. The phylogenetic studies of Lamiaceae family deduced T. grandis to be an early diverging lineage.4 The time estimated in the present study confirmed the early origin and divergence of the genera around 21.4508 Mya [95% highest posterior density (HPD): 10.11–34.52 Mya] (Fig. 4). Most of the genera of the family Lamiaceae were found to have a Miocene origin. Recently, two new subfamilies have been proposed to Lamiaceae viz. Callicarpoideae and Tectonoideae with Tectona as a monotypic taxon.77 Accordingly, assigning Tectona to a monogeneric subfamily Tectonoideae need to be considered, however it demands an extensive sampling within Lamiaceae and multigene phylogeny analysis to provide an appropriate taxonomical position.
Figure 4.
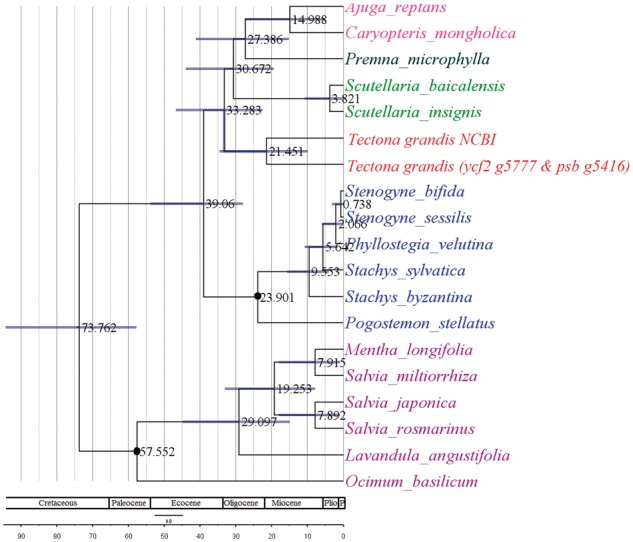
Chronogram of Lamiaceae (genus Tectona) based on two plastid sequences psb and ycf2, estimated from secondary calibration strategies as implemented in BEAST. Calibration points are indicated with black dots. Node bar indicates 95% HPD interval for node ages. Geological time scale is given in Mya.
4. Conclusion
Shrinkage of natural populations of teak in its native locations and worldwide increase of managed plantations demand conservation of native forests, which are critical in providing the best possible alleles to maintain genetic diversity. Captive plantations inherently have narrow genetic base, limited gene flow, and exist in non-native environments, and these characteristics often significantly alter the evolutionary trajectory leading to decrease in population fitness. Conservation and maintenance of wild progenitors are increasingly important for genetic improvement programmes. Thus, full genome sequence of teak was developed and an attempt for the functional analyses of genome components was carried out to use it as a tool for conservation and tree breeding programmes. The polymorphic DNA markers developed in this study will propel the genetic and genomic research in teak, hitherto unavailable for the highly valuable timber yielding tropical hardwood species. The draft genome of teak along with a large number of markers will benefit various explorative studies including, genetic basis of wood properties, pest tolerance, adaptive traits, germplasm movement and genetic resource conservation.
Supplementary Material
Acknowledgements
We are thankful to the State Forest Departments of Kerala, Tamil Nadu and Karnataka for their permission to collect teak samples. The financial support received from Department of Biotechnology, Government of India (No. BT/PR7143/PBD/16/1011/2012) is gratefully acknowledged.
Conflict of interest
None declared.
References
- 1. Tewari D.N. 1992, A Monograph on Teak (Tectona grandis Linn. f.), Dehra Dun, India: International Book Distributors
- 2. Thorne R. F. 1992, Classification and geography of the flowering plants, Bot. Rev., 58, 225–327. [Google Scholar]
- 3. Harley R. M., Atkins S., Budantsey A. L.. 2004, Labiatae In: Kubitzki K., Kadereit J. W. (eds) Families and Genera of Vascular Plants. Flowering Plants. Dicotyledons ∼ Lamiales (except Acanthaceae Including Avicenniaceae, vol. 7 Berlin: Springer, pp. 167–275. [Google Scholar]
- 4. Li B., Cantino P. D., Olmstead R. G.. 2016, A large-scale chloroplast phylogeny of the Lamiaceae sheds new light on its subfamilial classification, Sci. Rep., 6, 3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kollert W., Kleine M., eds. 2017, The Global Teak Study. Analysis, Evaluation and Future Potential of Teak Resources, IUFRO World Series, 36. Vienna. pp 108.
- 6. Kollert W., Cherubini L.. 2012, Teak resources and market assessment 2010, FAO Planted Forests and Trees Working Paper, FP/47/E, Rome.
- 7. Deb J. C., Phinn S., Butt N., Mcalpine C. A.. 2017, Climatic-induced shifts in the distribution of teak (Tectona grandis) in tropical Asia: implications for forest management and planning, Environ. Manag., 60, 422–35. [DOI] [PubMed] [Google Scholar]
- 8. Hansen O. K., Changtragoon S., Ponoy B., Lopez J., Richard J., Kjær E. D.. 2017, Worldwide translocation of teak—origin of landraces and present genetic base, Tree Genet. Genomes, 13, 87. [Google Scholar]
- 9. Keiding H., Wellendorf H., Lauridsen E. B.. 1986, Evaluation of an International Series of Teak Provenance Trials. Humlebaek: Danida Forest Seed Centre. [Google Scholar]
- 10. Kjaer E. D., Lauridsen E. B., Wellendorf H.. 1995, Second Evaluation of an International Series of Teak Provenance Trails. Humlebaek: Danida Forest Seed Centre. [Google Scholar]
- 11. Chaix G., Monteuuis O., Garcia C., et al. 2011, Genetic variation in major phenotypic traits among diverse genetic origins of teak (Tectona grandis L.f.) planted in Taliwas, Sabah, East Malaysia, Ann. For. Sci., 68, 1015–26. [Google Scholar]
- 12. Monteuuis O., Goh D. K. S., Garcia C., Alloysius D., Gidiman J., Bacilieri R., Chaix G.. 2011, Genetic variation of growth and tree quality traits among 42 diverse genetic origins of Tectona grandis planted under humid tropical conditions in Sabah, East Malaysia, Tree Genet. Genomes, 7, 1263–75. [Google Scholar]
- 13. Callister A. N. 2013, Genetic parameters and correlations between stem size, forking, and flowering in teak (Tectona grandis), Can. J. For. Res., 43, 1145–50. [Google Scholar]
- 14. Monteuuis O., Goh D. K. S.. 2015, Field growth performances of teak genotypes of different ages clonally produced by rooted cuttings, in vitro microcuttings, and meristem culture, Can. J. For. Res., 45, 9–14. [Google Scholar]
- 15. Gunaga R. P., Vasudeva R.. 2002, Variation in flowering phenology in a clonal seed orchard of teak, J. Tree Sci., 21, 1–10. [Google Scholar]
- 16. Nicodemus A., Nagarajan B., Narayanan C.. 2005, RAPD variation in Indian teak populations and its implications for breeding and conservation In: Bhat K.M., Nair K.K.N., Bhat K.V., Muralidharan E.M., Sharma J.K. (eds.), Quality Timber Products of Teak from Sustainable Forest Management. Kerala Forest Research Institute, Yokohama: India and International Tropical Timber Organization, pp. 321–30. [Google Scholar]
- 17. Shrestha M. K., Volkaert H., Straeten D. V. D.. 2005, Assessment of genetic diversity in Tectona grandis using amplified fragment length polymorphism markers, Can. J. For. Res., 35, 1017–22. [Google Scholar]
- 18. Sreekanth P. M., Balasundaran M., Nazeem P. A., Suma T. B.. 2012, Genetic diversity of nine natural Tectona grandis L.f. populations of the Western Ghats in Southern India, Conserv. Genet., 13, 1409–19. [Google Scholar]
- 19. Fofana I. J., Ofori D., Poitel M., Verhaegen D.. 2009, Diversity and genetic structure of teak (Tectona grandisL.f.) in its natural range using DNA microsatellite markers, New Forests, 37, 175–95. [Google Scholar]
- 20. Hansen O. K., Changtragoon S., Ponoy B., et al. 2015, Genetic resources of teak (Tectona grandis Linn. f.)—strong genetic structure among natural populations, Tree Genet. Genomes, 11, 802. [Google Scholar]
- 21. Galeano E., Vasconcelos T. S., Vidal M., Mejia-Guerra M. K., Carrer H.. 2015, Large-scale transcriptional profiling of lignified tissues in Tectona grandis, BMC Plant Biol., 15, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Diningrat D. S., Widiyanto S. M., Pancoro A., et al. 2015, Transcriptome of teak (Tectona grandis, L.f) in vegetative to generative stages development, J. Plant Sci., 10, 1–14. [Google Scholar]
- 23. Doyle J. J., Doyle J. L.. 1990, Isolation of plant DNA from fresh tissue, Focus, 12, 13–5. [Google Scholar]
- 24. Kajitani R., Toshimoto K., Noguchi H., et al. 2014, Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads, Genome Res., 24, 1384–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zimin A., Marçais G., Puiu D., Roberts M., Salzberg S., Yorke J.. 2013, The MaSuRCA genome assembler, Bioinformatics, 29, 2669–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Boetzer M., Henkel C. V., Jansen H. J., Butler D., Pirovano W.. 2011, Scaffolding pre-assembled contigs using SSPACE, Bioinformatics, 27, 578–9. [DOI] [PubMed] [Google Scholar]
- 27. Simpson J. T., Durbin R.. 2012, Efficient de novo assembly of large genomes using compressed data structures, Genome Res., 22, 549–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stanke M., Diekhans M., Baertsch R., Haussler D.. 2008, Using native and syntenically mapped cDNA alignments to improve de novo gene finding, Bioinformatics, 24, 637–44. [DOI] [PubMed] [Google Scholar]
- 29. Cantarella C., D’Agostino N.. 2015, PSR: polymorphic SSR retrieval, BMC Res. Notes, 8, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Thompson J. D., Gibson T. J., Plewniak F., Jeanmougin F., Higgins D. G.. 1997, The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools, Nuc. Acids Res., 25, 4876–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hall T. A. 1999, BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT, Nucleic Acids Symp. Ser., 41, 95–8. [Google Scholar]
- 32. Darriba D., Taboada G. L., Doallo R., Posada D.. 2012, JModelTest 2: more models, new heuristics and parallel computing, Nat. Methods, 9, 772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Akaike H. 1974, A new look at statistical model identification, IEEE Trans. Automat. Contr., 19, 716–23. [Google Scholar]
- 34. Ronquist F., Huelsenbeck J. P.. 2003, MRBAYES 3: bayesian phylogenetic inference under mixed models, Bioinformatics, 19, 1572–4. [DOI] [PubMed] [Google Scholar]
- 35. Drummond A. J., Suchard M. A., Xie D., Rambaut A.. 2012, Bayesian phylogenetics with BEAUti and the BEAST 1.7, Mol. Biol. Evol., 29, 1969–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Roy T., Lindqvist C.. 2015, New insights into evolutionary relationships within the subfamily Lamioideae (Lamiaceae) based on pentatricopeptide repeat (PPR) nuclear DNA sequences, Amer. J. Bot., 102, 1721–35. [DOI] [PubMed] [Google Scholar]
- 37. Drummond A. J., Ho S. Y. W., Phillips M. J., Rambaut A.. 2006, Relaxed phylogenetics and dating with confidence, PLoS Biol., 4, e88–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yamamura Y., Kurosaki F., Lee J. B.. 2017, Elucidation of terpenoid metabolism in Scoparia dulcis by RNA-seq analysis, Sci. Rep., 7, 43311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Holliday J. A., Aitken S. N., Cooke J. E. K., et al. 2017, Advances in ecological genomics in forest trees and applications to genetic resources conservation and breeding, Mol. Ecol., 26, 706–17. [DOI] [PubMed] [Google Scholar]
- 40. Ohri D., Kumar A.. 1986, Nuclear DNA amounts in some tropical hardwoods, Caryologia, 39, 303–7. [Google Scholar]
- 41. Ling H. Q., Zhao S., Liu D., Wang J., et al. 2013, Draft genome of the wheat A-genome progenitor Triticum urartu, Nature, 496, 87–90. [DOI] [PubMed] [Google Scholar]
- 42. Britten R. J. 2010, Transposable element insertions have strongly affected human evolution, Proc. Natl. Acad. Sci. U. S. A., 107, 19945–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mehrotra S., Goyal V.. 2014, Repetitive sequences in plant nuclear DNA: types, distribution, evolution and function, Genomics Proteomics Bioinformatics, 12, 164–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dover G. A. 1986, Molecular drive in multigene families: how biological novelties arise, spread and are assimilated, Trends Genet., 2, 159–65. [Google Scholar]
- 45. Xu H., Song J., Luo H., et al. 2016, Analysis of the genome sequence of the medicinal plant, Salvia Miltiorrhiza, Mol. Plant, 9, 949–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vining K. J., Johnson S. R., Ahkami A., et al. 2017, Draft genome sequence of Mentha longifolia and development of resources for mint cultivar improvement, Mol. Plant., 10, 323–39. [DOI] [PubMed] [Google Scholar]
- 47. Rastogi S., Kalra A., Gupta V., et al. 2015, Unravelling the genome of Holy basil: an “incomparable” “elixir of life” of traditional Indian medicine, BMC Genomics, 16, 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Upadhyay A. K., Chacko A. R., Gandhimathi A., et al. 2015, Genome sequencing of herb Tulsi (Ocimum tenuiflorum) unravels key genes behind its strong medicinal properties, BMC Plant Biol., 15, 212., [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Weiss-Schneeweiss H., Leitch A. R., Jamie J., Jang T. S., Macas J.. 2015, Employing next generation sequencing to explore the repeat landscape of the plant genome In: Hörandl E., Appelhans M. (eds). Next Generation Sequencing in Plant Systematic, Regnum Vegetabile 157. Königstein, Germany: Koeltz Scientific Books, pp. 1–25. [Google Scholar]
- 50. Neale D. B., Wegrzyn J. L., Stevens K. A., et al. 2014, Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies, Genome Biol. , 15, R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Parween S., Nawaz K., Roy R., et al. 2015, An advanced draft genome assembly of a desi type chickpea (Cicer arietinum L.), Sci. Rep., 5, 12806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Refulio-Rodriguez N. F., Olmstead R. G.. 2014, Phylogeny of Lamiidae, Am. J. Bot., 101, 287–99. [DOI] [PubMed] [Google Scholar]
- 53. Francisco A. M., Rodne Y. L., Rosa M. V., Clara N., Jose M. G. M.. 2008, Bioactive apocarotenoids from Tectona grandis, Phytochemistry, 69, 2708–15. [DOI] [PubMed] [Google Scholar]
- 54. Lacret R., Varela R. M., Molinillo J. M. G., Nogueiras C., Macías F. A.. 2012, Tectonoelins, new norlignans from a bioactive extract of Tectona grandis, Phytochem. Lett., 5, 382–6. [Google Scholar]
- 55. Celedon J. M., Bohlmann J.. 2018, An extended model of heartwood secondary metabolism informed by functional genomics, Tree Physiol., 14, 1–9. [DOI] [PubMed] [Google Scholar]
- 56. Toth G., Gaspari Z., Jurka J.. 2000, Microsatellites in different eukaryotic genome: survey and analysis, Genome, Res., 10, 967–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vieira M. L. C., Santini L., Diniz A. L., Munhoz C. D. F.. 2016, Microsatellite markers: what they mean and why they are so useful, Genet. Mol. Biol., 39, 312–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pullaiah T., Sri Rama Murthy K., Karuppusamy S.. 2007, Flora of Eastern Ghats, Vol. 3 New Delhi: Regency Publications. [Google Scholar]
- 59. Cantino P. D., Harley R. M., Wagstaff S. J.. 1992, Genera of Labiatae: status and classification In: Harley R.M., Reynolds T. (eds). Advances in Labiatae Science, Kew, UK: Key Botanic Garden, pp. 511–22. [Google Scholar]
- 60. Cantino P. D., Olmstead R. G., Wagstaff S. J.. 1997, A comparison of phylogenetic nomenclature with the current system: a botanical case study, Syst. Biol., 46, 313–31. [Google Scholar]
- 61. Wagstaff S. J., Olmstead R. G.. 1997, Phylogeny of Labiatae and Verbenaceae inferred from rbcL sequences, Syst. Bot., 22, 165–79. [Google Scholar]
- 62. Wagstaff S. J., Hickerson L., Spangler R., Reeves P. A., Olmstead R. G.. 1998, Phylogeny in Labiatae s. l., inferred from cpDNA sequences, Plant Syst. Evol., 209, 265–74. [Google Scholar]
- 63. Alvarenga S. A. V., Gastmans J. P., Rodrigues G. D. V., Moreno P. R. H., Emerenciano V. D. P.. 2001, A computer-assisted approach for chemotaxonomic studies - diterpenes in Lamiaceae, Phytochemistry, 56, 583–95. [DOI] [PubMed] [Google Scholar]
- 64. Bramley G. L. C., Forest F., De Kok R. P. J.. 2009, Troublesome tropical mints: re-examining generic limits of Vitex and relations (Lamiaceae) in South East Asia, Taxon, 58, 500–10. [Google Scholar]
- 65. Morton B. R., Levin J. A.. 1997, The atypical codon usage of the plant psbA gene may be the remnant of an ancestral bias, Proc. Nat. Aca. Sci. U.S.A., 94, 11434–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bendiksby M., Brysting A. K., Thorbek L., Gussarov G., Ryding G.. 2011, Molecular phylogeny and taxonomy of genus Lamium L (Lamiaceae): Disentangling origins of presumed allotetraploids, Taxon, 60, 986–1000., [Google Scholar]
- 67. Xiang C.-L., Zhang Q., Scheen A.-C., Cantino P. D., Funamoto T., Peng H.. 2013, Molecular phylogenetics of Chelonopsis (Lamiaceae: gomphostemmateae) as inferred from nuclear and plastid DNA and morphology, Taxon, 62, 375–86. [Google Scholar]
- 68. Jenks A. A., Walker J. B., Kim S. C.. 2011, Evolution and origins of the Mazatec hallucinogenic sage, Salvia divinorum (Lamiaceae): a molecular phylogenetic approach, J. Plant Res., 124, 593–600. [DOI] [PubMed] [Google Scholar]
- 69. Conn B. J., Wilson T. C., Henwood M. J., Proft K.. 2013, Circumscription and phylogenetic relationships of Prostanthera densa and P. marifolia (Lamiaceae), Telopea, 15, 149–64. [Google Scholar]
- 70. Neubig K. M., Whitten W. M., Carlsward B. S., Blanco M. A., Endara L., Williams N. H., Moore M.. 2009, Phylogenetic utility of ycf1 in orchids: a plastid gene more variable than matK, Plant Syst. Evol., 277, 75–84. [Google Scholar]
- 71. Dong W., Xu C., Li C., et al. 2015, ycf1, the most promising plastid DNA barcode of land plants, Sci. Rep., 5, 8348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Dong W., Liu J., Yu J., Wang L., Zhou S.. 2012, Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding, PLoS One, 7, e35071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Briquet J. 1897, Verbenaceae In: Engler A., Prantl K. (eds). Die Naturlichen Pflanzenfamilien Teil 4. Leipzig: Engelmann, pp. 132–182. [Google Scholar]
- 74. Melchior H. 1964, A. Engler’s Syllabus Der Pflanzenfamilien, vol. 2 Berlin: Borntraeger, pp. 666. [Google Scholar]
- 75. Moldenke H. N. 1975, Notes on new and noteworthy plants LXXVII, Phytologia, 31, 28. [Google Scholar]
- 76. Takhtajan A. 1983, Outline of the classification of flowering plants (Magnoliophyta), Brittonia, 35, 254–359. [Google Scholar]
- 77. Li B., Olmstead R.. 2017, Two new subfamilies in Lamiaceae, Phytotaxa, 313, 222–6. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.