

Abstract
High ortho selectivity for Ir-catalyzed C–H borylations (CHBs) of anilines results when B2eg2 (eg = ethylene glycolate) is used as the borylating reagent in lieu of B2pin2, which is known to give isomeric mixtures with anilines lacking a blocking group at the 4-position. With this modification, high selectivities and good yields are now possible for various anilines, including those with groups at the 2- and 3-positions. Experiments indicate that ArylN(H)Beg species are generated prior to CHB and support the improved ortho selectivity relative to B2pin2 reactions arising from smaller Beg ligands on the Ir catalyst. The lowest-energy transition states (TSs) from density functional theory computational analyses have N–H···O hydrogen-bonding interactions between PhN(H)Beg and O atoms in Beg ligands. Ir-catalyzed CHB of PhN(H)Me with B2eg2 is also highly ortho-selective. 1H NMR experiments show that N-borylation fully generates PhN(Me)Beg prior to CHB. The TS with the lowest Gibbs energy was the ortho TS, in which the Beg unit is oriented anti to the bipyridine ligand.
Keywords: C–H activation, hydrogen bonding, computational chemistry, aniline, ortho functionalization
INTRODUCTION
Anilines are chemicals with important dye, pharmaceutical, agrochemical, and polymer applications.1 Aniline is commercially prepared by benzene nitration followed by hydrogenation of the nitrobenzene intermediate. Most commercially available substituted anilines are prepared by derivatization of aniline, often through electrophilic aromatic substitution (EAS).2 The NH2 group is classified as a strong ortho/para director,3 even though traditional nitration conditions give 32–49% m-nitroaniline along with the major para isomer.4 The best selectivities for EASs are typically C4 functionalizations, and most ortho-selective examples are for anilines substituted at C4. For anilines that are unsubstituted at C4, EASs generate significant quantities of para isomers even in some of the most ortho-selective methods.5 The best traditional synthetic method for aniline ortho functionalization is directed ortho metalation (DoM) of carbamate derivatives followed by addition of an electrophile to the ortho carbanion.6 This approach requires conversion of the aniline to the carbamate, which is removed after the reaction if aniline products are desired.
Even though DoM reactions are remarkably powerful,7 catalytic methods can exhibit complementary selectivities and functional group tolerance.8 There are several examples of catalytic ortho functionalizations of aniline. Most require the installation of a directing group prior to C–H functionalization. Removal of the directing group is required to restore the nitrogen functionality to that in the aniline starting material.9–11 While the NH2 group would be untouched in ideal catalytic ortho functionalization of primary anilines, the next most desirable process is one where there is no trace in the product of any in situ modification of the amino group during catalysis.
Traceless Ir-catalyzed C–H borylation (CHB) of primary anilines has been described in the literature.12 C–H borylation is a synthetic method where sp2, sp3, and sp C–H bonds are converted to C–B bonds.13–15 With few exceptions,16,17 most examples require a catalyst, and metal-catalyzed CHBs have been reported for a number of transition metals.13,18–20 Some of the earliest reports of metal-catalyzed CHBs of arene C(sp2)–H bonds indicated that the least hindered C–H bonds were generally more reactive.21–23 This feature became a hallmark of Ir-catalyzed CHBs because the regioselectivities generally complement those found in EAS and DoM as well as the regioselectivities in early examples of catalytic intra- and intermolecular C–H functionalizations that are ortho-selective.24,25
The first report of catalytic ortho CHBs relied on classic chelate-directed mechanisms where a substrate functional group binds to a vacant metal site.26–28 Directing effects of this type have been branded as “inner-sphere”.29 Other inner-sphere approaches, like relay-directed ortho CHBs of silylated phenols and anilines, where reversible Si–H oxidative addition to Ir was proposed to direct borylation, were also developed.30
Mechanisms for ortho-directed CHBs where the metal center is not a directing element have been proposed.31,32 In line with Taube’s definition, these are defined as “outer-sphere” mechanisms.29,33 Examples of outer-sphere direction in ortho CHBs include Lewis acid–base,34–36 hydrogen bonding,12,33,37 electrostatic interactions,38 and an example where both inner-and outer-sphere mechanisms are plausible.39 The electrostatic mechanism is a more subtle variant of the ion-pairing mechanisms proposed by Phipps and co-workers in recently designed meta-selective CHBs,40 which complements other meta-selective CHBs where outer-sphere mechanisms are proposed to account for selectivity.34,37,41
Kanai and Kuninobu recently disclosed ortho CHBs of aniline and phenol derivatives (Scheme 1).36 The bipyridine with the best selectivity is not commercially available. It has an electron-withdrawing aryl group at the bipyridine 5-position and was synthesized from commercially available precursors.42 To achieve ortho CHB of primary anilines, thiomethyl methylene (CH2SMe) and acyl groups must be attached to N. While this approach provides excellent ortho selectivities, ligand synthesis and the additional synthetic steps to add and subsequently remove functional groups at N are unappealing if ortho-borylated primary anilines are the desired product.
Scheme 1.

Comparison of the Kanai–Kuninobu CHB of N-Acylated Anilines with This Work36,43,44
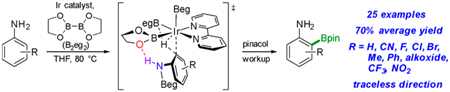
The previously reported traceless CHBs of primary anilines built on an initial report of ortho CHBs of N-Boc-anilines.12,33 For these aryl carbamates, experiment and theory were consistent with an outer-sphere mechanism involving N–H··· O hydrogen bonding between the aniline substrate and an Ir–Bpin ligand giving rise to the ortho selectivity.33 As shown in Figure 1, previous CHBs required C4 substituents larger than H to achieve high ortho selectivity.12,33 Additionally, substitution at C2 was deleterious to ortho selectivity. Given the fact that C–B bonds can be readily converted to a host of functional groups and the aforementioned limited scope of previous ortho-directed CHBs of anilines, a method overcoming these shortcomings would be highly desirable.
Figure 1.

Proposed transition states for ortho borylations of anilines and phenols.
Traceless ortho-directed CHB of phenols was also recently described.38 The initial CHB substrates were phenol O-boronate esters (ArOBpin, pin = pinacolate). Experimental and computational studies pointed to transition state (TS) stabilization arising from electrostatic interactions between the bipyridine bound to Ir and OBpin of the phenol boronate ester. Like previous aniline borylations, 4-substituents larger than H were necessary to achieve synthetically useful ortho selectivity.
An in silico redesign of the catalyst predicted that the ortho CHB transition state could be significantly stabilized if the Bpin groups on Ir and the phenol boronate ester were replaced with Beg (eg = ethylene glycolate). Indeed, this led to exquisite ortho selectivities for Ir-catalyzed CHBs of phenols when the diboron reagent B2eg2 was used in lieu of HBpin. This raised the question of whether ortho selectivities for aniline CHBs could be similarly improved using B2eg2. If the ortho selectivities indeed improve, is the transition state stabilization due to electrostatic interactions or enhanced hydrogen bonding? This paper addresses these questions using experiment and theory synergistically.
RESULTS AND DISCUSSION
Ir-catalyzed CHB of aniline with B2eg2 was used to optimize the reaction conditions. First, a tetrahydrofuran (THF) solution of aniline, 0.5 equiv of B2eg2, and 0.5 mol % [Ir(OMe)(cod)]2 was briefly heated to generate PhN(H)Beg, which was verified by 11B/1H NMR spectroscopy. Then NEt3, 4,4′-di-tBu-2,2′-bipyridine (dtbpy), and additional B2eg2 and [Ir(OMe)(cod)]2 were added, and the resulting solution was heated at 80 °C until borylation ceased. The best results were obtained with a 2.5 mol % loading of [Ir(OMe)(cod)]2, 5 mol % dtbpy, 2.0 equiv of B2eg2, and 2.0 equiv of NEt3. When CHB was complete, the eg group was transesterified by treating the reaction mixture with 3.0 equiv of pinacol, and the more stable Bpin product was purified and isolated in 67% yield. Conversion to products suffered at lower catalyst loadings; however, high regioselectivity was achieved. The regioselectivities for aniline CHBs with B2pin2 and B2eg2 are compared in Scheme 2. To avoid significant diborylation, the control reaction used less B2pin2 and a shorter reaction time. Notably, the 2.7:1.8:1 ortho:meta:para isomer ratio for CHB with B2pin2 is similar to the ratio previously reported for Ir-catalyzed aniline CHB with HBpin (ortho:meta:para = 2.3:1.5:1).12 While the major regioisomer is the ortho product, which suggests some favorable interactions for ortho CHB with B2pin2, substantial quantities of meta and para CHB products dampen the synthetic utility. In contrast, B2eg2, which is easily prepared from commercially available (OH)2B–B(OH)2 and ethylene glycol, provides exquisite ortho selectivity.
Scheme 2.

Aniline CHBs with B2pin2 and B2eg2
aAt a 0.5 mmol scale, 67% yield was obtained. At a 5 mmol scale with a 1 mol % loading of [Ir(OMe)(cod)]2 (0.25 mol % in step 1 and 0.75 mol % in step 2), the yield increased to 75%.
We next assessed the substrate scope for ortho selectivity. Table 1 lists the results for 24 substrates. The catalyst loadings in Table 1 are (6 mol % Ir) higher than we usually use for borylations because the reactions were run with 0.5 mmol of aniline substrates using weighed amounts of the [Ir(OMe)-(cod)]2. When CHB of aniline was performed on a 5 mmol scale with 0.25 and 0.75 mol % loadings of the precatalyst (2 mol % Ir) in steps 1 and 2, respectively, in Scheme 2, orthoborylated product 2a was isolated in 75% yield. The average isolated yield is 71 ± 4% for substrates in Table 1. For substrate 1q, 20% diborylation contributed to the low yield of monoborylated product. The only diborylated isomers detected were 2,6-regioisomers. The reaction with substrate 1s had the lowest yield, but it is the first metal-catalyzed CHB of a nitro-containing substrate that gives more than trace quantities. In crude reaction mixtures, CHB was detected only at sites ortho to NH2.
Table 1.
Ortho Borylation of Substituted Anilines with B2eg2a
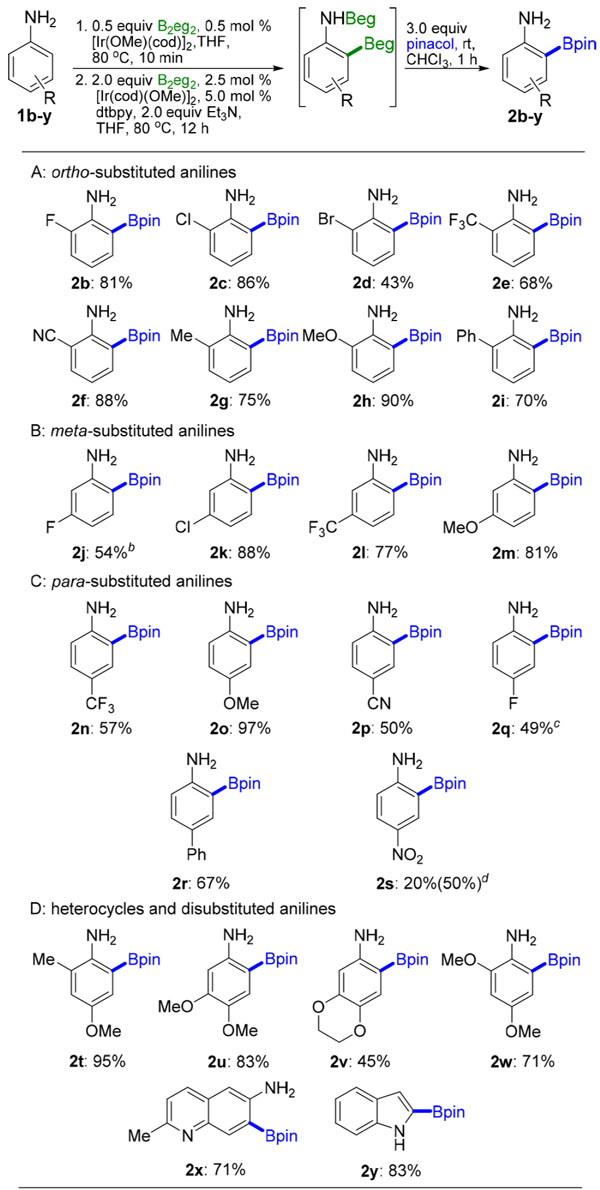 |
All reactions were carried out on a 0.5 mmol scale, and yields are reported for isolated materials after column chromatographic separation.
The other ortho-borylated isomer was observed but underwent rapid protodeboronation.
20% ortho, ortho diborylation was observed.
Isolated yield: 20%. GC conversion: 50%.
Gratifyingly, CHBs of meta-substituted anilines 1j–m did not generate 5-borylated products, as had been found for Ircatalyzed CHBs of anilines 1k–m with HBpin.12 The yields of ortho-borylated products from CHB of anilines 1k, 1l, and 1m increased over those previously reported from CHBs with HBpin by 46%, 24%, and 55%, respectively. Furthermore, this methodology outperforms previously reported CHB of N-Bocanilines. No CHBs of 2-substituted N-Boc-anilines have been reported, and the ortho selectivity eroded for substrates that lacked blocking groups at the 4-position. For example, CHB of 3-chloro-N-Boc-aniline provided 92% yield but exhibited an ortho:meta selectivity of only 2:1.33 In addition, this yield does not include removal of the Boc group; however, substrate 1k provided only the ortho-borylated product 2k, which was isolated in 88% yield. Substrates 1b–i underwent CHB with B2eg2 at C6 exclusively, yielding ortho-borylated products 2b–i. This stands in sharp contrast to previously reported CHBs of 2-substituted anilines with HBpin, where ortho borylation was not observed.12 Borylation of quinoline 1x proceeded smoothly, providing the 7-borylated product 2x in 71% isolated yield. Indole CHB with either B2eg2 or B2pin2 gives the 2-borylated products in comparable yields.45 Compounds 2c–i and 2t–x are new. Of these, 2g is the only structure whose boronic acid has been reported in the primary literature.46 Significantly, the transformations in Table 1 do not require installation and removal of a directing group and use dtbpy, the most commonly used ligand in Ir-catalyzed CHBs.
The improved selectivity raises the interesting question of whether its molecular origin arises from ligand–substrate electrostatic interactions or hydrogen bonding (Figure 2). To tackle this question, we turned to theory. Density functional theory (DFT) calculations used the M06 functional with the 6–31G* basis set for light atoms and the SDD basis set and core potential for Ir. The polarizable continuum model was the self-consistent reaction field method applied for the THF solvent. Compared with other systems that we have studied, transition state location was challenging. Low-energy imaginary frequencies associated with Me group rotations in the dtbpy ligands plagued calculations on the full system. Replacing the bipyridine tBu groups with Me groups made the problem more manageable, but Me rotations still generated multiple imaginary rotational frequencies when software default values for step sizes and integration grids were used. Ultimately, TSs with a single imaginary frequency corresponding to C–H scission were located. The maximum atom displacement in all of the calculated TSs exceeded software convergence thresholds. In addition, the root-mean-square (RMS) displacement exceeded the software convergence defaults in approximately half of the calculated TSs. For two of these TSs, the maximum displacement was 10 times greater than the convergence default. We mention them only in passing (vide infra). Atomic Cartesian coordinates and energies for these TSs are included in the Supporting Information (SI).
Figure 2.

Proposed transition states for ortho borylations of anilines and phenols.
Four transition states (TS1–4) were located for ortho borylation. Starting points for TS location included syn and anti orientations of the PhN(H)Beg moiety with respect to the bipyridine ligand (Figure 2) and two additional geometries with close contacts between the aniline N–H and O atoms of a Beg ligated to Ir. Figure 3 depicts the TSs and their relative Gibbs energies. TS1 is analogous to the lowest-energy transition state for phenol ortho borylation. In the other three TSs, the PhN(H)Beg H is hydrogen-bonded to a Beg O. The H···O distances in TS2, TS3, and TS4 are 2.07, 2.28, and 2.49 Å, respectively.
Figure 3.

Computed transition states for Ir-catalyzed CHB of PhN(H)Beg with B2eg2. The N–H hydrogen and the C–H hydrogen in the bond being cleaved are yellow. Dashed orange lines indicate hydrogen-bonding interactions. Grel and Hrel are ΔΔG⧧ and ΔΔH⧧ values relative to TS1, respectively. DFT calculations were performed using the M06 functional with the 6–31G* basis set for light atoms and the SDD basis set and core potential for Ir.
The ΔΔG⧧ values for TS2–4 relative to TS1 are given as Grel in Figure 3. The hydrogen-bonded TSs TS2, TS3, and TS4 are stabilized by 1.9, 2.6, and 1.7 kcal·mol−1, respectively, relative to TS1. Notably, the starting geometry for TS4 was similar to that for TS1 except that the Beg moiety was syn to the 4,4′-dimethylpyridine ligand (Figure 2). The syn PhOBeg TS has short (~3.0 Å) contacts between the Beg group and the bipyridine ligand that are reminiscent of π stacking.38 The analogous TS was not found for PhN(H)Beg. Instead, the N(H)Beg group rotated about the N–Cipso bond to engage in hydrogen bonding between the aniline proton and an Ir–Beg oxygen. A TS analogous to the syn geometry for PhOBeg was located for CHB of PhN(Me)Beg (TS8, vide infra).
The highest-energy hydrogen-bonded TS (TS4) has the longest H···O distance, but it is only 0.2 kcal·mol−1 less stable than TS2, where the H···O distance is 0.42 Å shorter. The number of heavy atoms in TS1–4 is too large to apply the level of theory that is typically used to quantify stabilization from hydrogen bonding.47 The νN–H values for N–H vibrations of the N(H)Beg group in TS2 (3533 cm−1), TS4 (3541 cm−1), and TS3 (3557 cm−1) do not correlate with distance, which is not surprising since Beg O lone pair interactions with the aniline H differ with the Beg ligand orientation. The νN–H values in TS2–4 are 62–38 cm−1 lower than that calculated for PhN(H)Beg (ν – N–H = 3595 cm−1) at the same level of theory. On the basis of the infrared shift, H···O distance, and N–H lengthening (see the SI), the hydrogen-bonding interaction is classified as a weak hydrogen bond, which is mostly electrostatic in nature.48
The TS for para CHB (TS5) was also located. Its Gibbs energy was higher than those of all of the hydrogen-bonding TSs, and it was separated from the lowest-energy TS (TS3) by 1.0 kcal·mol−1. Attempts to locate the TS for meta CHB yielded a structure with one imaginary frequency (TSmeta; see pp S43–S44 in the SI), but the respective RMS and maximum displacements were 10 and 34 times greater than the software default convergence criteria. The theoretical ΔΔG⧧ value of 1.0 kcal·mol−1 between TS3 and TS5 predicts an ortho:para ratio of 8.2:1. While the ortho:para ratio of 26:1 predicted from the calculated ΔΔH⧧ values matches experiment more closely, both the ΔΔG⧧ and ΔΔH⧧ predictions fall short of the observed experimental selectivity. This is not uncommon in combined experimental/computational studies.
The computational results predict that Beg outperforms Bpin because the N(H)Beg substituent and Beg ligands can adopt optimal hydrogen-bonding configurations with minimal steric interference. CHBs of PhN(H)-Bpin with B2eg2 and PhN(H)-Beg with B2pin2 were performed as an experimental test of this hypothesis (Scheme 3).
Scheme 3.

Aniline CHBs of PhN(H)Bpin with B2eg2 and PhN(H)Beg with B2pin2
CHB of PhN(H)Bpin exhibits the same high ortho selectivity when B2eg2 is the borylating agent as is observed for CHB of aniline with B2eg2. When the NBeg in the structure of TS3 is converted to NBpin and the syn bipyridine Me is converted to tBu, the closest C···C contact (4.31 Å) is longer than the closest C···C contact (3.96 Å) in the crystal structure of Ir-(Bpin)3(dtbpy)(coe) (coe = cyclooctene).23 Consequently, retention of high ortho selectivity for the NBpin/B2eg2 combination is not surprising.
In contrast, the ortho selectivity erodes when PhN(H)Beg is borylated with B2pin2, although the ortho:meta:para ratio of 11:1.2:1 is better than the 2.5:1.5:1 ratio for Ir-catalyzed CHB of aniline with HBpin.12 The TSs for the PhN(H)Beg/Ir–Bpin structures were not calculated. However, it is not unreasonable to expect that the calculated steric destabilization from changing Beg to Bpin in phenol ortho CHB transition states would translate to aniline CHBs.38 The experiments in Scheme 3 demonstrate that the B substituents on the boryl ligands, and thus the CHB reagent, have the greater influence on the ortho selectivity.
Meta and para CHB of N-methylaniline would further support the hypothesis that hydrogen bonding is responsible for the high ortho selectivity of B2eg2 in aniline CHBs since PhN(Me)Beg lacks an NH moiety. Remarkably, CHB of PhN(H)Me yields only the ortho isomer, albeit with only 24% conversion. In operando NMR spectroscopy shows that PhN(H)Me is fully converted to PhN(Me)Beg before CHB ensues (eq 1). Even though the conversion of PhN(Me)Beg to borylated products is low, the ortho product is the only CHB isomer detected.
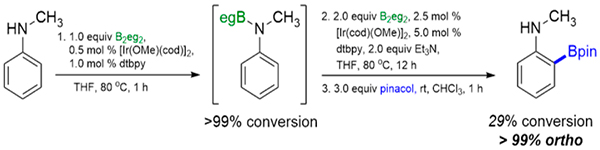 |
(1) |
This surprising result raises an obvious question. Would calculations also favor ortho CHB when hydrogen bonding is not an option? The calculated TS structures for meta CHB (TS6) and ortho CHB with anti (TS7) and syn (TS8) dispositions of the Beg group relative to the bipyridine ligand are shown in Figure 4.
Figure 4.

Computed transition states for Ir-catalyzed CHB of PhN(CH3)Beg with B2eg2. The hydrogen in the C–H bond being cleaved is shown in pale yellow. Grel and Hrel are ΔΔG⧧ and ΔΔH⧧ values relative to TS6.
In line with phenol ortho CHBs, the Gibbs energy of the syn TS (TS8) is higher than that of the anti TS (TS7). Other shared features of TS8 with the syn phenol TS include short contacts between the NBeg N and O atoms and the bipyridine N and flanking C atoms and a distortion of the pyridine Ir–N bond. This distortion reduces the σ overlap of the pyridine N lone pair with Ir and is best quantified by ∠σ, which is defined in eq 2:
| (2) |
When the Ir atom lies in the pyridine plane, the σ overlap is maximized, and ∠σ = 0. The Npy–BBeg distances, Cpy–O distances, and ∠σ values for the phenol and aniline TSs are summarized in Table 2. The N–B and C–O distances in TS8 are 0.05–0.13 Å longer than the corresponding values in the phenol ortho borylation TS. The negative charge on the EBeg group (E = NMe, O) is smaller for NMe than for O. Consequently, the electrostatic interaction is weaker, which is consistent with the elongation of the EBeg contacts with the syn pyridine of the bipyridine ligand when E = N(Me)Beg. The 6.5° decrease in ∠σ when E is switched from NMe to O also supports weakening of the electrostatic interaction between N(Me)Beg and the syn pyridine.
Table 2.
Structural Comparisons between Syn TSs in 4-C6H4OBeg and C6H5NMe(Beg) CHBs
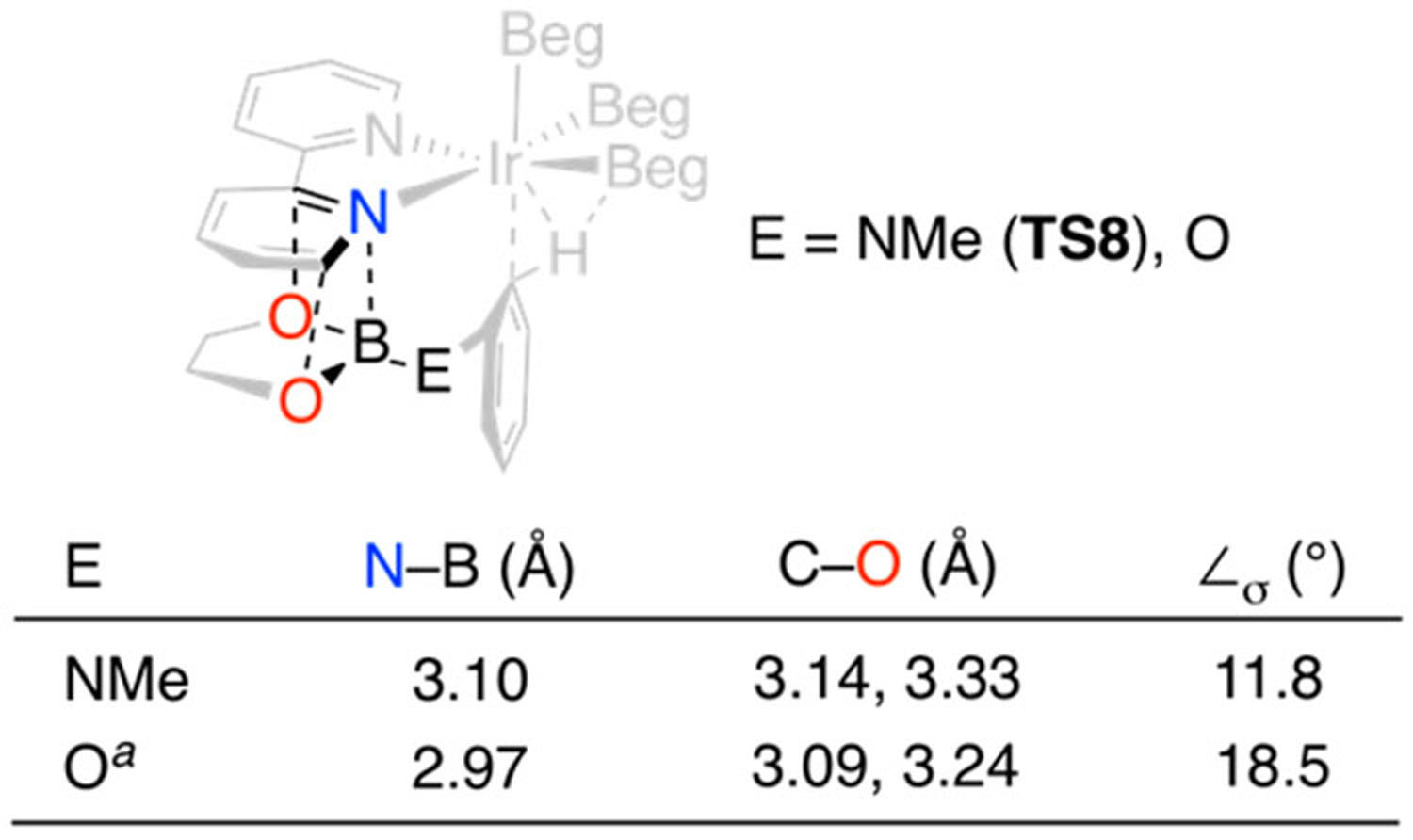 |
Data for TS5-OBegsyn‑dtbpy in Figure 3 of ref 36.
The Grel and Hrel values (ΔΔG⧧ and ΔΔH⧧ values relative to TS6) in Figure 4 predict that the ortho CHB TS7, where the Beg group is anti to the bipyridine ligand, has the lowest energy. The theoretical ΔΔG⧧ value of −0.3 kcal·mol−1 predicts an ortho:meta ratio of 1.5:1, and the ΔΔH⧧ value predicts a higher ortho:meta ratio of 4.1:1. Although the ortho isomer is predicted to be the major one, the actual selectivity is much higher. While the trend in the computed ratio correlates with the experimental results, future studies at a higher level of theory are warranted. Overall, the experimental findings show that hydrogen bonding is not required for ortho selectivity for N-methylaniline when the CHB reagent is switched from B2pin2 to B2eg2.
In addition to removing previous limitations for ortho CHBs of anilines with B2pin2 and HBpin, CHBs with B2eg2 can complement the selectivities for aniline CHBs with commonly used boron reagents. Scheme 4 shows two examples highlighting the most dramatic differences in CHB selectivities.
Scheme 4.

Regiochemical Consequences of Beg and Bpin Reagents in Aniline CHBs
CONCLUSIONS
The important findings of this study are listed below:
By changing the boron reagent from HBpin or B2pin2 to bis(ethylene glycolato)diboron (B2eg2), ortho CHBs can now be accomplished with a wide variety of anilines. Substrates whose previously poor (or altered) regioselectivity is now overcome include (i) anilines with no substituents at the 4-position, (ii) 2-substituted anilines, (iii) 3-substituted anilines, and (iv) N-methylaniline.
CHB ortho to NH2 in 2-methylquinolin-6-amine is possible.
The substituents on the Ir boryl ligands have the greatest impact on the selectivity.
The ortho-borylated isomer is the only product observed in the Ir-catalyzed CHB of N-methylaniline with B2eg2. 1H NMR studies show that PhN(H)Me is fully converted to PhN(Beg)Me before CHB ensues. Thus, hydrogen bonding in the TS cannot account for the ortho selectivity.
For Ir-catalyzed CHB of PhN(H)Beg, computational studies revealed three NH···O hydrogen-bonding transition states where the aniline N(Beg)H interacted with an Ir Beg ligand. The NH···O TSs have lower Gibbs energies than an ortho CHB TS that lacks hydrogen bonding.
The Gibbs energies of the three NH···O TSs are also lower than that for the para CHB TS of PhN(H)Beg
Of three TSs calculated for Ir-catalyzed CHB of PhN(Me)Beg, the ortho TS where the Beg moiety is anti to the bipyridine ligand has the lowest Gibbs energy. This TS closely resembled the favored TS for CHB ortho to the OBeg of 4-FC6H4OBeg, where the selectivity is proposed to arise from electrostatic interactions between the OBeg unit and the proximal pyridine ring of the bipyridine ligand.
In summary, the diboron reagent B2eg2 lifts the limitations seen in Ir-catalyzed CHBs of anilines with HBpin. Experiment and theory are consistent with stabilization of hydrogen-bonding TSs when Bpin Ir boryl ligands are replaced by less sterically encumbered Beg ligands. Further exploration of other diboron reagents and the synthetic utility of the associated CHB products is underway in our laboratories.
Supplementary Material
ACKNOWLEDGMENTS
B.C. thanks DST-SERB, New Delhi, for a Ramanujan Grant (SB/S2/RJN-45/2013) and a Young Scientist Research Grant (SB/FT/CS-141/2014) to execute this project and the Director, CBMR, for providing research facilities. R.B. thanks DST-SERB, New Delhi, for the SRF Fellowship under the Ramanujan Grant, and C.H. thanks the Young Scientist Research Grant for a Junior Research Fellowship. R.E.M. and M.R.S. thank the NIH (GM63188) for support and the Institute for Cyber-Enabled Research (ICER) at MSU for computational resources. We also thank the teams in the NMR and HRMS facilities at CBMR and MSU.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.8b00641.
Experimental details, product characterizations, computational methods, energies, and Cartesian coordinates (PDF)
The authors declare the following competing financial interest(s): R.E.M. and M.R.S. own a percentage of BoroPharm, Inc.
REFERENCES
- (1).Travis AS In The Chemistry of Anilines; John Wiley & Sons: Chichester, U.K., 2007; pp 715–782. [Google Scholar]
- (2).Berliner E Electrophilic Aromatic Substitution Reactions. Prog. Phys. Org. Chem 2007, 2, 253–321. [Google Scholar]
- (3).Clayden J; Greeves N; Warren S Organic Chemistry; Oxford University Press: Oxford, U.K., 2012. [Google Scholar]
- (4).Holleman AF; Hartogs JC; Van der Linden T Quantitative Untersuchungen über Die Nitrierung Des Anilins. Ber. Dtsch. Chem. Ges 1911, 44, 704–728. [Google Scholar]
- (5).Shen H; Vollhardt KPC Remarkable Switch in the Regiochemistry of the Iodination of Anilines by N-Iodosuccinimide: Synthesis of 1,2-Dichloro-3,4-Diiodobenzene. Synlett 2012, 2012, 208–214 [Google Scholar]
- (6).Takagishi S; Katsoulos G; Schlosser M Fluorine and Trifluoromethyl Substituted Anilines: Site Selective Metalation and Electrophilic Substitution. Synlett 1992, 1992, 360–362. [Google Scholar]
- (7).Whisler MC; MacNeil S; Snieckus V; Beak P Beyond Thermodynamic Acidity: A Perspective on the Complex-Induced Proximity Effect (CIPE) in Deprotonation Reactions. Angew. Chem., Int. Ed 2004, 43, 2206–2225. [DOI] [PubMed] [Google Scholar]
- (8).Hurst TE; Macklin TK; Becker M; Hartmann E; Kügel W; Parisienne-La Salle J-C; Batsanov AS; Marder TB; Snieckus V Iridium-Catalyzed C-H Activation versus Directed Ortho Metalation: Complementary Borylation of Aromatics and Heteroaromatics. Chem. - Eur. J 2010, 16, 8155–8161. [DOI] [PubMed] [Google Scholar]
- (9).Tischler MO; Tóth MB; Novák Z Mild Palladium Catalyzed Ortho C-H Bond Functionalizations of Aniline Derivatives. Chem. Rec 2017, 17, 184–199. [DOI] [PubMed] [Google Scholar]
- (10).Fang H; Dou Y; Ge J; Chhabra M; Sun H; Zhang P; Zheng Y; Zhu Q Regioselective and Direct Azidation of Anilines via Cu(II)-Catalyzed C-H Functionalization in Water. J. Org. Chem 2017, 82, 11212–11217. [DOI] [PubMed] [Google Scholar]
- (11).Allu S; Ravi M; Kumara Swamy KC Rhodium(III)-Catalysed Carbenoid C(sp2)–H Functionalisation of Aniline Substrates with α-Diazo Esters: Formation of Oxindoles and Characterisation/Utility of an Intermediate-Like Rhodacycle. Eur. J. Org. Chem 2016, 2016, 5697–5705. [Google Scholar]
- (12).Preshlock SM; Plattner DL; Maligres PE; Krska SW; Maleczka RE Jr.; Smith MR III. A Traceless Directing Group for C-H Borylation. Angew. Chem., Int. Ed 2013, 52, 12915–12919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Mkhalid IAI; Barnard JH; Marder TB; Murphy JM; Hartwig JF C-H Activation for the Construction of C-B Bonds. Chem. Rev 2010, 110, 890–931. [DOI] [PubMed] [Google Scholar]
- (14).Hartwig JF Borylation and Silylation of C-H Bonds: A Platform for Diverse C-H Bond Functionalizations. Acc. Chem. Res 2012, 45, 864–873. [DOI] [PubMed] [Google Scholar]
- (15).Lee C-I; Zhou J; Ozerov OV Catalytic Dehydrogenative Borylation of Terminal Alkynes by a SiNN Pincer Complex of Iridium. J. Am. Chem. Soc 2013, 135, 3560–3566. [DOI] [PubMed] [Google Scholar]
- (16).Del Grosso A; Singleton PJ; Muryn CA; Ingleson MJ Pinacol Boronates by Direct Arene Borylation with Borenium Cations. Angew. Chem., Int. Ed 2011, 50, 2102–2106. [DOI] [PubMed] [Google Scholar]
- (17).Légaré M-A; Courtemanche M-A; Rochette É; Fontaine F-G Metal-Free Catalytic C-H Bond Activation and Borylation of Heteroarenes. Science 2015, 349, 513–516. [DOI] [PubMed] [Google Scholar]
- (18).Mazzacano TJ; Mankad NP Base Metal Catalysts for Photochemical C-H Borylation That Utilize Metal-Metal Cooperativity. J. Am. Chem. Soc 2013, 135, 17258–17261. [DOI] [PubMed] [Google Scholar]
- (19).Obligacion JV; Semproni SP; Chirik PJ Cobalt-Catalyzed C–H Borylation. J. Am. Chem. Soc 2014, 136, 4133–4136. [DOI] [PubMed] [Google Scholar]
- (20).Furukawa T; Tobisu M; Chatani N Nickel-Catalyzed Borylation of Arenes and Indoles via C-H Bond Cleavage. Chem. Commun 2015, 51, 6508–6511. [DOI] [PubMed] [Google Scholar]
- (21).Cho J-Y; Iverson CN; Smith MR III. Steric and Chelate Directing Effects in Aromatic Borylation. J. Am. Chem. Soc 2000, 122, 12868–12869. [Google Scholar]
- (22).Cho J-Y; Tse MK; Holmes D; Maleczka RE Jr.; Smith MR III. Remarkably Selective Iridium Catalysts for the Elaboration of Aromatic C–H Bonds. Science 2002, 295, 305–308. [DOI] [PubMed] [Google Scholar]
- (23).Ishiyama T; Takagi J; Ishida K; Miyaura N; Anastasi NR; Hartwig JF Mild Iridium-Catalyzed Borylation of Arenes. High Turnover Numbers, Room Temperature Reactions, and Isolation of a Potential Intermediate. J. Am. Chem. Soc 2002, 124, 390–391. [DOI] [PubMed] [Google Scholar]
- (24).Jones WD; Kosar WP Carbon-Hydrogen Bond Activation by Ruthenium for the Catalytic Synthesis of Indoles. J. Am. Chem. Soc 1986, 108, 5640–5641. [Google Scholar]
- (25).Murai S; Kakiuchi F; Sekine S; Tanaka Y; Kamatani A; Sonoda M; Chatani N Efficient Catalytic Addition of Aromatic Carbon-Hydrogen Bonds to Olefins. Nature 1993, 366, 529. [Google Scholar]
- (26).Kawamorita S; Ohmiya H; Hara K; Fukuoka A; Sawamura M Directed Ortho Borylation of Functionalized Arenes Catalyzed by a Silica-Supported Compact Phosphine-Iridium System. J. Am. Chem. Soc 2009, 131, 5058–5059. [DOI] [PubMed] [Google Scholar]
- (27).Ishiyama T; Isou H; Kikuchi T; Miyaura N Ortho-C–H Borylation of Benzoate Esters with Bis(pinacolato)diboron Catalyzed by Iridium–phosphine Complexes. Chem. Commun 2010, 46, 159–161. [DOI] [PubMed] [Google Scholar]
- (28).Ros A; Fernández R; Lassaletta JM Functional Group Directed C–H Borylation. Chem. Soc. Rev 2014, 43, 3229–3243. [DOI] [PubMed] [Google Scholar]
- (29).Taube H; Posey FA The Study of a System Involving Equilibrium Between Inner Sphere and Outer Sphere Complex Ions - Co(NH3)5H2O+++ and SO4=. J. Am. Chem. Soc 1953, 75, 1463–1467. [Google Scholar]
- (30).Boebel TA; Hartwig JF Silyl-Directed, Iridium-Catalyzed Ortho-Borylation of Arenes. A One-Pot Ortho-Borylation of Phenols, Arylamines, and Alkylarenes. J. Am. Chem. Soc 2008, 130, 7534–7535. [DOI] [PubMed] [Google Scholar]
- (31).Davis HJ; Phipps RJ Harnessing Non-Covalent Interactions to Exert Control over Regioselectivity and Site-Selectivity in Catalytic Reactions. Chem. Sci 2017, 8, 864–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Haldar C; Emdadul Hoque M; Bisht R; Chattopadhyay B Concept of Ir-Catalyzed CH Bond Activation/borylation by Non-covalent Interaction. Tetrahedron Lett 2018, 59, 1269–1277. [Google Scholar]
- (33).Roosen PC; Kallepalli VA; Chattopadhyay B; Singleton DA; Maleczka RE Jr; Smith MR III. Outer-Sphere Direction in Iridium C-H Borylation. J. Am. Chem. Soc 2012, 134, 11350–11353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Bisht R; Chattopadhyay B Formal Ir-Catalyzed Ligand-Enabled Ortho and Meta Borylation of Aromatic Aldehydes via in Situ-Generated Imines. J. Am. Chem. Soc 2016, 138, 84–87. [DOI] [PubMed] [Google Scholar]
- (35).Li HL; Kuninobu Y; Kanai M Lewis Acid-Base Interaction-Controlled Ortho-Selective C-H Borylation of Aryl Sulfides. Angew. Chem., Int. Ed 2017, 56, 1495–1499. [DOI] [PubMed] [Google Scholar]
- (36).Li H-L; Kanai M; Kuninobu Y Iridium/Bipyridine-Catalyzed Ortho-Selective C-H Borylation of Phenol and Aniline Derivatives. Org. Lett 2017, 19, 5944–5947. [DOI] [PubMed] [Google Scholar]
- (37).Davis HJ; Genov GR; Phipps RJ Meta-Selective C–H Borylation of Benzylamine-, Phenethylamine-, and Phenylpropyl-amine-Derived Amides Enabled by a Single Anionic Ligand. Angew. Chem., Int. Ed 2017, 56, 13351–13355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Chattopadhyay B; Dannatt JE; Andujar-De Sanctis IL; Gore KA; Maleczka RE Jr; Singleton DA; Smith MR III. Ir-Catalyzed Ortho-Borylation of Phenols Directed by Substrate-Ligand Electrostatic Interactions: A Combined Experimental/in Silico Strategy for Optimizing Weak Interactions. J. Am. Chem. Soc 2017, 139, 7864–7871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Yamamoto T; Ishibashi A; Suginome M Regioselective Synthesis of O-Benzenediboronic Acids via Ir-Catalyzed O-C-H Borylation Directed by a Pyrazolylaniline-Modified Boronyl Group. Org. Lett 2017, 19, 886–889. [DOI] [PubMed] [Google Scholar]
- (40).Davis HJ; Mihai MT; Phipps RJ Ion Pair-Directed Regiocontrol in Transition-Metal Catalysis: A Meta-Selective C-H Borylation of Aromatic Quaternary Ammonium Salts. J. Am. Chem. Soc 2016, 138, 12759–12762. [DOI] [PubMed] [Google Scholar]
- (41).Kuninobu Y; Ida H; Nishi M; Kanai M A Meta-Selective CH Borylation Directed by a Secondary Interaction between Ligand and Substrate. Nat. Chem 2015, 7, 712–717. [DOI] [PubMed] [Google Scholar]
- (42). The ligand for the Kanai–Kuninobu ortho CHB of aniline is prepared via a Suzuki cross-coupling of 5-bromo-2,2′-bipyridine ($14000/mol) with 1-Bpin-4-(trifluoromethyl)benzene ($870/mol) in 73% yield. B2pin2 ($50/mol) is the borylating agent. The prices shown are the lowest per-mole values of all commercial sources listed by SciFinder Scholar on 02/01/18.
- (43). The yields for deprotection are given in an end note in ref 35. The products are not specified, and experimental details were not reported. We presume that the deprotected compounds are N-acylated since all of the ArN(Ac)(CH2SMe) reactants were prepared from acylated anilines.
- (44). The Ir ligand used in this work is dtbpy ($2300/mol). B2eg2 is prepared from B2(OH)4 ($51/mol) and ethylene glycol ($0.22/mol) in 91% yield.
- (45).Robbins DW; Hartwig JF A C-H Borylation Approach to Suzuki-Miyaura Coupling of Typically Unstable 2-Heteroaryl and Polyfluorophenyl Boronates. Org. Lett 2012, 14, 4266–4269. [DOI] [PubMed] [Google Scholar]
- (46).Bartlett RK; Turner HS; Warne RJ; Young MA; Lawrenson IJ The Reaction of Primary Amines with Boron Halides. Part II. Arylamines. The Effect of Ortho-Substitution: Formation of Di-B-Halogenoborazoles. J. Chem. Soc. A 1966, 479–500. [Google Scholar]
- (47).Thanthiriwatte KS; Hohenstein EG; Burns LA; Sherrill CD Assessment of the Performance of DFT and DFT-D Methods for Describing Distance Dependence of Hydrogen-Bonded Interactions. J. Chem. Theory Comput 2011, 7, 88–96. [DOI] [PubMed] [Google Scholar]
- (48).Parthasarathi R; Subramanian V Characterization of Hydrogen Bonding: From van der Waals Interactions to Covalency In Hydrogen Bonding—New Insights; Grabowski SJ, Ed.; Springer: Dordrecht, The Netherlands, 2006; pp 1–50. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.