

Abstract
Objective
Observations of microbial dysbiosis in patients with RA have raised interest in studying microbial-mucosal interactions as a potential trigger of RA. Using the murine collagen-induced arthritis (CIA) model, we hypothesized that microbiota modulate immune responses leading to autoimmune arthritis.
Methods
CIA was induced by immunization of mice with type II collagen (CII) in adjuvant on days 0 and 21, with arthritis appearing at days 23–24. Intestinal microbiota were profiled by 16S rRNA sequencing every 7 days during the course of CIA, and intestinal mucosal changes evaluated on days 14 and 35. Then, microbiota were depleted either in early (7 days preceding) or late (after day 21) CIA by administration of broad spectrum antibiotics. Disease severity, autoantibody and systemic cytokine production, and intestinal mucosal responses were monitored in the setting of microbial reduction.
Results
Significant dysbiosis and mucosal inflammation occurred early in CIA, prior to visible arthritis, and continued to evolve during the course of disease. Depletion of the microbiota prior to the induction of CIA resulted in ~40% reduction in disease severity and significantly reduced serum inflammatory cytokines and anti-CII antibodies. In intestinal tissue, IL-17A and IL-22 production were delayed. Unexpectedly, microbial depletion during the late phase of CIA resulted in >90% decrease in disease severity. Anti-CII antibodies were mildly reduced, but significantly impaired in their ability to activate complement likely due to altered glycosylation profiles.
Conclusions
These data support a model in which intestinal dysbiosis triggers mucosal immune responses that stimulate T and B cells that are key for the development of inflammatory arthritis.
Introduction
Rheumatoid arthritis (RA) is a systemic autoimmune disease characterized by chronic and progressive joint pain, articular destruction, deformity and disability. The pathogenesis of RA is thought to be influenced by a combination of genetic and environmental factors [1–3]. Genetic studies of individuals with rheumatoid arthritis have identified multiple risk loci, but fail to predict with sufficient precision those who ultimately will develop disease [4, 5], an observation that underscores the importance of environmental triggers in RA pathogenesis. Intestinal dysbiosis, the alteration of the community of commensal bacteria within the small intestine (SI) and colon [6], is one such environmental factor that has been associated with individuals with new-onset and established RA [7, 8]. However, whether dysbiosis is merely a consequence of the disease process or is itself pathogenic remains unknown.
Collagen-induced arthritis (CIA) is a commonly used model for probing the pathogenesis of RA (reviewed by Brand et al.) [9]. In this model, genetically susceptible DBA/1J mice are immunized twice (day 0 and day 21) with bovine collagen type II (CII) emulsified in complete Freund’s adjuvant (CFA). Initially, there is a preclinical period of disease development in which T and B cell autoreactivity towards CII is generated without overt signs of arthritis. With time these result in a proliferative synovitis, which is first clinically observed 2–3 days following the second immunization, featuring polymorphonuclear and mononuclear cellular infiltrates, formation of pannus, degradation of cartilage, fibrosis, and bone erosion [9]. Not only is the model dependent upon generation of T and B cell reactivity to CII, but also complement activation and the generation of systemic cytokines IL-1β, IL-6, IL-17, IFN-γ, and TNF-α; the absence or inhibition of any of these factors significantly attenuates disease development [10–13].
Recently, others have described intestinal dysbiosis at the terminal time point of this model in mice that developed disease compared with those that were immunized but did not develop observable joint swelling [14]. Transfer of fecal material from arthritic but not from non-arthritic, immunized mice into germ free mice followed by CII immunization resulted in disease induction [14]. However, questions from this study regarding microbial changes over time and whether a time-dependent role for microbiota in pathogenesis exists remain unanswered. Furthermore, this study did not address the mucosal or systemic effects of the microbial changes or the possible mechanism(s) by which microbiota modulate susceptibility to CIA.
Given the outstanding questions regarding the influence of dysbiosis at various stages during the development of inflammatory arthritis, we hypothesized that intestinal microbial communities would be dynamic during the course of CIA, and would demonstrate multiple phases of dysbiosis. In turn, dysbiosis would be expected to modulate local intestinal mucosal immune factors that promote systemic inflammation, autoantibody generation, and disease severity. Furthermore, we hypothesized that microbiota would affect disease development differently at discrete time points in the development of disease. Using the CIA model, we assessed microbial changes in the intestine throughout the course of disease and identified the critical time points at which dysbiosis modulated disease activity. We characterize the mucosal and systemic immune responses that correlate with dysbiosis, and propose that microbiota alters disease in CIA through modulation of cytokines and autoantibody production.
Methods
Collagen-Induced Arthritis (CIA)
6 week old male DBA/1J mice (The Jackson Laboratory), were injected intradermally with 200 μg bovine CII (Chondrex®) emulsified in 50μl CFA (Sigma) on days 0 and 21. As controls, a subset of mice were immunized with 50μl CFA only on days 0 and 21. Disease severity was measured as a mean clinical score for each of the animal’s 4 paws, where 0 = normal, 1 = erythema, 2 = swelling, and 3 = ankylosis. At the specified time points, feces, serum, and tissues were harvested from mice. All animal experiments were approved by the University of Colorado School of Medicine Institutional Animal Care and Use Committee.
Collagen Antibody-Induced Arthritis (CAIA)
6 week old male DBA/1J mice, were given antibiotic or control drinking water for 1 week prior to and throughout the study. Mice were injected with 1.5 mg of 5-clone anti-CII antibody cocktail (Chondrex) by intraperitoneal (IP) injection at day 0. On day 3, 50 μg of LPS was injected IP. Arthritis scoring was done every day starting at day 4 through day 10. Disease severity was assessed as described above for CIA.
Microbiome Analysis
Fresh fecal samples were collected at 7 day intervals throughout the course of CIA. Total genomic DNA was extracted using the UltraPure Fecal DNA kit (MolBio) according to the manufacturer’s protocol. Harvested DNA was PCR amplified with broad-range bacterial primers targeting the 16S rRNA gene V1V2 region and pooled amplicons subjected to Illumina MiSeq sequencing, as previously described [15–17]. Assembled sequences were aligned and classified with SINA (1.3.0-r23838) [18] using the 418,497 bacterial sequences in Silva 115NR99 [19] as reference configured to yield the Silva taxonomy. Operational taxonomic units (OTUs) were produced by clustering sequences with identical taxonomic assignments. OTUs with >0.01% abundance in any sample and observed in >5% of the samples were included in further analyses. Analyses of OTU relative abundance and biodiversity were conducted using Explicet software [20]. All samples had a Good’s coverage index >99%, indicating excellent depth of sequencing coverage.
FITC-Dextran Flux
Mice were orally gavaged with 0.6 mg/kg body weight 4 kDa dextran labeled with fluorescein (Sigma) and serum was collected 4 hours later. The amount of fluorescence was measured with a fluorimeter (Promega) at 485/530 nm. A standard curve was generated to calculate the amount of dextran that was present in the sera.
Histopathology
Whole colon tissue was harvested from mice, flushed with 1X PBS, dissected longitudinally, and pinned in wax for fixation in formaldehyde overnight. Tissues were then embedded in paraffin. Sections of 5 μm were cut from paraffin embedded tissues and stained with hematoxylin and eosin (H & E). Four high-powered fields of well-oriented colon tissue per mouse were analyzed at 40X magnification for quantification of inflammatory cells in the lamina propria, numbers of mitotic figures, and length of crypts.
Cytokine Assay
A multi-analyte ELISA (Meso Scale Diagnostics) was used to measure the levels of IFN-γ (0.04 pg/ml lower limit of detection - LLD), IL-12p70 (9.95 pg/ml LLD), IL-1β (0.11 pg/ml LLD), IL-6 (0.61 pg/ml LLD), IL-10 (0.95 pg/ml LLD), IL-22 (1.2 pg/ml LLD), IL-23 (4.9 pg/ml LLD), IL-17A (0.3 pg/ml LLD), and TNF-α (0.13 pg/ml LLD) in tissue lysates and serum, following manufacturer’s instructions. In tissues, cytokines were normalized to total protein content of the tissue lysate, which was determined using a BSA assay (Pierce).
Anti-CII ELISA
96-well plates (Costar, Corning) were coated overnight at 4°C with 5 μg/mL ELISA grade mouse CII (Chondrex®). The plates were then washed and blocked with 0.01% BSA in 1X PBS for 4 hours at 4°C. Again plates were washed three times. Serum samples were diluted 1:10,000 in 1X PBS. Samples were added to wells and incubated overnight at 4°C. The plates were then washed, and horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (Jackson ImmunoResearch Laboratories), goat anti mouse IgG1, IgG2a or IgG2b (Invitrogen) diluted 1:1000 in 1X PBS was added for 4 hours at 4°C. Following washing, 1X TMB ELISA substrate solution (eBioscience) was added to wells for 30 min at room temperature. The reaction then was stopped with 2N H2SO4 and read at 450 nM wavelength. Pooled serum from several mice with severe CIA was used to generate a standard curve in which the top standard was diluted 1:2000 and considered 250 U/ml [21].
Antibiotic Treatment
Ampicillin (1 g/L), Neomycin (1 g/L), Vancomycin (0.5 g/L), Metronidazole (0.5 g/L) (Sigma) and sugar-sweetened grape Kool-Aid (20 g/L) (Kraft Foods) were given in drinking water to mice starting at day −7 (before immunization on day 0) and stopped at day 0 for the short course or continued throughout the study for the long course. For the late course of antibiotics, mice were provided the antibiotic drinking water beginning on day 21 through the end of the study. Control drinking water consisted of (20 g/L) Kool-Aid.
qPCR
Bacterial DNA from feces was extracted as described for microbiome analysis. To determine total bacterial load, qPCR reactions consisted of 1 μl bacterial DNA or cDNA, 0.6 μM each forward and reverse primers, 1X SYBR® Green PCR Master Mix (Applied Biosystems), and water for a total volume of 17 μl. Samples were denatured at 95°C for 2 minutes, cycled 40 times through 95°C for 20 seconds, 58°C for 20 seconds, and 72°C for 30 seconds, and then denaturation curves determined from 58°C through 95°C. All qPCR assays were conducted in an Applied Biosystems 7500 real time PCR system.
Bacterial 16S universal primers F (TCCTACGGGAGGCAGCAGT) and R (GGACTACCAGGGTATCTAATCCTGTT) [22] were used for fecal microbial DNA quantification. Both control water treated and antibiotic-treated mice were assayed simultaneously within each reaction as well as a bacterial DNA standard curve for calculation of bacteria concentration in ng/μL. Concentrations were normalized to fecal weight. In each reaction, individual samples were compared to the mean concentration of bacteria in control water treated mice to report the percent of control microbiota.
C3 Activation ELISA
96-well plates (Costar, Corning) were coated overnight at 4°C with 5 μg/mL, ELISA grade mouse CII (Chondrex®). The ELISA plates were then washed and blocked with 0.01% BSA in 1X PBS for 4 hours at 4°C. The ELISA plates were washed three times. Serum samples were diluted 1:10,000 in 1X PBS. Samples were added to wells and incubated overnight at 4°C. Next day, the wells were washed five times with 1X PBS, then incubated with 15% normal mouse serum diluted in DPBS+0.9 mM CaCl2+ 0.5 mM MgCl2 for 30 min at 37 °C. The plates then washed five times with 1X PBS and incubated with cappel HRP goat IgG to mouse C3 (MP Biomedicals) in 1:2500 dilution, for one hour at room temperature with rocker shaking. The plates were washed with 1X PBS and developed with TMB1X (eBioscience) for 10 min, stopped with H2SO4 and read at 450 nM.
Glycosylation Studies
Serum total IgG was purified by using Pierce™ Protein G Agarose (Thermo Fisher Scientific) following manufacturer’s instructions. To concentrate the eluted total IgGs, 100K Ultracel centrifugal filters (Amicon) were used. Total N-linked glycan was released from glycoproteins using PNGaseF (New England Biolabs, Inc.), according to manufacturer’s instruction. Deglycosylation reactions were carried out at 37°C overnight to ensure effective release of glycans. Glycans were purified from the reaction using GlykoClean™ G Cartridges (Prozyme), dried, and fluorescently labeled with 2-AB (2-aminobenzamide) (Sigma-Aldrich). Labeled glycans were cleaned with GlykoClean™ S-plus Cartridges (Prozyme), dried, and subjected to HPLC analysis. Glycan samples were dissolved in 100mM ammonium formate (pH4.5) and separated using Agilent 1260 Infinity Quaternary LC system, outfitted with AdvanceBio Glycan Mapping column 2.1 × 150mm, 2.7 μm and a fluorescent detector. Resulting peaks were analyzed in OpenLAB software (Agilent) and assigned glycoforms by comparing peaks of commercially available human IgG N-linked glycan library.
Statistical Analysis
Microbiome data were analyzed using Explicet (v2.10.5) [20] and R statistical software, including the vegan package [23]. Tests of overall community composition used a non-parametric multiple analysis of variance (PERMANOVA) test of Morisita-Horn dissimilarity scores, with p-values estimated through 1,000,000 permutations. Alpha-diversity indices were estimated through 1,000 resamplings at the rarefaction point of 90,000 sequences. Between-group differences in alpha-diversity were assessed by ANOVA. Both PERMANOVA and ANOVA tests included mouse identification numbers to factor out animal-to-animal variability, as might occur through cage effects. Other data were analyzed with GraphPad Prism version 7.01; specific statistical tests for comparisons are referenced in the figure legends.
Results
Microbial communities and mucosal inflammation continuously change through the course of CIA
Because dysbiosis is observed in individuals with symptomatic RA [7, 24], and mice with clinical arthritis [14, 25], we queried when dysbiosis developed during the CIA disease process. CIA was induced by immunization of male 6-week old DBA/1J mice with CII in CFA on days 0 and 21, with arthritis first observed at days 23–24. We collected fecal samples on day 0 and every 7 days during the course of CIA until the end of the study on day 35. DNA from fecal pellets was isolated and subjected to broad-range bacterial 16S ribosomal RNA gene amplification (V1V2 region) and high-throughput sequencing. Compared with fecal samples collected at day 0 (Fig. 1a), appreciable differences in overall microbiota composition were observed at day 14 (PERMANOVA p = 0.032) and day 21 (PERMANOVA p = 0.0063). Significant differences also were observed between day 7 and days 14 (PERMANOVA p = 0.040) and 21 (PERMANOVA p = 0.010). Although microbiota composition at day 35 did not differ significantly from previous time points, the smaller number of animals sampled at this time point (N = 6, compared with N = 11 for all other time points), likely limited statistical power in the PERMANOVA tests. Nevertheless, two measures of microbial biodiversity, evenness and Shannon complexity, progressively declined over the course of CIA treatment and were significantly lower at day 35, compared with days 0 and 7. (Fig. 1b). Comparison of bacterial families over time revealed that Lactobacillaceae significantly expanded while S24-7 was significantly reduced at days 14 and 21, and Lachnospiraceae was significantly increased at day 35 compared to baseline (Fig. 1c). It is difficult to determine if the changes in microbial communities observed in mice with CIA are due solely to an innate immune response generated by the adjuvant, as the microbiome profile of CFA immunized mice also dynamically changed over time, but in a different pattern compared to those with CIA (Supplemental Fig. S1). Thus, significant changes in the composition and diversity of the fecal microbiota appeared to occur during the early, preclinical stages of CIA prior to arthritis being observed.
Figure 1. Microbial dysbiosis occurs early during the development of CIA.
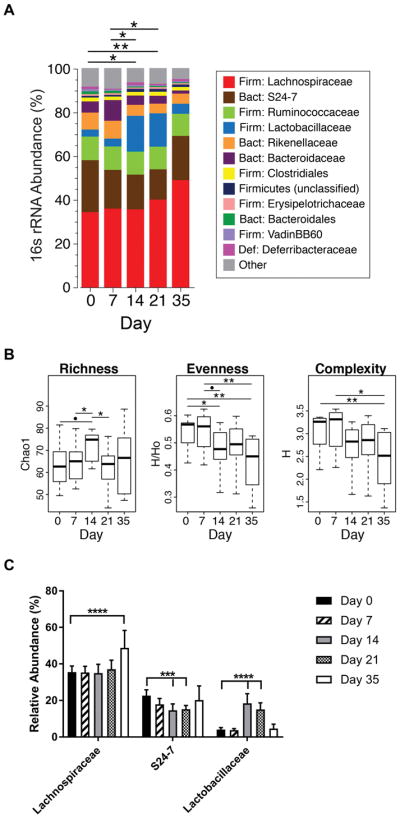
Six week old male DBA/1J mice were immunized with CII in CFA on days 0 and 21. Fecal pellets were collected from mice every 7 days from day 0 through 21 and on day 35 after the initial immunization. Fecal bacterial DNA was sequenced for 16S rRNA and analyzed. N=11 for time points 0–21 and N=6 for day 35; data are from two independent experiments. (A) The percent abundance of the top operational taxonomic units (OTU) families were compared across time points. Differences in the overall composition of microbial communities were determined by PERMANOVA. *P<0.05, **P<0.01. (B) Alpha-diversity measures for richness (Chao1), evenness, and complexity (ShannonH) across time points are shown as box-and-whisker plots. Statistical analysis was performed with ANOVA as described in Methods. *P<0.05, **P<0.01 and •P<0.001. (C) Statistically significant changes in family level OTU mean relative abundance ± SEM in were determined by Kruskal-Wallis with Dunn’s post-test. ***P<0.001; ****P<0.0001.
We next examined whether the observed dysbiosis was accompanied by intestinal mucosal changes during the development of CIA. To address this, we first evaluated intestinal barrier integrity, as assessed by the permeability of fluorescently-labeled (FITC) dextran, a non-digestible sugar that paracellularly permeates through an impaired intestinal barrier and can be detected in serum. During the development of CIA, elevated dextran was first detected in the serum at day 14, and further increased significantly at day 35 in mice with CIA but not CFA controls (Fig. 2a), suggesting intestinal barrier impairment occurring at these time points during the development of CIA. The cause of impairment was suggested to be secondary to inflammation, as histologic analysis of colon tissue from mice with CIA at day 35 compared to untreated DBA/1J and CFA control mice demonstrated significantly increased inflammatory cell infiltration in the lamina propria, increased crypt cell mitosis, and crypt hyperplasia (Fig. 2b–e). Furthermore, as another indication of intestinal mucosal inflammation, cytokines in whole tissue homogenates from the SI, colon, and mesenteric lymph nodes (MLNs) from mice with CIA at days 14 and 35, but not CFA controls, demonstrated early significant increases in IL-17A, IL-22, and a trend in IL-23 (Fig. 2f–h), while cytokines IFN-γ, IL-12p70, IL-1β, IL-6, IL-10 and TNF-α were not significantly affected in mice with CIA (Supplemental Fig. S2). Taken together, these data strongly suggest a pattern of intestinal dysbiosis correlating with a Th17 pattern of mucosal inflammation that begins before arthritis is clinically observed in CIA.
Figure 2. Mucosal inflammation parallels dysbiosis during the development of CIA.

6 week old male DBA/1J mice were immunized with CII in CFA on days 0 and 21. (A) Barrier integrity was assessed by permeability of FITC-dextran orally gavaged and detected in serum 4 hours later. Data are the mean ±SEM of the serum concentration of FITC-dextran. N=10 in each group. ***, p<0.001; ****, p<0.0001 as determined by repeated-measures ANOVA. (B) Representative histology from control DBA/1J, CFA controls, and CIA mice at day 35 are shown at 40X. Bar=20 μm. (C–E) Four high-powered fields of well-oriented colon tissue per mouse were analyzed at 40X magnification for quantification of (C) inflammatory cells in the lamina propria, (D) epithelial mitotic figures, and (E) crypt length. N=3–6 mice per group. Data are the mean ± SEM; statistical analyses were performed with Mann-Whitney test *, p<0.05. (F–H) IL-17A, IL-22, and IL-23 in SI, colon, and MLNs were measured by ELISA in DBA/1J unimmunized controls, CFA controls, and in mice with CIA at days 14 and 35 after initial immunization. Cytokine concentrations were normalized to total protein and reported as pg/ml/mg protein. N=3–6 in each group. Data are the mean ±SEM and statistical analysis was performed using a Kruskal-Wallis with Dunn’s post-test. *, p<0.05; **, p<0.01.
Microbiota influence disease severity in CIA through modulation of mucosal and systemic cytokines and autoantibodies
To clarify the role of microbiota for the development of CIA, we used broad spectrum antibiotics (ampicillin, metronidazole, neomycin and vancomycin) in drinking water to deplete the microbiota. Three treatment regimens were utilized (Fig.3a): oral antibiotic-treatment or control water starting 7 days prior to induction of CIA and continued throughout the study (early long course); antibiotic-treatment or control water for only 7 days prior to induction of CIA followed by control water (early short course); and control water from 7 days prior to induction of CIA until day 21, followed by antibiotic-treatment or control water until the end of the study (late course; day 21–35). To confirm microbial depletion in antibiotic-treated mice, the microbial load in fecal samples was measured by qPCR using a universal 16S rRNA primer set [22]. The bacterial concentration in each mouse sample was compared to the mean bacterial concentration of the control group, and those that were not below the 10% confidence interval for the median normalized bacterial abundance in mice on control drinking water were excluded from further analyses (Supplemental Fig. S3a). No differences in the total microbial abundance of both early antibiotic treatment courses (early short vs. early long) at day 35 were detected; thus, the two treatment groups were combined in our analyses.
Figure 3. Depletion of microbiota by administration of antibiotics reduces CIA severity.

Six week old male DBA/1J mice were treated with broad-spectrum antibiotics in drinking water starting a week before CII immunization and stopped at day 0 (early short course) or continued until euthanasia at day 35 (early long course), or mice were treated with antibiotics from days 21 through 35 (late course). All groups were paired with control water-treated mice. All antibiotic and control-water treated groups underwent immunization with CII in CFA at days 0 and 21. (A) Diagram of experimental treatments is shown. (B) Arthritis scores were assessed every other day starting after day 21, N= 6–14 mice/group. Data are the mean arthritis score ± SEM. *, p<0.05; **, p<0.01; ****, p<0.0001 as determined by a two-way ANOVA with Bonferroni’s multiple comparisons test. (C) Serum anti-CII IgG was measured by ELISA. N=6–11 mouse/group; data are the mean U/ml of antibodies ±SEM. *, p<0.05; ****, p<0.0001 as determined by a two-way ANOVA with Bonferroni’s multiple comparisons test.
Following immunization of antibiotic-treated and control mice at days 0 and 21, mice were assessed for the severity of arthritis every other day until day 35, at which time they were euthanized. Mice given early antibiotic-treatment (early-short or early-long course; Fig. 3 and Supplemental Fig. S3b) demonstrated a 40% reduction in the severity of arthritis at day 35 while, surprisingly, mice treated with antibiotics after the second immunization (late course) developed only a slight, transient arthritis that was >95% reduced in severity (Fig. 3b). The prevalence of arthritis at day 35 with early antibiotic treatment remained 100%, the same as mice with CIA on control water; however, the prevalence of arthritis was significantly reduced to ~50% (p<0.0001 by log rank test) with late antibiotic treatment compared to controls (Supplemental Fig. S4a). Supporting the reduced disease severity, the levels of four important inflammatory cytokines for the development of CIA (IFN-γ, IL-1β, IL-6 and TNF-α) [26, 27] were significantly decreased in both early and late antibiotic-treated mice with CIA (Supplemental Fig. S4d), noting a more profound reduction in the late antibiotic-treated mice. The development of serum anti-CII IgG antibodies was significantly delayed in mice given early antibiotics, but late antibiotic treatment mildly reduced, although not significantly (Fig. 3c). Intriguingly, while arthritis scores positively correlated with microbial abundance in both the early and late antibiotic-treated groups (Supplemental Fig. S4b), serum CII autoantibodies only correlated with early but not late antibiotic treatment (Supplemental Fig. S4c).
Because we observed day 14 as the earliest time point for dysbiosis and mucosal inflammation characterized by elevated IL-17A, IL-22, and IL-23 in our initial experiments, we measured the levels of these three cytokines in SI, colon, and MLN of mice treated with antibiotics or control water during CIA. IL-17A and IL-22 significantly decreased in the SI and colon of mice with CIA after depleting microbiota with the early antibiotic treatment, while IL-23 remained unaffected (Fig. 4a). However, at day 35, all three cytokines were significantly increased in the early antibiotic treated mice compared to controls. (Fig. 4b). Intriguingly, IL-17A and IL-22 were significantly decreased in intestinal tissues from mice with CIA and late antibiotic treatment compared to CIA with control treatment while IL-23 was significantly reduced in the colon and MLNs (Fig. 4c). Thus, it appears that microbiota may have differing, time-dependent impacts on the development of both mucosal and systemic immune responses that result in CIA. IL-17A, IL-22, and IL-23 were unchanged at the draining inguinal lymph nodes for the CIA immunization site in control and antibiotic treated mice (Supplemental Fig. S5), suggesting that the Th17-derived cytokines originate in the intestinal tissues and associated MLNs rather than at the site of immunization.
Figure 4. Microbiota are required for the development of mucosal Th17 cytokines during CIA.

Six week old male DBA/1J mice were treated as in Figure 3. Intestinal tissues (SI and colon) in addition to MLN were collected either at day 14 or day 35, homogenized and tested for cytokines by multi-analyte ELISA. The final cytokine concentrations in each tissue were normalized to total protein and reported as pg/ml/mg protein. N=6–11 in each group. Data are the mean ± SEM of the tissue concentrations of cytokine. *, p<0.05, **, p<0.01 as determined by a Kruskal-Wallis test with Dunn’s post-test. (A) IL-17A, IL-22, and IL-23 in tissues from mice with CIA and CIA + early Abx at day 14. (B) IL-17A, IL-22, and IL-23 in tissues from mice with CIA and CIA + early Abx at day 35. (C) IL-17A, IL-22, and IL-23 in tissues from mice with CIA and CIA + late Abx at day 35.
To determine whether antibiotic treatment may have anti-inflammatory properties during the effector phase of arthritis, we utilized the collagen antibody-induced arthritis (CAIA) model. In this model, arthritis is induced in DBA/1J mice by transferring a cocktail of 5 murine monoclonal antibodies to CII, bypassing the need to develop an adaptive immune response. DBA/1J mice were treated with antibiotics or control drinking water for one week prior to anti-CII antibody transfer and throughout the study. Arthritis severity was unaffected by treatment of mice with antibiotics (Supplemental Fig. S6), demonstrating that antibiotics themselves did not modulate inflammation, but rather, microbiota modulated the developing immune responses that lead to arthritis. This conclusion is further supported by our finding that mice which did not have microbial depletion while on antibiotics developed CIA disease severity similar to controls (Supplemental Fig. S3c).
Microbial depletion impairs autoantibody function in CIA without affecting IgG isotypes
One striking feature of our data was the profound reduction in arthritis incidence and severity when microbiota were depleted beginning at day 21 after the induction of CIA, particularly since total IgG anti-CII antibodies were only slightly reduced (Fig. 3c). To explore the potential mechanism for disease protection, we assessed the functionality of CII autoantibodies in the serum from the mice by examining their ability to activate complement C3. Strikingly, anti-CII antibodies from the serum of mice treated with antibiotics late in the course of CIA failed to activate C3 complement compared to controls or early antibiotic-treated mice with CIA (Fig. 5a–b and Supplemental Figure S7). While these findings may be due in part to a decrease in IgG2b isotype anti-CII antibodies in the late antibiotic treatment group, the same trends in CII autoantibodies were observed in the early antibiotic treatment group (Supplemental Fig. S8), which did not demonstrate reduced complement activity. Since autoantibody glycosylation could significantly impact the immune reactivity and complement-dependent cytotoxicity at the effector phase, we measured and analyzed glycosylation patterns of total serum IgG in mice with CIA treated with control water, early antibiotics, and late antibiotics. While control water treated and early antibiotic treated mice with CIA showed similar glycosylation profiles, mice with CIA that were treated with antibiotics late at day 21 demonstrated lower galactose levels (Fig. 5c–e). Thus, we conclude that the antibiotic-induced alteration of microbiota at day 21 after induction of disease led to significant glycosylation changes accompanied by reduction in anti-CII antibody activation of complement.
Figure 5. Microbiota alter the ability of serum autoantibodies to activate complement.

Activation of the classical complement pathway by day 35 serum anti-CII antibodies from mice with CIA or mice with CIA treated with antibiotics was assessed by a modified ELISA described in Methods. C3 activation is shown as the mean U/ml ± SEM. N=6–11 per group. Statistical significance was determined by a two-tailed Student’s t-test. *, p<0.05; ns, not significant. (A) CIA and CIA+ early Abx. (B) CIA and CIA+late Abx. (C) Agalactosylated, (D) galactose, and (E) sialic acid content in purified serum immunoglobulins were quantified by high-performance liquid chromatography (HPLC).
Discussion
There is an increasing appreciation of the role of the microbial-mucosal imbalance in the development of autoimmune diseases [7, 8, 28–30]. Using the CIA murine model, we investigated the importance of microbiota in developing inflammatory arthritis. Our results suggest that significant shifts in the composition and diversity of microbial communities occurred during the first 21 days of the CIA model. In parallel, mucosal barrier impairment was observed with an initial insult at day 14 that stabilized through day 21 but worsened by day 35. Early after induction of CIA by day 14, we observed a reduction in the abundance of the Bacteroidetes class S24-7 and an increase in Lactobacillaceae; however, by day 35 Lactobacillaceae and S24-7 returned to day 0–7 levels, while Lachnospiraceae significantly increased in abundance. These are in agreement with a previous study showing the same trends in Lactobacillaceae and Lachnospiraceae with advanced CIA compared to unimmunized mice [14].
Mucosal inflammation in the intestine as determined by colon histology and elevation of tissue cytokines IL-17A and IL-22 paralleled dysbiosis during the development of CIA. Intriguingly, despite robust inflammatory responses that are elicited by CFA, the significant changes in intestinal microbial communities of these mice did not result in the generation of local Th17 cytokine responses, suggesting that the developing autoimmune response to CII is an element of the mucosal changes. We found that mucosal IL-17A and IL-22, particularly in the small intestine, were significantly increased at day 14 of CIA when the S24-7 were low and Lactobacillaceae were increased. Both cytokines then decreased by day 35 of CIA to near day 0 levels, just as the abundance of S24-7 and Lactobacillaceae returned to near day 0 levels. These two bacterial groups, S24-7 and Lactobacillaceae, are well described to be inhabitants of the gastrointestinal tract of humans and animals [31, 32]. S24-7 is an IgA-coated, normal commensal of the gut in humans and murine models and involved in carbohydrate fermentation and utilization [32–34] while Lactobacillaceae are thought to have a regulatory effect on host immunity [35]. For example, L. salivarius and L. plantarum transferred to mice prior to the induction of CIA reduced Th17 and increased regulatory T cells and resulted in reduced arthritis severity [36]. The precise mechanism by which these microbial changes during the development of CIA modulate host immunity, though, requires further study. However, using a known oral pathogen, Sandal et al. demonstrated that oral infection with Porphyromonas gingivalis could transiently expand Th17 cells in the draining cervical lymph nodes and circulation and exacerbated disease in HLA-DRβ1 transgenic mice with CIA, even in those that had previously resisted development of disease [37]. Another study showed that treatment by oral gavage with Prevotella histicola attenuates CIA in HLA-DQ8 mice by increasing IL-10–producing regulatory T cells and reducing Th17 cells in the gut [38]. These provocative findings along with those in the K/BxN model [25] and ours presented herein suggest specific bacteria that stimulate mucosal Th17 drive disease development.
Depletion of microbiota through administration of broad-spectrum antibiotics resulted in decreased disease severity, reduced autoantibody generation, and impaired Th17-derived cytokine responses. The impact of intestinal microbes in the development of inflammatory arthritis was previously proposed in the K/BxN mouse model. This is a spontaneous model of inflammatory arthritis that is due to a transgenic TCR recognizing the ubiquitous autoantigen glucose-6-phosphate isomerase (GPI). The well-described pathology is similar to that in RA and begins around 4 weeks of age requiring T cell help for the generation of autoreactive B cells producing anti-GPI autoantibodies [39, 40]. In a germ free environment, mice develop delayed onset of arthritis with reduced severity. Segmented filamentous bacteria (SFB) were shown to modulate autoantibodies to GPI through the development of Th17 and Tfh cells within the intestines [25, 41]. Although we did not assess in this study the presence of T cell subsets in the intestines of mice with CIA, we did find increased intestinal tissue IL-17A and IL-22, two products of Th17 cell activation, which supports the development of these cells during CIA. Similarly, microbial depletion of mice with CIA through use of broad-spectrum antibiotics resulted in an initial significant decrease in IL-17A and IL-22 in intestinal tissues, suggesting a decrease in Th17 cell presence or activation. In conjunction, we observed a positive correlation between microbial abundance and the production of autoantibodies, which is in agreement with the K/BxN studies.
Whether commensal bacteria are necessary for the development of CIA remains unknown. Use of broad-spectrum antibiotic in our study allowed depletion of the microbiota but not complete removal of all bacteria, as noted by others previously [42]. Depleting microbiota early with either one week of antibiotic administration or continuous administration throughout the study resulted in the same effect on all disease assessments. This observation excludes the possibility that our results are solely due to direct antibiotic modulation of mucosal immunity, confirmed by our CAIA studies in which antibiotic treatment did not affect disease severity. To avoid the confounding effects of antibiotics and residual bacteria, germ free studies will be critical in establishing direct effects of specific bacteria in modulating immune responses and disease development in CIA.
In the setting of a reduced microbial load, we note significantly decreased CIA disease prevalence and severity. Most surprising, though, was our finding that the reduction in microbial burden at the time of the second, booster immunization had a more profound effect on disease severity. Our further investigations showed that, although total and IgG2b subclass anti-CII antibodies were only mildly reduced in antibiotic-treated mice, these antibodies were significantly impaired in their ability to activate complement. Autoantibody function has been linked to decreased glycosylation of the Fc segment in CIA [43, 44] and individuals with RA [45, 46]. An important effect of glycosylation of the Fc portion of autoantibodies is the modulation of complement activation [47], which is required for CIA [21, 43, 48]. In our study, mice treated with late antibiotics exhibited a distinctive glycosylation profile, with lower galactose levels. Importantly, terminal galactose residues increase antibody binding to C1q, resulting in C3b deposition more proinflammatory IgG effector functions [44, 47]. Another recent report linked the activity of pathogenic IgG in both the CIA and K/BxN models to IL-23 regulated Fc glycosylation, in particular sialylation [50]. Although in our studies IL-23 in the MLNs of mice in the late antibiotic treated group were reduced during late antibiotic treatment, we did not observe an alteration in the sialylation of immunoglobulins from these mice. Clearly additional studies are needed to solidify the role of the microbiota in driving CIA via modulation of antibody glycosylation, but our data together with that of others [25, 51, 52] supports the mechanism proposed in Fig. 6.
Figure 6. Proposed mechanism for microbial modulation of CIA.

During the early, preclinical phase of CIA, changes in the intestinal microbiome occur, which are associated with a more permeable barrier and mucosal inflammation. Mucosal inflammation is characterized by Th17 responses, as reflected by high levels of IL-17A and IL-22 in tissue, and downstream activation of B cells and mucosal autoantibody production. Circulation of mucosa generated Th17 and B cells leads to systemic autoimmunity. Microbiota also influence autoimmune arthritis later in the disease course, by promoting complement activation by autoantibodies possibly via altered glycosylation.
In our studies, we examined how both the microbiota and mucosal immune response change over the course of CIA development, which has not been the focus of prior reports that link microbiota with the development of autoimmune arthritis [14, 25, 51]. We were intrigued to observe that modification of the microbiota through administration of broad spectrum antibiotics at different time points during disease resulted in different patterns of mucosal and systemic immune responses. Depletion of the microbiota prior to the induction of CIA resulted in an initial decrease in mucosal IL-17A and IL-22 and anti-CII autoantibodies by day 14; however, by day 35 mucosal IL-17A, IL-22, and IL-23 were significantly increased compared to mice with CIA on control water, and anti-CII autoantibodies appeared to be trending towards similar levels between the two groups. Although the antibiotic treated mice had >50% reduction in the total microbial burden compared to controls, there may be shifts in specific bacterial operational taxonomic unit (OTU) during the course of CIA with antibiotics that may explain this later “catching up” of the Th17 response. Similarly, when mice with CIA were given antibiotics after the induction of disease, we found significantly reduced mucosal IL-17A and IL-22 as well as IL-23 in the MLNs that we propose link to reduced autoantibody effector function. Again, this may possibly be related to shifts in specific bacterial taxa without effect of the overall microbial abundance. Such shifts occurring after a local Th17 response has developed, at day 14 as suggested by our studies, could modify the local and systemic immune milieu much more differently than microbial shifts would affect immunity prior to the Th17 response, at day 0. Thus, we suggest that there are differing, time-dependent effects of the microbiota on the developing immune responses during CIA.
In summary, we characterize the intestinal microbiome and mucosal changes that occur during CIA. We demonstrate that in the absence of an intact microbiota, mice with CIA fail to develop robust disease. We propose that microbial dysbiosis in the preclinical phase of CIA are associated with significant mucosal changes. These changes provoke a mucosal Th17 immune response, educate mucosal lymphoid tissues to produce autoantibodies, and modulate the effector functions of autoantibodies. Autoreactive B cells that develop at mucosal surfaces expand, circulate, and result in systemic autoimmunity (Fig. 6). While additional studies are needed to more specifically identify causative bacteria and validate our proposed mechanism, targeting the microbiota during the preclinical phase of RA may have potential for disease prevention.
Supplementary Material
Acknowledgments
The authors would like to acknowledge V. Michael Holers and Sean P. Colgan for their helpful discussions regarding the project and manuscript. Funding for these studies was provided through NIAMS 5T32AR007534 (WKJ and JDH), NIAMS AR51749 (NKB and V. Michael Holers), and NIDDK K08DK107905 and Pfizer ASPIRE grants (KAK).
Footnotes
Disclosure: The authors declare no commercial or financial conflict of interest.
Author Contributions
WKJ, JDH and KAK designed the study, performed experiments, analyzed data, and wrote the manuscript. SA, HMS, ELS, and NKB assisted with the CIA and IgG experimental procedures and data acquisition. JP and RA performed glycosylation studies and analysis. DI, CER, and DNF performed microbiome sequencing, alignment, and analysis. All authors reviewed the manuscript and approved its final, submitted version.
References
- 1.Tobon GJ, Youinou P, Saraux A. The environment, geo-epidemiology, and autoimmune disease: Rheumatoid arthritis. J Autoimmun. 2010;35(1):10–14. doi: 10.1016/j.jaut.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 2.Silman AJ, Pearson JE. Epidemiology and genetics of rheumatoid arthritis. Arthritis Res. 2002;4(Suppl 3):S265–272. doi: 10.1186/ar578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Padyukov L, Silva C, Stolt P, Alfredsson L, Klareskog L. A gene-environment interaction between smoking and shared epitope genes in HLA-DR provides a high risk of seropositive rheumatoid arthritis. Arthritis Rheum. 2004;50(10):3085–3092. doi: 10.1002/art.20553. [DOI] [PubMed] [Google Scholar]
- 4.van Gaalen FA, Linn-Rasker SP, van Venrooij WJ, de Jong BA, Breedveld FC, Verweij CL, Toes RE, Huizinga TW. Autoantibodies to cyclic citrullinated peptides predict progression to rheumatoid arthritis in patients with undifferentiated arthritis: a prospective cohort study. Arthritis Rheum. 2004;50(3):709–715. doi: 10.1002/art.20044. [DOI] [PubMed] [Google Scholar]
- 5.Shi J, Knevel R, Suwannalai P, van der Linden MP, Janssen GM, van Veelen PA, Levarht NE, van der Helm-van Mil AH, Cerami A, Huizinga TW, et al. Autoantibodies recognizing carbamylated proteins are present in sera of patients with rheumatoid arthritis and predict joint damage. Proc Natl Acad Sci U S A. 2011;108(42):17372–17377. doi: 10.1073/pnas.1114465108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frank DN, Zhu W, Sartor RB, Li E. Investigating the biological and clinical significance of human dysbioses. Trends Microbiol. 2011;19(9):427–434. doi: 10.1016/j.tim.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, Rostron T, Cerundolo V, Pamer EG, Abramson SB, et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife. 2013;2:e01202. doi: 10.7554/eLife.01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D, Wu X, Li J, Tang L, Li Y, et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nature medicine. 2015;21(8):895–905. doi: 10.1038/nm.3914. [DOI] [PubMed] [Google Scholar]
- 9.Brand DD, Latham KA, Rosloniec EF. Collagen-induced arthritis. Nat Protoc. 2007;2(5):1269–1275. doi: 10.1038/nprot.2007.173. [DOI] [PubMed] [Google Scholar]
- 10.Madhok R, Crilly A, Murphy E, Smith J, Watson J, Capell HA. Gold therapy lowers serum interleukin 6 levels in rheumatoid arthritis. J Rheumatol. 1993;20(4):630–633. [PubMed] [Google Scholar]
- 11.Taylor PC, Feldmann M. Anti-TNF biologic agents: still the therapy of choice for rheumatoid arthritis. Nat Rev Rheumatol. 2009;5(10):578–582. doi: 10.1038/nrrheum.2009.181. [DOI] [PubMed] [Google Scholar]
- 12.Burger D, Dayer JM, Palmer G, Gabay C. Is IL-1 a good therapeutic target in the treatment of arthritis? Best Pract Res Clin Rheumatol. 2006;20(5):879–896. doi: 10.1016/j.berh.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 13.Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. Journal of immunology. 2003;171(11):6173–6177. doi: 10.4049/jimmunol.171.11.6173. [DOI] [PubMed] [Google Scholar]
- 14.Liu X, Zeng B, Zhang J, Li W, Mou F, Wang H, Zou Q, Zhong B, Wu L, Wei H, et al. Role of the Gut Microbiome in Modulating Arthritis Progression in Mice. Sci Rep. 2016;6:30594. doi: 10.1038/srep30594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Son JS, Khair S, Pettet DW, 3rd, Ouyang N, Tian X, Zhang Y, Zhu W, Mackenzie GG, Robertson CE, Ir D, et al. Altered Interactions between the Gut Microbiome and Colonic Mucosa Precede Polyposis in APCMin/+ Mice. PloS one. 2015;10(6):e0127985. doi: 10.1371/journal.pone.0127985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frank DN, Bales ES, Monks J, Jackman MJ, MacLean PS, Ir D, Robertson CE, Orlicky DJ, McManaman JL. Perilipin-2 Modulates Lipid Absorption and Microbiome Responses in the Mouse Intestine. PloS one. 2015;10(7):e0131944. doi: 10.1371/journal.pone.0131944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alkanani AK, Hara N, Lien E, Ir D, Kotter CV, Robertson CE, Wagner BD, Frank DN, Zipris D. Induction of diabetes in the RIP-B7. 1 mouse model is critically dependent on TLR3 and MyD88 pathways and is associated with alterations in the intestinal microbiome. Diabetes. 2014;63(2):619–631. doi: 10.2337/db13-1007. [DOI] [PubMed] [Google Scholar]
- 18.Pruesse E, Peplies J, Glockner FO. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics. 2012;28(14):1823–1829. doi: 10.1093/bioinformatics/bts252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glockner FO. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic acids research. 2007;35(21):7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robertson CE, Harris JK, Wagner BD, Granger D, Browne K, Tatem B, Feazel LM, Park K, Pace NR, Frank DN. Explicet: graphical user interface software for metadata-driven management, analysis and visualization of microbiome data. Bioinformatics. 2013;29(23):3100–3101. doi: 10.1093/bioinformatics/btt526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Banda NK, Kraus D, Vondracek A, Huynh LH, Bendele A, Holers VM, Arend WP. Mechanisms of effects of complement inhibition in murine collagen-induced arthritis. Arthritis Rheum-Us. 2002;46(11):3065–3075. doi: 10.1002/art.10591. [DOI] [PubMed] [Google Scholar]
- 22.Nadkarni MA, Martin FE, Jacques NA, Hunter N. Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology. 2002;148(Pt 1):257–266. doi: 10.1099/00221287-148-1-257. [DOI] [PubMed] [Google Scholar]
- 23.Oksanen J, Kindt R, Legendre P, O’Hara B, Simpson GL, Solymos P, Stevens HH, Wagner S. Vegan: Community Ecology Package. R package version 1.15–1 2008 [Google Scholar]
- 24.Henriksson AE, Blomquist L, Nord CE, Midtvedt T, Uribe A. Small intestinal bacterial overgrowth in patients with rheumatoid arthritis. Ann Rheum Dis. 1993;52(7):503–510. doi: 10.1136/ard.52.7.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, Littman DR, Benoist C, Mathis D. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity. 2010;32(6):815–827. doi: 10.1016/j.immuni.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Annu Rev Immunol. 1996;14:397–440. doi: 10.1146/annurev.immunol.14.1.397. [DOI] [PubMed] [Google Scholar]
- 27.Kokkonen H, Soderstrom I, Rocklov J, Hallmans G, Lejon K, Rantapaa Dahlqvist S. Up-regulation of cytokines and chemokines predates the onset of rheumatoid arthritis. Arthritis Rheum. 2010;62(2):383–391. doi: 10.1002/art.27186. [DOI] [PubMed] [Google Scholar]
- 28.Frank DN, Robertson CE, Hamm CM, Kpadeh Z, Zhang T, Chen H, Zhu W, Sartor RB, Boedeker EC, Harpaz N, et al. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm Bowel Dis. 2011;17(1):179–184. doi: 10.1002/ibd.21339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(34):13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li E, Hamm CM, Gulati AS, Sartor RB, Chen H, Wu X, Zhang T, Rohlf FJ, Zhu W, Gu C, et al. Inflammatory bowel diseases phenotype, C. difficile and NOD2 genotype are associated with shifts in human ileum associated microbial composition. PloS one. 2012;7(6):e26284. doi: 10.1371/journal.pone.0026284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dethlefsen L, McFall-Ngai M, Relman DA. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature. 2007;449(7164):811–818. doi: 10.1038/nature06245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ormerod KL, Wood DL, Lachner N, Gellatly SL, Daly JN, Parsons JD, Dal’Molin CG, Palfreyman RW, Nielsen LK, Cooper MA, et al. Genomic characterization of the uncultured Bacteroidales family S24-7 inhabiting the guts of homeothermic animals. Microbiome. 2016;4(1):36. doi: 10.1186/s40168-016-0181-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Salzman NH, de Jong H, Paterson Y, Harmsen HJ, Welling GW, Bos NA. Analysis of 16S libraries of mouse gastrointestinal microflora reveals a large new group of mouse intestinal bacteria. Microbiology. 2002;148(Pt 11):3651–3660. doi: 10.1099/00221287-148-11-3651. [DOI] [PubMed] [Google Scholar]
- 34.Palm NW, de Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L, Degnan PH, Hu J, Peter I, Zhang W, et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell. 2014;158(5):1000–1010. doi: 10.1016/j.cell.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Forbes JD, Van Domselaar G, Bernstein CN. The Gut Microbiota in Immune-Mediated Inflammatory Diseases. Frontiers in Microbiology. 2016:7. doi: 10.3389/fmicb.2016.01081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu X, Zhang J, Zou Q, Zhong B, Wang H, Mou F, Wu L, Fang Y. Lactobacillus salivarius Isolated from Patients with Rheumatoid Arthritis Suppresses Collagen-Induced Arthritis and Increases Treg Frequency in Mice. J Interferon Cytokine Res. 2016;36(12):706–712. doi: 10.1089/jir.2016.0057. [DOI] [PubMed] [Google Scholar]
- 37.Sandal I, Karydis A, Luo J, Prislovsky A, Whittington KB, Rosloniec EF, Dong C, Novack DV, Mydel P, Zheng SG, et al. Bone loss and aggravated autoimmune arthritis in HLA-DRbeta1-bearing humanized mice following oral challenge with Porphyromonas gingivalis. Arthritis Res Ther. 2016;18(1):249. doi: 10.1186/s13075-016-1143-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marietta EV, Murray JA, Luckey DH, Jeraldo PR, Lamba A, Patel R, Luthra HS, Mangalam A, Taneja V. Suppression of Inflammatory Arthritis by Human Gut-Derived Prevotella histicola in Humanized Mice. Arthritis Rheumatol. 2016;68(12):2878–2888. doi: 10.1002/art.39785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsumoto I, Staub A, Benoist C, Mathis D. Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science. 1999;286(5445):1732–1735. doi: 10.1126/science.286.5445.1732. [DOI] [PubMed] [Google Scholar]
- 40.Korganow AS, Ji H, Mangialaio S, Duchatelle V, Pelanda R, Martin T, Degott C, Kikutani H, Rajewsky K, Pasquali JL, et al. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity. 1999;10(4):451–461. doi: 10.1016/s1074-7613(00)80045-x. [DOI] [PubMed] [Google Scholar]
- 41.Block KE, Zheng Z, Dent AL, Kee BL, Huang H. Gut Microbiota Regulates K/BxN Autoimmune Arthritis through Follicular Helper T but Not Th17 Cells. J Immunol. 2016;196(4):1550–1557. doi: 10.4049/jimmunol.1501904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hill DA, Hoffmann C, Abt MC, Du Y, Kobuley D, Kirn TJ, Bushman FD, Artis D. Metagenomic analyses reveal antibiotic-induced temporal and spatial changes in intestinal microbiota with associated alterations in immune cell homeostasis. Mucosal immunology. 2010;3(2):148–158. doi: 10.1038/mi.2009.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Banda NK, Wood AK, Takahashi K, Levitt B, Rudd PM, Royle L, Abrahams JL, Stahl GL, Holers VM, Arend WP. Initiation of the Alternative Pathway of Murine Complement by Immune Complexes Is Dependent on N-Glycans in IgG Antibodies. Arthritis Rheum-Us. 2008;58(10):3081–3089. doi: 10.1002/art.23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raju TS. Terminal sugars of Fc glycans influence antibody effector functions of IgGs. Curr Opin Immunol. 2008;20(4):471–478. doi: 10.1016/j.coi.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 45.Ercan A, Cui J, Chatterton DE, Deane KD, Hazen MM, Brintnell W, O’Donnell CI, Derber LA, Weinblatt ME, Shadick NA, et al. Aberrant IgG galactosylation precedes disease onset, correlates with disease activity, and is prevalent in autoantibodies in rheumatoid arthritis. Arthritis Rheum. 2010;62(8):2239–2248. doi: 10.1002/art.27533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scherer HU, van der Woude D, Ioan-Facsinay A, el Bannoudi H, Trouw LA, Wang J, Haupl T, Burmester GR, Deelder AM, Huizinga TW, et al. Glycan profiling of anti-citrullinated protein antibodies isolated from human serum and synovial fluid. Arthritis Rheum. 2010;62(6):1620–1629. doi: 10.1002/art.27414. [DOI] [PubMed] [Google Scholar]
- 47.Quast I, Keller CW, Maurer MA, Giddens JP, Tackenberg B, Wang LX, Munz C, Nimmerjahn F, Dalakas MC, Lunemann JD. Sialylation of IgG Fc domain impairs complement-dependent cytotoxicity. J Clin Invest. 2015;125(11):4160–4170. doi: 10.1172/JCI82695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Banda NK, Levitt B, Glogowska MJ, Thurman JM, Takahashi K, Stahl GL, Tomlinson S, Arend WP, Holers VM. Targeted inhibition of the complement alternative pathway with complement receptor 2 and factor H attenuates collagen antibody-induced arthritis in mice. J Immunol. 2009;183(9):5928–5937. doi: 10.4049/jimmunol.0901826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu L. Antibody glycosylation and its impact on the pharmacokinetics and pharmacodynamics of monoclonal antibodies and Fc-fusion proteins. J Pharm Sci. 2015;104(6):1866–1884. doi: 10.1002/jps.24444. [DOI] [PubMed] [Google Scholar]
- 50.Pfeifle R, Rothe T, Ipseiz N, Scherer HU, Culemann S, Harre U, Ackermann JA, Seefried M, Kleyer A, Uderhardt S, et al. Regulation of autoantibody activity by the IL-23-TH17 axis determines the onset of autoimmune disease. Nat Immunol. 2017;18(1):104–113. doi: 10.1038/ni.3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Teng F, Klinger CN, Felix KM, Bradley CP, Wu E, Tran NL, Umesaki Y, Wu HJJ. Gut Microbiota Drive Autoimmune Arthritis by Promoting Differentiation and Migration of Peyer’s Patch T Follicular Helper Cells. Immunity. 2016;44(4):875–888. doi: 10.1016/j.immuni.2016.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kubinak JL, Petersen C, Stephens WZ, Soto R, Bake E, O’Connell RM, Round JL. MyD88 Signaling in T Cells Directs IgA-Mediated Control of the Microbiota to Promote Health. Cell Host & Microbe. 2015;17(2):153–163. doi: 10.1016/j.chom.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.