

Abstract
Ischemic postconditioning is increasingly being investigated as a therapeutic approach for cerebral ischemia. However, the majority of studies are focused on the acute protection of neurons per se. Whether and how postconditioning affects multiple cells in the recovering neurovascular unit remains to be fully elucidated. Here, we asked whether postconditioning may modulate help-me signaling between injured neurons and reactive microglia. Rats were subjected to 100 min of focal cerebral ischemia, then randomized into a control versus postconditioning group. After 3 days of reperfusion, infarct volumes were significantly reduced in animals treated with postconditioning, along with better neurologic outcomes. Immunostaining revealed that ischemic postconditioning increased expression of VEGF in neurons within peri-infarct regions. Correspondingly, we confirmed that VEGFR2 was expressed on Iba1-positive microglia/macrophages, and confocal microscopy showed that in postconditioned rats, these cells were polarized to a ramified morphology with higher expression of M2-like markers. Treating rats with a VEGF receptor 2 kinase inhibitor negated these effects of postconditioning on microglia/macrophage polarization. In vitro, postconditoning after oxygen-glucose deprivation upregulated VEGF release in primary neuron cultures, and adding VEGF to microglial cultures partly shifted their M2-like markers. Altogether, our findings support the idea that after postconditioning, injured neurons may release VEGF as a “help me" signal that promotes microglia/macrophage polarization into potentially beneficial phenotypes.
Keywords: neuroprotection, postconditioning, neurovascular unit, microglia, stroke
Graphical abstract
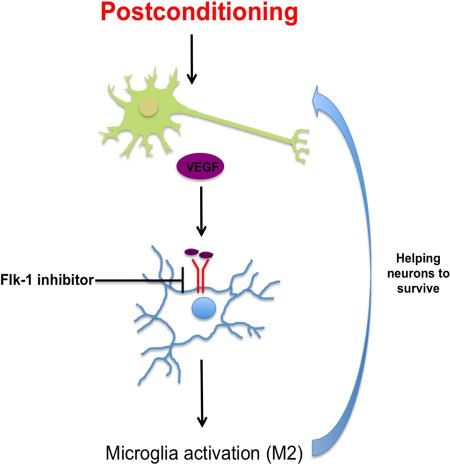
Our hypothesis is that ischemic postconditioning can promote neuroprotection by increasing the ability of the brain to “help itself” via cell-cell interactions. We proposed that postconditioning may induce neurons to release VEGF as a “help-me signal” that shifts microglia/macrophage into beneficial forms (M2-like), and that this event can have a neuroprotective effect. Treatments with Flk-1 inhibitor to block VEGF signaling annuls the M2-like microglia polarization and the neuroprotection after postconditioning.
INTRODUCTION
Initially studied in the field of myocardial ischemia, ischemic postconditioning has been increasingly investigated in cerebral ischemia. Many experimental studies suggest that ischemic postconditioning decreases neuronal death during the acute phase after stroke, and more recently this neuroprotection has been confirmed for longer periods of recovery as well (Esposito et al., 2015; Zhao, 2009). However, almost all postconditioning studies thus far, are focused on neuroprotection per se, i.e. molecular mechanisms that are activated “inside” neurons that reduce cell death or increase pro-survival mediators (Pignataro et al., 2008; Zhao et al., 2012).
It is now recognized that stroke affects not only neurons but all cells within the entire neurovascular unit. Dynamic interactions between endothelial cells, glia, neurons and matrix may underlie the balance between acute injury and endogenous repair (Maki et al., 2013). To date, a few studies suggest that postconditioning may also affect non-neuronal cells (Teo et al., 2015; Yin et al., 2014), however these studies do not analyze how those cells are rescued and if they play any role in improving neuronal recovery. Therefore, here we ask if it is possible that ischemic postconditioning not only acutely protects neurons but also improves outcomes by augmenting endogenous mechanisms of cell-cell signaling as the entire neurovascular unit attempts to recover.
VEGF is an important growth factor best known for its effects on endothelial cells to promote the formation of new vessels. Although originally described as an angiogenic factor in vascular systems, it is now well established that VEGF also plays a crucial role in the nervous system (Zhang and Chopp, 2002). In fact, during brain ischemia, VEGF can mediate recovery through three major mechanisms: stimulating angiogenesis and modulating vascular permeability, direct neuroprotection, and promoting neurogenesis (Ruiz de Almodovar et al., 2009). However, more recently, VEGF is also known as a signal that can regulate neuroinflammation. Neural progenitor cells regulate microglia function and activity by releasing VEGF (Mosher et al., 2012). Moreover, VEGF is known to be involved in the neuroprotection achieved by preconditioning (Koch et al., 2014; Park et al., 2014). Hence, in this study, we explored the idea that postconditoning may also upregulate neuronal VEGF, and the beneficial effects of this pathway may involve the polarization of microglia/macrophages into pro-recovery modes.
MATERIALS AND METHODS
Focal Cerebral Ischemia in Rats
Male Sprague–Dawley (RRID:RGD_5508397) rats (Charles River) weighting 320 to 340 g were housed in groups of 4/cage under diurnal lighting conditions (12 hours darkness/light) with free access to water and food. All experiments were performed following protocol no.2016N000528 approved and preregistered by Massachusetts General Hospital Institutional Animal Care and Use Committee in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and following Animals in Research: Reporting In vivo Experiments (ARRIVE) guidelines. The rats were randomly assigned (simple randomization with computer-generated random numbers), before starting the surgery, to two groups of stroke animals, one group of 28 rats (plus 5rats for Flk1 inhibitor injection) exposed to postconditioning, and the other group of 41 rats exposed to standard post-stroke care. Ischemic postconditioning was induced as described previously (Esposito et al., 2015). Briefly, after 100 minutes of MCAO, reperfusion was established for 10 minutes after which the MCA was reoccluded for 10 minutes, and then animals were recovered for 72 hours (Figure 2A). Transient focal ischemia was induced, as described previously (Pignataro et al., 2008), by suture occlusion of the MCA in male rats. Before surgery, rats were anesthetized using 5% isofluorane, 70% N2O, and 30% O2 for 1 to 2 min until they became completely anesthetized (deep breathing, toe and tail pinch to tail pinch), and then isoflurane was switch to 1.5% to maintain the animals under anesthesia during the all procedures. Pain management after surgery was performed by Buprenorphine 0.05 mg/kg IP q12h for the first 3 days post-op. Ischemia was induced by introducing a 5-O surgical monofilament nylon suture (Doccol Corporation, Cat# 503856PK10) from the external carotid artery into the internal carotid artery and advancing it into the circle of Willis to the branching point of the MCA, thereby occluding the MCA (Longa et al., 1989). Achievement of ischemia was confirmed by monitoring regional cerebral blood flow in the area of the right MCA. Adequate ischemia was confirmed by continuous laser Doppler flowmetry (LDF) (Perimed, North Royalton, OH, U.S.A.) (Pignataro et al., 2011b). For placement of the LDF probe, a burr hole 2 mm to 3 mm in diameter was created in the right parietal bone (2 mm posterior and 6 mm lateral to bregma). After 100-minutes MCA occlusion, the monofilament suture was gently withdrawn in order to restore blood flow, and LDF values were recorded for 30 minutes after reperfusion. Rats that did not demonstrate a significant reduction to less than 30% baseline LDF values during MCAO and MCAO plus postconditioning, or rapid restoration of the LDF signal during reperfusion were excluded, for a total of 25 rats (10 rats were excluded in MCAO group, 10 in postconditioning and 5 in postconditioning plus Flk-1 inhibitor), we did not observe any bleeding and none of the rat died so the LDF measurement was the only criteria we used for exclusion. The rats included were used for Nissl staning and behavioral (9rats for MCAO, 8 rats for postconditioning and 5 rats for postconditioning plus inhibitor), western blot (3–4 rats per group), immunostaining (5–6 rats per group). Rectal temperature was maintained at 37°C±0.5°C with a thermostatically controlled heating pad and lamp. The sham-operated groups (n=6) received the same procedure without inserting the filament into the MCA. All procedures and measurements were performed in a blinded and randomized fashion as follows: EE performed MCAO or postconditioning surgery. A second investigator (SC) performed the i.c.v. injections. A third investigator (BA) performed the in vitro studies, VEGF or media without VEGF were provided by KH, who also blinded all animal treatments after surgery. EE performed the behavioral tests, immunostaining and western blot. Each investigator performed a different step of the study, in this way the investigator was completely unaware of the other steps (surgery, injections, etc.). All the procedures were performed in our laboratories.
Figure 2. Ischemic postconditioning neuroprotection.

(A) After100 minutes of MCAO postconditioning was induced by 10 minutes reperfusion after which the MCA was re-occluded for 10 minutes, then animals were recovered for 72 hours (B) Ischemic volume was significantly smaller in postconditioning (POSTC n=8 rats) group compared to controls (CTL; 100 min ischemia n=9 rats). (C) Neurological score and forelimb placing test showed a significant improvement (with a lower score) after postconditioning treatment compared to control group. *P<0.05. Data are mean±SD.
A Flowchart diagram showing an overview of the study design for all the experimental procedures used in this study is presented in Figure 1. For in vivo studies, after the animals were tested for behavioral outcomes, ischemia and protein expression were analyzed by Nissl staining, western blot and immunostaining. To prove loss of function, animals after postconditioning were randomized in postconditioning plus vehicle or postconditioning plus Flk1 inhibitor.
Figure 1. Flowchart diagram.

Overview of the study design for all the experimental procedures used in this study.
Evaluation of Infarct Volume and Neurological Tests
All rats were assessed blindly at 3 days using two tests: forelimb placement test and a 5-point neuroscore scale, 9 rats for MCAO and 8 rats for postconditioning, For the forelimb-placing test, the animals were held close to a tabletop and scored for the ability to place the forelimb on the tabletop in response to whisker, visual, tactile, or proprioceptive stimulation, they were scored as 0 = normal, and 8 = maximally impaired. The Neurological Score was graded on a scale of 0 to 4, with a higher score indicating more severe sensory-motor deficits (Esposito et al., 2013). Then animals underwent transcardial perfusion with 40mL of phosphate-buffered saline (PBS) under deep (5%) isoflurane anesthesia. Brains were quickly removed and frozen, and coronal sections of 20µm thickness were prepared from frozen rat brains. Infarction volumes were quantified on Nissl-stained sections, 9 rats for MCAO and 8 rats for postconditioning, using the “indirect” morphometric method (Lin et al., 1993) with Image J software.
Primary Neuronal Cultures
Primary neuron cultures (4 cultures in triplicate) were prepared from cerebral cortices of Sprague–Dawley rat embryos (E)17. In brief, cortices were dissected, and the isolated cells were plated onto plates coated with poly-d-lysine (Sigma, P7886) and cultured in DMEM (NBM, Life Technology, 11965-084) containing 25 mM glucose, 4 mM glutamine, 1 mM sodium pyruvate, and 5% FBS at a density of 5 × 105 cells/mL. After 24 h DMEM medium was substituted with Neurobasal medium (Invitrogen, 21103-049) supplemented with B-27 (Invitrogen, 17504044) and 0.5 mM glutamine. 7 to 10 days later, OGD or OGD plus postconditioning was performed.
To perform OGD, the cell medium was replaced with deoxygenated, glucose-free DMEM (Life Technology, 11966-025). The cell cultures were introduced in a specialized, humidified chamber (Heidolph, incubator 1000, Brinkmann Instruments) kept at 37 °C, which contained an anaerobic gas mixture (90% N2, 5% H2, and 5% CO2). After 60 mins challenge, cultures were removed from the anaerobic chamber, and for the OGD samples the cultures was replaced with Neurobasal medium.
Postconditioning in vitro was performed by 60 minutes of OGD plus 6 minutes of reoxygenation (by substitution of glucose-free DMEM with Neurobasal) plus 6 minutes of OGD (by substitution of Neurobasal with glucose-free DMEM and replacing the cells in the chamber). After that, media was replaced with fresh Neurobasal medium in both, OGD and postconditioning samples. After 3 hours reoxygenation media was collected and ELISA was performed (Rat VEGF Quantikine ELISA Kit, R&D Systems). See flowchart in Fig 1.
Primary Microglial Cultures
Primary mixed glial cultures (4 cultures in triplicate) were prepared from postnatal day 0–2 Sprague-Dawley rat pups. After removing the meninges, isolated cortices were minced and digested for 15 min in 5 mL Hank's balanced salt solution (Gibco) containing 190 µL DNase and 50 µL 2.5% Trypsin. The mixed cortical cells were passed through a 70-µm nylon mesh cell strainer and seeded in T75 flasks in DMEM/F12 with 20% FBS, the medium was completely replaced every other day. After achieving confluency at about 14 days in vitro, the microglia were isolated from mixed glial cultures via shaking. Confluent mixed glial cultures were placed on an orbital shaker at 220 rpm for 1 h. The supernatant containing the detached microglial cells was collected and centrifuged for 5 min at 1000rpm. Cell pellet was obtained and resuspended in DMEM with 10% FBS. Then cells were seeded in a cell density of 2×105 cells/mL for 1 h to allow microglial attachment. After 1 h, the nonadherent cells were removed. Microglia was allowed to rest overnight prior treatments. Human VEGF (Sigma-Aldrich, Cat# V7259) was added to the media 24 hours later at a concentration of 5ng/ml, 10ng/ml or 100ng/ml. See flowchart in Fig 1.
Intracerebroventricular administration
A total of 5 rats were positioned on a stereotaxic frame and a 23-g stainless steel guide cannula was implanted into the right lateral ventricle using the stereotaxic coordinates from the bregma: −0.9 mm caudal, 1.4 mm lateral and 3.8 mm from the skull. VEGF Receptor 2 (Flk-1) Kinase Inhibitor I (5µL, 2µM; Millipore, Cat# 676480) and respective vehicle (PBS) were injected 10 min after postconditioning in a randomized fashion.
Immunohistochemistry
Immunohistochemistry was performed on 6 sham, 6 MCAO, 5 postconditioning and 5 postconditioning plus FLK-1 inhibitor rats, using primary antibodies anti-VEGF (1:500, Santa Cruz Biotechnology, Cat# sc-152, RRID:AB_2212984) and anti-VEGFR2 (1:200, Abcam, Cat# ab45010, RRID:AB_883436); anti-GFAP (1:200, Invitrogen, Scientific Cat# 13-0300, RRID:AB_2532994) as a marker of activated astrocytes; anti-Iba1 (1:200, Wako, Cat# 019-19471, RRID:AB_2665520) as marker of microglia; anti-CD68 (1:100, Abcam, Cat# ab955, RRID:AB_307338) as a general microglia activation marker; anti-CD206 (1:200, Santa Cruz Biotechnology, Cat# sc-34577, RRID:AB_2144904) as an M2-like marker; anti-CD86 (1:200, Abcam, Cat# ab53004, RRID:AB_869050) as an M1-like marker, and anti-NeuN (1:100, Millipore, Cat# MAB377, RRID:AB_2298772) as a marker of neurons. To investigate increase in angiogenesis, immunostaining was performed by quantifying microvessels that were double-positive for collagen-IV (1:10, SouthernBiotech) for vascular remodeling and Ki67 (1:500, Abcam, Cat# ab16667, RRID:AB_302459) a general cell proliferation marker. Three non-overlapping areas were chosen in the boundary zone of the ischemic core to analyze the peri-infarct area. Immunostaining was analyzed with a fluorescence microscope (Nikon ECLIPSE Ti-S) interfaced with a digital charge-coupled device camera and an image analysis system.
The fluorescent-stained sections for anti-Neurofilament (1:100, Millipore, Cat# MAB1615, RRID:AB_94285), as marker of neurons and Iba1 were analyzed on a Leica DMRB/Bio-Rad 1024 confocal microscope with krypton–argon laser.
Western Blotting
For in vivo studies: at 3 days after ischemia animals (sham n=4, MCAO n=3, postconditioning n=3) underwent transcardial perfusion with 40 mL of phosphate-buffered saline (PBS) under deep (5%) isoflurane anesthesia. Brains were quickly removed and frozen. Coronal brain sections 100µm thick were cut in the cryostat at −20°C (Leica Biosystems Buffalo Grove, IL). For each section, by using a 5mm tip knife, we were able to cut and collect exactly 5mm around the ischemic boundary. These samples were pulled together in a tube for further western blot analysis. With this method we precisely cut 5mm around the ischemic boundary for the all brain. With conventional techniques the collection of a specific area is not as precise as ours because of the brain shape. Only cutting slide by slide and collecting 5 mm from the ischemic boundary we were sure to study the exact location of our interest (Figure 3A). The samples were homogenized using an18-gauge needle with a Protein Extraction Kit (iNtRON PRO-PREP(TM) containing protease inhibitors (PMSF, EDTA, Pepstatin A, Leupeptin, Aprotininl), 600ml solution per 10–20mg tissue. For in vitro studies (3n per group): microglial cells were treated with the same protein extraction kit, 400ml solution per 5×10(6) cells. Then, the samples were centrifuged at 14000rpm for 20 min. Supernatant was collected. The protein concentrations of cell lysates were determined using a protein assay kit (Protein Quantification Kit, Dojindo Molecular Technologies, Cat# PQ01). Samples containing equal amounts (20µg) of protein were resolved by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (Nu-PAGE 4–12% Bis-Tris-Gel, Invitrogen) and transferred to nitrocellulose membranes using I-Blot 2 (Invitrogen). The membranes were blocked with 5% skim milk for one hour followed by overnight incubation at 4°C with specific primary antibodies against VEGF (RRID:AB_2212663), CD206 (Santa Cruz Biotechnology, Cat# sc-34577, RRID:AB_2144904) and β-actin (1:2000; Sigma-Aldrich Cat# A5441, RRID:AB_476744). The day after, the membranes were washed 3 times with Tris-buffered saline containing 0.05% Tween-20 (TBST) for 10 min each time and then processed using a horseradish peroxidase (HRP)-conjugated anti-goat or anti-mouse antibody for one hour at room temperature (Jackson Immunoresearch laboratories) followed by enhanced chemiluminescence (ECL) detection (Thermo Fisher Scientific). The results from the Western blots were quantified with an image analyzer (Bio-Rad, Quantity One).
Figure 3. Ischemic postconditioning increases VEGF in neurons.

(A) The total amount of VEGF protein levels did not change in the area 5mm around the ischemic infarct (n=4 brains for sham group, n=3 brains for CTL group, n=3 brains for POST group). (B) The expression of VEGF in neurons about 500µm around the peri-infarct area increased after ischemic postconditioning compared to sham and control group, control group expression was higher than in sham group. (C) Higher magnification of VEGF/NeuN cell positive number (n=6 brains for sham group, n=6 brains for CTL group, n=6 brains for POST group) (D) ELISAs showed that in neuronal cultures after postconditioning (protocol showed in the top part) VEGF release was significantly higher compared to OGD alone. *P<0.05 versus sham; ^P<0.05 versus postconditioning. Data are mean±SD.
Statistical analysis
Values are expressed as mean ± SD. Infarct volumes and angiogenesis were assessed with Student's t-test or one-way ANOVA followed by Tukey’s test. For western blot analysis and cell counts of immunopositive cells one-way ANOVA followed by Tukey’s test was performed. Neurological outcomes were analyzed using Mann Whitney test. P values of p<0.05 were considered statistically significant. Statistical analysis was performed with GraphPad Prism 6.01. All studies were randomized and blinded. Animal sample size was predetermine using the software available online: https://www.danielsoper.com/statcalc/calculator.aspx?id=47. The calculation was based on Cohen’s d value where SD and average were estimated from our historical and preliminary data. Drawing on the collaborative expertise of the MGH stroke program, my lab could begin to estimate power α = 0.05, 1 − β = 0.8. For in vivo infarct volumes, coefficients of variation usually range between 25–45%, so we planned to use n=8–9 per group to detect effects of 50%. For in vivo immunohistochemistry, we usually get coefficients of variation of 30%, so n=5–6 per group should allow us to detect 50–70% effect sizes. Coefficients of variation for our battery of behavior tests during stroke recovery in rats are usually around 30–40%, so n=8–9 per group should allow us to be sensitive to effect sizes of approximately 50–55%. For cell culture assays, we expected coefficients of variation around 20–25%, so we planned to use 3–4 cultures in triplicate to detect effect sizes of approximately 10–20%.
RESULTS
Ischemic postconditioning reduced infarction and improved neurologic outcomes at 3 days post-stroke
By 3 days after stroke onset, Nissl staining revealed well-defined infarcts in control animals subjected to 100 minutes of transient MCAO (171.8±46.19 mm3, n=9). With ischemic postconditioning, infarct volumes were markedly reduced (84.9±40.31 mm3, n=8, P=0.0009) (Figure 2B). Postconditioning led to significantly better outcomes in the forelimb placement test (3.25 ± 1.5) versus controls (5.5 ± 1.6, P=0.0309). Rats that received postconditioning (0.87±0.6, n=8) also had significantly better outcomes in neurological score compared to controls (2.0±1.0, n=9, P=0.0329) (Figure 2C).
Ischemic postconditioning increases VEGF expression in peri-infarct neurons and in neuronal cultures
Western blot analysis of total VEGF protein levels within peri-infarct areas (approx. 5 mm rim around the core) did not show any difference between postconditioning and control groups P=0.1664 (Figure 3A). However, in spite of this lack of whole tissue protein response, immunostaining suggested that VEGF signals might have been affected at the cellular level (Figure 3B). Within a smaller peri-infarct perimeter comprising a 500µm boundary around the infarct (Figure 3B, area A, C, D), co-expression of VEGF and NeuN at 3 days post-ischemia was significantly increased in the MCAO group (n=6; 93.5±29.33 positive cells/mm2) compared to sham operated group (n=6; 32.67±9.20 positive cells/mm2). However, when the rats were subjected to ischemic postconditioning the VEGF specifically expressed by neurons was even higher (n=6; 211.37±63.31 positive cells/mm2, P< 0,0001; Figure 3C). Moreover, to compare the same identical area in all groups we also analyzed area A, B and C for control group. In ischemic rats subjected to 100 minutes of MCAO area B was affected by strong infarct and mostly of the cells were dead. The total cell count resulted still significantly increased in postconditioning group compared to ischemia alone (n=6; 63.33±20.10 positive cells/mm2, Figure 3B, area A, B, C) or sham groups. There was no significant difference between control group when we analyzed A, B, C and control group when we analyzed A, C, D. For sham group the area analyzed were A, B, C.
We confirmed this idea in vitro. We performed OGD and OGD plus postconditioning on neuronal cultures, and then used ELISAs to quantify the VEGF released into the conditioned media that was collected 3 hours after reoxygenation. Our analysis showed that after postconditioning, VEGF (n=4; 408.28±26.39 pg/mL) was significantly (P=0.0163) increased compared to the OGD alone group (n=4; 343.28±29.16 pg/mL) (Figure 3D).
Because VEGF is usually responsible for angiogenesis, we assessed the effect of postconditioning at 3 days after stroke on new microvessel formation. The density of collagen-IV-Ki67 microvessels was not significantly higher in the postconditioning group (n=4; 49.17±17.32 positive cells/mm2) compared with controls (n=4; 37.22±15.54 positive cells/mm2, P=0.3443) (Figure 4). To evaluate if other cell types increased VEGF as well, immunohistochemistry was performed to look for responses in GFAP, a general marker of astrocytes, and Iba1, a general marker of microglia/macrophages. Co-expression of VEGF and GFAP at 3 days post-ischemia did not increase after postconditioning compared with controls or sham in the peri-infarct area (Figure 5A). However, co-expression of VEGF and Iba1 in the peri-infarct area after postconditioning seemed slightly increased compared with controls (Figure 5B).
Figure 4. Ischemic postconditioning does not increase angiogenesis at 3 days.

The cell numbers coexpressing collagen IV and anti-Ki67/mm2 in the peri-infarct area were not increased in postconditioning group compared to control group. (n=4 brains for CTL group, n=4 brains for POST group). Data are mean±SD.
Figure 5. Ischemic postconditioning does not increase VEGF in microglia/macrophages or astrocytes.

No evident difference was observed in coexpression of VEGF and GFAP (A) or VEGF and Iba1 (B) indicating that VEGF levels do not change in microglia or astrocytes after ischemic postconditioning (n=3 brains for CTL group, n=3 brains for POST group).
Next, we asked whether these subsets of microglia/macrophages might possess the appropriate receptors that allow them to respond to VEGF after cerebral ischemia. As expected, Iba-1-positive cells co-localized with the VEGFR2 receptor, compared to Neu-N-positive cells (Figure 6A–B).
Figure 6. VEGFR2 expression after ischemic postconditioning.

(A) The co-expression of VEGFR2 and Iba1 was detected in sham and MCAO groups as well as in postconditioning. (B) VEGFR2 was also detected on neuronal surface.
Ischemic Postconditioning in vivo and VEGF cell treatment in vitro alter microglia/macrophage polarization
Since neurons appeared to increase VEGF and Iba1-positive cells express the VEGF receptor VEGFR2, we asked whether this pathway may represent a potential type of help me signaling that allows injured neurons to shift microglia/macrophage polarization.
First, we directly exposed primary rat microglia cultures to VEGF and assessed the effects on M2-like phenotype. Addition of exogenous VEGF (100ng/ml) to primary microglia resulted in an increased number of cells expressing the M2-like marker CD206 by immunocytochemistry. This result was confirmed by western blot analysis where VEGF increased CD206 protein expression (n=3; 1.0±0.16% control vs 1.13±0.1% VEGF added group; P=0.045) (Figure 7A)
Figure 7. Ischemic postconditioning increased microglia expression with a ramified morphology.

(A) In vitro cell culture experiments showed that the number of microglia cells expressing CD206 increased when VEGF was added to the cell cultures compared with untreated (control) cells in immunostaing and western blot analysis (n=3 cultures in triplicate for controls, n=3 cultures in triplicate for VEGF treated cells). (B) After 100 minutes MCAO, microglia/macrophages showed unramified/amoeboid morphology different from the brains exposed to postconditioning that presented a ramified morphology. (C) The co-expression of Iba1 and CD68 increased in postconditioning group compared with controls in the peri-infarct area but it did not increased when postconditioning rats were treated with Flk-1 inhibitor (n=3 brains for CTL group, n=3 brains for POST group). *P<0.05 versus control; ^P<0.05 versus postconditioning or OGD. Data are mean±SD.
Second, we asked whether these microglial effects could be detected in vivo. After cerebral ischemia, activated Iba1-positive cells tended to assume a typical unramified/amoeboid morphology. Confocal analysis suggested that in our rat focal ischemia model, postconditioning altered the morphology of these activated microglia/macrophages into a ramified morphology (Figure 7B).
Third, we attempted a loss-of-function approach to ask whether blocking the VEGF receptor could interfere with these hypothesized effects of postconditioning on Iba1-positive cells. Immunohistochemistry was performed to assess markers of microglia/macrophage polarization, focusing on CD68, CD206 and CD86 as representative markers of various activation states. CD68 is known as a pro-phagocytosis marker that is associated with general activation. Iba1 and CD68 double staining suggested an overall increase in activated microglia/macrophage density after MCAO when compared to sham. But, after postconditioning the expression significantly increased compared to the MCAO only control group. This effect of postconditioning was prevented when the rats were treated with Flk-1 inhibitor to block VEGF signaling (Figure 7C). The co-expression of Iba1 and the M2-like marker CD206 was significantly increased within the peri-infarct area in MCAO group (n=6; 65.83±20.74 positive cells/mm2) compared with sham brains (n=6; 17.33±8.94 positive cells/mm2). After postconditioning the rat brains showed an increase of Iba1/CD206 positive cells (n=5; 125.2±24.26 positive cells/mm2) compared to both sham and MCAO only groups. However, after i.c.v. Flk-1 inhibitor injections, CD206 was no longer increased in postconditioning group (n=5; 46.8±30.49 positive cells/mm2, P<0,0001) (Figure 8B). Along, with the M2-like marker results, as expected, the co-expression of Iba1 and the general M1-like activation marker CD86 increased after MCAO (n=6; 76±34.66 positive cells/mm2) but not in postconditioning group (n=5; 34±16 positive cells/mm2) when compared to sham (n=6; 36.17±7.36 positive cells/mm2 P=0.0171). Postconditioning plus Flk-1 inhibitor (n=5; 60.6±23.18 positive cells/mm2) did not revel any significant difference (Figure 8D).
Figure 8. After postconditioning, microglia/macrophages presents M2-like markers.

(A) Schematic of postconditioning regulation on microglia/ macrophage activation. (B) In vivo co-expression of Iba1 and CD206 was increased within the peri-infarct area in MCAO group compared with sham brains. After postconditioning the rat brains showed an increase of Iba1/CD206 positive cells compared to both sham and MCAO groups. The increase was not present anymore when postconditioning rats were treated with Flk-1 inhibitor (n=6 brains for sham group, n=6 brains for CTL group, n=5 brains for POST group, n=5 brains for POST + Flk1inh group). (C) Flk-1 inhibitor reverts in part the neuroprotection induced by postconditioning (CTL n=9 rats, POSTC n=8 rats, POSTC + Flk1 inh n=5 rats). (D) The co-expression of Iba1 and CD86 increased after MCAO but not in postconditioning group when compared to sham. Postconditioning plus Flk-1 inhibitor did not revel any significant difference (n=6 brains for sham group, n=6 brains for CTL group, n=5 brains for POST group, n=5 brains for POST + Flk1inh group). *P<0.05 versus sham (panel B) or CTL (panel C); ^P<0.05 versus postconditioning. Data are mean±SD.
Finally, to prove that inhibition of VEGF activity can truly block the protective effects of ischemic postconditioning, we performed loss of function studies by analyzing the effect of Flk-1 inhibitor on the ischemic injury volume after postconditioning. Our data show that the inhibition of VEGFR2 partially reverts (155.2±34.49 mm3, n=5) the neuroprotection induced by postconditioning (P=0.0012) (Figure 8C).
DISCUSSION
Clinical trials of pharmacological neuroprotection in stroke have been disappointing. So attention has turned to the brain’s own endogenous strategies for protection. Ischemic postconditioning is defined as a series of brief non-injurious mechanical occlusions and reperfusions subsequent to a prolonged episode of harmful ischemia (Pignataro et al., 2008; Zhao et al., 2006). Rapid ischemic postconditioning, applied immediately after the harmful event, is able to modify reperfusion-induced adverse events (Gao et al.; Leconte et al., 2009; Wang et al., 2008; Xing et al., 2008). One immediate effect is probably associated with changes in cerebral blood flow profiles during reperfusion. Subsequent events such as free radical production, BBB integrity, inflammation and endothelial function have also been shown to be involved (Gao et al., 2008; Guo et al., 2014; Joo et al., 2013; Zhao et al., 2012).
So far, the majority of experimental studies have been focused on the neuronal effects of ischemic postconditioning; it is now well know that ischemic postconditioning can increase neuron survival (Pignataro et al., 2011a; Pignataro et al., 2011b; Ren et al., 2008). However, many gaps in knowledge still exist. Most importantly, how postconditioning affects non-neuronal cells remains largely unknown. For example, it is known that some of the benefits of preconditioning may involve indirect effects on astrocytes (Narayanan et al., 2015). Our goal is to define the molecular mechanisms of postconditioning that involve interactions between different cell types during initial injury and subsequent recovery. Is it possible that non-neuronal signaling pathways may also underlie some of the benefits of postconditioning? So far, there is no knowledge about different cell type interactions after postconditioning, and whether these cross-talk mechanisms may be eventually beneficial for further recovery. Our idea is based in part on the concept of the neurovascular unit, i.e. dynamic interactions between endothelial cells, glia, neurons and matrix. Our hypothesis is that ischemic postconditioning can promote neuroprotection by increasing the ability of the brain to “help itself” via cell-cell interactions. In the present study, our focus was to analyze the effects of ischemic postconditioning on the potential links between neuronal VEGF and microglia/macrophage polarization.
VEGF plays a crucial role in the nervous system and its neuroprotection can be dose-dependent. For instance, low doses of systemic VEGF promote neuroprotection of ischemic brains without inducing angiogenesis, whereas a high dose of VEGF induces angiogenesis but does not protect ischemic brains (Manoonkitiwongsa et al., 2004). Recently, it has also been shown that neural progenitor cells regulate microglia functions and activity by releasing VEGF (Mosher et al., 2012). Moreover, VEGF increase is known to be involved in the preconditioning neuroprotection (Park et al., 2014). These prior studies hence provide a rationale for the idea that postconditioning increases VEGF release in neurons and may affect the ability of endogenous VEGF to polarize microglia.
After cerebral ischemia, microglia release neurotoxic factors and free radicals that amplify secondary neuronal death. However, under some conditions reactive microglia may polarize to a beneficial phenotype as the brain tries to recover (Hu et al., 2012). Microglia modulate their environment and respond to injury by secreting cytokines and chemokines. These biphasic effects have led to analogies with peripheral macrophages, where damaging M1-like phenotypes are contrasted against beneficial M2-like phenotypes (Patel et al., 2013) (Zhao et al., 2015). Our data suggest that (a) postconditioning in a rat model of focal ischemia reduced infarct size and improved neurological outcomes at 3 days; (b) after ischemic postconditioning neurons increased VEGF levels only around the peri-infarct area with no effect on angiogenesis; (c) postconditioning shifted microglia into potentially beneficial ramified morphology with M2-like markers; and (d) treatment with Flk-1 inhibitor to block VEGF signaling annulled the M2-like microglia polarization and the neuroprotection after postconditioning.
Taken together, these data suggest that postconditioning may induce neurons to release VEGF as a “help-me signal” that shifts microglia/macrophage into beneficial forms, and that this event can have a neuroprotective effect (Figure 8A). These results may be consistent with previous studies reporting that another factor, lipocalin-2, is released by injured neurons as a help me distress signal that activates microglia and astrocytes into potentially pro-recovery phenotypes (Xing et al., 2014). Postconditioning may be a practically feasible way to promote neuroprotection after stroke. But many obstacles prevent clinical translation. All the studies investigated so far are only focused on neuronal cells. But the brain contains many other non-neuronal cells. Additional investigation of postconditioning effects in different cell types and their interactions is needed. Understanding these mechanisms may eventually lead to enhanced ways to improve stroke recovery with postconditioning.
However, there are several caveats to keep in mind. First, our primary focus here is on neuron-to-microglia signaling but of course after stroke, injury and recovery mechanisms will surely involve cell–cell signaling between all neuronal, glial and vascular elements (Arai and Lo, 2010; Ji et al., 2013; Saini et al., 2011). How postconditioning affects the entire neurovascular unit should be further dissected. Second, microglia modulate their environment and respond to injury by secreting cytokines and chemokines. Beyond VEGF per se, other cytokines and growth factors (i.e. BDNF, TNFα etc.) (Cekanaviciute et al., 2014; Eltzschig and Eckle, 2011; Gomez-Pinilla et al., 2002; Iadecola and Anrather, 2011; Low et al., 2014) should also be investigated in the context of postconditioning therapies. Third, Iba-1 is the most commonly used microglial marker for immunostaining, but Iba-1 may also bind various cells of monocytic lineage. So our Iba-1-based data may also include contributions from peripheral macrophage infiltration, and we are careful to acknowledge that our crosstalk hypothesis includes activated and potentially overlapping subsets of both microglia and macrophages. How postconditioning may affect central-peripheral crosstalk will be carefully examined in future studies.
From a clinical perspective, ischemic postconditioning appears to be attractive as it can be potentially translated, in particular for patients subjected to surgery and to endovascular therapies associated with blood vessel occlusion and revascularization. A large body of data supports its beneficial effects in neurons. The present study provides proof-of-concept that postconditoning may also modify neuronal-microglial crosstalk during stroke recovery. Future studies are warranted to dissect how postconditioning affects help-me signals within the entire neurovascular unit as the ischemic brain transitions from injury into repair.
Acknowledgments
SOURCES OF FUNDING
This work was supported in part by grants from the National Institutes of Health and the Rappaport Foundation.
Abbreviations
- VEGF
Vascular endothelial growth factor
- VEGFR2
Vascular endothelial growth factor receptor 2
- Flk-1
Fetal Liver Kinase 1
- LDF
laser doppler flowmetry
- PBS
phosphate-buffered saline
- GFAP
glial fibrillary acidic protein
- DMEM
Dulbecco's modified eagle medium
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- ELISA
enzyme-linked immunosorbent assay
- RRID
Research Resource Identifier
Footnotes
DISCLOSURES
None.
References
- Arai K, Lo EH. Astrocytes protect oligodendrocyte precursor cells via MEK/ERK and PI3K/Akt signaling. J Neurosci Res. 2010;88:758–763. doi: 10.1002/jnr.22256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cekanaviciute E, Fathali N, Doyle KP, Williams AM, Han J, Buckwalter MS. Astrocytic transforming growth factor-beta signaling reduces subacute neuroinflammation after stroke in mice. Glia. 2014;62:1227–1240. doi: 10.1002/glia.22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltzschig HK, Eckle T. Ischemia and reperfusion--from mechanism to translation. Nat Med. 2011;17:1391–1401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito E, Mandeville ET, Hayakawa K, Singhal AB, Lo EH. Effects of normobaric oxygen on the progression of focal cerebral ischemia in rats. Experimental neurology. 2013;249:33–38. doi: 10.1016/j.expneurol.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito E, Hayakawa K, Maki T, Arai K, Lo EH. Effects of Postconditioning on Neurogenesis and Angiogenesis During the Recovery Phase After Focal Cerebral Ischemia. Stroke; a journal of cerebral circulation. 2015;46:2691–2694. doi: 10.1161/STROKEAHA.115.009070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Jiang T, Guo J, Liu Y, Cui G, Gu L, Su L, Zhang Y. Inhibition of autophagy contributes to ischemic postconditioning-induced neuroprotection against focal cerebral ischemia in rats. PLoS One. 2012;7:e46092. doi: 10.1371/journal.pone.0046092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Zhang H, Takahashi T, Hsieh J, Liao J, Steinberg GK, Zhao H. The Akt signaling pathway contributes to postconditioning's protection against stroke; the protection is associated with the MAPK and PKC pathways. Journal of neurochemistry. 2008;105:943–955. doi: 10.1111/j.1471-4159.2008.05218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Pinilla F, Ying Z, Roy RR, Molteni R, Edgerton VR. Voluntary exercise induces a BDNF-mediated mechanism that promotes neuroplasticity. Journal of neurophysiology. 2002;88:2187–2195. doi: 10.1152/jn.00152.2002. [DOI] [PubMed] [Google Scholar]
- Guo Q, Du X, Zhao Y, Zhang D, Yue L, Wang Z. Ischemic postconditioning prevents renal ischemia reperfusion injury through the induction of heat shock proteins in rats. Molecular medicine reports. 2014;10:2875–2881. doi: 10.3892/mmr.2014.2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, Gao Y, Chen J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke; a journal of cerebral circulation. 2012;43:3063–3070. doi: 10.1161/STROKEAHA.112.659656. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji K, Miyauchi J, Tsirka SE. Microglia: An Active Player in the Regulation of Synaptic Activity. Neural Plast. 2013;2013:627325. doi: 10.1155/2013/627325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo SP, Xie W, Xiong X, Xu B, Zhao H. Ischemic postconditioning protects against focal cerebral ischemia by inhibiting brain inflammation while attenuating peripheral lymphopenia in mice. Neuroscience. 2013;243:149–157. doi: 10.1016/j.neuroscience.2013.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch S, Della-Morte D, Dave KR, Sacco RL, Perez-Pinzon MA. Biomarkers for ischemic preconditioning: finding the responders. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2014;34:933–941. doi: 10.1038/jcbfm.2014.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leconte C, Tixier E, Freret T, Toutain J, Saulnier R, Boulouard M, Roussel S, Schumann-Bard P, Bernaudin M. Delayed hypoxic postconditioning protects against cerebral ischemia in the mouse. Stroke; a journal of cerebral circulation. 2009;40:3349–3355. doi: 10.1161/STROKEAHA.109.557314. [DOI] [PubMed] [Google Scholar]
- Lin TN, He YY, Wu G, Khan M, Hsu CY. Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke; a journal of cerebral circulation. 1993;24:117–121. doi: 10.1161/01.str.24.1.117. [DOI] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke; a journal of cerebral circulation. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- Low PC, Manzanero S, Mohannak N, Narayana VK, Nguyen TH, Kvaskoff D, Brennan FH, Ruitenberg MJ, Gelderblom M, Magnus T, et al. PI3Kdelta inhibition reduces TNF secretion and neuroinflammation in a mouse cerebral stroke model. Nature communications. 2014;5:3450. doi: 10.1038/ncomms4450. [DOI] [PubMed] [Google Scholar]
- Maki T, Hayakawa K, Pham LD, Xing C, Lo EH, Arai K. Biphasic mechanisms of neurovascular unit injury and protection in CNS diseases. CNS Neurol Disord Drug Targets. 2013;12:302–315. doi: 10.2174/1871527311312030004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoonkitiwongsa PS, Schultz RL, McCreery DB, Whitter EF, Lyden PD. Neuroprotection of ischemic brain by vascular endothelial growth factor is critically dependent on proper dosage and may be compromised by angiogenesis. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2004;24:693–702. doi: 10.1097/01.WCB.0000126236.54306.21. [DOI] [PubMed] [Google Scholar]
- Mosher KI, Andres RH, Fukuhara T, Bieri G, Hasegawa-Moriyama M, He Y, Guzman R, Wyss-Coray T. Neural progenitor cells regulate microglia functions and activity. Nature neuroscience. 2012;15:1485–1487. doi: 10.1038/nn.3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan SV, Dave KR, Saul I, Perez-Pinzon MA. Resveratrol Preconditioning Protects Against Cerebral Ischemic Injury via Nuclear Erythroid 2-Related Factor 2. Stroke; a journal of cerebral circulation. 2015;46:1626–1632. doi: 10.1161/STROKEAHA.115.008921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park YS, Cho JH, Kim IH, Cho GS, Cho JH, Park JH, Ahn JH, Chen BH, Shin BN, Shin MC, et al. Effects of ischemic preconditioning on VEGF and pFlk-1 immunoreactivities in the gerbil ischemic hippocampus after transient cerebral ischemia. Journal of the neurological sciences. 2014;347:179–187. doi: 10.1016/j.jns.2014.09.044. [DOI] [PubMed] [Google Scholar]
- Patel AR, Ritzel R, McCullough LD, Liu F. Microglia and ischemic stroke: a double-edged sword. International journal of physiology, pathophysiology and pharmacology. 2013;5:73–90. [PMC free article] [PubMed] [Google Scholar]
- Pignataro G, Meller R, Inoue K, Ordonez AN, Ashley MD, Xiong Z, Gala R, Simon RP. In vivo and in vitro characterization of a novel neuroprotective strategy for stroke: ischemic postconditioning. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2008;28:232–241. doi: 10.1038/sj.jcbfm.9600559. [DOI] [PubMed] [Google Scholar]
- Pignataro G, Cuomo O, Esposito E, Sirabella R, Di Renzo G, Annunziato L. ASIC1a contributes to neuroprotection elicited by ischemic preconditioning and postconditioning. International journal of physiology, pathophysiology and pharmacology. 2011a;3:1–8. [PMC free article] [PubMed] [Google Scholar]
- Pignataro G, Esposito E, Cuomo O, Sirabella R, Boscia F, Guida N, Di Renzo G, Annunziato L. The NCX3 isoform of the Na+/Ca2+ exchanger contributes to neuroprotection elicited by ischemic postconditioning. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2011b;31:362–370. doi: 10.1038/jcbfm.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren C, Gao X, Niu G, Yan Z, Chen X, Zhao H. Delayed postconditioning protects against focal ischemic brain injury in rats. PloS one. 2008;3:e3851. doi: 10.1371/journal.pone.0003851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz de Almodovar C, Lambrechts D, Mazzone M, Carmeliet P. Role and therapeutic potential of VEGF in the nervous system. Physiological reviews. 2009;89:607–648. doi: 10.1152/physrev.00031.2008. [DOI] [PubMed] [Google Scholar]
- Saini MG, Pinteaux E, Lee B, Bix GJ. Oxygen-glucose deprivation and interleukin-1alpha trigger the release of perlecan LG3 by cells of neurovascular unit. J Neurochem. 2011;119:760–771. doi: 10.1111/j.1471-4159.2011.07484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo JD, Morris MJ, Jones NM. Hypoxic postconditioning reduces microglial activation, astrocyte and caspase activity, and inflammatory markers after hypoxia-ischemia in the neonatal rat brain. Pediatric research. 2015;77:757–764. doi: 10.1038/pr.2015.47. [DOI] [PubMed] [Google Scholar]
- Wang JY, Shen J, Gao Q, Ye ZG, Yang SY, Liang HW, Bruce IC, Luo BY, Xia Q. Ischemic postconditioning protects against global cerebral ischemia/reperfusion-induced injury in rats. Stroke. 2008;39:983–990. doi: 10.1161/STROKEAHA.107.499079. [DOI] [PubMed] [Google Scholar]
- Xing B, Chen H, Zhang M, Zhao D, Jiang R, Liu X, Zhang S. Ischemic postconditioning inhibits apoptosis after focal cerebral ischemia/reperfusion injury in the rat. Stroke. 2008;39:2362–2369. doi: 10.1161/STROKEAHA.107.507939. [DOI] [PubMed] [Google Scholar]
- Xing C, Wang X, Cheng C, Montaner J, Mandeville E, Leung W, van Leyen K, Lok J, Wang X, Lo EH. Neuronal production of lipocalin-2 as a help-me signal for glial activation. Stroke; a journal of cerebral circulation. 2014;45:2085–2092. doi: 10.1161/STROKEAHA.114.005733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J, Li H, Feng C, Zuo Z. Inhibition of brain ischemia-caused notch activation in microglia may contribute to isoflurane postconditioning-induced neuroprotection in male rats. CNS & neurological disorders drug targets. 2014;13:718–732. doi: 10.2174/1871527313666140618110837. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Chopp M. Vascular endothelial growth factor and angiopoietins in focal cerebral ischemia. Trends in cardiovascular medicine. 2002;12:62–66. doi: 10.1016/s1050-1738(01)00149-9. [DOI] [PubMed] [Google Scholar]
- Zhao H, Sapolsky RM, Steinberg GK. Interrupting reperfusion as a stroke therapy: ischemic postconditioning reduces infarct size after focal ischemia in rats. J Cereb Blood Flow Metab. 2006;26:1114–1121. doi: 10.1038/sj.jcbfm.9600348. [DOI] [PubMed] [Google Scholar]
- Zhao H. Ischemic postconditioning as a novel avenue to protect against brain injury after stroke. J Cereb Blood Flow Metab. 2009;29:873–885. doi: 10.1038/jcbfm.2009.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Ren C, Chen X, Shen J. From rapid to delayed and remote postconditioning: the evolving concept of ischemic postconditioning in brain ischemia. Current drug targets. 2012;13:173–187. doi: 10.2174/138945012799201621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Wang H, Sun G, Zhang J, Edwards NJ, Aronowski J. Neuronal Interleukin-4 as a Modulator of Microglial Pathways and Ischemic Brain Damage. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2015;35:11281–11291. doi: 10.1523/JNEUROSCI.1685-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]